ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth (original) (raw)
Abstract
Tumor cell adaptation to hypoxic stress is an important determinant of malignant progression. While much emphasis has been placed on the role of HIF-1 in this context, the role of additional mechanisms has not been adequately explored. Here we demonstrate that cells cultured under hypoxic/anoxic conditions and transformed cells in hypoxic areas of tumors activate a translational control program known as the integrated stress response (ISR), which adapts cells to endoplasmic reticulum (ER) stress. Inactivation of ISR signaling by mutations in the ER kinase PERK and the translation initiation factor eIF2α or by a dominant-negative PERK impairs cell survival under extreme hypoxia. Tumors derived from these mutant cell lines are smaller and exhibit higher levels of apoptosis in hypoxic areas compared to tumors with an intact ISR. Moreover, expression of the ISR targets ATF4 and CHOP was noted in hypoxic areas of human tumor biopsy samples. Collectively, these findings demonstrate that activation of the ISR is required for tumor cell adaptation to hypoxia, and suggest that this pathway is an attractive target for antitumor modalities.
Keywords: apoptosis, ATF4, endoplasmic reticulum, hypoxia, PERK
Introduction
The development of fluctuating hypoxic and anoxic regions in tumors has profound consequences for malignant progression, response to therapy and overall patient survival (Hockel and Vaupel, 2001). Elucidating the processes that enable tumor cells to adapt to the unfavorable conditions of hypoxia is critical for understanding malignant progression and for developing more effective antitumor modalities. The hypoxia-inducible factor 1 (HIF-1) plays a key role in tumor cell adaptation to hypoxia by regulating the expression of over 60 genes involved in angiogenesis, anaerobic glycolysis and cell survival (Ratcliffe et al, 1998; Semenza, 2000). Loss-of-function studies indicate that, in most cases, HIF-1 promotes overall tumor growth (Maxwell et al, 1997; Ryan et al, 2000). However, the role of HIF-1 is complex and may be dependent on the tumor microenvironment (Carmeliet et al, 1998; Blouw et al, 2003).
The hypoxic cell also elicits additional, HIF-1-independent adaptive responses that contribute to increased survival under low oxygen conditions. An immediate reaction to hypoxia is a reduction in the rates of global protein synthesis that is thought to reduce energy demands when oxygen and ATP levels are low (Hochachka et al, 1996). This translational inhibition appears to occur in distinct phases and to involve multiple pathways. We have previously shown that hypoxia rapidly increases phosphorylation of the translation initiation factor eIF2α through activation of the endoplasmic reticulum (ER) kinase PERK (Koumenis et al, 2002; Blais et al, 2004). These events serve two major functions in a cell experiencing ER stress. The first is to rapidly downregulate protein synthesis, which reduces the ER protein load and leads to lower energy expenditure, since both protein synthesis and protein folding are ATP-requiring processes (Dorner et al, 1990; Shi et al, 1998; Harding et al, 2000b). The second is to upregulate genes that promote amino-acid sufficiency and redox homeostasis (Harding et al, 2003), thereby further promoting cell survival. Some, but not all of the effects of PERK are mediated by the transcription factor ATF4, which is translationally upregulated by ER stress in an eIF2α phosphorylation-dependent manner (Harding et al, 2000a, 2003). This pathway, known as the integrated stress response (ISR) (Ron, 2002), constitutes one arm of a larger coordinated program called the unfolded protein response (UPR), which facilitates the cell to adapt to ER stress. Cells with compromised PERK and eIF2α signaling are substantially more sensitive to ER-induced cell death than wild-type cells (Harding et al, 2000b, 2001; Scheuner et al, 2001; Zhang et al, 2002).
Recently, it was reported that hypoxia upregulates ATF4 in a PERK-dependent manner (Blais et al, 2004) and that ATF4 levels are increased by anoxic stress and in human tumors (Ameri et al, 2004). These findings prompted us to investigate the physiological role for the ISR in cellular adaptation to hypoxia in vitro and in tumor development in vivo. We report that hypoxia/anoxia activates the ISR pathway in vitro and in vivo, and that several proteins previously implicated in the UPR are also upregulated by hypoxia. Expression of ATF4 and CHOP colocalizes with hypoxic areas of primary human tumors and animal xenografts, and inactivation of the ISR compromises cell survival during hypoxia in vitro and in vivo and significantly impairs tumor growth. These findings establish the ISR as an important mediator of hypoxia tolerance and tumor growth, and raise the possibility that the ISR may be an attractive target for antitumor modalities.
Results
Hypoxic/anoxic stress induces expression of ISR genes in a PERK- and eIF2_α_-phosphorylation-dependent manner
To investigate the effects of hypoxia on the expression of ISR-related genes, mouse embryonic fibroblasts (MEFs) from PERK+/+ and PERK−/− animals were exposed to extreme hypoxia (⩽0.02%) or were treated with thapsigargin, an inhibitor of the SERCA (Ca2+-ATPase) pump and potent ER stressor (Note: for brevity, hypoxia in the text denotes extreme hypoxia unless otherwise noted). Consistent with previous findings (Koumenis et al, 2002), hypoxia and thapsigargin caused a time-dependent increase in PERK autophosphorylation and eIF2α phosphorylation in PERK+/+ MEFs (results not shown). Phosphorylation of eIF2α stimulates translational upregulation of ATF4, which activates the expression of downstream targets involved in the UPR, such as CHOP. As shown in Figure 1A, ATF4 levels increased by hypoxia and thapsigargin in the PERK+/+ MEFs, whereas this increase was transient and considerably attenuated in PERK−/− MEFs. Induction of CHOP did not occur until after 16 h of hypoxia, reflecting a requirement for ATF4 accumulation. BiP was not significantly increased by hypoxia. Hypoxia-induced ATF4 accumulation occurred primarily at the post-transcriptional level, because ATF4 mRNA levels were not upregulated in hypoxic PERK+/+ MEFs (Figure 1B). CHOP and BiP mRNA levels were substantially increased by hypoxia and by thapsigargin in PERK+/+ and PERK−/− MEFs. The increase in CHOP was higher after 16 h of hypoxia in the PERK+/+ cells, consistent with the higher levels of ATF4 protein in these cells, but PERK status had no effect on the increase in BiP mRNA levels.
Figure 1.
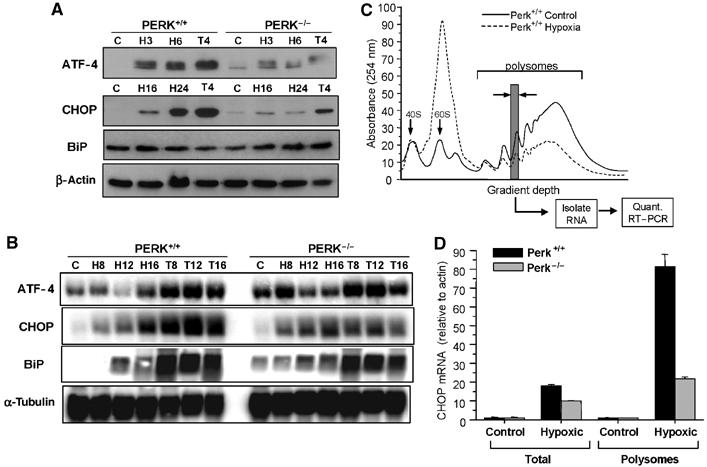
Extreme hypoxia induces ATF4 and CHOP upregulation in a PERK-dependent manner. PERK+/+ and PERK−/− MEFs were exposed to ⩽0.02% O2 or treated with 1 μM thapsigargin for the times indicated. (A) Immunoblots with anti-ATF4, anti-CHOP, anti-BiP and anti-β-actin (loading control). Different time points are used since induction of ATF4 occurs much earlier than CHOP or BiP. (B) Northern blot analysis of UPR mRNAs following hypoxia and thapsigargin treatments. PERK+/+ and PERK−/− MEFs were treated as in (A). Blotting for α-tubulin was used as a loading control. (C) Translation efficiency of CHOP mRNA following hypoxia. PERK+/+ and PERK−/− MEFs were treated as in (A) and lysates were subjected to sucrose gradient sedimentation. Polysome profiles shown are from cell lysates of aerobic and hypoxic PERK+/+ cells. The positions of the 40S and 60S subunits as well as the polysomal RNA are indicated. (D) The amounts of CHOP, β-actin and 18S mRNA were determined by real-time quantitative PCR in each of the polysome fractions. The gray column indicates the polysome fraction used for quantitative PCR. Values plotted represent the total amount of CHOP mRNA found within the polysomes relative to β-actin. Also shown is the total cellular amount of CHOP found in the PERK+/+ and PERK−/− MEFs normalized to β-actin as determined by quantitative RT–PCR.
Although hypoxia induced a largely PERK-independent increase in CHOP mRNA, there was a strong PERK dependence on CHOP protein accumulation, suggesting that the translational efficiency of CHOP mRNA might be regulated in a PERK-dependent manner. Thus, we analyzed the levels of both overall and gene-specific translation in the PERK+/+ and PERK−/− cell lines. We have previously shown that inhibition of global translation during hypoxia is dependent on PERK at early times after hypoxia (<8 h), but, after longer periods of hypoxia translation, is inhibited in both PERK+/+ and PERK−/− cells (Koumenis et al, 2002). As shown in Figure 1C, 16 h of hypoxia caused a strong inhibition of translation initiation in the PERK+/+ cells, as evident by a reduction in polysomal RNA and an increase in free monosome and ribosome subunits. A similar inhibition was observed in PERK−/− cells after 16 h of hypoxia; however, following 2 h hypoxia, translation was inhibited much more strongly in the PERK+/+ compared to PERK−/− cells (data not shown). To explore potential PERK-dependent differences in specific CHOP mRNA translation following prolonged hypoxia, polysome fractions were collected and individually analyzed by quantitative RT–PCR for the levels of 18S, actin and CHOP. In both PERK+/+ and PERK−/− cells, the levels of actin associated with polysomes decreased during hypoxia in accordance with the drop in overall translation. In contrast, CHOP mRNA levels increased substantially within the polysomes. Notably, the relative increase in CHOP mRNA within the polysomes was approximately four times higher in the PERK+/+ cells than in the PERK−/− cells (Figure 1D). Thus, in addition to an increase in CHOP mRNA, there is a significant PERK dependence on polysome association of CHOP mRNA during hypoxia.
Similarly, by coupling ribosome fractionation with microarray gene analysis, we identified changes in steady-state gene expression and translation-mediated changes in HeLa cells following 16 h of extreme hypoxia. We found that the mRNA levels of genes associated with the UPR (including ORP150, heme oxygenase; HO-1 and MIF1) were not only substantially induced by hypoxia but also remained efficiently translated, as indicated by their association with high-molecular-weight polysomes (Figure S1). Conversely, the free total and polysome-bound mRNA levels of genes associated with other types of stress, such as the heat-shock response (HSF2, HSP70) or of constitutively synthesized genes (ribosomal protein S20 and HSC70), were either only weakly upregulated or substantially downregulated by hypoxia.
Prolonged hypoxia induces apoptosis in a PERK-dependent manner
Cells with a compromised UPR are more susceptible to ER stress-induced apoptosis compared to cells with an intact UPR (Kaufman, 2002; Ma and Hendershot, 2004). We previously reported that PERK−/− cells exhibit reduced clonogenic survival under extreme hypoxia compared to PERK+/+ cells (Koumenis et al, 2002). To determine whether the difference in clonogenic survival was due to differential apoptotic sensitivity, we assayed for cleavage of caspase-12, caspase-3 and PARP following exposure to extreme hypoxia. Caspase-12, associated with the ER, is cleaved during ER stress and this event can be used as an indicator of UPR activation (Nakagawa et al, 2000; Rao et al, 2001). Exposure of PERK−/− cells to hypoxia resulted in higher levels of caspase-12 cleavage (as evident by the disappearance of the procaspase-12 form) by 9 h of hypoxia compared to PERK+/+ cells (Figure 2A). However, caspase-12−/− MEFs were as sensitive as caspase-12+/+ MEFs under hypoxia (results not shown), indicating that, while caspase-12 processing is a marker of hypoxia-induced ER stress, it cannot account for the higher sensitivity of PERK−/− cells to hypoxia. Activation of caspase-3, a terminal caspase and proteolytic processing of PARP, a marker for late-stage apoptosis, followed a similar pattern with higher levels of cleaved caspase-3 and PARP in the PERK−/− compared to PERK+/+ cells (Figure 2B and C) following treatments with hypoxia and thapsigargin. These results indicate that PERK contributes to cellular survival under prolonged hypoxia.
Figure 2.
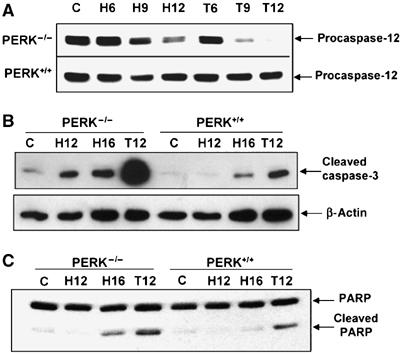
PERK confers resistance to hypoxia-induced apoptosis. (A–C) PERK+/+ and PERK−/− MEFs were exposed to ⩽0.02% O2 or treated with 500 nM thapsigargin for the indicated times before immunoblotting with antibodies specific for procaspase-12 (A), cleaved caspase-3 or anti-β-actin (B), or cleaved and uncleaved PARP (C).
PERK contributes to tumor growth in vivo
The increased sensitivity of PERK−/− cells to hypoxic stress in vitro suggested that PERK might play a role in tumor growth in vivo. To investigate this possibility, PERK+/+ and PERK−/− MEFs were immortalized with SV40 large/small T-antigen and transformed with oncogenic Ki-RasV12. This transformation did not cause any overt morphological differences between the two cell lines (data not shown). Phosphorylation of ERK1 and ERK2 was analyzed in PERK+/+ and PERK−/− MEFs with or without Ki-RasV12. As expected, expression of Ki-RasV12 increased ERK phosphorylation without affecting the levels of total ERK1 and ERK2. The extent of this phosphorylation was comparable in PERK+/+ and PERK−/− MEFs (Figure 3A), suggesting that PERK does not influence Ki-RasV12 signaling. Further experiments demonstrated that PERK status did not affect the in vitro growth kinetics or the ability of the Ki-RasV12-transformed cell lines to grow colonies in soft agar (Figures 3B and C).
Figure 3.
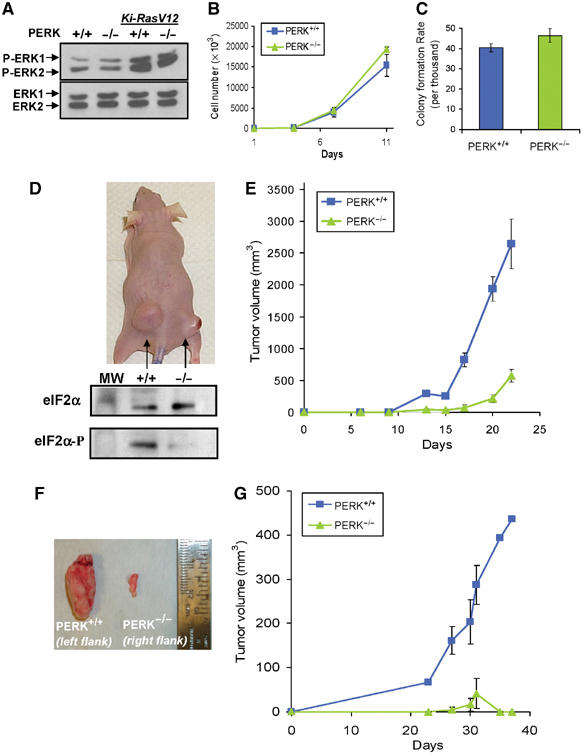
PERK affects the rate of tumor growth in vivo. (A) Immunoblot of untransformed PERK+/+ and PERK−/− MEFs, and MEFs stably transformed with Ki-RasV12, using a phospho-specific anti-ERK1/2 antibody (top), or an antibody against total ERK1 and ERK2 (bottom). (B) Growth rates of Ki-RasV12-transformed PERK+/+ and PERK−/− MEFs in vitro. (C) PERK+/+.Ki-RasV12 and PERK−/−.Ki-RasV12 MEFs form colonies in soft agar at similar rates. (D) Top, picture of a representative nude mouse injected with PERK+/+.Ki-RasV12 MEFs (left flank) and PERK−/−.Ki-RasV12 MEFs (right flank) with 3 × 106 cells/site at 22 days following injection. The bottom panel shows eIF2α phosphorylation levels from the excised tumors shown in the top panel. Tumor sections were homogenized and immunoblotted for phospho-eIF2α and total-eIF2α. (E) PERK+/+ (blue line and squares) and PERK−/− (green line and triangles) tumor volumes from seven animals monitored over a period of 22 days following inoculation (mean±s.e.). (F) Tumors from a mouse injected with clone #2 PERK+/+.Ki-RasV12 MEFs (left) or clone #2 PERK−/−.Ki-RasV12 MEFs (right). (G) Growth of PERK+/+.Ki-RasV12#2 (blue line and rectangles) and PERK−/−.Ki-RasV12#2 (green line and triangles) tumors (_N_=4) monitored over a period of 37 days (mean±s.e.).
Equal numbers of these transformed MEFs were injected in the flanks of nude mice and tumor growth was monitored over a period of 3–4 weeks. Tumors from PERK+/+.RasV12 MEFs grew faster and larger compared to those from the PERK−/−.RasV12 MEFs (Figures 3D and E). Several PERK−/− tumors displayed an opaque morphology with apparent necrosis of the skin when the experiment was terminated (Figure 3D, top). Analysis of eIF2α phosphorylation status in homogenized tissue revealed markedly higher levels of phosphorylated eIF2α in PERK+/+ tumors compared to PERK−/− tumors, indicating PERK-dependent eIF2α phosphorylation caused by the tumor microenvironment (Figure 3D, bottom). On average, PERK+/+ tumors grew approximately six times larger than PERK−/− tumors after 22 days (average 2647±573 versus 391±101 mm3, _N_=7) (Figure 3E). Similar results were obtained when Ha-RasV12 instead of Ki-RasV12 was used to transform PERK+/+ and PERK−/− MEFs (data not shown).
To minimize the effect of random clonal variation in this experimental system, we also examined the in vivo growth of individual clones of Ki-RasV12-transfected cells. Although the growth kinetics of PERK+/+.Ki-RasV12.#2 and PERK−/−. Ki-RasV12.#2 MEFs were indistinguishable in vitro (results not shown), the PERK−/− tumors failed to grow beyond a few mm in size (Figures 3F and G). Collectively, these results indicate that PERK status affects tumor growth in a manner that is independent of in vitro growth characteristics, suggesting that the differential response of these cells to the tumor microenvironment must play a critical role in tumor development.
PERK confers resistance to hypoxia-induced apoptosis in vivo
The compromised ability of PERK−/− cells to tolerate hypoxic conditions in vitro and the slower growth of PERK−/− tumors suggested that the presence of PERK increased the ability of transformed MEFs to survive tumor hypoxia, resulting in tumors with larger hypoxic areas. We therefore studied the patterns of cell death in relation to areas of hypoxia within PERK+/+ and PERK−/− tumor sections. The hypoxia marker EF-5 is metabolized and forms macromolecular adducts preferentially in hypoxic areas that can be visualized using a Cy3-labeled anti-EF5 adduct antibody. Nude mice with PERK+/+.Ki-RasV12 and PERK−/−.Ki-RasV12 flank tumors were injected with EF-5 and sections were co-stained with Cy3-labeled anti-EF5 antibody and with an anti-cleaved caspase-3 antibody. The patterns of EF-5 and caspase-3 staining in the PERK+/+ and PERK−/− tumors were quite distinct. In PERK−/− tumors, areas of hypoxia were fewer and smaller compared to the PERK+/+ tumors (Figure 4A). Image analysis revealed that ∼70% of apoptotic cells in four distinct fields colocalized with hypoxic areas (Figure 4B). In contrast, PERK+/+ tumors exhibited more extensive areas of hypoxia that were largely devoid of apoptotic cells, with only ∼22% apoptotic cells being within the hypoxic areas. Strong staining for cleaved caspase-3 was observed only in areas surrounded by a ‘hypoxic ring', which is indicative of a necrotic tumor area. Similar results were obtained from two independent tumors of each cell line. These results suggest that PERK+/+.Ki-RasV12 MEFs can better tolerate hypoxia in vivo, resulting in larger tumors with extensive hypoxic areas. In contrast, PERK−/−.Ki-RasV12 MEFs, which are more sensitive to hypoxia, form smaller tumors with fewer hypoxic areas. Since hypoxic areas in vivo are usually accompanied by conditions of low pH and/or growth factor levels, it is possible that the sensitivity of the PERK−/− cells in vivo may be due not only to low oxygen levels but also to these factors as well.
Figure 4.
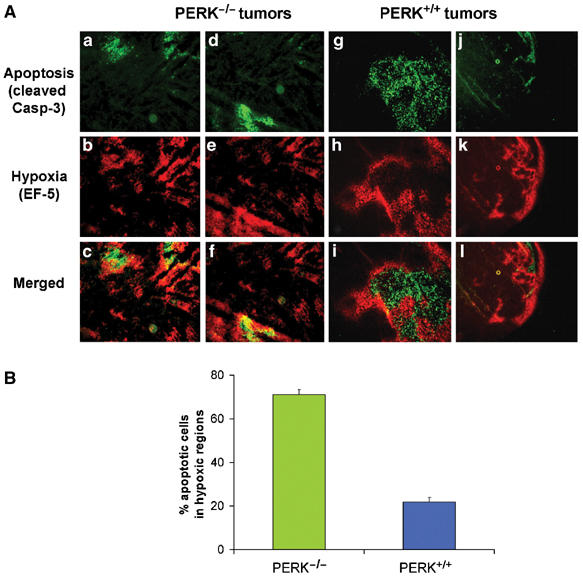
Hypoxic areas in PERK−/− but not in PERK+/+ tumors overlap with areas of apoptosis. (A) Sections of PERK−/− tumors (a−f) or PERK+/+ tumors (g−l) were incubated with a Cy3-conjugated antibody to EF-5 adducts (hypoxia, red) (b, e and h, k), followed by incubation with a FITC-conjugated anti-cleaved caspase-3 antibody (apoptosis, green) (a, d and g, j). Panels c, f and i, l are merged images showing both apoptotic and hypoxia areas. Two different PERK−/− (a−c and d−f) and PERK+/+ (g−i and j−l) tumor sections are shown. Bar=15 μm (a−i) and 60 μm (j−l). (B) Quantitation of four independent field images from PERK+/+ or PERK−/− tumors. The number of apoptotic cells in hypoxic (EF-5 positive) and normoxic (EF-5 negative) areas of each image were automatically counted and expressed as a percentage of the total number of apoptotic cells in the image. Error bars represent s.e. values.
Cells with a nonphosphorylatable ‘knock-in' mutation of eIF2_α_ display attenuated induction of ISR genes under hypoxia, and are sensitive to hypoxic stress
Although eIF2α phosphorylation is a primary target of activated PERK, it has been suggested that PERK phosphorylation may activate additional targets (Cullinan et al, 2003). To further study the role of the ISR in tumor cell adaptation to hypoxic stress, we used MEFs in which the endogenous eIF2α gene has been genetically replaced by a nonphosphorylatable (S51A) allele. These cells display dramatically reduced downstream effector responses to ER stress and other stresses that induce eIF2α phosphorylation (Scheuner et al, 2001). Exposure of these cells to hypoxia failed to induce eIF2α phosphorylation (Figure 5A) and induction of ISR proteins ATF4 and CHOP by hypoxia was significantly attenuated (Figure S2A). At the mRNA level, hypoxia again failed to induce a significant upregulation of ATF4 (Figure S2B), further confirming that the induction of ATF4 by hypoxia is post-transcriptional (Blais et al, 2004).
Figure 5.
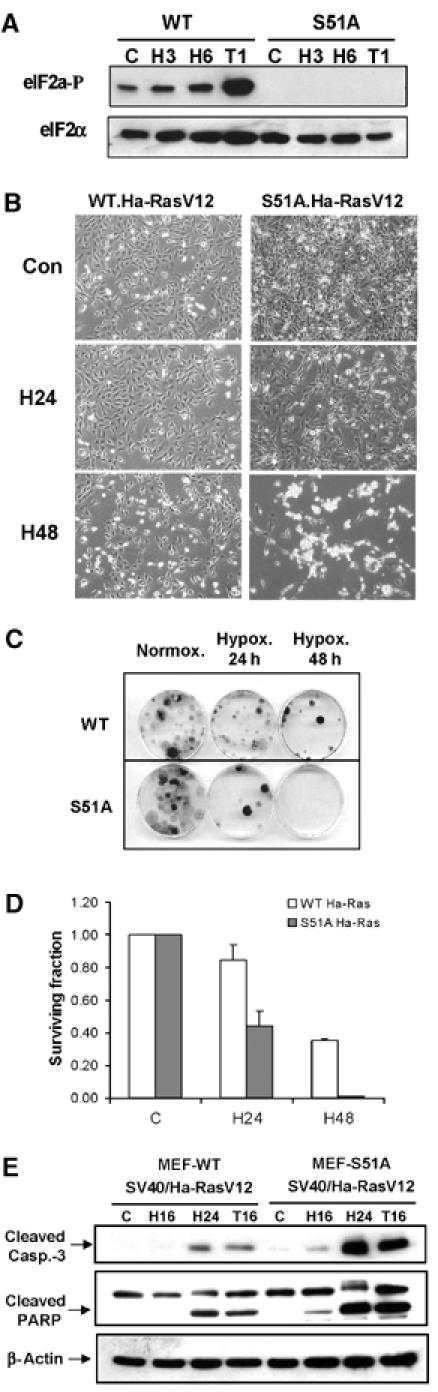
MEFs with nonphosphorylatable eIF2α mutant are more sensitive to extreme hypoxia than WT MEFs. (A) WT and S51A MEFs were exposed to ⩽0.02% O2 or treated with 1 μM thapsigargin before immunoblotting with anti-phospho-eIF2α or anti-total-eIF2α antibodies. (B) WT and S51A MEFs transformed with SV40/Ha-RasV12 were exposed to extreme hypoxia (⩽0.02% O2) and then photographed using phase-contrast microscopy. (C) Reduced clonogenic survival of S51A MEFs after hypoxia compared to WT MEFs. (D) Reduced survival of S51A MEFs after hypoxia compared to WT MEFs, as measured by clonogenic survival assay. Cells were treated as in (C). Experiments were performed in triplicate and error bars represent standard errors. (E) WT and S51A MEFS transformed with SV40/Ha-RasV12 were exposed to hypoxia or treated with 500 nM thapsigargin before immunoblotting for cleaved caspase-3, PARP or β-actin.
The S51A knock-in mice display striking phenotypic similarities with the PERK knockout mice, particularly in the sensitivity of cells with specialized and increased secretory capacity to endogenous ER stress (Harding et al, 2001; Scheuner et al, 2001). To determine whether similar parallels exist in the response of PERK−/− and S51A cells to hypoxic stress, we transformed S51A and wild-type MEFs (WT-MEFs) with SV40 large/small T-antigen and mutant Ha-RasV12 and tested their survival under hypoxia. S51A cells exhibited progressively higher apoptotic sensitivity to hypoxia compared to WT-MEFS (Figure 5B). Furthermore, exposure of WT-MEFs to 24 and 48 h of hypoxia resulted in 16 and 60% decreases in clonogenic survival, respectively, whereas S51A-transformed MEFs were significantly more sensitive, exhibiting 60 and 100% decreases in survival, respectively (Figure 5C and D). The increased levels of cell death in the S51A cells were accompanied by higher levels of cleaved caspase-3 and PARP at 16 and 24 h of hypoxia compared to WT-MEFs (Figure 5E). Thus, the higher sensitivity of S51A cells to hypoxia compared to WT cells is similar to that of the PERK−/− cells and parallels the sensitivity of S51A cells to other ER stressors like thapsigargin (Harding et al, 2000b).
Phosphorylation of eIF2_α_ affects tumor growth and ISR signaling in vivo
As was the case with PERK−/− and PERK+/+ transformed MEFs, in vitro growth rates of SV40/Ha-RasV12-transformed WT and S51A MEFs were very similar (Figure S2C). However, when injected into nude mice, the S51A tumors grew slower during the first 14 days following inoculation, and, between days 14 and 21, the majority of S51A tumors developed areas of apparent necrosis (Figure 6A) and the animals had to be euthanized. At that time, the average weight of S51Á tumors was approximately half of that of WT tumors (179±26 versus 318±77 mg, respectively; P<0.05%) (Figure 6B). Similar to the PERK−/− tumors, the S51A tumors exhibited extensive colocalization of apoptotic and hypoxic areas (Figure S3A, a–f and m, n), whereas hypoxic areas of WT tumors were largely devoid of apoptotic cells and apoptosis was confined to necrotic areas (Figure S3A, g–l and o, p). Quantitative image analysis showed that in WT tumors only 17% of apoptotic cells were in hypoxic regions, compared to 80% in tumors from the S51A cells (Figure S3B). Furthermore, the difference in tumor weight was not due to differences in the angiogenic response to hypoxia, since secretion of VEGF was not statistically different between the two cell types (Figure 6C). To determine whether the induction of the ISR in vivo is eIF2α-dependent, we analyzed the induction of ATF4 in the two tumor types. WT tumors displayed areas of ATF4 expression that colocalized with hypoxia, whereas the S51A tumors were devoid of any significant staining for ATF4 (Figure 6D). We failed to detect any significant expression of ATF4 in 6/6 S51A sections obtained from three different mouse tumors. These results indicate that eIF2α phosphorylation is required for hypoxic induction of ATF4 in vivo. Thus, the phenotype of S51A tumors paralleled that of the PERK−/− tumors, indicating that the PERK-eIF2α component of the ISR is required for in vivo resistance to hypoxic stress and tumor growth.
Figure 6.
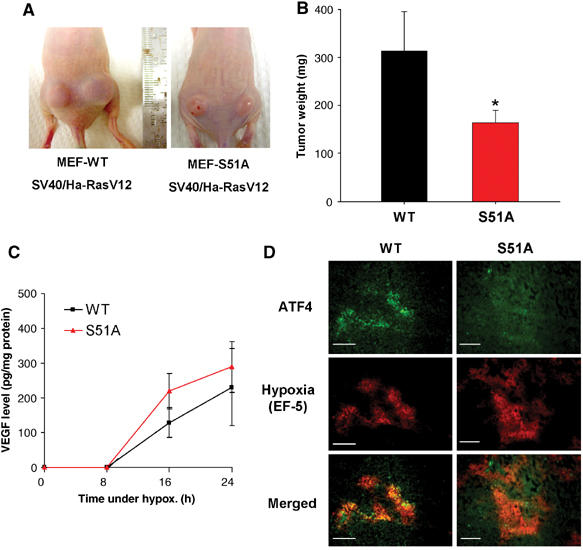
Inhibition of eIF2α phosphorylation affects the rate of tumor growth in vivo. (A) Nu/Nu mice were injected on each side with 1 × 106 cells/site, with either WT.Ha-RasV12 MEFs (left panel) or S51A.Ha-RasV12 MEFs (right panel). Necrotic areas on the skin and opaque morphology are evident in the S51A.Ha-RasV12 tumors. (B) Tumor growth after 3 weeks. At the end of the experiment, tumors were excised and weighed (mean±s.e., _N_=7, *P<0.05, one-tailed Student's _t_-test). (C) VEGF levels following hypoxia in WT and S51A MEFs. Ras-transformed MEFs were treated as in Figure 5A and secreted VEGF levels in the media were analyzed at the times indicated. Experiments were performed in triplicate and error bars represent s.e. values. (D) ATF4 upregulation in hypoxic areas is dependent on eIF2α phosphorylation. Tumor xenografts from Ha-RasV12-transformed WT and S51A cells were stained for ATF4 expression. Staining for ATF4 was observed only in WT tumors and localized within hypoxic regions. No significant staining was seen in 6/6 sections from three different S51A tumors. Bar=15 μm.
Expression of a dominant-negative PERK (dn-PERK) allele in human tumor cells reduces hypoxia tolerance and inhibits tumor growth
The results presented so far were obtained using minimally transformed MEFs, a useful and informative isogenic system, but one that may not be representative of human tumor cells of epithelial origin. To examine the role of the PERK-eIF2α pathway in vitro and in vivo, we used a dn-PERK construct to block the activity of endogenous PERK in an established human cancer cell line. This dn-allele, PERKΔC, lacks the C-terminal kinase domain, has a myc-tag epitope and was shown to inhibit the ability of endogenous PERK to phosphorylate eIF2α following various stresses, including hypoxia (Brewer and Diehl, 2000; Koumenis et al, 2002). HT29 colorectal carcinoma cells stably expressing the dn-PERK construct (HT29.PERKΔC), or the empty vector (HT29.Puro) were exposed to normoxia or 6 h of extreme hypoxia. HT29.PERKΔC cells failed to increase eIF2α phosphorylation after 6 h of hypoxia, confirming that this construct exerts a dominant-negative function by inhibiting endogenous PERK activity (Figure 7A). We next tested the apoptotic sensitivity of these cells to hypoxia and thapsigargin. After 32 h of extreme hypoxia, substantially higher levels of cleaved PARP were observed in the HT29.PERKΔC cells compared to the HT29.Puro cells (Figure 7B, top). The HT29.PERKΔC cells also displayed morphology consistent with swollen ER, indicative of ER stress (Figure 7B, bottom). The in vitro growth of the control and PERKΔC cell lines was not significantly different over a period of 12 days in culture (data not shown) and HIF-1α was induced to comparable levels in both cell lines (Figure 7C), confirming that HIF-1 regulation is not dependent on PERK status. To test the role of PERK in HT29 tumor growth, cells were injected into the flanks of nude mice and tumor volume was followed for up to 25 days. Tumors obtained from HT29.PERKΔC cells grew significantly smaller compared to those obtained from the HT29.Puro cells (Figure 7D and E), providing additional evidence that inhibition of PERK activity hinders tumor growth in vivo. Immunoblot analysis from homogenized tumors showed that expression of the dn-PERKΔC allele was retained for the duration of tumor growth and correlated with inhibition of eIF2α phosphorylation in the HT29.PERKΔC tumors (Figure 7F). Also, similar to the PERK−/− MEF tumors, the HT29.PERKΔC tumors displayed extensive colocalization of apoptosis with areas of hypoxia. In contrast, the HT29.Puro tumors had larger hypoxic areas that were largely devoid of apoptotic cells (Figure 7G). Collectively, these results provide further evidence that inhibition of PERK activity compromises hypoxia tolerance of tumor cells in vitro and in vivo.
Figure 7.
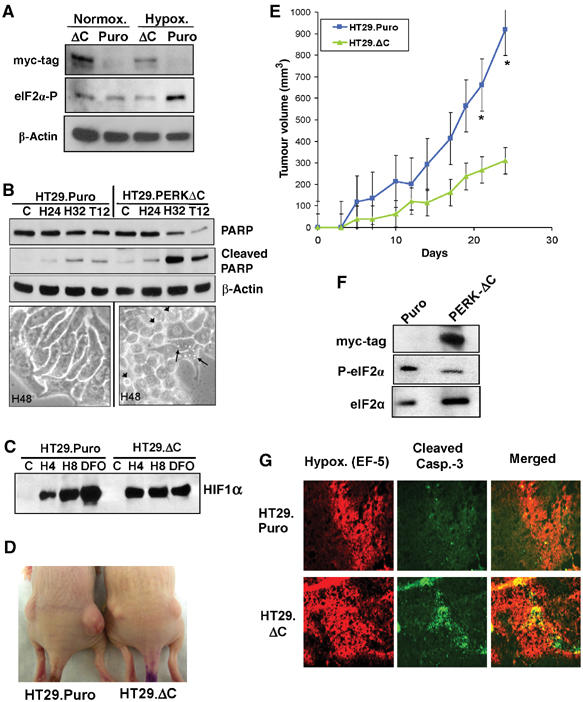
Expression of a dn-PERK allele in human tumor cells decreases hypoxia tolerance and inhibits tumor growth. (A) Immunoblot of myc-tagged PERKΔC, eIF2α phosphorylation and β-actin (loading control). Exposure of HT29.Puro cells to extreme hypoxia induces an increase in eIF2α phosphorylation, which is blocked in HT29.PERKΔC cells. (B) Immunoblot of uncleaved and cleaved PARP in HT29.Puro and HT29.PERKΔC cells following hypoxia or 12 h treatment with 500 nM thapsigargin (top). β-Actin was used as a loading control. Apoptotic morphology following extreme hypoxia for 48 h at × 40 magnification (bottom). Arrowheads indicate cells with membrane blebbing and arrows indicate cells exhibiting vacuoles, a sign of ER stress. (C) PERK status does not affect induction of HIF-1α levels. HT29.Puro and HT29.PERKΔC cells were treated with extreme hypoxia or DFO. HIF-1α levels were determined by immunoblotting using nuclear extracts. (D) Tumor xenografts from HT29.Puro cells (left mouse) grow larger compared to tumors from HT29.PERKΔC cells (right mouse). (E) Growth measurements of HT29.Puro xenografts (blue line and squares) and HT29.PERKΔC xenografts (green line and triangles). Growth from six tumors was monitored over a period of 25 days following injection (mean±s.e.) (*P<0.05). (F) Immunoblot analysis of PERKΔC expression, eIF2α phosphorylation status or total eIF2α levels from excised and homogenized tumors shown in (D, E). (G) Sections from HT29.Puro and HT29.PERKΔC xenografts were stained for regions of hypoxia using a Cy-3-labeled anti-EF-5 antibody and apoptosis using an anti-cleaved caspase-3, FITC-labeled antibody. The merged images are shown on the right.
Cells lacking ATF4 are sensitive to hypoxic stress and expression of the ISR proteins ATF4 and CHOP is upregulated in hypoxic areas of primary human tumors
Since both PERK−/− and S51A MEFs were more sensitive to hypoxia compared to the corresponding WT-MEFs, we wished to determine whether lack of the downstream target ATF4 would result in a similar phenotype. Extreme hypoxia resulted in significant levels of apoptosis in the ATF4−/− MEFs but not in ATF4+/+ MEFs, which was evident both morphologically (Figure 8A) and by cleaved caspase-3 and cleaved PARP analysis (Figure 8B). The sensitivity of ATF4−/− cells to hypoxia also occurred at more moderate hypoxic levels. Exposure of the MEFs to 1% O2 resulted in a 55% decrease in survival in ATF4−/− cells compared to 25% in ATF4+/+ cells as assayed by MTT assay, whereas the sensitivity of the two cell types to a different stress, ionizing radiation, was the same (Figure 8C).
Figure 8.
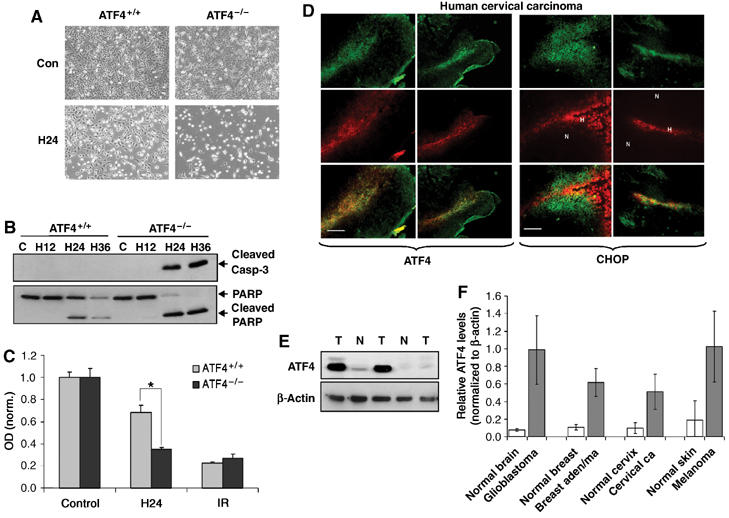
ATF4 contributes to hypoxia tolerance, and ATF4 and CHOP are upregulated in hypoxic areas of primary human tumors. (A) Cells were exposed to extreme hypoxia for 24 h and photographed for apoptotic morphology at × 100 magnification. (B) Immunoblot of cleaved caspase-3 (top) or uncleaved and cleaved PARP (bottom) following hypoxia. (C) MTT assay of ATF4+/+ and ATF4−/− MEFs following exposure to moderate hypoxia (1% O2) or treatment with a 4 Gy dose of IR. Experiments were performed in triplicate and results are normalized to control levels. Error bars represent s.e. values (*P<0.05). No significant difference in survival was observed between untreated ATF4+/+ and ATF4−/− MEFs. (D) Immunofluorescence for ATF4 and hypoxia (a−f), or CHOP and hypoxia (g−l) in primary human cervical tumors. Sections were stained with anti-ATF4 (a, d) or anti-CHOP (g, j) polyclonal antibodies followed by a FITC-conjugated secondary antibody, and with a Cy-3-labeled anti-Pimonidazole antibody (b, e, h, k). Panels c, f, i and l are combined images of (a+b), (d+e), (g+h), (j+k), respectively. Bar=15 μm (a−c, g−i) and 30 μm (d−f, j−i). (E) Immunoblot of ATF4 in lysates from human GBMs and normal human brain tissue. (F) Analysis of ATF4 levels (normalized to levels of β-actin) in normal and malignant tissues obtained from human patients. Error bars represent s.e. values. Brain, _N_=6 normal and 6 glioblastoma; breast, _N_=3 normal and 3 adenocarcinoma; cervix, _N_=4 normal and 6 carcinoma; skin, _N_=2 normal and 2 melanoma.
To investigate whether the induction of the ISR also occurs in primary human tumors, we analyzed ATF4 and CHOP levels in human cervical tumor sections obtained from patients that had been injected with the hypoxia-sensitive dye pimonidazole (Varia et al, 1998). Immunohistochemical staining showed significant intra- and intertumoral variations of ATF4 and CHOP expression. However, expression of both proteins colocalized with either hypoxic areas or areas adjacent to hypoxic regions. ATF4 expression showed stronger association with highly hypoxic areas (pimonidazole-positive staining, Figure 8D), whereas CHOP expression was associated with highly hypoxic and surrounding areas (Figure 8D). Similar results were obtained in four different sections obtained from two distinct cervical tumors (Figure S4A). We also examined ATF4 expression in several normal and malignant tissues obtained from patients with brain, breast, cervical and skin cancers. The expression of ATF4 was significantly higher in all malignant tissues compared to the corresponding normal tissues (Figures 8E and F and Figure S4B). The results with ATF4 are in agreement with a previously reported study in which ATF4 expression was found to be higher in breast tumors than in normal breast tissue, and to be present near necrotic areas (Ameri et al, 2004).
Discussion
We have presented evidence that the ISR arm of the UPR is activated by hypoxic/anoxic stress in vitro and in hypoxic/anoxic areas of solid tumors, and that failure of tumor cells to mount an effective ISR in response to low oxygen results in increased apoptosis, reduced overall cell survival and inhibition of tumor growth. Recently, a study by Chen et al showed that XBP1, the target of IRE1, is essential for cell survival under hypoxic conditions and that XBP1 is required for tumor growth (Romero-Ramirez et al, 2004). Together, these studies suggest that the full UPR is activated under hypoxia/anoxia and contributes to cellular adaptation to this stress.
The induction of gene expression under conditions of global translational repression is reminiscent of other stress responses (e.g., heat-shock response), during which select groups of genes are upregulated in a background of repressed gene expression to help in the recovery from the stress. Thus, it is noteworthy that several genes with important roles in the hypoxic response, including VEGF and HIF-1α, harbor unusually long 5′ UTRs with extensive secondary structures that presumably enable them to be efficiently translated under hypoxic conditions (Akiri et al, 1998; Lang et al, 2002). Conversely, our screen for genes whose mRNAs are efficiently translated under hypoxic conditions revealed that several mRNAs of UPR genes, including BiP, CHOP and ORP150, are among the most efficiently translated mRNAs (Figure S1). It is tempting to speculate that the hypoxic/anoxic response and the UPR may share common post-translational regulation mechanisms.
The complex picture of tumor hypoxia, composed of significant intra- and intertumoral variations in oxygen levels that can change dynamically in time, prompted the investigation of UPR gene expression in hypoxic areas of tumors. We show that at least two ISR target genes, ATF4 and CHOP, are highly expressed in hypoxic areas of tumors. Previously, we demonstrated that induction of eIF2α phosphorylation (a requirement for significant ATF4 and CHOP accumulation as shown in Figure S2) can occur at more moderate hypoxic levels—as high as 1% O2—albeit with slower kinetics (Koumenis et al, 2002), suggesting that the ISR can be elicited over a wide range of hypoxic conditions.
What is the physiological role for these proteins in hypoxia tolerance? ATF4 activates the induction of downstream UPR genes, but has also been implicated in antioxidant cellular defense processes (Fawcett et al, 1999; Harding et al, 2003; Lu et al, 2004). Recent mechanistic studies on HIF-1α induction appear to support a model of increased oxidative stress in the cytosol due to release of reactive oxygen species from the mitochondria (Guzy et al, 2005; Mansfield et al, 2005). Furthermore, reoxygenation following hypoxia produces oxidative stress severe enough to activate DNA damage response pathways (Hammond et al, 2003). We hypothesize that induction of ATF4 by hypoxia serves to upregulate antioxidant-related genes to ameliorate oxidative stress. CHOP, itself a target of ATF4, is a transcription factor with proapoptotic properties (Zinszner et al, 1998; McCullough et al, 2001). While CHOP may promote apoptosis under prolonged/severe hypoxia, it is evident—as is also the case for induction of CHOP by pharmacological agents (Harding et al, 2000a)—that the consequences of PERK ablation are significantly more severe in terms of apoptosis than an reduced CHOP expression. Thus, the significance of CHOP induction by hypoxia at present remains unclear and will have to be elucidated by using cells with abrogated CHOP expression.
Cells with a compromised ISR pathway show significant sensitivity to ER stress due to the deleterious effects of accumulated misfolded proteins in the ER (Harding et al, 2000b; Scheuner et al, 2001). Our results showing PERK-dependent activation of caspase-12 provide further evidence of ER stress under hypoxic stress. However, caspase-12−/− MEFs display equal sensitivity to prolonged hypoxia compared to their WT-MEFs (data not shown), suggesting that caspase-12 is not required for hypoxia-induced cell death. Recently, the BH3-only proapoptotic proteins Bax and Bak have been localized in the ER membrane and shown to mediate stress-induced release of ER Ca2+ (Scorrano et al, 2003; Zong et al, 2003), raising the possibility that they could play a critical role in hypoxia-induced, ER-dependent apoptosis.
An interesting question not addressed by these studies is the nature of the signal for induction of the UPR by hypoxia/anoxia. Activation of PERK, IRE1 and ATF6 by ER stress is negatively regulated by BiP. Under physiological conditions, BiP binds to the ER-lumenal portion of the proteins and keeps them in an inactive state (Bertolotti et al, 2000; Shen et al, 2002), and this binding is perturbed during ER stress. One can envision a scenario in which low oxygen perturbs the pro-oxidant environment of the ER lumen causing misfolding of labile proteins, thereby triggering BiP-dependent PERK activation. This model is supported by the findings that (a) in yeast, molecular oxygen is the final electron acceptor in protein disulfide isomerase-dependent protein folding, (b) BiP expression is upregulated by hypoxia (Gazit et al, 1999) and (c) downregulation of BiP by antisense cDNA decreases the survival of tumor cells (Koong et al, 1994). Further validation of this model will require careful examination of the interactions between BiP and the initiators of the UPR under normoxic and hypoxic conditions. Based on the findings presented here, we propose that, in addition to HIF-1-dependent processes, the induction of the UPR by hypoxia represents an encompassing and critical aspect of hypoxia resistance and tumor development. Since stringent hypoxic conditions do not normally exist outside a tumor microenvironment, targeting UPR processes could provide an alternative or complementary approach for therapeutic exploitation of tumor hypoxia.
Materials and methods
Cell culture and generation of the stable cell lines
PERK+/+, PERK−/−, S51A, ATF4−/− and WT MEFs, and HeLa cells were cultured in DMEM. HT29 colorectal carcinoma cells (ATCC) were cultured in McCoy's 5A medium. All media were supplemented with penicillin, streptomycin, 10% fetal calf serum and L-glutamine. S51A mutant and ATF4−/− MEFs and HT.29PERKΔC cells were also supplemented with 20% FBS, 1 × MEM nonessential amino-acid mix and 55 μM 2-mercaptoethanol. Cell transformation and establishment of HT29.PERKΔC cells are described in Supplementary data.
Hypoxia treatments
Cells were placed in either a Bactron 1 Anaerobic Chamber (Sheldon Manufacturing) or an InVivo2 400 Hypoxia Workstation (Biotrace, Inc.). for the time(s) indicated. The oxygen concentration in the anaerobic chamber was maintained at ⩽0.02% and monitored with a polarographic, membrane-covered oxygen sensor (Animas Corp.).
Animals
Athymic Nu/Nu mice (Charles River Labs) were subcutaneously injected with 3 × 106 transformed PERK+/+ and PERK−/− MEFs or transformed MEF-WT and S51A MEFs or 1.5 × 106 HT29.Puro or HT.29PERKΔC cells in 150 μl of PBS. Tumors were monitored daily until they became cumbersome or necrotic. Tumor volumes were measured every other day based on the formula _V_=length2 × width, where length was always the longest dimension. For hypoxia visualization, mice were injected with 300 μl of 3 mg/ml EF-5 (University of Pennsylvania) i.p. 1 h before euthanization, tumors were immediately excised, frozen and embedded in OCT freezing medium.
Human tumors
Human cervical squamous cell carcinoma tumors were obtained at the UNC-CH School of Medicine. Patients were injected with pimonidazole prior to surgical resection of tumors. Tumors were flash frozen in liquid nitrogen, embedded in OCT and stored at −80°. Human tumor and normal tissues were obtained through the WFU Comprehensive Cancer Center Tissue Procurement Core Lab. Collection and processing of human specimens was performed in accordance to UNC-CH and WFUSM IRB regulations.
Immunohistochemistry
Frozen tissue was sectioned in 5 μm and fixed to glass slides in 4% formaldehyde solution for 10 min. For detection of hypoxic areas using EF-5, sections were further fixed with 100% methanol for 10 min, blocked in 3% BSA in PBS for 30 min and incubated with a Cy3-conjugated ELK 3–51 (75 μg/ml) monoclonal antibody (University of Pennsylvania) that recognizes EF-5 adducts, followed by incubation with a FITC-conjugated anti-cleaved caspase-3 antibody (Cell Signaling). Fluorescence was detected using Olympus IX70 Epifluoroscope and photographed at several magnifications. For detection of hypoxia/ATF4 and hypoxia/CHOP colocalization, mouse and human cervical tumor sections were fixed in 95, 75 and 50% ethanol, blocked in 3% BSA in PBS and hypoxia adducts detected with either ELK 3-51 antibody for mouse tumors or Cy3-conjugated anti-pimonidazole monoclonal antibody (2 μg/ml) (UNC-CH) for human tumors. Sections were incubated with anti-CHOP or anti-ATF4 polyclonal antibodies, followed with a FITC-conjugated secondary antibody.
Immunoblotting
Immunoblotting was performed as previously described (Koumenis et al, 2002). For primary antibodies used, please see Supplementary data.
Supplementary Material
Figure S1
Figure S2
Figure S3
Figure S4
Acknowledgments
We thank Chris Counter and Peter Howley for the Ha-RasV12 and Ki-RasV12 expression plasmids, Alan Diehl for the PERKΔC expression vector, Andrew Thorburn and John Connor for critically reading the manuscript, Albert Koong for helpful discussions and Lori Hart, Dawn Eads and Jack Smallwood for expert technical help. This work was supported by NIH grants CA94214 (CK), ES08681 (DR) and DK42394 (RJK).
References
- Akiri G, Nahari D, Finkelstein Y, Le SY, Elroy-Stein O, Levi BZ (1998) Regulation of vascular endothelial growth factor (VEGF) expression is mediated by internal initiation of translation and alternative initiation of transcription. Oncogene 17: 227–236 [DOI] [PubMed] [Google Scholar]
- Ameri K, Lewis CE, Raida M, Sowter H, Hai T, Harris AL (2004) Anoxic induction of ATF-4 through HIF-1-independent pathways of protein stabilization in human cancer cells. Blood 103: 1876–1882 [DOI] [PubMed] [Google Scholar]
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2: 326–332 [DOI] [PubMed] [Google Scholar]
- Blais JD, Filipenko V, Bi M, Ron D, Koumenis C, Wouters BG, Bell JC (2004) Transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 24: 7469–7482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouw B, Song H, Tihan T, Bosze J, Ferrara N, Gerber HP, Johnson RS, Bergers G (2003) The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell 4: 133–146 [DOI] [PubMed] [Google Scholar]
- Brewer JW, Diehl JA (2000) PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc Natl Acad Sci USA 97: 12625–12630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E, Keshet E (1998) Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394: 485–490 [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23: 7198–7209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner AJ, Wasley LC, Kaufman RJ (1990) Protein dissociation from GRP78 and secretion are blocked by depletion of cellular ATP levels. Proc Natl Acad Sci USA 87: 7429–7432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ (1999) Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J 339 (Part 1): 135–141 [PMC free article] [PubMed] [Google Scholar]
- Gazit G, Hung G, Chen X, Anderson WF, Lee AS (1999) Use of the glucose starvation-inducible glucose-regulated protein 78 promoter in suicide gene therapy of murine fibrosarcoma. Cancer Res 59: 3100–3106 [PubMed] [Google Scholar]
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT (2005) Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 1: 401–408 [DOI] [PubMed] [Google Scholar]
- Hammond EM, Dorie MJ, Giaccia AJ (2003) ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J Biol Chem 278: 12207–12213 [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000a) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099–1108 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D (2001) Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell 7: 1153–1163 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000b) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5: 897–904 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633 [DOI] [PubMed] [Google Scholar]
- Hochachka PW, Buck LT, Doll CJ, Land SC (1996) Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proc Natl Acad Sci USA 93: 9493–9498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockel M, Vaupel P (2001) Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst 93: 266–276 [DOI] [PubMed] [Google Scholar]
- Kaufman RJ (2002) Orchestrating the unfolded protein response in health and disease. J Clin Invest 110: 1389–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koong AC, Chen EY, Lee AS, Brown JM, Giaccia AJ (1994) Increased cytotoxicity of chronic hypoxic cells by molecular inhibition of GRP78 induction. Int J Radiat Oncol Biol Phys 28: 661–666 [DOI] [PubMed] [Google Scholar]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG (2002) Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol 22: 7405–7416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang KJ, Kappel A, Goodall GJ (2002) Hypoxia-inducible factor-1alpha mRNA contains an internal ribosome entry site that allows efficient translation during normoxia and hypoxia. Mol Biol Cell 13: 1792–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP (2004) Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J 23: 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM (2004) The role of the unfolded protein response in tumor development: friend or foe? Nat Rev Cancer 4: 966–977 [DOI] [PubMed] [Google Scholar]
- Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC (2005) Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-[alpha] activation. Cell Metab 1: 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, Hankinson O, Pugh CW, Ratcliffe PJ (1997) Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA 94: 8104–8109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ (2001) Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21: 1249–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403: 98–103 [DOI] [PubMed] [Google Scholar]
- Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE (2001) Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem 276: 33869–33874 [DOI] [PubMed] [Google Scholar]
- Ratcliffe PJ, O'Rourke JF, Maxwell PH, Pugh CW (1998) Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J Exp Biol 201 (Part 8): 1153–1162 [DOI] [PubMed] [Google Scholar]
- Romero-Ramirez L, Cao H, Nelson D, Hammond E, Lee AH, Yoshida H, Mori K, Glimcher LH, Denko NC, Giaccia AJ, Le QT, Koong AC (2004) XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res 64: 5943–5947 [DOI] [PubMed] [Google Scholar]
- Ron D (2002) Translational control in the endoplasmic reticulum stress response. J Clin Invest 110: 1383–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM, Johnson RS (2000) Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res 60: 4010–4015 [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ (2001) Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell 7: 1165–1176 [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139 [DOI] [PubMed] [Google Scholar]
- Semenza GL (2000) Surviving ischemia: adaptive responses mediated by hypoxia-inducible factor 1. J Clin Invest 106: 809–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, Prywes R (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 3: 99–111 [DOI] [PubMed] [Google Scholar]
- Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC (1998) Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol 18: 7499–7509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varia MA, Calkins-Adams DP, Rinker LH, Kennedy AS, Novotny DB, Fowler WC Jr, Raleigh JA (1998) Pimonidazole: a novel hypoxia marker for complementary study of tumor hypoxia and cell proliferation in cervical carcinoma. Gynecol Oncol 71: 270–277 [DOI] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR (2002) The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol 22: 3864–3874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12: 982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB (2003) Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 162: 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Figure S4