Long-Term Enhancement of Central Synaptic Transmission by Chronic Brain-Derived Neurotrophic Factor Treatment (original) (raw)
Abstract
Acute effects of neurotrophins on synaptic plasticity have recently received much attention, but the roles of these factors in regulating long-lasting changes in synaptic function remain unclear. To address this issue we studied the long-term (days to weeks) and short-term (minutes to hours) effects of brain-derived neurotrophic factor (BDNF) on excitatory synaptic transmission in autaptic cultures of hippocampal CA1 neurons. We found that BDNF induced long-term enhancement of the strength of non-NMDA receptor-mediated glutamatergic transmission. This upregulation of EPSC amplitude occurred via an increase in the size of unitary synaptic currents, with no significant contribution from other aspects of neuronal electrical and synaptic function including cell size, voltage-gated sodium and potassium current levels, the number and size of synaptic contacts, and the frequency of spontaneous neurotransmitter release. Chronic BDNF treatment also decreased the degree of synaptic depression measured in response to paired stimuli. Thus, BDNF induced long-term synaptic enhancement of both basal and use-dependent synaptic transmission via specific changes to the synapse rather than through generalized potentiation of neuronal growth and differentiation. Finally, we showed that the long-term effects of BDNF are functionally and mechanistically distinct from its acute effects on synaptic transmission, suggesting that, in vivo, BDNF activation of Trk receptors can have different functional effects depending on the time course of its action.
Keywords: neurotrophins, BDNF, Trk receptors, TrkB, hippocampal neurons, synaptic plasticity, excitatory synaptic transmission, autapses, microcultures
In contrast to the many known extracellular signaling molecules that are involved in short-term neuromodulation on the time scale of seconds to hours [e.g., purines and neuropeptides (for review, see Kaczmarek and Levitan, 1987; Nicoll et al., 1990)], little is known about signaling molecules that modulate longer-lasting forms of plasticity that, once induced, persist for days, weeks, or more. In recent years the neurotrophins have been associated with the regulation of neuronal activity on an acute time scale in vitro (Kim et al., 1994; Lessmann et al., 1994;Kang and Schuman, 1995; Levine et al., 1995; Figurov et al., 1996;Patterson et al., 1996; Gottschalk et al., 1998; Lessmann and Heumann, 1998; Li et al., 1998). However, increasing evidence indicates that neurotrophins also regulate synaptic plasticity in the long term, particularly in areas of the nervous system such as hippocampus and neocortex that exhibit robust activity-dependent neuronal plasticity (for review, see McAllister et al., 1998).
Compelling in vivo evidence of a long-term role for neurotrophins in modulating synaptic plasticity is suggested by experiments demonstrating involvement of TrkB ligands in establishing proper connectivity between neurons of the lateral geniculate nuclei and layer IV of the visual cortex (Cabelli et al., 1995, 1997). This activity-dependent refinement of connections occurs over a developmental period of days to weeks. Consistent with this time frame,in vivo and in vitro experiments indicate that activity can regulate neurotrophin expression with relatively slow kinetics. In response to physiological levels of stimuli such as LTP induction (Patterson et al., 1992; Castren et al., 1993; Bramham et al., 1996), brain-derived neurotrophic factor (BDNF) mRNA levels do not increase for at least 2 h, but then can remain enhanced for 24 hr or more. Even in cases of extreme activity such as limbic seizures, BDNF mRNA levels do not peak until 6–12 hr after seizure in the hippocampus and neocortex (Isackson et al., 1991). Changes at the protein level are slower still; hippocampal BDNF protein levels peak at 16 hr after seizure activity and are maintained for several days (Nawa et al., 1995).
Neurotrophin action in target neurons, mediated in part through the Trk family of tyrosine kinase receptors, also occurs on a relatively slow time scale. Although Trk activation can induce rapid post-translational modification of neuronal proteins (Jovanovic et al., 1996; Suen et al., 1997), additional signal transduction pathways lead to the regulation of gene expression over a period of hours to days (Halegoua et al., 1991; Lewin and Barde, 1996; Segal and Greenberg, 1996). The slow time course of these latter actions suggests that long-term effects of neurotrophins on synaptic transmission, even if initiated rapidly, may require prolonged induction periods for their full manifestation. Regulation at the level of gene expression also suggests that many effects of neurotrophins will persist beyond the initial period of Trk receptor activation. Thus, it is probable that neurotrophin regulation of neuronal function, like its promotion of neuronal survival and differentiation (Davies, 1994), includes important components that occur over the time scale of days or more.
Studies in several systems have suggested that long-term treatment with neurotrophins can alter synaptic transmission indirectly, by regulating neuronal morphology or excitability (Dichter et al., 1977; Mandel et al., 1988; Purves et al., 1988; McAllister et al., 1995; Causing et al., 1997; Gonzalez and Collins, 1997; Lesser et al., 1997), or directly, by modulating unitary synaptic properties (Wang et al., 1995;Liou et al., 1997; Wang and Poo, 1997; Rutherford et al., 1998). Here, we have addressed the long-term versus short-term neurotrophin effects on synaptic transmission in excitatory central neurons from the hippocampal CA1 region and compared the cellular mechanisms underlying these effects. Using isolated autaptic neurons, we examined glutamatergic synaptic transmission after chronic (>1 week) or acute (<2.5 hr) treatment with BDNF, focusing in particular on non-NMDA-type glutamate receptor currents, which initiate postsynaptic depolarization in these neurons. We found that chronic BDNF treatment increased basal evoked current amplitudes by 70%. This regulation occurred via specific and selective enhancement of unitary synaptic strength, rather than as a consequence of generalized cell growth, synaptogenesis, or changes in neuronal excitability. Neurons treated chronically with BDNF also showed reduced synaptic depression during repetitive firing. In comparison, the rapid effects of BDNF were very different, both mechanistically and functionally, from its long-term effects on synaptic transmission.
MATERIALS AND METHODS
Electrophysiology. Neurons growing in isolation formed electrophysiologically detectable “autaptic” synaptic connections onto themselves by 7 d after plating and were used in whole-cell patch-clamp experiments up to 16 d after plating. Cultures of matched ages were used for short-term and long-term experiments, averaging 9–10 d in vitro at the time of recording. Short-term experiments were performed by two independent methods: either by using single excitatory neurons sequentially recorded in the untreated and +BDNF conditions or by comparing the synaptic properties of control populations with those treated briefly with BDNF (10–150 min). Recordings for long-term experiments were performed on parallel populations of untreated neurons and neurons treated with BDNF or TrkB–IgG for >6 d. Extracellular solutions contained (in mm): 137 NaCl, 5 KCl, 1 MgCl2, 3 CaCl2, 10 glucose, 5 HEPES, adjusted to ∼350 mOsm, pH 7.3. To ensure that EPSCs consisted only of non-NMDA-type glutamate receptor currents, APV (100 μm) was added to the extracellular solution just before recording. Potassium currents were isolated and recorded using an extracellular solution of 145 mm_n_-methyl-glucamine, 2.8 mm KCl, 2 mm MgCl2, 10 mm HEPES. Intracellular solutions contained (in mm): either 150 potassium gluconate, 10 NaCl, 10 HEPES, 10 EGTA, 3 Mg-ATP, adjusted to pH 7.2 with KOH, or 100 gluconic acid, 0.6 EGTA, 5 MgCl2, 2 Na2-ATP, 0.3 Na-GTP, 40 HEPES, adjusted to pH 7.2 with CsOH. Voltage-clamp recordings were obtained with an Axopatch-1D patch clamp (Axon Instruments, Foster City, CA) and acquired using WinClamp3.7, a Visual Basic program written in-house. Borosilicate patch pipettes (VWR, West Chester, PA; Sutter Instrument Co., Novato, CA) were pulled to resistances of 3–5 mΩ. Leak currents were generally 20–50 pA; recordings with leak currents ≥ 100 pA or series resistances exceeding 20 mΩ were discarded. Cell capacitance measurements were made by integration of capacitive transients. Data were sampled at 100 μsec intervals for sodium currents, 200 μsec intervals for synaptic currents, and 500 μsec intervals for potassium currents; recordings were filtered at 1–5 kHz. Neurons were confirmed to be excitatory by the reversal of evoked synaptic currents near 0 mV. Cells were voltage-clamped such that spontaneous action potentials were rarely observed when the cell was held at its resting potential (_V_hold = −70 mV); when they did occur, action potential-driven spontaneous events were preceded by a rapid, high-amplitude spike and were easily distinguished from miniature EPSCs (mEPSCs). In all cases, no differences in spontaneous event frequencies were observed after the addition of TTX (1 μm); thus, the measured spontaneous events reflected true action potential-independent mEPSCs. mEPSC amplitudes and kinetics were measured via an automatic detection software program (MiniMAD) written in-house. Detection criteria were set such that recognized spontaneous events had peak amplitudes at least twofold greater than baseline noise; such events were confirmed by the user to have rise times under 5 msec and decay kinetics that were well fitted by single exponential functions. Spontaneous and evoked synaptic current rise times were measured from the start to the peak of the currents; decay time constants were determined by fitting single exponential functions to the falling phase of the currents. Quantal content, the number of unitary release events occurring in response to an action potential (and thus a measure of the number of synapses contributing to an EPSC), was calculated for individual cells as (peak EPSC amplitude)/(mean mEPSC amplitude), where the mean mEPSC amplitude was taken to represent the average amplitude of a unitary event. Mean values are shown ± SE; statistical significance was tested by ANOVA.
Cell culture. All long-term experiments were performed in CF-1 murine cultures. Short-term experiments used both Sprague Dawley rats (single-cell experiments) and mice (single-cell and population experiments); no differences in the rapid effects of BDNF were observed between the two species. Autaptic cultures (Bekkers and Stevens, 1991; Pan et al., 1993) were made using hippocampal neurons dissected from postnatal day 0 rat pups or embryonic day 17 mice. We used only CA1 neurons to reduce the likelihood of intrinsic differences in the responsiveness of different cell types since levels of BDNF and TrkB expression (as well as the other neurotrophins and Trks) are differentially regulated among hippocampal regions (Maisonpierre et al., 1990; Ernfors et al., 1991). Briefly, glass coverslips or plastic tissue culture dishes (VWR; Corning, Corning NY) were coated with 0.15% agarose, then sprayed with an atomizer containing poly-d-lysine and collagen (Collaborative Research, Bedford, MA) to yield “microdots” of growth substrate. Cells were plated at low density to yield single neurons growing on islands of glia, confined by the agarose to the microdots. Media contained MEM (Life Technologies, Gaithersburg, MD) with 5% non-heat-inactivated FBS (Hyclone, Logan, UT), 20 mm glucose, 1% Mito-Serum Extender (Collaborative Research), and 1% sodium pyruvate (Life Technologies). For chronic treatment experiments, BDNF or TrkB–IgG (generous gifts of Regeneron Pharmaceuticals, Tarrytown, NY) was added to cultures the day after plating at 100 ng/ml or 10 μg/ml, respectively. Cultures were maintained in 5% CO2and fed BDNF or TrkB–IgG every 3–4 d. Proliferation of non-neuronal cells was controlled by the addition of mitotic inhibitors (5-fluoro-2-deoxyuridine and uridine) to the media.
Immunocytochemistry and quantification of fluorescent staining. Autaptic cultures were fixed at 7–9 d in vitro in 4% paraformaldehyde and 5% sucrose for 10 min, then incubated at 4°C overnight with a polyclonal antibody directed against synapsin Ia,b (1:500; generous gift of P. De Camilli, Yale University, New Haven, CT) and a monoclonal antibody directed against glutamic acid decarboxylase (GAD) (1:750; Boehringer Mannheim, Indianapolis, IN). Primary antibodies were visualized using Texas Red-conjugated goat anti-rabbit IgG (Vector, Burlingame, CA; Molecular Probes, Eugene, OR) and fluorescein- or Oregon Green-conjugated goat anti-mouse IgG (Molecular Probes), each incubated at 1:750 for 1 hr. Immunostained neurons were imaged for uniform exposure times using a 25×, 0.8 NA oil immersion objective with a 12-bit cooled CCD camera (Princeton Instruments, Trenton, NJ) and were analyzed with IPLabs software (Signal Analytics, Vienna, VA; courtesy of D. Chikaraishi, Duke University). Analysis of anti-synapsin staining was performed by imaging GAD-negative autaptic neurons on islands not significantly larger than the visual field, manually tracing individual fluorescently stained anti-synapsin punctae that occurred along neuronal processes for each imaged neuron, and then quantifying number and area of punctae using IPLabs software. The resolution of the imaging system was such that one image pixel represented 0.06 μm2.
RESULTS
Because of the complexity of neural circuits even in conventional dissociated cultures, we studied BDNF regulation of synaptic transmission in microdot cultures of rodent CA1 hippocampal neurons (Furshpan et al., 1976; Segal and Furshpan, 1990; Bekkers and Stevens, 1991; Pan et al., 1993). Neurons in this system are confined to grow in isolation and form synapses only on themselves (autapses). Autaptic synaptic transmission is functionally indistinguishable from that found in conventional dissociated culture (Bekkers and Stevens, 1991), and the simplicity of these cultures has been critical for the investigation of several fundamental properties of synaptic transmission (Landis, 1976; Furshpan et al., 1986; Pan et al., 1993;Mennerick and Zorumski, 1995a, 1996; Mennerick et al., 1995;Stevens and Tsujimoto, 1995; Tong et al., 1996; Kimura et al., 1997). We identified excitatory autaptic neurons by the reversal of their postsynaptic currents at ∼0 mV in electrophysiology experiments or by the absence of GAD immunoreactivity in morphological experiments. In combination with whole-cell patch-clamping, immunocytochemistry, and image analysis, this system enabled clear determination of BDNF-induced changes in excitability, morphology, synaptic transmission, and short-term synaptic plasticity at the level of individual excitatory neurons and synapses.
Chronic enhancement of basal synaptic transmission by BDNF
We studied the long-term actions of BDNF by measuring non-NMDA receptor-mediated EPSCs (Fig.1A) elicited in autaptic neurons grown under each of three experimental conditions for 6–15 d: untreated, +TrkB–IgG to block potential activity of endogenous BDNF (10 μg/ml) (Shelton et al., 1995; Binder et al., 1999), or +BDNF (100 ng/ml). Consistent with other neurotrophin studies in hippocampal systems, neither addition of BDNF nor of TrkB-IgG affected neuronal survival (Ip et al., 1993; Ohsawa et al., 1993; M. Bolton and D. Lo, unpublished observations). The number of surviving neurons after 1 week of BDNF or TrkB–IgG treatment, normalized to survival in untreated plates, was 1.06 ± 0.07 and 1.25 ± 0.28, respectively (mean ± SE; p > 0.38 and 0.42 by ANOVA; n = 3 separate experiments). Because TrkB–IgG-treated neurons did not differ from untreated neurons in any parameter measured, these treatment groups were combined (“control”) (Table 1).
Fig. 1.

Chronic BDNF treatment increased the amplitude of evoked non-NMDA-type postsynaptic currents. A,Top, EPSCs were elicited by depolarizing CA1 autaptic neurons from a prepulse potential of −120 mV to +10 mV for 1.6 msec and returning to −70 mV, causing a rapid inward sodium spike (arrow) and subsequent EPSC (arrowhead). Recorded EPSCs could be completely blocked by NBQX, leaving only a residual Na+ current (center). The NBQX-sensitive component of such traces (bottom; difference of top and center traces) defined non-NMDA-type glutamate receptor-mediated synaptic currents that reversed at or near 0 mV. B, C, Chronic BDNF treatment (100 ng/ml; n = 61) increased average EPSC peak amplitude by 1.7 ± 0.1-fold (mean ± SE) compared with those recorded from controls (i.e., untreated or 10 μg/ml TrkB–IgG-treated neurons;n = 109; p < 8.8 × 10−8 by ANOVA; statistically significant difference denoted by the asterisk). Representative EPSCs are shown in B. Mean EPSC amplitudes (C) ranged from ∼1 to 4 nA and were normalized to controls within each experiment to compare results from different platings.
Table 1.
TrkB–IgG had no effect on CA1 excitatory neurons
| Untreated (n) | +TrkB–IgG (n) | p value (ANOVA) | |
|---|---|---|---|
| EPSC amplitude | 1.0 ± 0.1 (31) | 0.9 ± 0.1 (34) | >0.5 |
| Cell capacitance | 1.0 ± 0.1 (22) | 1.0 ± 0.0 (28) | >0.9 |
| Sodium current | 1.0 ± 0.1 (15) | 1.0 ± 0.1 (21) | >0.9 |
| Synapse number | 446 ± 63 (6) | 558 ± 93 (7) | >0.3 |
| Synapse size (μm2) | 0.38 ± 0.02 (6) | 0.32 ± 0.02 (7) | >0.05 |
| %PPD | 33 ± 7 (8) | 41 ± 6 (5) | >0.4 |
| mEPSC rise time (msec) | 2.7 ± 0.3 (16) | 2.7 ± 0.2 (24) | >0.9 |
| mEPSC τdecay(msec) | 2.8 ± 0.3 (16) | 2.2 ± 0.2 (24) | >0.1 |
| EPSC rise time (msec) | 4.2 ± 0.3 (17) | 4.5 ± 0.4 (16) | >0.5 |
| EPSC τdecay(msec) | 7.9 ± 0.7 (19) | 7.4 ± 0.7 (16) | >0.6 |
In contrast, neurons grown in the continuous presence of BDNF showed basal excitatory postsynaptic responses that were on average 1.7-fold greater than controls (Fig. 1B,C) (p < 8.8 × 10−8; combination of five separate experiments). These potentiating effects of BDNF were maintained for at least several hours in the absence of added BDNF or TrkB–IgG in the recording salines. The possible cellular mechanisms underlying this potentiation fall into one or more of the following categories: (1) changes in neuronal excitability; (2) an increase in synaptogenesis; and (3) changes in the function of individual synapses. We thus experimentally examined each of these areas in turn.
Intrinsic excitability
Several observations suggested that changes in passive membrane properties and action potential propagation were not significant factors in the synaptic potentiation induced by BDNF. First, the rise times for both evoked and spontaneous synaptic currents were unchanged by BDNF treatment, indicating an absence of significant changes in passive membrane properties (Table 2). Second, cell membrane area, as measured by electrical capacitance, was not affected by BDNF (Fig.2A) (p > 0.15).
Table 2.
BDNF had no effect on the rise or decay times of synaptic currents
| mEPSC rise time, msec (n) | mEPSC τdecay, msec (n) | EPSC rise time, msec (n) | EPSC τdecay, msec (n) | |
|---|---|---|---|---|
| Control | 2.7 ± 0.2 (40) | 2.5 ± 0.2 (40) | 4.3 ± 0.3 (33) | 7.7 ± 0.5 (35) |
| +BDNF | 2.8 ± 0.3 (34) | 2.5 ± 0.2 (32) | 4.4 ± 0.4 (32) | 8.8 ± 0.8 (34) |
Fig. 2.
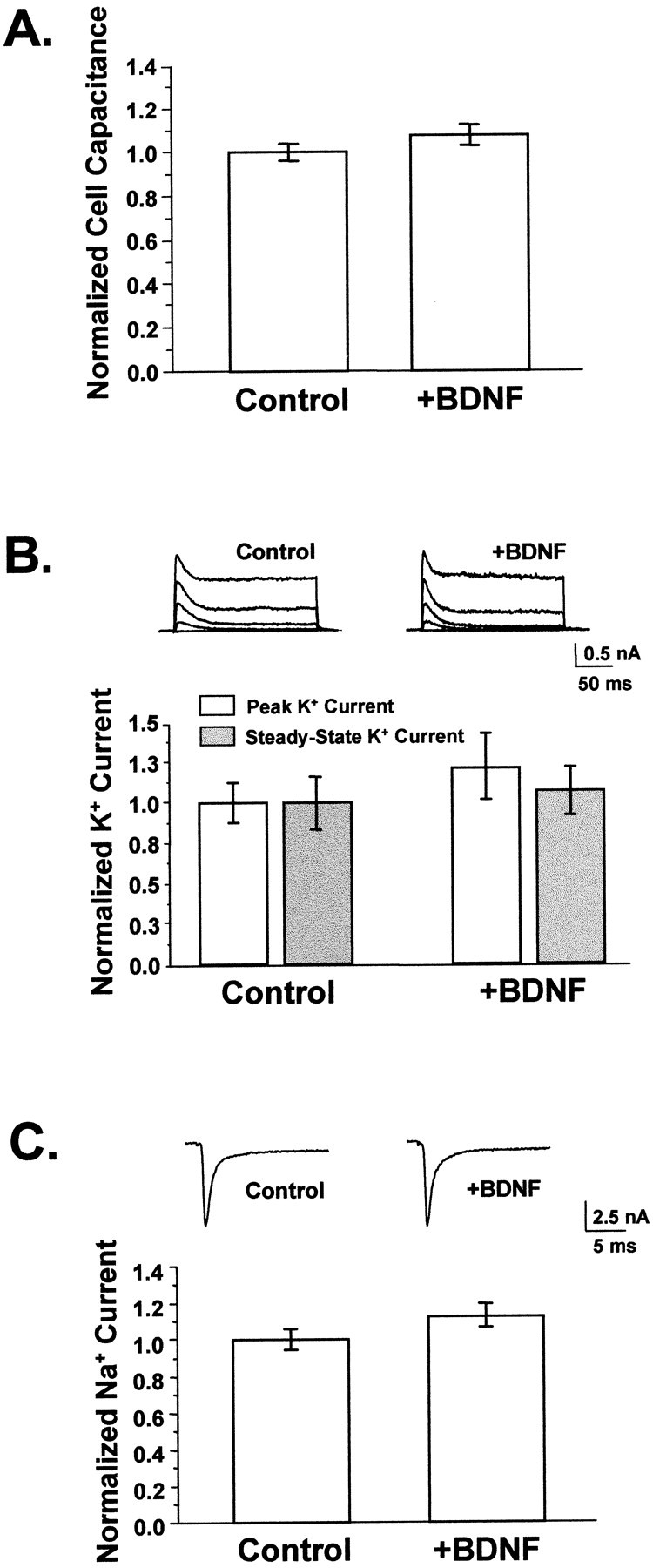
BDNF did not appreciably affect neuronal size or membrane excitability. A, BDNF did not affect cell size. Cell capacitance was used to measure total membrane area, which did not differ between neurons grown in the presence or absence of added BDNF (p > 0.15). Average capacitances ranged from 40 to 60 pF (Control, n = 85;+BDNF, n = 41). B, BDNF did not affect potassium currents. Voltage-gated potassium currents were elicited by depolarizing neurons from a 20 msec prepulse of −120 mV to between −40 and +40 mV in 20 mV increments for 200 msec; representative traces are shown for control and BDNF-treated cells. Currents at +40 mV were measured at peak and at steady state to reflect the contributions of transient and sustained potassium current components. No differences were observed between untreated controls and neurons grown in BDNF for either of these measurements. Average potassium currents were ∼2 nA. Control,n = 10; +BDNF, _n_= 9. p > 0.35 for both peak and steady-state current comparisons. C, BDNF did not alter sodium currents. Whole-cell sodium currents were recorded by depolarizing neurons to −10 mV from a brief hyperpolarizing pulse of −120 mV; representative traces are shown. Sodium currents activated and inactivated within 5 msec after the start of the depolarizing pulse. Average peak sodium currents were equivalent in BDNF-treated neurons and controls (n = 41 and 60, respectively;p > 0.09) and ranged from 4 to 8 nA.
We also measured functional levels of whole-cell voltage-gated potassium and sodium currents. Neither peak nor steady-state potassium current component amplitudes were affected by BDNF (Fig.2B) (p > 0.35). Similarly, BDNF treatment did not alter the amplitudes or kinetics of voltage-gated sodium currents measured concurrently with evoked synaptic currents (Fig. 2C) (p > 0.09). Thus, neither changes in the passive nor the active electrical characteristics of neuronal membranes appeared to be the mechanisms by which EPSC amplitudes were increased.
Synaptogenesis
We next examined whether BDNF could, on this time scale, be acting simply as a “growth factor” for synaptogenesis. We addressed this directly by quantifying the number and size of synapses in BDNF-treated and control cultures. Synapses were visualized by immunostaining cultures with a polyclonal antibody directed against synapsin Ia and Ib (gift of Dr. P. De Camilli) (Fig.3A). Localized in mature presynaptic terminals, these proteins are important in synaptogenesis and the regulation of neurotransmitter release (Greengard et al., 1993). Quantitation of anti-synapsin immunostaining in isolated glutamatergic neurons showed that BDNF affected neither the number of synapses per neuron (Fig. 3B) (p > 0.16) nor their average size (Fig. 3C) (p > 0.56). This provided direct evidence that BDNF did not alter the rate of synaptogenesis in these cultures and thus that the increase in evoked current amplitude was not caused by differences in synapse number or size.
Fig. 3.
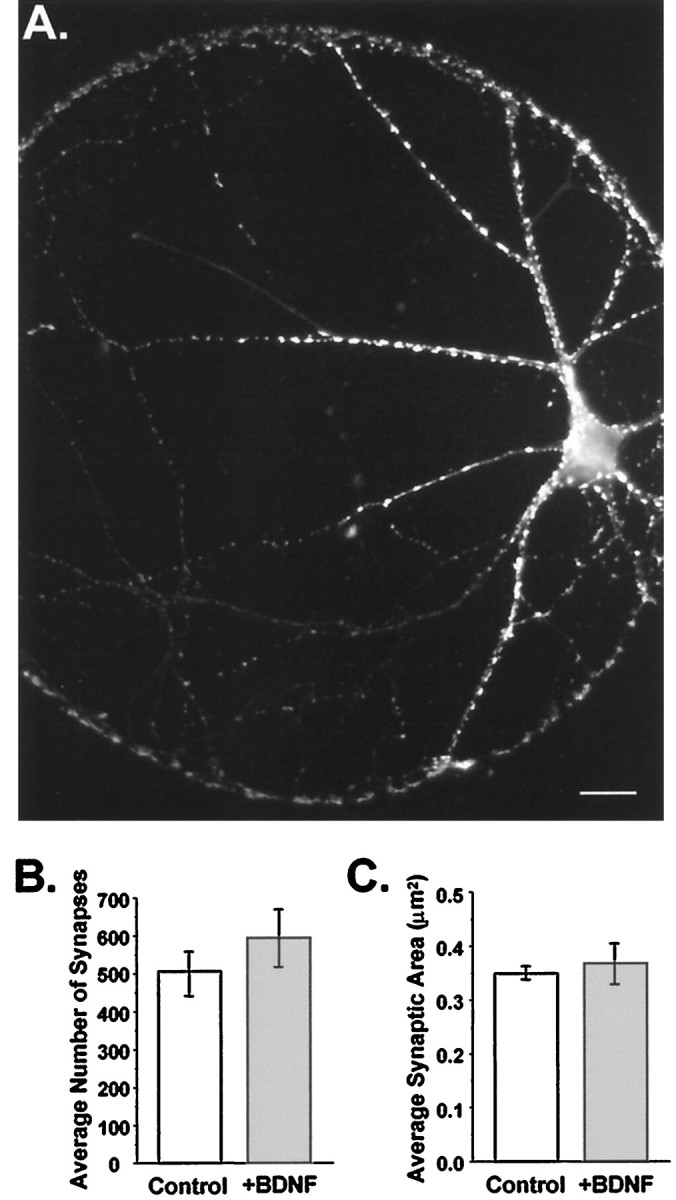
Synaptogenesis was not affected by chronic BDNF treatment. A, Fluorescence image of an autaptic neuron labeled with an antibody directed against synapsin Ia,b and visualized using a Texas Red-conjugated secondary antibody. Each bright puncta represents a presynaptic specialization. Scale bar, 20 μm.B, The number of synapses per neuron did not differ between treatment groups. Average numbers of synapses per cell were (mean ± SE) 506 ± 58 in controls and 594 ± 74 in BDNF-treated neurons (n = 13 and 7, respectively;p > 0.37). C, The average size of synapses was unaffected by BDNF. Mean synapse size, with each synapse defined as the pixel area of an isolated puncta, was 0.35 ± 0.01 and 0.37 ± 0.04 μm2 per synapse for control (n = 13) and +BDNF (n = 7) cells, respectively; p > 0.56.
Synaptic function
Because the preceding experiments suggested that the principal regulatory effects of BDNF were not on neuronal excitability, cell growth, or synaptogenesis, we examined the remaining possibility that BDNF modified the unitary properties of synaptic transmission. Because mEPSCs occur independently of action potentials, we could directly examine changes localized at synapses by measuring the amplitudes and kinetics of these spontaneous events.
Autaptic neurons are unique in that mEPSCs and EPSCs arise from the same neuron and thus the same set of synapses. Because only one set of synapses is involved, direct correspondences can be drawn between the properties of evoked versus miniature synaptic currents. To address most rigorously the possibility that changes in unitary synaptic properties underlie the BDNF-induced increase in EPSC amplitude, we first focused on the set of experiments that had the greatest number of mEPSC and EPSC recordings from the same cells. In these experiments, evoked postsynaptic current amplitudes increased on average by 1.8-fold in BDNF-treated cultures relative to controls (Fig.4A,B) (p < 0.02). Strikingly, the amplitude of spontaneous events in BDNF-treated neurons increased proportionally with the increase in evoked current amplitudes, by 1.8-fold relative to control neurons (Fig. 4_A,B_) (_p_ < 0.03). No differences in rise or decay kinetics were observed for EPSCs or mEPSCs recorded from control versus BDNF-treated neurons (Fig. 4_A_, Table 2), nor did BDNF affect the frequency of mEPSCs (Fig. 4_C_) (_p_ > 0.25).
Fig. 4.

BDNF increased EPSC and mEPSC amplitudes proportionally but did not affect mEPSC frequency. A, EPSC (left) and mEPSC (right) traces were averaged for control (dashed traces) and +BDNF–treated (solid traces) cells and superimposed. BDNF increased EPSCs and mEPSCs to similar extents. Scaling the averaged control traces to their respective +BDNF traces (dotted lines) showed that the kinetics of activation and decay were indistinguishable between treatment groups for both evoked and spontaneous responses.B, EPSC amplitudes (shaded bars) were 1.8 ± 0.03-fold greater in BDNF-treated cells compared with controls (p < 0.02; _+BDNF_,_n_ = 13; _Control_,_n_ = 16). BDNF increased the amplitudes of mEPSCs to the same extent (1.8 ± 0.02-fold; _p_ < 0.03;_+BDNF_, _n_ = 10;_Control_, _n_ = 12). Mean EPSC amplitudes were 3.6 and 2.0 nA for +BDNF and control cells, respectively, whereas mEPSC amplitudes averaged 34.2 and 18.6 pA. Values normalized to control cells are shown; _asterisks_denote statistical significance. _C_, In contrast, BDNF did not affect the average frequency of mEPSCs (_+BDNF_,_n_ = 10; _Control_,_n_ = 12; _p_ > 0.25). mEPSCs were measured from 51.2 sec of continuous recording while cells were held at −70 mV.
These data suggested that at the level of individual neurons, increases in evoked EPSC amplitude induced by BDNF were quantitatively accounted for by a parallel increase in mEPSC size. We further tested this idea by comparing EPSC amplitudes versus mEPSC amplitudes for a larger population of neurons, combining multiple experimental trials. Even across different experiments, EPSC and mEPSC amplitudes were strongly correlated and covaried (Fig. 5) (r = 0.57, p < 0.0001). In contrast, a plot of EPSC amplitudes versus mEPSC frequencies for individual cells showed no correlation (Fig. 5) (_r_ = 0.01,_p_ > 0.9).
Fig. 5.
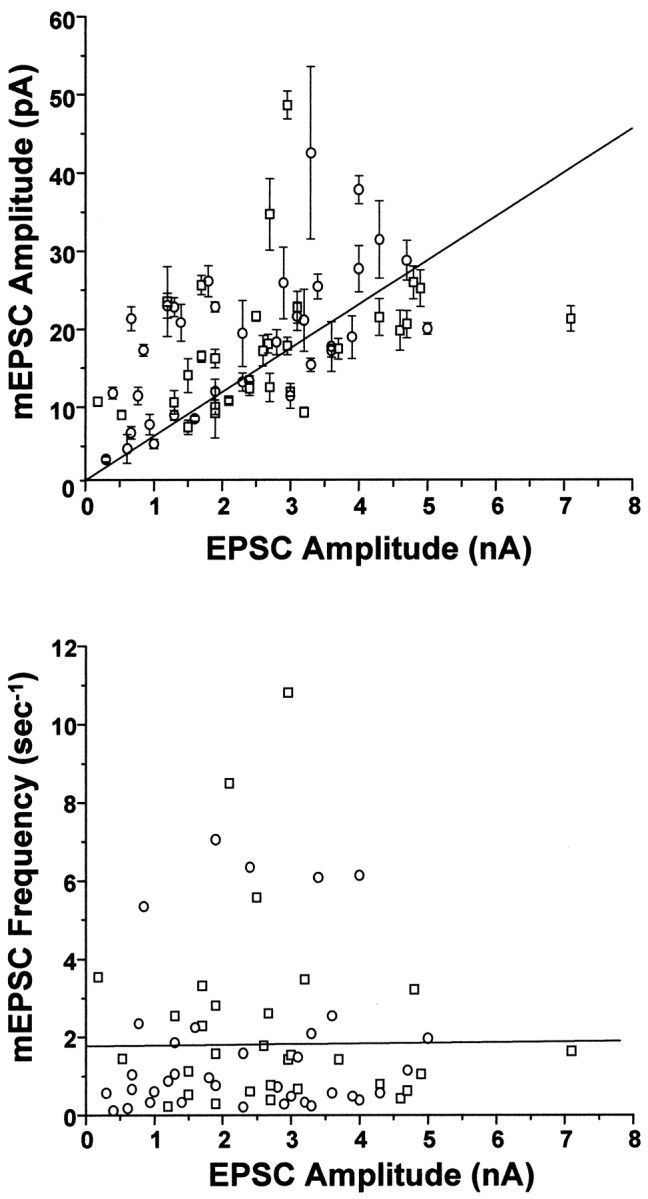
mEPSC amplitude, but not frequency, covaried with EPSC amplitude in populations of BDNF-treated and control neurons.Top, Amplitudes of spontaneous and evoked postsynaptic currents covaried with a correlation coefficient of_r_ = 0.57 (p < 1 × 10−4); best linear least-squares fit is shown (_solid line_). Each point represents recordings from a single neuron; mEPSC amplitudes are the mean ± SE of all events occurring within the 51.2 sec recording period. Two sources of variation in these data points were evident. (1) Limitations in the signal-to-noise of some recordings reduced the detectability of mEPSCs smaller than ∼3 pA, leading to an overestimation of average mEPSC amplitudes in these neurons. Hence, there is more scatter above the linear fit than below it. (2) Although BDNF never altered the quantal content of neurons within a given cell culture plating, there was some variation in overall quantal content from plating to plating (122–225). Thus, the slope of the linear fit here reflects the weighted average of the quantal contents of all neurons in all platings (178). It is noteworthy that the strong correlation between mEPSCs and EPSC amplitudes remained clear despite these significant sources of experimental variation. _Bottom_, In contrast, a plot of mEPSC frequency versus EPSC amplitude for individual cells revealed no correlation: _r_ = 0.01, _p_ > 0.9. n = 36 and 30 for control (○) and BDNF-treated (■) cells, respectively.
The above findings thus indicated that the BDNF-induced increase in EPSC amplitudes arose from a proportional increase in average mEPSC amplitude. Furthermore, if a change in mEPSC amplitude completely accounted for the change in EPSC amplitude, these results predicted that quantal content, the average number of quanta contributing to an EPSC [= (EPSC amplitude)/(mEPSC amplitude); see Materials and Methods], should be unaffected by BDNF treatment. In fact this was observed: 211 ± 36 and 225 ± 43 quanta were released per EPSC (control vs BDNF-treated neurons, n = 12 and 9, respectively; p > 0.8). Thus, chronic BDNF treatment increased basal evoked synaptic current amplitudes via regulation of the amplitudes of unitary synaptic currents and not by increasing the probability of neurotransmitter release.
Chronic BDNF enhances use-dependent synaptic transmission
In the above experiments, synaptic currents were elicited at very low frequencies (≤0.05 Hz). However, neurons _in vivo_normally respond to repetitive stimuli at substantially higher frequencies. To determine whether BDNF also regulated the efficacy of synaptic transmission in response to higher frequency stimuli, we examined a simple form of use-dependent synaptic plasticity in these cells, synaptic depression, using a paired-pulse depression (PPD) paradigm (Zucker, 1989).
Pairs of evoked currents were elicited 1 sec apart, and the resultant degree of synaptic depression was determined by measuring the amplitude of the second EPSC relative to the first. Autaptic neurons treated chronically (>1 week) with BDNF were much less depressed by successive firing than were controls. While EPSCs elicited by the second stimulus were 36% smaller than the initial EPSC in control cells (i.e., 36% PPD), BDNF-treated neurons showed only 8% PPD (p < 0.005) (Fig.6A,B). Chronic treatment with BDNF thus significantly enhanced synaptic efficacy in response to repetitive stimuli as evidenced by a four- to fivefold reduction in PPD at a 1 sec interpulse interval.
Fig. 6.
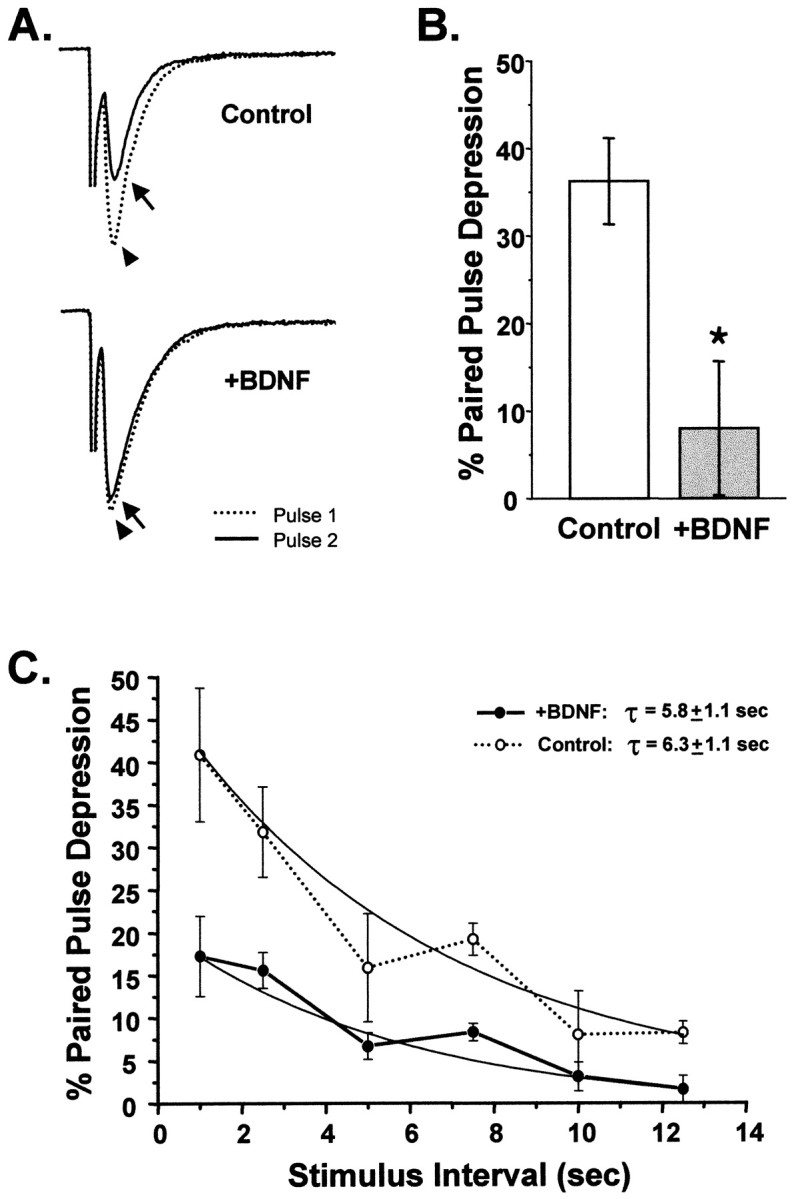
Chronic BDNF treatment attenuated paired-pulse depression. A, Sample traces of EPSCs recorded using a paired-pulse depression (PPD) paradigm for cells grown in the presence or absence of exogenous BDNF. The dotted traces(arrowhead) show the first evoked postsynaptic current.Solid traces show the second EPSC (arrow) evoked 1 sec later. In the BDNF-treated neuron the two traces are nearly overlapping, indicating that little PPD occurred. In contrast, in the control neuron the second EPSC was substantially depressed relative to the first. B, On average, control neurons showed 36 ± 5% PPD at an interpulse interval of 1 sec (n = 13). In contrast, BDNF-treated cells showed only 8 ± 8% PPD (n = 12;p < 0.005; statistical significance denoted by_asterisk_). C, The rate of recovery from PPD was not affected by BDNF. PPD was measured for a range of interpulse intervals ranging from 1 to 12.5 sec. The plot of PPD versus interpulse interval shows that control neurons were more depressed than BDNF-treated neurons at all intervals. The time courses of recovery from PPD, however, were fitted well by single exponential functions with similar time constants (Control, 6.3 ± 1.1 sec, n = 12; +BDNF, 5.8 ± 1.1 sec, n = 8; fits constrained to be asymptotic to zero).
In several systems, including hippocampal autapses, PPD has been attributed to the depletion of the readily releasable pool of synaptic vesicles (Mennerick and Zorumski, 1995a; Dobrunz and Stevens, 1997). Thus, one simple mechanism by which the observed decrease in PPD could have occurred is via an increase in the rate at which this pool is replenished. We examined the possibility that BDNF increased the refilling rate of the readily releasable pool in the range of stimulation frequencies that cause PPD (1–12.5 sec) (Fig.6C). We found that recovery rates from PPD did not differ between BDNF-treated and BDNF-deprived neurons at any of these stimulus intervals (τBDNF = 5.8 ± 1.1 sec; τcontrol = 6.3 ± 1.1 sec). This suggested that although BDNF attenuated synaptic depression by several-fold, this effect did not occur via alterations in the rate at which synapses recover from PPD.
Acute effects of BDNF on synaptic transmission
Recent work from several laboratories has provided evidence for rapid effects of BDNF on non-NMDA receptor-mediated glutamatergic transmission in hippocampal neurons (Kim et al., 1994; Lessmann et al., 1994; Kang and Schuman, 1995, 1996; Levine et al., 1995; Figurov et al., 1996; Patterson et al., 1996; Tanaka et al., 1997; Gottschalk et al., 1998; Lessmann and Heumann, 1998; Li et al., 1998). We were interested in whether the chronic effects on synaptic transmission described above—–increased amplitudes of both evoked and spontaneous EPSCs—–were related to potential effects of brief BDNF exposure on synaptic transmission, or whether BDNF regulates synaptic function in distinct ways over acute versus prolonged time scales.
We examined the acute effects of BDNF on basal synaptic transmission using two independent methods. First, non-NMDA receptor-mediated EPSC and mEPSC amplitudes were measured in continuous recordings from single neurons before and after bath application of BDNF. In contrast to the effects of chronic BDNF treatment described above, no appreciable change in the amplitude of EPSCs was detected for the duration of these recordings, which lasted from 40 min to >2 hr (Figs.7,8A, bottom). This is consistent with results observed in hippocampal slices (Figurov et al., 1996; Korte et al., 1996; Patterson et al., 1996; Tanaka et al., 1997; Frerking et al., 1998; Gottschalk et al., 1998; but see alsoKang and Schuman, 1995, 1996). The amplitudes of mEPSCs were also unaffected by BDNF addition for at least 1.5 hr after BDNF addition (Figs. 7, 8A, center). However, the frequency of spontaneous events increased significantly within 10 min after the addition of BDNF, by 2.5-fold on average (Figs. 7,8A, top), similar to other findings in conventional dissociated hippocampal cultures (Lessmann et al., 1994;Lessmann and Heumann, 1998; Li et al., 1998). These events were insensitive to TTX, indicating that BDNF increased the frequency of true “minis” (Fig. 7).
Fig. 7.
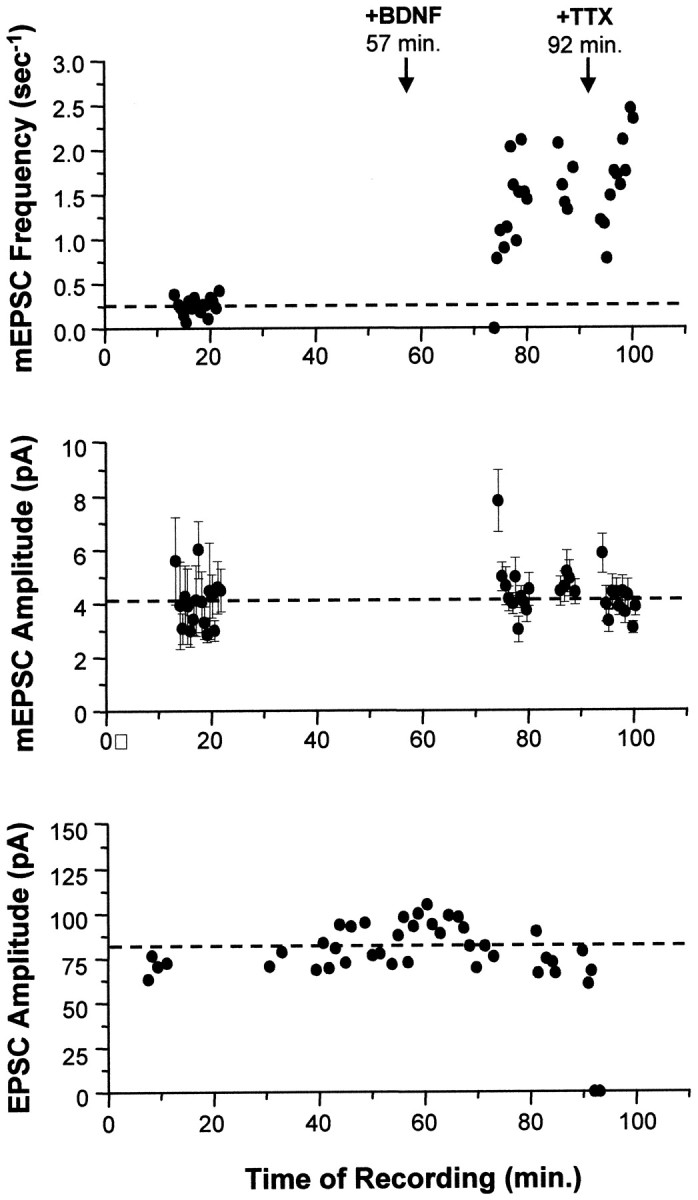
Acute application of BDNF increased only the frequency of mEPSCs. In this example, synaptic currents from a single autaptic neuron were first recorded in the absence of BDNF to establish baseline synaptic transmission properties (mEPSC frequency, mEPSC amplitude, and EPSC amplitude). Each mEPSC point represents 25.6 sec of recording. BDNF was then added to the bath (100 ng/ml at_t_ = 57 min), and these synaptic properties were monitored for 35 min. Finally, TTX was added at t = 92 min to confirm that the mEPSCs measured were true action potential-independent minis. Top, The frequency of mEPSCs increased 5.2-fold (p < 2 × 10−9) in response to BDNF addition. This effect was seen within minutes and was not affected by the subsequent addition of TTX (_p_ > 0.1, comparing average post-BDNF frequencies measured before and after TTX).Center, No differences were observed in average mEPSC amplitudes (±SE) throughout the recording; p > 0.9 for mEPSC amplitudes measured before and after BDNF addition. Blank periods in this and the graph above are due to EPSCs being recorded during these intervals. Bottom, Similarly, EPSCs were unaffected by the addition of BDNF (p > 0.8, comparing pre-BDNF and post-BDNF measurements). Note that the last two EPSC amplitudes are zero, confirming that sodium channels were blocked by TTX, eliminating all action potentials and EPSCs.
Fig. 8.
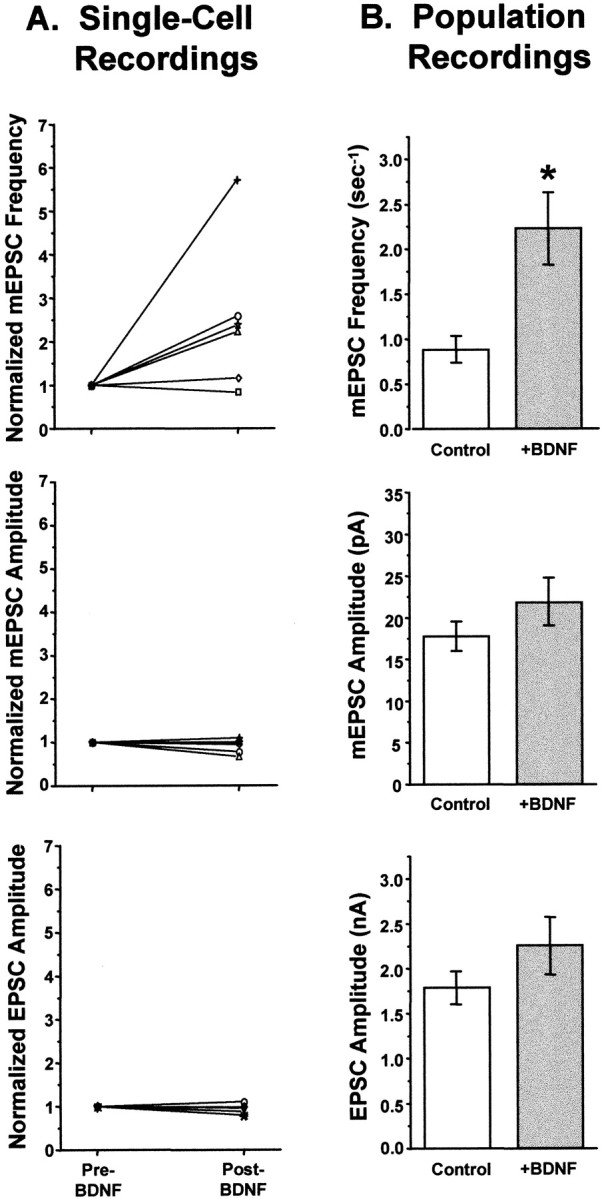
Acute application of BDNF increased mEPSC frequency but not mEPSC or EPSC amplitudes. A, Summary of single-cell neuron recordings. Each symbol represents an individual neuron recorded continuously, first in control salines and then for up to 80 min after the addition of BDNF. Values are normalized to the control (pre-BDNF) measurement for each neuron.Top, mEPSC frequency increased significantly by 10 min after the addition of BDNF, by an average of 2.5-fold (n = 6; p < 0.006). This increase was insensitive to TTX, indicating that BDNF increased the frequency of true minis (Fig. 7 and data not shown)._Center_, mEPSC amplitudes were unchanged by BDNF addition (_n_ = 6). _Bottom_, EPSC amplitudes were also unaffected by short-term BDNF treatment (_n_ = 7). _B_, Summary of data taken from populations of neurons untreated or treated with BDNF for <2.5 hr. _Top_, As in the single cell experiments, BDNF increased mEPSC frequency by 2.5-fold (_Control_, 0.88 ± 0.14 sec−1,_n_ = 24; _+BDNF_, 2.23 ± 0.39 sec−1, _n_ = 18; _p_ < 0.001; significance denoted by_asterisk_). _Center_, mEPSC amplitudes were indistinguishable between control and BDNF-treated cells (_Control_, _n_ = 26;_+BDNF_, _n_ = 19; _p_> 0.1). Bottom, Similarly, evoked postsynaptic currents did not differ significantly in amplitude between control and BDNF-treated neurons (Control, n = 27; +BDNF, n = 22;p > 0.17).
Because of the difficulty in recording stable currents from autaptic neurons for prolonged periods of time, we confirmed these results in a larger number of neurons by recording from neuronal populations that were either untreated or treated briefly with BDNF (<2.5 hr, with BDNF present throughout the recording period). The results of these experiments were identical to those observed in the continuous recordings: neither EPSC nor mEPSC amplitudes were affected by brief BDNF treatment (Fig. 8_B_, _bottom_ and_center_) (EPSC, _p_ > 0.17; mEPSC,p > 0.10), but the average frequency of mEPSCs increased by 2.5-fold (Fig. 8B, top) (p < 0.001).
Finally, we asked whether BDNF had rapid effects on short-term synaptic depression by measuring PPD. We found that untreated neurons and neurons treated briefly (<1 hr) with BDNF were equivalently depressed at a 1 sec interpulse interval, by 28 ± 2% and 32 ± 5% (control and BDNF-treated, respectively; _p_ > 0.37). Thus, on a rapid time scale, BDNF had no effect on the use dependence of evoked synaptic transmission as assessed by PPD. Rather, the predominant effect of BDNF treatment on the time scale of minutes to hours was an increase in the frequency of mEPSCs. All of the effects of brief BDNF treatment were thus distinct from its long-term actions: the selective increase in the size but not the frequency of mEPSCs and the enhancement of both basal and use-dependent evoked synaptic transmission.
DISCUSSION
Although much evidence has accumulated in recent years of a role for neurotrophins in the rapid modulation of neuronal activity, the slow time courses of neurotrophin regulation and action, as well as the known long-term effects of neurotrophins on neuronal morphology and excitability, suggest that these growth factors also regulate neuronal activity on longer time scales. We addressed this issue directly by using isolated excitatory hippocampal neurons to investigate the long- versus short-term effects of BDNF on central excitatory synaptic transmission.
We found that chronic treatment (1–2 weeks) of autaptic CA1 neurons with BDNF led to a 70% enhancement of action potential-evoked synaptic currents mediated through non-NMDA-type glutamate receptors. This effect was unlikely to be caused by selection for a population of neurons with larger EPSCs, because no effects of BDNF or TrkB–IgG were observed on neuronal survival. Long-term treatment of autaptic neurons with BDNF also regulated a form of use-dependent, transient synaptic plasticity, paired-pulse depression. BDNF-treated neurons showed substantially less PPD than controls in that successive firing only marginally depressed EPSC amplitudes in these neurons. Notably, these long-term effects of BDNF were directly on synaptic function, rather than occurring as a secondary consequence of enhanced synaptogenesis or neuronal excitability. Our results are particularly interesting in light of observations in hippocampal neurons of mice chronically deficient in BDNF [BDNF heterozygous and homozygous knockouts (Patterson et al., 1996)]. These animals show a gene dosage-dependent reduction in levels of basal synaptic transmission, which can be partially rescued only after at least 12 hr of BDNF incubation.
In contrast to the long-term effects of BDNF, brief BDNF application (<2.5 hr) affected neither the amplitude nor the extent of paired-pulse depression of evoked EPSCs. Rather, BDNF acutely regulated the frequency of mEPSCs, a target that was not affected in long-term studies. Thus, although BDNF directly regulated synaptic function in both the short term and long term, it did so on these two time scales in distinct ways, via distinct cellular mechanisms. The clear differences between these rapid versus long-term effects of BDNF suggest that, in vivo, BDNF can have very different consequences on synaptic transmission and plasticity depending on the time course of BDNF availability and action.
Cellular mechanisms of chronic BDNF regulation
Of the three cellular mechanisms by which BDNF could have increased EPSC amplitudes—regulation of membrane excitability, synapse number, or synapse strength—only one mechanism was observed in CA1 neurons: an increase in synaptic strength. BDNF’s long-term effects on basal evoked synaptic transmission were caused by the upregulation of quantal size, as evidenced by the proportional increase in mEPSC amplitudes that quantitatively accounted for the change in EPSC amplitude.
Because mEPSCs increased in amplitude but not frequency, our evidence was most consistent with a postsynaptic locus of regulation by BDNF. Changes in mEPSC amplitude typically arise from postsynaptic modifications (Korn and Faber, 1991; Bekkers and Stevens, 1995); for example, changes in the number, localization, or phosphorylation state of AMPA-type glutamate receptors could increase quantal amplitudes without affecting kinetics (Craig et al., 1993; Smart, 1997). Changes indicative of a presynaptic site of modulation were notably absent; namely, BDNF did not increase the probability of neurotransmitter release because no differences were observed either in the frequency of mEPSCs or in quantal content. However, it is also possible that the amount of neurotransmitter packaged per quantum was the target of BDNF regulation, as has been observed for the regulation of midbrain dopamine neurons by GDNF (Pothos et al., 1998). Such a scenario would require that postsynaptic glutamate receptors are not saturated by basal levels of neurotransmitter release per quantum (Bekkers and Stevens, 1990; Liu and Tsien, 1995) (but see Clements et al., 1992;Tong and Jahr, 1994; Forti et al., 1997).
Interestingly, Rutherford et al. (1998) have reported very different effects of BDNF on quantal size at excitatory synapses in mixed cultures of visual cortical neurons. In their studies, long-term treatment with BDNF decreased, rather than increased, the amplitude of mEPSCs onto excitatory neurons. Several experimental differences could account for this discrepancy in BDNF’s long-term effects, including brain region and cell type. Indeed, these differences underscore the diversity of actions that, depending on the specific cell context of a particular target neuron, may result from activation of a given receptor (Sherwood et al., 1997).
Our observation of a BDNF-mediated reduction in PPD in the absence of a concomitant change in the probability of release (_P_r) may seem initially surprising, because it is well known that changing overall_P_r (for instance, by changing the extracellular Ca2+ concentration) results directly in a change in PPD. However, the converse of this relationship may not be obligatory. Because PPD is a function not only of initial_P_r(_P_r1), but also of_P_r during the second stimulus (_P_r2), our observation that PPD was modified in the absence of a change in_P_r1 suggests that BDNF induced a change in _P_r2 only. The cellular mechanism by which PPD occurs in most systems, including hippocampal autaptic cultures, is thought to be via depletion of neurotransmitter vesicles available for release at individual presynaptic terminals [the readily releasable pool (Zucker, 1989; Mennerick and Zorumski, 1995a; Dobrunz and Stevens, 1997)]. Because our experiments ruled out an increased refilling rate of this pool, a potential mechanism for the decrease in PPD caused by chronic BDNF treatment is a change in the ability of vesicles to be released in response to the second stimulus. Such selective changes in “fusion efficiency” (Stevens and Wesseling, 1999) could occur, for example, if vesicular release in response to the second pulse occurs at sites distinct from initial release (Jiang and Abrams, 1998).
Rapid effects of BDNF
The above mechanisms of chronic regulation by BDNF were distinct from its rapid effects: a selective increase in the frequency of spontaneous events with no change in evoked EPSCs, mEPSC size, or paired-pulse depression. The dissociation between mEPSC frequency and EPSC amplitude in the rapid action of BDNF is notable because these two properties of synaptic transmission are usually linked. However, because “true” mEPSCs are independent of action potentials, it is possible to regulate mEPSC frequency and EPSC amplitude independently. In cortical autapses, for example, Kimura et al. (1997) have found that not all synapses that are capable of spontaneous release also release neurotransmitter in response to depolarization. Cash et al. (1996) have observed that heterosynaptic depression of EPSC amplitude in neuromuscular cultures occurs without a corresponding change in the frequency of spontaneous events. Similarly, Deitcher et al. (1998) andYoshihara et al. (1999) have reported dissociation of these properties at the Drosophila neuromuscular junction. In their neuronal synaptobrevin (n-syb) mutants, evoked synaptic currents are eliminated but spontaneous release still occurs in a Ca2+-dependent manner, demonstrating that synaptobrevin mechanistically distinguishes evoked from spontaneous vesicular release.
In contrast to our findings, several recent studies have reported rapid enhancement of EPSC amplitude by BDNF in dissociated hippocampal neurons (Levine et al., 1995; Lessman and Heumann, 1998; Li et al., 1998). Such discrepancies may have arisen, at least in part, from differences in analysis and in the preparation of hippocampal cultures. For example, the acute effects of BDNF in mixed cultures of whole hippocampi reported in these studies may have arisen from subpopulations of neurons not represented in our CA1 autaptic cultures. Furthermore, Lessmann and Heumann (1998) have suggested the intriguing possibility that the responsiveness of neurons to BDNF is dependent on previous levels of activity, a parameter that may have varied considerably among these different culture systems.
Rapid versus chronic effects of BDNF
The relationship between the rapid and chronic effects of BDNF that we observed is of interest. One possibility is that the differences between acute versus chronic BDNF treatment represent functionally independent effects, elicited by the same initial signaling event but manifested along different time courses. Wang and Poo (1997) have suggested, for example, that NT-4/5 has immediate postsynaptic regulatory actions on Xenopus neuromuscular acetylcholine receptors but that it also elicits presynaptic effects on neurotransmitter secretion that are longer lasting (≥ 1 hr) and do not require the constant presence of the neurotrophin. Similarly, the short- and long-term effects of BDNF may be mechanistically and functionally distinct in the mammalian central excitatory neurons studied here.
Alternatively, the differences we observed between rapid and chronic BDNF treatment may reflect different time points in a linear series of cellular changes initiated by TrkB activation. In developing neurons in culture, spontaneous events can be detected well before evoked currents (Basarsky et al., 1994; Gottmann et al., 1994), suggesting that the increases in mEPSC frequency that we observed may represent an early, obligatory phase of the later, chronic regulatory effects of BDNF on synaptic transmission. Such increases in mEPSC frequency are also characteristic of developing neuromuscular junctions (Hume et al., 1983; Young and Poo, 1983) and, indeed, have been observed shortly after neurotrophin application to embryonic neuromuscular cultures (Lohof et al., 1993; Wang et al., 1995; Wang and Poo, 1997). In either case, our results indicate that the action of a single signaling molecule such as BDNF can elicit multiple types of synaptic regulation depending on (1) the temporal characteristics of its availability and (2) the time window in which its synaptic effects are observed.
Classical studies of long-term nerve growth factor effects in the peripheral nervous system have established the requirement for this prototypic neurotrophin in promoting the survival and morphology of specific neuronal cell types (for review, see Purves et al., 1988). More recent studies in visual cortex have provided strong evidence that neurotrophins play an essential role in the activity-dependent establishment of synaptic connectivity between the LGN and visual cortex during development (Bonhoeffer, 1996; Cellerino and Maffei, 1996; Riddle et al., 1996). Intriguingly, BDNF has most recently been implicated in the establishment of L-LTP (Korte et al., 1998), a transcription-dependent form of synaptic potentiation that can last for days (Bailey et al., 1996). Our results indicate that neurotrophins such as BDNF can play important roles in regulating synaptic function on such prolonged time scales through the direct modulation of synaptic strength and reliability. Because the production, release of, and response to BDNF are intimately tied to levels of neuronal activity (Thoenen, 1995; McAllister et al., 1996), findings such as these further strengthen the idea that neurotrophins are key regulators of synaptic plasticity in the CNS in the long term as well as the short term.
Footnotes
This work was supported by the Bryan Scholars Fund and National Research Service Award Grant 5F31MH11058 (N.T.S.), and the Alfred P. Sloan Foundation, the McKnight Endowment for the Neurosciences, and National Institutes of Health Grant NS32742 (D.C.L.). We also thank Regeneron Pharmaceuticals for BDNF and TrkB-IgG; P. De Camilli and O. Mundigl for anti-synapsin I; C. Jahr, C. Stevens, J. Wesseling, and C. Boyer for autapse and other advice; D. Chikaraishi for imaging equipment; and T. Blanpied, M. Bolton, T. Yacoubian, and D. Sherwood for stimulating discussions.
Correspondence should be addressed to Dr. Donald C. Lo, Department of Neurobiology, Box 3209, Duke University Medical Center, Durham, NC 27710.
Dr. Sherwood’s present address: Division of Biology, California Institute of Technology, Pasadena, CA, 91125.
REFERENCES
- 1.Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1996;93:13445–13452. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basarsky TA, Parpura V, Haydon PG. Hippocampal synaptogenesis in cell culture: developmental time course of synapse formation, calcium influx, and synaptic protein distribution. J Neurosci. 1994;14:6402–6411. doi: 10.1523/JNEUROSCI.14-11-06402.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- 4.Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci USA. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekkers JM, Stevens CF. Quantal analysis of EPSCs recorded from small numbers of synapses in hippocampal cultures. J Neurophysiol. 1995;73:1145–1156. doi: 10.1152/jn.1995.73.3.1145. [DOI] [PubMed] [Google Scholar]
- 6.Binder DK, Routbort MJ, Ryan TE, Yancopoulos GD, McNamara JO. Selective inhibition of kindling development by intraventricular administration of TrkB receptor body. J Neurosci. 1999;19:1424–1436. doi: 10.1523/JNEUROSCI.19-04-01424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonhoeffer T. Neurotrophins and activity-dependent development of the neocortex. Curr Opin Neurobiol. 1996;6:119–126. doi: 10.1016/s0959-4388(96)80017-1. [DOI] [PubMed] [Google Scholar]
- 8.Bramham CR, Southard T, Sarvey JM, Herkenham M, Brady LS. Unilateral LTP triggers bilateral increases in hippocampal neurotrophin and trk receptor mRNA expression in behaving rats: evidence for interhemispheric communication. J Comp Neurol. 1996;368:371–382. doi: 10.1002/(SICI)1096-9861(19960506)368:3<371::AID-CNE4>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Cabelli RJ, Hohn A, Shatz CJ. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 10.Cabelli RJ, Shelton DL, Segal RA, Shatz CJ. Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- 11.Cash S, Zucker RS, Poo M-M. Spread of synaptic depression mediated by presynaptic cytoplasmic signaling. Science. 1996;272:998–1001. doi: 10.1126/science.272.5264.998. [DOI] [PubMed] [Google Scholar]
- 12.Castren E, Pitkanen M, Sirvio J, Parsadanian A, Lindholm D, Thoenen H, Riekkinen PJ. The induction of LTP increases BDNF and NGF mRNA but decreases NT-3 mRNA in the dentate gyrus. NeuroReport. 1993;4:895–898. doi: 10.1097/00001756-199307000-00014. [DOI] [PubMed] [Google Scholar]
- 13.Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron. 1997;18:257–267. doi: 10.1016/s0896-6273(00)80266-4. [DOI] [PubMed] [Google Scholar]
- 14.Cellerino A, Maffei L. The action of neurotrophins in the development and plasticity of the visual cortex. Prog Neurobiol. 1996;49:53–71. doi: 10.1016/0301-0082(96)00008-1. [DOI] [PubMed] [Google Scholar]
- 15.Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- 16.Craig AM, Blackstone CD, Banker G. The distribution of glutamate receptors in cultured rat hippocampal neurons: postsynaptic clustering of AMPA-selective subunits. Neuron. 1993;10:1055–1068. doi: 10.1016/0896-6273(93)90054-u. [DOI] [PubMed] [Google Scholar]
- 17.Davies AM. The role of the neurotrophins in the developing nervous system. J Neurobiol. 1994;25:1334–1348. doi: 10.1002/neu.480251103. [DOI] [PubMed] [Google Scholar]
- 18.Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL. Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene, neuronal-synaptobrevin. J Neurosci. 1998;18:2028–2039. doi: 10.1523/JNEUROSCI.18-06-02028.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diamond JS, Jahr CE. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron. 1995;15:1097–1107. doi: 10.1016/0896-6273(95)90098-5. [DOI] [PubMed] [Google Scholar]
- 20.Dichter MA, Tischler AS, Greene LA. Nerve growth factor-induced increase in electrical excitability and acetylcholine sensitivity of a rat pheochromocytoma cell line. Nature. 1977;268:501–504. doi: 10.1038/268501a0. [DOI] [PubMed] [Google Scholar]
- 21.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 22.Ernfors P, Bengzon J, Kokaia Z, Persson H, Lindvall O. Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron. 1991;7:165–176. doi: 10.1016/0896-6273(91)90084-d. [DOI] [PubMed] [Google Scholar]
- 23.Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 24.Forti L, Bossi M, Bergamaschi A, Villa A, Malgaroli A. Loose-patch recordings of single quanta at individual hippocampal synapses. Nature. 1997;388:874–878. doi: 10.1038/42251. [DOI] [PubMed] [Google Scholar]
- 25.Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. J Neurophysiol. 1998;80:3383–3386. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- 26.Furshpan EJ, MacLeish PR, O’Lague PH, Potter DD. Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: evidence for cholinergic, adrenergic, and dual-function neurons. Proc Natl Acad Sci USA. 1976;73:4225–4229. doi: 10.1073/pnas.73.11.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furshpan EJ, Landis SC, Matsumoto SG, Potter DD. Synaptic functions in rat sympathetic neurons in microcultures. I. Secretion of norepinephrine and acetylcholine. J Neurosci. 1986;6:1061–1079. doi: 10.1523/JNEUROSCI.06-04-01061.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonzalez M, Collins WF. Modulation of motoneuron excitability by brain-derived neurotrophic factor. J Neurophysiol. 1997;77:502–506. doi: 10.1152/jn.1997.77.1.502. [DOI] [PubMed] [Google Scholar]
- 29.Gottmann K, Pfrieger FW, Lux HD. The formation of glutamatergic synapses in cultured central neurons: selective increase in miniature synaptic currents. Dev Brain Res. 1994;81:77–88. doi: 10.1016/0165-3806(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 30.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 32.Halegoua S, Armstrong RC, Kremer NE. Dissecting the mode of action of a neuronal growth factor. Curr Top Microbiol Immunol. 1991;165:119–170. doi: 10.1007/978-3-642-75747-1_7. [DOI] [PubMed] [Google Scholar]
- 33.Hume RI, Role LW, Fischbach GD. Acetylcholine release from growth cones detected with patches of acetylcholine receptor-rich membranes. Nature. 1983;305:632–634. doi: 10.1038/305632a0. [DOI] [PubMed] [Google Scholar]
- 34.Ip NY, Li Y, Yancopoulos GD, Lindsay RM. Cultured hippocampal neurons show responses to BDNF, NT-3, and NT-4, but not NGF. J Neurosci. 1993;13:3394–3405. doi: 10.1523/JNEUROSCI.13-08-03394.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- 36.Jiang X-Y, Abrams TW. Use-dependent decline of paired-pulse facilitation at Aplysia sensory neuron synapses suggests a distinct vesicle pool or release mechanism. J Neurosci. 1998;18:10310–10319. doi: 10.1523/JNEUROSCI.18-24-10310.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaczmarek LK, Levitan IB. Neuromodulation: the biochemical control of neuronal excitability. Oxford UP; New York: 1987. [Google Scholar]
- 39.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 40.Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 41.Kim HG, Wang T, Olafsson P, Lu B. Neurotrophin 3 potentiates neuronal activity and inhibits gamma-aminobutyratergic synaptic transmission in cortical neurons. Proc Natl Acad Sci USA. 1994;91:12341–12345. doi: 10.1073/pnas.91.25.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kimura F, Otsu Y, Tsumoto T. Presynaptically silent synapses: spontaneously active terminals without stimulus-evoked release demonstrated in cortical autapses. J Neurophysiol. 1997;77:2805–2815. doi: 10.1152/jn.1997.77.5.2805. [DOI] [PubMed] [Google Scholar]
- 43.Korn H, Faber DS. Quantal analysis and synaptic efficacy in the CNS. Trends Neurosci. 1991;14:439–445. doi: 10.1016/0166-2236(91)90042-s. [DOI] [PubMed] [Google Scholar]
- 44.Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci USA. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Korte M, Kang H, Bonhoeffer T, Schuman E. A role for BDNF in the late-phase of hippocampal long-term potentiation. Neuropharmacology. 1998;37:553–559. doi: 10.1016/s0028-3908(98)00035-5. [DOI] [PubMed] [Google Scholar]
- 46.Landis SC. Rat sympathetic neurons and cardiac myocytes developing in microcultures: correlation of the fine structure of endings with neurotransmitter function in single neurons. Proc Natl Acad Sci USA. 1976;73:4220–4224. doi: 10.1073/pnas.73.11.4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lesser SS, Sherwood NT, Lo DC. Neurotrophins differentially regulate voltage-gated ion channels. Mol Cell Neurosci. 1997;10:173–183. doi: 10.1006/mcne.1997.0656. [DOI] [PubMed] [Google Scholar]
- 48.Lessmann V, Heumann R. Modulation of unitary glutamatergic synapses by neurotrophin-4/5 or brain-derived neurotrophic factor in hippocampal microcultures: presynaptic enhancement depends on pre-established paired-pulse facilitation. Neuroscience. 1998;86:399–413. doi: 10.1016/s0306-4522(98)00035-9. [DOI] [PubMed] [Google Scholar]
- 49.Lessmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 50.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- 52.Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liou JC, Yang RS, Fu WM. Regulation of quantal secretion by neurotrophic factors at developing motoneurons in Xenopus cell cultures. J Physiol (Lond) 1997;503:129–139. doi: 10.1111/j.1469-7793.1997.129bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu G, Tsien RW. Properties of synaptic transmission at single hippocampal synaptic boutons. Nature. 1995;375:404–408. doi: 10.1038/375404a0. [DOI] [PubMed] [Google Scholar]
- 55.Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 56.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 57.Mandel G, Cooperman SS, Maue RA, Goodman RH, Brehm P. Selective induction of brain type II Na+ channels by nerve growth factor. Proc Natl Acad Sci USA. 1988;85:924–928. doi: 10.1073/pnas.85.3.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McAllister AK, Lo DC, Katz LC. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15:791–803. doi: 10.1016/0896-6273(95)90171-x. [DOI] [PubMed] [Google Scholar]
- 59.McAllister AK, Katz LC, Lo DC. Neurotrophin regulation of cortical dendritic growth requires activity. Neuron. 1996;17:1057–1064. doi: 10.1016/s0896-6273(00)80239-1. [DOI] [PubMed] [Google Scholar]
- 60.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1998;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 61.Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol (Lond) 1995a;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mennerick S, Zorumski CF. Presynaptic influence on the time course of fast excitatory synaptic currents in cultured hippocampal cells. J Neurosci. 1995b;15:3178–3192. doi: 10.1523/JNEUROSCI.15-04-03178.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mennerick S, Zorumski CF. Postsynaptic modulation of NMDA synaptic currents in rat hippocampal microcultures by paired-pulse stimulation. J Physiol (Lond) 1996;490:405–407. doi: 10.1113/jphysiol.1996.sp021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mennerick S, Que J, Benz A, Zorumski CF. Passive and synaptic properties of hippocampal neurons grown in microcultures and in mass cultures. J Neurophysiol. 1995;73:320–332. doi: 10.1152/jn.1995.73.1.320. [DOI] [PubMed] [Google Scholar]
- 65.Nawa H, Carnahan J, Gall C. BDNF protein measured by a novel enzyme immunoassay in normal brain and after seizure: partial disagreement with mRNA levels. Eur J Neurosci. 1995;7:1527–1535. doi: 10.1111/j.1460-9568.1995.tb01148.x. [DOI] [PubMed] [Google Scholar]
- 66.Nicoll RA, Malenka RC, Kauer JA. Functional comparison of neurotransmitter receptor subtypes in mammalian central nervous system. Physiol Rev. 1990;70:513–565. doi: 10.1152/physrev.1990.70.2.513. [DOI] [PubMed] [Google Scholar]
- 67.Ohsawa F, Widmer HR, Knusel B, Denton TL, Hefti F. Response of embryonic rat hippocampal neurons in culture to neurotrophin-3, brain-derived neurotrophic factor and basic fibroblast growth factor. Neuroscience. 1993;57:67–77. doi: 10.1016/0306-4522(93)90112-s. [DOI] [PubMed] [Google Scholar]
- 68.Pan ZZ, Tong G, Jahr CE. A false transmitter at excitatory synapses. Neuron. 1993;11:85–91. doi: 10.1016/0896-6273(93)90273-t. [DOI] [PubMed] [Google Scholar]
- 69.Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- 70.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 71.Pothos EN, Davila V, Sulzer D. Presynaptic recording of quanta from midbrain dopamine neurons and modulation of the quantal size. J Neurosci. 1998;18:4106–4118. doi: 10.1523/JNEUROSCI.18-11-04106.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Purves D, Snider WD, Voyvodic JT. Trophic regulation of nerve cell morphology and innervation in the autonomic nervous system. Nature. 1988;336:123–128. doi: 10.1038/336123a0. [DOI] [PubMed] [Google Scholar]
- 73.Riddle DR, McAllister AK, Lo DC, Katz LC. Neurotrophins in cortical development. Cold Spring Harb Symp Quant Biol. 1996;61:85–93. [PubMed] [Google Scholar]
- 74.Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitudes of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–530. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 75.Segal MM, Furshpan EJ. Epileptiform activity in microcultures containing small numbers of hippocampal neurons. J Neurophysiol. 1990;64:1390–1399. doi: 10.1152/jn.1990.64.5.1390. [DOI] [PubMed] [Google Scholar]
- 76.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 77.Shelton DL, Sutherland J, Gripp J, Camerato T, Armanini MP, Phillips HS, Carroll K, Spencer SD, Levinson AD. Human trks: molecular cloning, tissue distribution, and expression of extracellular domain immunoadhesins. J Neurosci. 1995;15:477–491. doi: 10.1523/JNEUROSCI.15-01-00477.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sherwood NT, Lesser SS, Lo DC. Neurotrophin regulation of ionic currents and cell size depends on cell context. Proc Natl Acad Sci USA. 1997;94:5917–5922. doi: 10.1073/pnas.94.11.5917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Silver RA, Traynelis SF, Cull-Candy SG. Rapid-time-course miniature and evoked excitatory currents at cerebellar synapses in situ. Nature. 1992;355:163–166. doi: 10.1038/355163a0. [DOI] [PubMed] [Google Scholar]
- 80.Smart TG. Regulation of excitatory and inhibitory neurotransmitter-gated ion channels by protein phosphorylation. Curr Opin Neurobiol. 1997;7:358–367. doi: 10.1016/s0959-4388(97)80063-3. [DOI] [PubMed] [Google Scholar]
- 81.Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proc Natl Acad Sci USA. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stevens CF, Wesseling JF. Augmentation is a potentiation of the exocytotic process. Neuron. 1999;22:139–146. doi: 10.1016/s0896-6273(00)80685-6. [DOI] [PubMed] [Google Scholar]
- 83.Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-d-aspartate receptor subunit 1. Proc Natl Acad Sci USA. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 86.Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 87.Tong G, Malenka RC, Nicoll RA. Long-term potentiation in cultures of single hippocampal granule cells: a presynaptic form of plasticity. Neuron. 1996;16:1147–1157. doi: 10.1016/s0896-6273(00)80141-5. [DOI] [PubMed] [Google Scholar]
- 88.Trussell LO, Fishbach GD. Glutamate receptor desensitization and its role in synaptic transmission. Neuron. 1989;3:209–218. doi: 10.1016/0896-6273(89)90034-2. [DOI] [PubMed] [Google Scholar]
- 89.Vyklicky L, Patneau DK, Mayer ML. Modulation of excitatory synaptic transmission by drugs that reduce desensitization at AMPA/kainate receptors. Neuron. 1991;7:971–984. doi: 10.1016/0896-6273(91)90342-w. [DOI] [PubMed] [Google Scholar]
- 90.Wang T, Xie K, Lu B. Neurotrophins promote maturation of developing neuromuscular synapses. J Neurosci. 1995;15:4796–4805. doi: 10.1523/JNEUROSCI.15-07-04796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang XH, Poo MM. Potentiation of developing synapses by postsynaptic release of neurotrophin-4. Neuron. 1997;19:825–835. doi: 10.1016/s0896-6273(00)80964-2. [DOI] [PubMed] [Google Scholar]
- 92.Yoshihara M, Ueda A, Zhang D, Deitcher DL, Schwarz TL, Kidokoro Y. Selective effects of neuronal-synaptobrevin mutations on transmitter release evoked by sustained versus transient Ca2+ increases and by cAMP. J Neurosci. 1999;19:2432–2441. doi: 10.1523/JNEUROSCI.19-07-02432.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Young SH, Poo MM. Spontaneous release of transmitter from growth cones of embryonic neurones. Nature. 1983;305:634–637. doi: 10.1038/305634a0. [DOI] [PubMed] [Google Scholar]
- 94.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]