FURIN AT THE CUTTING EDGE: FROM PROTEIN TRAFFIC TO EMBRYOGENESIS AND DISEASE (original) (raw)
. Author manuscript; available in PMC: 2007 Sep 4.
Published in final edited form as: Nat Rev Mol Cell Biol. 2002 Oct;3(10):753–766. doi: 10.1038/nrm934
Abstract
Furin catalyses a simple biochemical reaction – the proteolytic maturation of proprotein substrates in the secretory pathway. But the simplicity of this reaction belies furin's broad and important roles in homeostasis, as well as in diseases ranging from Alzheimer's disease and cancer to anthrax and Ebola fever. This review summarizes various features of furin – its structural and enzymatic properties, intracellular localization, trafficking, substrates, and roles in vivo.
Furin is a cellular endoprotease that was identified in 1990 (BOX 1); it proteolytically activates large numbers of proprotein substrates in secretory pathway compartments. As well as activating pathogenic agents (BOX 2), furin has an essential role in embryogenesis, and catalyses the maturation of a strikingly diverse collection of proprotein substrates. These range from growth factors and receptors to extracellular-matrix proteins and even other protease systems that control disease. Until recently, furin was thought to be an unglamorous housekeeping protein; however, furin's crucial role in so many different cellular events — and in diseases ranging from from anthrax and bird flu (BOX 2) to cancer, dementia and Ebola fever — has caused researchers to re-evaluate it. In this review, I summarize the various features of furin: its structural and enzymatic properties, autoactivation, intracellular localization and trafficking; its substrates; and its roles in vivo, including the requirement for furin in determining the pathogenicity of many viruses and bacteria.
Box 1 The discovery of furin.
In 1990, the identification of furin as the first bona fide mammalian proprotein convertase (PC) ended a nearly quarter-century-long search for the mammalian enzymes that catalyse the proteolytic maturation of prohormones and proproteins (for a review see refs 150,151). The quest for the PCs began with seminal studies by Donald Steiner in 1967 (refs 152,153), who showed for the first time that peptide hormones are post-translationally excised from larger prohormones by cleavage of the precursor at doublets, or clusters, of basic amino acids (for example, –Lys–Arg↓– and –Arg–Arg↓–, where ↓ identifies the cleavage site). These studies were as revolutionary as those by Krebs and Fischer, which showed that protein phosphorylation is a universal modification in signal transduction. Concurrent studies by Michel Chrétien and Choh Hoh Li on the structural relationships between β-melanocyte stimulating hormone (β-MSH), γ-lipotropin (γ-LPH) and β-lipotropin (β-LPH)154 — a subset of peptides derived from a complex pituitary prohormone, proopiomelanocortin (POMC) — provided the first clues to the greater generality of proprotein processing.
Together, these studies set the foundations of subsequent research in protein processing over the next 20 years, which showed that virtually all peptide hormones, numerous bioactive proteins (for example, growth factors, receptors and cell-adhesion molecules), and many bacterial toxins and viral envelope glycoproteins, follow this fundamental maturation scheme to generate the mature and biologically active molecule (for a review, see ref. 151). The enzymes that catalyse these vital reactions, however, were not identified until 1984, when the yeast endoprotease, kexin or Kex2, was isolated155.
Kex2 excises α-mating pheromone and killer toxin from their precursors in late Golgi compartments. Because Kex2 could also correctly process mammalian proproteins, one or more of the mammalian PCs were anticipated to share structural features with the yeast enzyme156. A database search identified a previously reported protein encoded by the FUR (‘fes/fps upstream region’) locus157, an open reading frame adjacent to the fes/fps proto-oncogene, as the first mammalian Kex2 homologue158. The product of this gene — furin — was soon shown to correctly process precursors for neurotrophic factors, serum proteins and pathogen molecules6,159,160. PCR strategies were then used to identify the remaining six members of the PC family (FIG. 1; TABLE 1). Together, the PCs catalyse the proteolytic maturation of an enormous collection of bioactive peptides and proteins that regulate virtually every process controlling homeostasis and disease.
Box 2 Furin and bioterrorism.
The bioterrorism plot following the World Trade Center tragedy on 11 September 2001 attempted to inflict countless deaths by disseminating Bacillus anthracis spores through the mail system. Twenty-two people were diagnosed with anthrax that was contracted from contact with contaminated mail — five died within days of exposure161,162. However, the affected sorting facilities processed 85 million pieces of mail after the contaminated letters were sent, reinforcing just how close we came to disaster. And anthrax is not alone in its capacity to ignite disaster. It is eerily reminiscent of the influenza pandemic that could have erupted in Hong Kong in 1997, when a renegade pathogenic avian influenza virus — able to jump directly from birds to humans — killed six of the 18 people who were clinically diagnosed as having ‘bird flu’ in a week. If it had not been for the attenuated infectivity of this H5N1 influenza virus, the death toll from the outbreak could have been far worse. As well as illustrating our vulnerability to deadly microbes, there is another link between these two close calls, and that link is furin, a host-cell endoprotease that activates the toxic agents of these pathogens (see text for more details).
ENDOPROTEASE.
An enzyme that hydrolyses peptide and protein substrates at specific internal peptide bonds. By contrast, exoproteases remove either the carboxy-terminal (carboxypeptidase) or amino-terminal (aminopeptidase) residues.
PROPROTEIN.
A precursor protein that is proteolytically cleaved, typically at sites formed by doublets or clusters of basic amino acids, to produce the mature and active protein or, in the case of prohormones, the mature peptide hormones.
PATHOGENICITY.
A measure of the ability of a pathogen to invade a host and cause disease. The degree of the pathogenicity is measured as its virulence.
The biochemical properties of furin
Domain structure
Furin is a ubiquitously expressed 794-amino-acid type-i transmembrane protein that is found in all vertebrates and many invertebrates1,2. Its large lumenal/extracellular region has an overall homology with the same regions of other members of the proprotein convertase (PC) family (FIG. 1; TABLE 1), which belongs to the subtilisin superfamily of serine endoproteases. The greatest sequence similarity resides in the subtilisin-like catalytic domain; the aspartate (Asp), histidine (His) and serine (Ser) residues that form the catalytic triad are rigorously conserved, and the catalytic domains of the other PCs are 54–70% identical in sequence to furin. In addition to the signal peptide, which directs translocation of the pro-enzyme into the endoplasmic reticulum (ER), furin and the other PCs contain prodomains that are flanked by the signal peptidase cleavage site on the amino-terminal side and by a conserved set of basic amino acids that comprise the autoproteolytic cleavage site on the carboxy-terminal side. This essential prodomain has a crucial role in the folding, activation and transport of PCs, and in the regulation of PC activity. Furin and the other PCs also share a conserved P domain, which is essential for enzyme activity and the modulation of pH and calcium requirements3; this P domain is absent from the related bacterial enzymes. The furin cytoplasmic domain controls the localization and sorting of furin in the _trans_-Golgi network (TGN)/endosomal system, and furin is an important model for understanding the regulation of protein trafficking in mammalian cells.
Figure 1.

Schematic diagram of the proprotein convertase (PC) family. Shown are schematics for furin and the six other PCs. Schematics of yeast Kex2 and the evolutionarily related bacterial subtilisin (which lacks the conserved P domain) are also shown. PC5/6 is expressed as either the A or B isoform. These isoforms are generated by alternative splicing, and the diagonal dashed line links the two halves of PC5/6B. The PC5/6B isoform contains all of PC5/6A except for a small part of its carboxyl terminus that is positioned after the splice site. The bold labels D, H and S highlight the active-site residues, whereas the non-bold labels N and D highlight the oxyanion-hole residues.
Table 1.
The proprotein convertase family
| Proprotein convertase | Size (amino acids) | Tissue distribution | Subcellular localization | Knockout phenotype |
|---|---|---|---|---|
| Furin | 794 | Broad | TGN/endosomal | Embryonic lethality (day 10.5), impaired axial rotation |
| PC7 | 785 | Broad | TGN/endosomal | Viable; misexpression causes thymic defects |
| PC5/6A* | 915 | Broad | Secretory granules | ND |
| PC5/6B* | 1877 | Broad | TGN/endosomal | ND |
| PACE4 | 963 | Broad | TGN/endosomal | Embryonic lethality (day 15.5), craniofacial and CNS defects |
| PC1/3 | 753 | Neuroendocrine system | Secretory granules | Mouse: severe growth defects, defective GHRH and POMC processing, hyperproinsulinaemia |
| ‡Humans: severe early obesity, adrenocortical insufficiency, hyperproinsulinaemia | ||||
| PC2 | 638 | Neuroendocrine system | Secretory granules | Hypoglycaemia, proinsulinaemia, glucagon deficiency, defects in opioid peptide processing |
| PC4 | 655 | Testicular and ovarian germ cells | ND | Males: infertility |
| Females: mild infertility |
pH and ion requirements
Furin has a broad pH optimum; it has more than 50% of its enzymatic activity between pH 5 and 8, depending on the substrate being cleaved. Like other members of the subtilisin superfamily, furin is strictly calcium dependent, requiring approximately 1 mM calcium for full activity. Modelling studies based on the alignment of furin with the solved structure of bacterial thermitase – a well-characterized subtilisin that shares 29% sequence identity with the furin catalytic domain — indicate that furin has two calcium-binding pockets: one with medium affinity and one with high affinity4. Furin also binds weakly to potassium, and 20 mM potassium increases furin activity by enhancing the rate of deacylation, which is important in furin's catalytic cycle5.
TYPE-I TRANSMEMBRANE PROTEIN.
Proteins that contain a single membrane-spanning domain, with the carboxyl terminus oriented towards the cytoplasm and the amino terminus oriented towards the lumen of membrane compartments or extracellularly.
PROPROTEIN CONVERTASE.
Members of the family of calcium-dependent, subtilisin-like serine endoproteases that are structurally related to Kex2 and furin and that cleave proprotein substrates at the carboxy-terminal side of doublets or clusters of basic amino acids.
α-MATING PHEROMONE.
A peptide pheromone that is secreted by yeast alpha-cells to stimulate mating with yeast a-cells. The α-mating pheromone is synthesized as a proprotein substrate that is cleaved by Kex2. Yeast alpha-cells lacking Kex2 are sterile.
Consensus cleavage site
The consensus site that furin cleaves, which is positioned after the carboxy-terminal arginine (Arg) residue in the sequence –Arg–X–Lys/Arg–Arg↓– (where Lys is lysine, X is any amino acid and ↓ identifies the cleavage site), was determined biochemically using two bona fide in vivo furin substrates — anthrax toxin protective antigen (PA) and avian influenza virus haemagglutinin (HA)6,7. Because anthrax toxin PA and influenza virus HA are cleaved at the cell surface and in the TGN/biosynthetic pathway, respectively, these studies provided the first indication that furin is active in several cellular compartments and is a key enzyme in the activation of diverse pathogens (FIG. 2). Arg residues at the p1 and p4 positions in this cleavage site are essential, whereas the P2 basic amino acid (Lys/Arg) is not, but it can greatly enhance processing efficiency. Therefore, –Arg–X–X–Arg↓– represents the minimal furin cleavage site, although favourable residues at P2 and P6 can compensate for less favourable ones at position P4 (ref. 8). Accordingly, in exceptional cases, –Lys/Arg–X–X–X–Lys/Arg–Arg↓– can be cleaved by furin.
Figure 2.

Furin-processing compartments of the _trans_-Golgi network (TGN)/endosomal system. At steady-state, furin (represented by scissors) is localized principally to the TGN, where it cycles between this sorting compartment, the cell surface and the early endosomes. In the TGN/biosynthetic pathway, furin cleaves many substrates including pro-β-nerve growth factor (pro-β-NGF), pro-bone morphogenetic protein-4 (pro-BMP-4), the insulin pro-receptor and Ebola Zaire pro-glycoprotein (pro-GP). At the cell surface, furin cleaves substrates such as anthrax protective antigen (PA), proaerolysin and Clostridium septicum α-toxin. In mildly acidic early endosomes (endocytic pathway), furin cleaves substrates including diphtheria toxins, shiga toxin and shiga-like toxin-1, and Pseudomonas exotoxin A. See text for more details.
As described below, the various permutations of the consensus furin cleavage site have important implications for the proprotein substrate, both in terms of the compartmental specificity and the sequential ordering of the furin cleavage events. A model of the furin catalytic domain, based on the structures of the bacterial subtilisins BPN' and thermitase, predicts that negatively-charged residues in the S1, S2 and S4 subsites of the binding pocket might interact with the basic amino acids in the substrate4,9. The lack of a three-dimensional furin structure, however, precludes any certainty about the substrate and cofactor binding sites.
Inhibitors
Furin's cleavage site requirements have been used to produce potent peptide- and protein-based inhibitors that block furin activity in vitro and in vivo10-12. Perhaps the two most widely used furin inhibitors are the stoichiometric peptidyl inhibitor decanoyl–Arg–Val–Lys–Arg–CH2Cl (where Val is valine) and α1-antitrypsin Portland (α1-PDX), a bioengineered variant of α1-antitrypsin. Decanoyl–Arg–Val–Lys–Arg–CH2Cl inhibits all PCs with a low nanomolar _K_i (ref. 13), although the alkylating properties of the reactive group limit the usefulness of this reagent. Nonetheless, in cell-culture studies, decanoyl–Arg–Val–Lys–Arg–CH2Cl blocks the processing of several furin substrates. The α1-PDX inhibitor was generated by mutating the reactive-site loop of α1-antitrypsin to contain the minimal consensus sequence for furin cleavage (–Arg–Ile–Pro–Arg–)14 (where Ile is isoleucine and Pro is proline), and it is highly selective for furin in vitro (_K_i = 600 pM), although at higher concentrations it will also inhibit other PCs13. In biochemical, cellular and animal studies, α1-PDX has been used to block furin activity and to prevent the production of pathogenic viruses, bacterial toxin activation, and cancer metastasis10 (see below).
Multistep furin autoactivation
The 83-amino-acid furin prodomain acts as an intramolecular chaperone propeptide that guides the folding and activation of the endoprotease15. The folding of furin's catalytic centre into its correct conformation is driven by a multistep, compartment-specific pair of cleavages in the prodomain, which yield the active enzyme (FIG. 3). To accomplish this task, furin exploits its own cleavage-site rules and cuts the prodomain twice. The first, and most rapid, cut (t½ = 10 mins) takes place in the neutral pH environment of the ER after Arg107 in the consensus furin cleavage site (–Arg–Thr–Lys–Arg107 ↓– (where Thr is threonine)), which is located at the border of the catalytic domain. The second, slower, cut (t½ < 2 hrs) is made after Arg75 of the pH-sensitive furin propeptide cleavage site in the prodomain (–Arg70–Gly–Val–Thr–Lys–Arg75↓– (where Gly is glycine)) during trafficking of the propeptide–furin complex within the mildly acidic TGN/endosomal system. Furin's internal-propeptide-cleavage-site pH sensitivity is again shown by the mildly acidic conditions that are needed for the cleavage of proalbumin and Pseudomonas exotoxin A at similar furin sites10 (FIG. 2; also see below).
Figure 3.
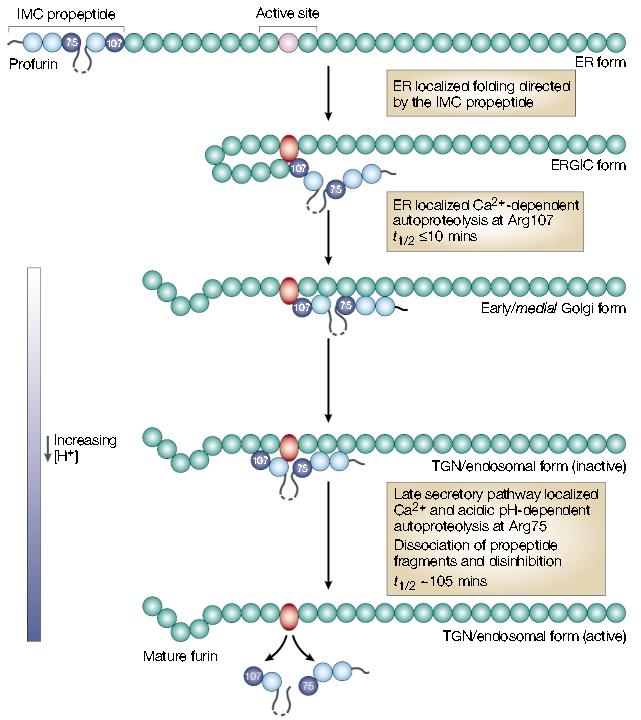
The furin autoactivation pathway. Following translocation and signal sequence removal, the furin prodomain acts as an intramolecular chaperone (IMC) to facilitate folding of the unstructured, inactive catalytic domain (pink circle) into the active conformation (red oval). After the initial endoplasmic reticulum (ER) folding events, furin undergoes autoproteolytic intramolecular excision of the propeptide at Arg107. The propeptide, however, remains associated with the mature domain and functions as a potent autoinhibitor in trans during transport to the late secretory pathway. Propeptide excision can be blocked by inactivating furin and results in the accumulation of an apparent folding intermediate in the ER–Golgi intermediate compartment (ERGIC)/_cis_-Golgi network. These data indicate that both the ER and ERGIC compartments participate in the initial steps of furin activation. Following propeptide excision, the inactive propeptide complex transits to late secretory compartments — _trans_-Golgi network (TGN)/endosomes — where the relatively acidic pH promotes autoproteolytic, intramolecular cleavage of the propeptide at a second, internal site (Arg75). The Arg75 cleavage is followed by the rapid dissociation of the propeptide fragments and disinhibition of furin. Adapted with permission from ref. 15. © (2002) American Society for Biochemistry and Molecular Biology.
SUBTILISIN SUPERFAMILY.
The serine proteases are composed of the subtilisin and chymotrypsin (including trypsin, thrombin and elastase) superfamilies. Surprisingly, the two superfamilies are evolutionarily distinct, yet the atoms that form the catalytic centre are in nearly identical positions — a remarkable example of convergent evolution.
CATALYTIC TRIAD.
In serine endoproteases, the catalytic triad comprises the three spatially-optimized active-site residues — serine, histidine and aspartate — that conspire to bring about peptide-bond hydrolysis.
BACTERIAL THERMITASE.
A thermostable bacterial subtilisin with a solved crystal structure.
P1 AND P4.
A broadly used nomenclature that identifies the amino-acid residues that flank the cleavage site (scissile bond) in protein and peptide substrates. The amino acid that is amino-terminal to the scissile bond is designated P1. P2 is the amino acid that is amino-terminal to P1. So, P4 is the amino acid that is four residues to the amino-terminal side of the scissile bond. The amino acids that are carboxy-terminal to the scissile bond are labelled P1′, P2′, and so on.
α1-ANTITRYPSIN.
The circulating protein inhibitor of neutrophil elastase. It inhibits serine proteases by acting as a slow tight-binding inhibitor or suicide substrate. Inhibitors that use this type of mechanism are called serpins. The reactive site, which lures the protease, can be engineered to target specific proteases.
_K_i.
The inhibitor constant that describes the dissociation of an enzyme (E)–inhibitor (I) complex, _K_i = [E][I]/[EI].
JUXTACRINE VERSUS PARACRINE.
Juxtacrine is communication between adjacent cells, whereas paracrine is communication between cells that are further apart.
The furin activation pathway shows evolutionary conservation with the activation pathways of bacterial subtilisin and α-lytic proteases that use their propeptide excision sites to guide the folding of the catalytic centre, which results in a structural reorganization of the folded pro-enzyme16-19. Furin's ‘measure once, cut twice’ method also seems to be a biochemical template that is used by members of the transforming growth factor-β (TGF-β) family to control juxtacrine versus paracrine signalling, and by paramyxoviruses to produce fusion-competent envelope glycoproteins.
Furin localization and trafficking
The release of furin's propeptide fragments unmasks its endoprotease activity and enables it to cleave substrates in trans. Furin localizes to the TGN — a late Golgi structure that is responsible for sorting secretory pathway proteins to their final destinations, including the cell surface, endosomes, lysosomes and secretory granules20,21. From the TGN, furin follows a highly regulated trafficking itinerary through several TGN/endosomal compartments and the cell surface10,22 (FIG. 4). This itinerary explains, in part, the ability of furin to process a diverse collection of proprotein substrates in vivo. Moreover, analysis of furin trafficking has uncovered novel roles for protein phosphorylation, the actin cytoskeleton and sorting adaptors in the regulation of protein traffic that controls cellular homeostasis and disease.
Figure 4.

Model of furin trafficking. Budding of furin from the _trans_-Golgi network (TGN) is mediated by the binding of the tyrosine-based or di-leucine-like hydrophobic sorting motifs to adaptor protein (AP)-1, which targets furin to endosomes, or binding to AP-4, which targets furin to the basolateral surface from either the TGN or possibly endosomes. In endocrine and neuroendocrine cells, AP-1 directs furin budding from the TGN into immature secretory granules (ISGs). Phosphofurin acidic cluster sorting protein-1 (PACS-1) connects the casein kinase 2 (CK2)-phosphorylated furin acidic cluster to AP-1/clathrin to retrieve furin to the TGN either from ISGs, prior to their progression to mature secretory granules (MSGs), or from endosomes. Furin molecules arriving at the cell surface can be tethered by the cytoskeletal protein filamin, which is also called actin-binding protein (ABP)-280. The dynamin/clathrin-dependent internalization of cell-surface furin is mediated principally by the tyrosine-based motif, which binds to AP-2. Once inside early endosomes, furin molecules that are dephosphorylated by specific protein phosphatase 2A (PP2A) isoforms are delivered to the TGN, apparently through a late endosomal compartment. By contrast, CK2-phosphorylated furin is recycled back to the plasma membrane in a PACS-1-dependent step. Hence, the TGN- and peripheral-cycling loops are essentially mirror images of each other. On the basis of the similar itinery of carboxypeptidase D (CPD), movement of furin from a post-TGN endosomal compartment to the cell surface might also require PP2A (dashed lines). Sorting nexin-15 (SNX-15), a PX-domain containing protein that binds phosphoinositides, modulates furin sorting through endosomes.
Localization to the trans**-Golgi network**
As with many itinerant type-I membrane proteins, both the TGN localization of furin and its dynamic cycling are controlled by sequences in its 56-amino-acid cytoplasmic domain (FIG. 5). Localization of furin to the TGN requires a bipartite motif that is composed of the casein kinase 2 (CK2)-phosphorylated acidic cluster (EECPpSDpSEEDE, where p highlights the serine residues that are phosphorylated) and a membrane-proximal segment containing two hydrophobic motifs (YKGL and LI)23-27 (FIG. 5). The bipartite motif controls two stages of a local cycling loop — the membrane-proximal segment is necessary for the efficient budding of furin from the TGN to endosomes, whereas the phosphorylated acidic cluster directs the efficient retrieval of endosomal furin to the TGN28.
Figure 5.

The sorting motifs of the furin cytoplasmic domain. Shown is the sequence of the human furin cytoplasmic domain. The various intracellular sorting motifs are indicated. See text for more details. ABP, actin-binding protein; AP, adaptor protein; CK2, casein kinase 2; PACS-1, phosphofurin acidic cluster sorting protein-1; PP2A, protein phosphatase 2A; TGN, _trans_-Golgi network. For a key to furin domain organization, see FIG. 1.
The steady-state localization of furin to the TGN has led to the supposition that this endoprotease cleaves proprotein substrates in this compartment, and several studies support this model29,30. However, recent studies indicate that the processing compartments might be formed by the fusion of endocytic furin-containing compartments with export vesicles that contain substrate proteins31,32. What, then, might be the role of the TGN in furin localization? Perhaps, in addition to housing the processing of some substrates, this compartment might also serve as a strategically located reservoir of furin molecules that are not active in proprotein processing. Such a mechanism would help to explain the control of furin processing in vivo.
Basolateral sorting
In polarized cells, furin targeting to the basolateral surface is similarly controlled by a bipartite signal that is composed of the carboxy-terminal part of the acidic cluster — EEDE — and the FI motif33 (FIG. 5). The FI motif seems to bind to the sorting adaptor protein (AP)-4, and AP-4 is required for the basolateral sorting of furin34 (FIG. 4). This role of the ubiquitously expressed AP-4 explains the lack of a function for the epithelial-specific basolateral sorting adaptor AP-1B in furin trafficking35,36.
Budding from the trans**-Golgi network**
Little is known about the cytoplasmic machinery that directs TGN budding of furin. Earlier studies showed that furin localizes to clathrin-coated regions of the TGN, which indicates a role for the sorting adaptor protein AP-1.Indeed,both the LI and YKGL motifs bind to the μ1a chain of AP-1 (ref. 37) (figs 4, 5). The recent identification of the Golgi-localized, γ-ear-containing, ADP-ribosylation-factor-binding (GGA) proteins, which control budding of the mannose-6-phosphate receptor from the TGN, raises the possibility of a second pathway through which furin could exit the TGN. Consistent with such a possibility, the furin LI motif is contained within a GGA3 consensus binding sequence (DXXLI)38-40 (FIG. 5).
CIS VERSUS TRANS CLEAVAGE.
cis cleavage refers to the autoproteolytic intramolecular cleavage of an endoprotease propeptide by the catalytic triad that is part of the same molecule. trans cleavage refers to the processing of other molecules.
ANTEROGRADE AND RETROGRADE.
Anterograde refers to trafficking from the endoplasmic reticulum (ER) towards the plasma membrane, whereas retrograde refers to trafficking from the plasma membrane towards the ER.
Retrieval from endosomes
Unlike the budding of furin from the TGN, the retrieval of furin from endosomes to the TGN is much better understood. The CK2-phosphorylated furin acidic cluster binds to the sorting protein PACS-1 (phosphofurin acidic cluster sorting protein-1) — a sorting connector that links furin to the AP-1 clathrin adaptor and transports furin from endosomes to the TGN28,41 (FIG. 4). The fact that binding of AP-1 to PACS-1 is crucial for endosome-to-TGN sorting concurs with the discovery that mutation of the AP-1 binding site in PACS-1 or genetic deletion of AP-1 both cause a similar mislocalization of furin to endosomal compartments36,41,42. PACS-1 is not exclusively dedicated to furin; it controls the endosome-to-TGN sorting of several membrane proteins that contain acidic cluster-sorting motifs. These include cellular proteins, such as the cation-independent mannose-6-phosphate receptor (CI-MPR), the furin homologue PC5/6B (FIG. 1), carboxypeptidase D (CPD), Sortilin, and several pathogen proteins, including HIV-1 Nef (for ‘negative factor’) and several herpes virus envelope glycoproteins, such as varicella zoster virus gE and human cytomegalovirus gB28,43-45 (L. Wan and G.T., unpublished observations). PACS-1 binding to HIV-1 Nef is required for immunoevasion through the downregulation of cell-surface major histocompatibility class-I (MHC-I) molecules, and PACS-1 binding to herpes virus envelope glycoproteins is involved in the production of infectious virus41,44 (C. M. Crump and G.T., unpublished observations). How AP-1 might contribute both to anterograde and retrograde transport between the TGN and the endosomes is not known, but reports showing that AP-1 links the CI-MPR to an anterograde kinesin — KIF13A— for delivery to the cell surface from the TGN indicate that sorting proteins might combine to provide transport directionality46.
Endocytosis
Many sorting motifs that localize furin to the TGN also direct its endocytic sorting itinerary. Endocytosis of furin is directed principally by the YKGL motif, which binds to the μ2 subunit of the AP-2 adaptor37. Recruitment of cell-surface furin molecules into endocytic compartments is regulated by tethering through its VY motif to filamin, a subcortical actin-binding protein that is involved in cell locomotion and signalling47,48 (figs 4, 5; G. Liu and G.T., unpublished observations). In addition, sorting nexin-15 (SNX-15) affects furin internalization (FIG. 4), and overexpression of this sorting protein impedes the internalization of furin-containing chimaeras49.
In early endosomes, furin can either be recycled to the cell surface or trafficked to the TGN. Recycling to the cell surface requires CK2 phosphorylation of the furin acidic cluster and PACS-1, whereas transport to the TGN requires dephosphorylation of the furin acidic cluster by specific isoforms of protein phosphatase 2A (PP2A)50, which apparently occurs before transit through a late endosome intermediate51 or through sorting/recycling endosomes (FIG. 4).
So, PACS-1 and CK2 seem to place furin in one of two local cycling loops — one at the TGN and one between the plasma membrane and early endosomes. Sorting between these two loops requires dephosphorylation by PP2A. The sorting of CPD is similarly controlled by the phosphorylation state of its acidic cluster. Moreover, PP2A binds directly to the CPD cytoplasmic domain and this binding is essential for the control of CPD transport between the TGN and the cell surface52. The highly coordinated sorting itineraries of furin and CPD correlate with their sequential roles in the processing of proprotein substrates in vivo.
The regulated secretory pathway
The study of furin trafficking in endocrine and neuroendocrine cells has challenged the long-held view regarding the separation of the regulated and constitutive pathways. In these cell types, furin buds into nascent immature secretory granules (ISGs), together with hormones and other molecules that are destined for dense core mature secretory granules (MSGs) (FIG. 4). ISGs are short-lived AP-1/clathrin-coated compartments that undergo homotypic fusions and extensive membrane remodelling during micro-tubule-based transport to the cell periphery53-55. At the cell periphery, furin is removed from ISGs, apparently during the brefeldin A (BFA)-sensitive, ADP ribosylation factor-1 (ARF1)-dependent remodelling of the ISG membrane56,57, and is returned to the TGN55,58. BFA blockage of ISG remodelling is likely to be due to inhibition of the ARF1-mediated recruitment of AP-1/clathrin and the subsequent membrane budding. Removal of furin from the ISGs requires a CK2-phosphorylated acidic cluster and PACS-1 (refs 41,58). Similar results have been reported for the retrieval of CPD and the vesicular monoamine transporter-2, which highlights important roles for CK2 and PACS-1 in granule maturation59-61. Several furin substrates are sorted to the regulated pathway 62-65, which indicates that furin and CPD have crucial roles in proprotein processing in ISGs.
REGULATED VERSUS CONSTITUTIVE PATHWAYS.
The regulated pathway in endocrine and neuroendocrine cells constitutes the anterograde pathway, leading from the _trans-_Golgi network (TGN) to the peptide-hormone-containing, mature secretory granules that can be stimulated to exocytose following an influx of extracellular calcium. Secretion by the constitutive pathway is not stimulated by calcium.
NEUROTROPHINS.
A class of molecules that control various aspects of neuronal survival and plasticity, and that include nerve growth factor, brain-derived neurotrophic factor, neurotrophin-3 and neurotrophin-4/5. The neurotrophins are synthesized as proproteins that require cleavage at consensus furin sites.
Furin in development, homeostasis and disease
The insights gained by the analysis of furin trafficking are equalled by those obtained from recent studies that indicate that furin has broad and important roles in embryogenesis, homeostasis and disease. Paradoxically, although furin is an enzyme that is essential for embryogenesis, its activity can also lead to fatal diseases in adults. Owing to space limitations, I will limit this discussion primarily to work reported during the past two years.
To cleave or not to cleave — neuronal innervation and dementia
The 16-kDa β-nerve growth factor (β-NGF) is the prototypic target-derived neurotrophin, and biochemical studies show that furin is the principal endoprotease that cleaves pro-β-NGF10,22 (FIG. 6a). Surprisingly, the furin-catalysed processing of pro-β-NGF controls whether the neurotrophin activates cell-survival or cell-death pathways within innervating neurons66. Processed β-NGF mediates cell survival through high-affinity binding to the Trk proto-oncogene receptor tyrosine kinases, which mediate the trophic effects of β-NGF and other neurotrophins. Conversely, secreted, unprocessed pro-β-NGF mediates apoptosis by high-affinity binding to the 75-kDa neurotrophin receptor (p75NTR). This receptor is a member of the tumour necrosis factor (TNF) receptor/FAS family that antagonizes the trophic signalling mediated by β-NGF and Trk receptors. Regulation of furin activity might therefore have a central role in determining which neurons form synaptic complexes and which neurons die (FIG. 6a).
Figure 6.
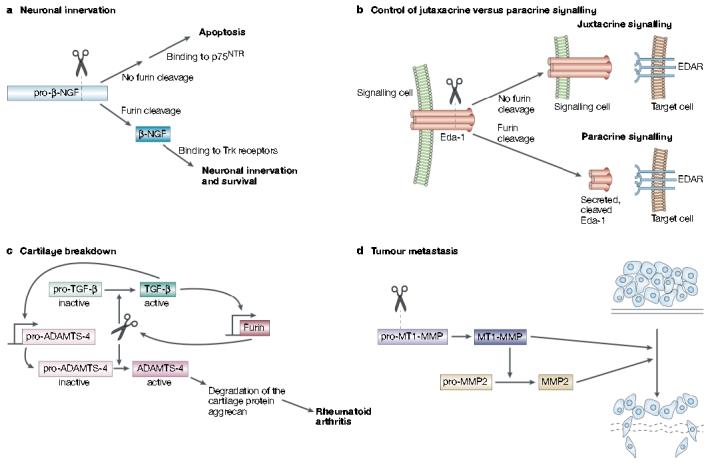
Furin in development, homeostasis and disease. a | Furin-mediated cleavage of pro-β-nerve growth factor (NGF) produces the 13-kDa β-NGF neurotrophin that binds to Trk receptors to promote synaptic innervation. By contrast, inhibition or sequestering of furin results in the secretion of pro-β-NGF that binds to the 75-kDa neurotrophin receptor (p75NTR) to promote cell-death pathways. b | Ectodysplasin-A (Eda-1) is a trimeric tumour necrosis factor family member that stimulates morphogenesis of ectodermal structures by activation of its receptor, EDAR, on target cells. Eda-1 can signal in a juxtacrine manner by binding to EDAR on adjacent cells. However, cleavage of membrane anchored Eda-1 by furin releases the ligand and enables it to signal through EDAR on distant cells in a paracrine manner. c | In synoviocytes, furin and transforming growth factor (TGF)-β participate in a positive feedback loop that results in elevated levels of ‘a disintegrin and metalloprotease with thrombospondin motifs-4’ (ADAMTS-4, or aggrecanase-1). Furin cleaves both pro-TGF-β and pro-ADAMTS-4 to yield the active growth factor and protease, respectively. The secreted mature form of TGF-β then binds to its receptor and, through a SMAD2 and mitogen-activated protein kinase (MAPK) convergent pathway, increases furin expression. The increased levels of furin lead to an increase in TGF-β, which creates a positive-feedback loop. In synoviocytes, TGF-β also stimulates the expression of pro-ADAMTS-4. So, because furin and TGF-β are in this positive loop, the levels of active ADAMTS-4 are greatly elevated, which leads to destruction of the cartilage protein aggrecan and hence to rheumatoid arthritis. d | Furin activates several membrane-type matrix metalloproteinases (MT-MMPs) that are involved in tumour formation and metastasis. Furin-activated MT-MMP1 activates MMP2 (gelatinase), which degrades the extracellular matrix, and MT-MMP1 also directly degrades extracellular matrix itself.
This control of furin activity might well extend to additional developmental programmes. For example, furin cleavage of the transmembrane receptor Notch is required for the release of the Notch intracellular domain by γ-secretase proteolysis. This intracellular domain then binds to the transcriptional regulator CSL (for ‘C promoter binding factor/Suppressor of Hairless/Lag-1’), which activates genes required for cell–cell communication during development67. By contrast, uncleaved Notch mediates a distinct signalling pathway that inhibits cell differentiation68. How furin activity is controlled to regulate the processing of these substrates is unknown.
Furin's role in the α-, β- and γ-secretase-mediated processing of the β-amyloid precursor protein (APP) helps to determine whether APP-derived peptides enhance NGF signalling to innervating neurons or cause the massive neurodegeneration that is associated with Alzheimer's disease69,70. The extracellular domain of APP is cleaved by α-secretase to produce soluble APPs that enhance the anti-apoptotic and neuroprotective activities of NGF71,72. By contrast, cleavage of APP by a combination of the β- and γ-secretases releases the amyloidogenic βAPP1–40, βAPP1–42 and related peptides, which form amyloid plaques and are responsible for the neurodegeneration that is suffered by individuals with Alzheimer's disease73.
AMYLOID DEMENTIA.
Neurological abnormality caused by deposits of proteinaceous matter.
ENAMEL KNOT.
Signalling centre in the dental epithelium that controls tooth morphogenesis.
Recent studies point to an essential role for furin in the activation of both α- and β-secretase. Two members of the ADAMs (for ‘a disintegrin and metalloproteinase-like’) family of zinc metalloproteinases — ADAM10 and ADAM17 — have been implicated as the α-secretase74,75. Characteristic of this protein family, both ADAM10 and ADAM17 contain propeptides that are linked to their catalytic domains by a consensus furin motif, and cleavage at this site is required for their activation76,77. Interestingly, PC7 might activate the α-secretase under basal conditions, whereas furin might activate it following protein kinase C activation, which increases the α-secretase-catalysed release of soluble APP77.The β-secretase, also called β-site APP-cleaving enzyme (BACE), is a type-I membrane protein that localizes to the TGN/endosomal system and requires proteolytic removal of its proregion by furin at a minimal furin site (–Arg–Leu–Pro–Arg–↓)78-80. The similarities in BACE and furin trafficking further support the idea that furin is the BACE-activating enzyme81,82.
Alzheimer's is only one type of amyloid dementia in which furin has a crucial role. Two separate mutations in the BRI gene that encodes a widely expressed type-II membrane protein cause either familial British dementia (FBD) or familial Danish dementia (FDD). In healthy individuals, cleavage of this protein by furin, or possibly PC7, at an atypical furin site, which contains a lysine at P6 (–Lys–Gly–Ile–Gln–Lys–Arg–↓ (where Gln is glutamine)), releases a 23-residue carboxy-terminal peptide with an unidentified function83,84. However, the nucleotide transversion or decamer duplication in FBD and FDD, respectively, causes aberrant 34-residue amyloidogenic peptides to be produced on furin cleavage83-85.
Recent studies also show a role for furin in both Finnish- and Danish-type familial amyloidoses. Both diseases are caused by mutations that disrupt the binding of calcium to plasma gelsolin, which is a circulating scavenger of extracellular actin31,86,87. The disrupted calcium binding causes aberrant cleavage by furin at the carboxy-terminal side of a cryptic –Arg–Val–Val–Arg–↓ site, which is normally buried in the core of the wild-type molecule. The furin-mediated cleavage initiates the release of a 70-amino-acid amyloidogenic peptide87.
Furin, the TNFs, and the TGF-βs — short- versus long-range signalling in development and disease
The proteolytic release of TNF-α from the plasma membrane by ADAM17 has long been regarded as a key mechanism that mediates juxtacrine- versus paracrine-signalling of this cytokine family88. Recently, however, furin has been shown to control the signalling range of another TNF family member — ectodysplasin-A (Eda-1; FIG. 6b). Eda-1 is a type II plasma membrane protein that controls the formation of several epithelial tissues, including hair, teeth and eccrine sweat glands. The earliest expression of Eda-1 and its receptor — EDAR — is in the partially overlapping regions of the thickened dental epithelium89. During proliferation of the epithelium into the underlying mesenchyme to form the tooth bud, EDAR expression accompanies the leading edge of the epithelial layer and is ultimately confined to the enamel knot, whereas Eda-1 remains several cell distances away in the outer epithelium. A furin-mediated switch from juxtacrine to paracrine signalling might accompany the spatial uncoupling of the receptor and ligand (FIG. 6b). Mutations in the furin cleavage site of Eda-1 account for ∼20% of all know mutations in X-linked hydrohidrotic ectodermal dysplasia, and block the ability of Eda-1 to signal in a paracrine fashion90,91.
The importance of furin for the signalling of two other TNF family members — B-cell activating factor (BAFF) and a proliferation-inducing ligand (APRIL) — indicates a broad role for furin in controlling TNF function92-94.
Furin's role in activating members of the TNF family is surpassed by its role in controlling TGF-β-family signalling. Inactivation of the furin gene in mice creates an embryonic lethal phenotype, with death occurring at an early embryonic stage95. Furin is required both in the extra-embryonic tissues and in the cardiogenic mesoderm to promote yolk sac vasculogenesis and ventral closure, heart-looping and axial rotation. The failure to maintain asymmetry in the embryo is likely to arise from a block in the furin-catalysed production of the TGF-β family members Nodal and Lefty-2 (ref. 96). Consistent with this model, furin cleaves several TGF-β members, including TGF- β1 and bone morphogenetic protein-4 (BMP-4)97,98. Moreover, disrupting pro-BMP-4 maturation in four-cell Xenopus laevis embryos results in a dorsalized phenotype that mimics the phenotype that is observed when the BMP-4 signalling pathway is disrupted98.
MORPHOGENS.
Secreted signalling molecules that govern developmental patterns and axis formation by producing a concentration gradient emanating from the cells in which they are synthesized.
GLIOBLASTOMAS.
Aggressive malignant brain tumours that are derived from astrocytes and that account for ∼30% of all primary brain tumours.
Furin's autoactivation method also seems to be used by BMP-4 to control its signalling strength and range during embryogenesis99. Furin first cleaves pro-BMP-4 at the consensus furin site that joins the pro- and BMP-4 domains (–Arg–Ser–Lys–Arg↓–), followed by a second cleavage at a minimal consensus furin site within the propeptide (–Arg–Ile–Ser–Arg↓–). The context of the two sites ensures the ordered processing of pro-BMP-4 and the correct activity of this morphogen. The presence of consensus and minimal furin sites in other BMP-4-related signalling molecules10,22,99 indicates that the ‘measure once, cut twice’ method is used to control signalling gradients in many organisms. Moreover, this method might extend to viral pathogenesis, in which generation of the correctly folded, respiratory-syncytical-virus (RSV) fusion protein requires sequential cleavage at two furin sites to produce infectious progeny100,101.
Although furin-catalysed TGF-β activation is essential for embryogenesis, this pathway causes disease in adults. Furin and TGF-β cooperate in a novel positive feedback loop that exacerbates rheumatoid arthritis (FIG. 6c). TGF-β can bind to its own receptor to stimulate furin gene transcription by a SMAD2 and mitogen-activated protein kinase (MAPK) convergent pathway102-104. In synoviocytes, which are fibroblast- and macrophage-like cells that line the synovium of joints, the amplified levels of furin and TGF-β combine to increase the levels of ADAMTS-4 (a disintegrin and metalloprotease with thrombospondin motifs-4). ADAMTS-4 (previously identified as aggrecanase-1) is a member of a new family of ADAMs proteases, and it degrades the cartilage protein aggrecan and causes rheumatoid arthritis105,106 (FIG. 6c).
Furin and tumour metastasis
Furin is upregulated in several cancers, including non-small-cell lung carcinomas, squamous-cell carcinomas of the head and neck, and glioblastomas107. Moreover, the increased levels of furin in tumours correlate both with the increased aggressiveness of head, neck and lung cancers and with an increase in the levels of one of its substrates — membrane type 1-matrix metalloproteinase (MT1-MMP)108,109. MT1-MMP activates extracellular pro-MMP2 (pro-gelatinase) to induce rapid tumour growth and neovascularization110 (FIG. 6d). Activation of MMPs classically uses a cysteine-switch mechanism, in which the catalytic-site zinc atom that is bound to a cysteine residue in the pro-region of the latent pro-enzyme switches to binding a water molecule in the active protease. However, activation of MT1-MMP and related family members seems decidedly more complex and requires furin-mediated cleavage of their pro-region111,112. Furin inhibitors, including α1-PDX, block the activation of MT1-MMP in head, neck and oral squamous-cell carcinomas, which leads to a block in both MMP2 activation and tumour metastasis in transplanted mice109,113. The fact that the MT1-MMP/MMP2 axis is essential for alveolization of the embryonic lung114 provides another example of a furin-activated cascade that is essential in embryogenesis but that is detrimental in adults.
A second furin substrate, insulin-like growth factor-1 (IGF1), is upregulated in colon, breast, prostate and lung cancers. Its receptor, IGF1R, which is also a furin substrate, is upregulated on the surface of the tumour cells115. IGF1 and IGF1R processing are catalysed by furin or PC5/6A, and inhibition of this processing by α1-PDX reduces the incidence, size and vascularization of tumour development in transplanted mice116.
NEOVASCULARIZATION.
De novo stimulation of new blood supplies to a growing tumour.
ALVEOLIZATION.
Encompasses the latter stages of lung development, which begin with bronchial and respiratory-tree development, and culminate in the formation of terminal saccules and alveoli to facilitate efficient gas exchange.
Furin is not the only PC that is associated with a poor prognosis for many cancers. The furin homologue, PACE4 (FIG. 1; TABLE 1), is upregulated in breast tumours117, and expression of this PC increases the invasiveness of mouse squamous-cell carcinomas by converting them to more aggressive, poorly differentiated, spindle-cell carcinomas118. Together, these studies indicate that inhibiting PCs might be a novel approach to combating various aggressive cancers.
Anthrax, AIDS, Ebola> — what next?
Early studies showing furin's role in both anthrax toxin activation and avian influenza virus HA maturation merely provided a glimpse into the devastating role of furin in the activation of various bacterial and viral pathogens. The analysis of bacterial toxin activation has further illuminated distinct roles for furin-catalysed proprotein processing at the cell surface or early endosomes, providing a single perspective for unravelling the regulation of protein trafficking in mammalian cells. Cell-surface furin activates the anthrax toxin6,119 — a now infamous weapon of bioterrorism120 (BOX 2) — as well as the aerolysin toxin, which is a causative agent in many food-borne illnesses121, and Clostridium septicum α-toxin, which causes gas gangrene122 (FIG. 2). Cleavage of each toxin by furin is an obligatory step in making the toxin able to form pores in cell membranes.
The anthrax toxin comprises three proteins: PA, protective antigen, so-called for its ability to educe immune protection against anthrax; and two toxic proteins — lethal factor (LF) or oedema factor (EF)123. LF is a metalloproteinase that cleaves MAPK kinases, whereas EF is a calmodulin-dependent adenylate cyclase123. The 83-kDa PA molecule that is secreted from the bacterium binds to the anthrax toxin receptor (ATR)124, and is then cleaved by cell-surface furin to generate a cell-associated 63-kDa PA and a free 20-kDa PA (FIG. 7). The cell-associated PA molecule heptamerizes, binds to either of the two toxic factors, and is then internalized into early endosomes125. In the early endosomal acid pH environment, the PA heptamer forms a membrane channel that shuttles the toxic factors into the host-cell cytoplasm, which results in oedema, systemic shock and death (FIG. 7). In the absence of furin, the toxin fails to assemble and is not lethal126. Moreover, mutation of the furin cleavage site results in a dominant-negative protein that binds to ATR but fails to oligomerize127. Both proaerolysin and Clostridium septicum α-toxin bind to glycosylphosphatidylinositol-anchored molecules, and, similar to PA, furin cleavage of both molecules is required for them to form ion-permeable heptameric pores in the host-cell plasma membrane, which leads to cell toxicity128,129.
Figure 7.

Furin activation of the anthrax toxin. Cleavage of anthrax protective antigen (PA) by furin leads to internalization and activation of lethal factor (LF), which is a zinc metalloproteinase that cleaves mitogen-activated protein kinase (MAPK) kinases, and oedema factor (EF), which is a calmodulin-dependent adenylate cyclase. EF causes eschar formation, which are black masses of scab-like necrotized tissue, and massive soft-tissue oedema123. In lung macrophages, phagocytosed LF leads to changes in the levels of tumour necrosis factor-α and interleukin-1β, as well as apoptosis. The altered cytokine levels coupled with the paralysed innate immune system rapidly result in anthrax-induced systemic shock and death by a complex, unresolved process. See text for more details. Modified with permission from REF. 163. © (2002) Birkhäuser Publishing Ltd.
Early endosomal furin activates other bacterial toxins, including Pseudomonas exotoxin A (PEA), shiga toxin (ST), shiga-like toxin-1 (ST-1) and diphtheria (DT) toxins10 (FIG. 2). Unlike the cell-surface-activated toxins, these toxins are all A/B-type toxins that contain an active domain (A) and a binding domain (B) that are joined by a furin cleavage site129. Following receptor binding, each toxin is endocytosed into early endosomes, where it is cleaved by furin. Cleavage of ST, ST-1 and PEA by furin requires the acidic pH that is characteristic of early endosomal compartments, whereas cleavage of DT does not10. As in the cancer models discussed, inhibition of furin activity by the extracellular delivery of α1-PDX protects cells from PEA and other bacterial toxins13 (F. Jean and G.T., unpublished observations). The sensitivity of cells to PEA is increased in the absence of filamin (FIG. 4), which normally tethers furin to the cell surface, indicating that filamin might control the formation of endosomal furin-processing compartments47. The crystal structure of PEA shows that exposure of the molecule to an acidic pH unmasks the furin cleavage site, which at least partially explains the requirement for acidic pH-dependent furin processing130.
Surprisingly, furin cleavage enables PEA, ST/ST-1 and DT to translocate to the cytosol through three distinct trafficking pathways. Following cleavage, the DT B domain forms a channel in the early endosomal membrane that shuttles the A fragment into the host-cell cytosol131. By contrast, cytosol delivery of both PEA and ST/ST-1 requires retrograde trafficking to the ER, where the toxins are apparently translocated to the cytosol through the sec61 channel132,133. The retrograde trafficking of PEA requires the kdel receptor, which binds to the processed PEA and retrieves the toxin to the ER132,133,whereas cleaved ST/ST-1 traffics to the ER through a pathway that is both KDEL-receptor- and copi-independent, which indicates that vesicle coats other than COPI direct their retrieval to the ER. However, retrieval of ST/ST-1 is dependent on RAB6, a small GTPase that controls intra-Golgi transport and the cycling of Golgi-resident glycosyltransferases through the ER132-134. As it has also been suggested that furin might be sorted to the ER to metabolize misfolded insulin receptors, it will be important to determine whether furin is also sorted through the ST/ST-1 pathway135.
SEC61 CHANNEL.
The main protein that forms the pore in the endoplasmic reticulum (ER) membrane that facilitates the translocation of nascently synthesized proteins into the secretory pathway. The SEC61 channel might also be a conduit for the reverse translocation (dislocation) of proteins from the ER into the cytoplasm.
KDEL RECEPTOR.
Golgi-localized membrane protein that binds to carboxy-terminal KDEL (–Lys–Asp–Glu–Leu–) motifs, which are present on many resident endoplasmic reticulum (ER) proteins that escape to the Golgi, and that retrieves them to the ER.
COPI.
(Coatomer protein complex I). A specific type of coat on vesicles that traffic principally between Golgi cisternae and from the Golgi to the endoplasmic reticulum. Also reported on early endosomes.
VIRULENCE.
The extent or degree to which a pathogen can cause disease.
The broad role of furin in activating bacterial toxins is exceeded by its role in activating numerous pathogenic viruses. Many pathogenic viruses, including avian influenza virus, HIV-1, measles virus and RSV, express envelope glycoproteins that must be cleaved at consensus furin sites to form the mature and fusogenic envelope glycoprotein10,22. For example, processing of HIV-1 gp160 unveils the amino-terminal gp41 fusogenic peptide that is contained within the trimeric gp120/g41 envelope complex. Whether furin or one of the other PCs (for example, PACE4, PC5/6B or PC7) is the in vivo gp160 convertase is unknown136. Lovo cells, which lack furin, process HIV-1 gp160 (ref. 137). Nonetheless, furin inhibitors block processing of HIV-1 gp160 and, in turn, the production of infectious HIV-1, as well as blocking other viruses that require processing of their envelope glycoproteins at consensus furin sites10. Furin cleaves HIV-1 gp160 at the carboxy-terminal side of the consensus sequence –Arg–Glu–Lys–Arg–↓. The P3 glutamate in this cleavage site reduces the efficiency of furin processing, and the conservation of this residue in several HIV isolates has raised doubts about furin's involvement in gp160 processing136. Indeed, mutation of this cleavage site to make a site containing all basic amino acids (–Arg–Arg–Lys–Arg–↓) enhances processing by furin138. Surprisingly, however, recombinant HIV containing this all-basic site is attenuated, which indicates a selective advantage for the inefficiently cleaved –Arg–Glu–Lys–Arg–↓ site in HIV-1.
HIV-1 seems to maintain a selective growth advantage by using a suboptimal furin site in its envelope glycoprotein, whereas the analysis of viral tropism — that is, the molecular determinants that enable a virus to spread throughout the body — shows that the virulence of many deadly viruses (including avian influenza virus, Newcastle Diseases virus and, potentially, Ebola virus) is directly correlated with the ability of these viruses to incorporate a consensus furin cleavage site within their envelope proteins139-142. For example, the pathogenicity of avian influenza viruses has long been recognized to correlate directly with the cleavability of its fusion protein precursor HA0, which is cut by furin to generate the fusion-competent HA1–HA2 complex. Similar to HIV-1 gp160, cleavage of HA0 exposes the fusogenic peptide located at the amino terminus of HA2, which can fuse with target-cell membranes.
Avirulent avian influenza viruses, which lack a consensus furin site in HA0, cause a localized infection in the intestinal tract. However, mutation of the HA0 cleavage site to a consensus furin site enables the virus to be activated by the ubiquitously expressed furin, invariably enabling the virus to spread systemically throughout the bird, including infection of the central nervous system139. This ability relates to the deadly flu outbreak in Hong Kong in 1997 (BOX 2). Analysis of the H5N1 influenza virus, which killed at least six people, showed that just two mutations were required to generate the lethal virus — a mutation in a subunit of the viral RNA polymerase PB2, together with the generation of a tandem furin site in the cleavage junction between HA1 and HA2 (–Arg–Glu–Arg–Arg–Arg–Lys–Lys–Arg–↓)143. Exactly how the tandem furin site and the PB2 mutation contribute to the increased virulence of influenza H5N1 is unknown. Fortunately, the attenuated infectivity of this virus, which is also poorly understood, impeded its spread through the population. Nonetheless, the propensity for the rapid mutation and reassortment rate in avian influenza and its documented ability to jump directly from birds to humans underscore our vulnerability to this pathogen144.
The importance of furin for pathogen virulence extends to other viruses, including Ebola virus. For example, the highly pathogenic Ebola Zaire and Ivory Coast strains — which cause a massive and sudden (fulminant) haemorrhagic fever that is characterized by massive internal and external bleeding and that kills 90% of the people who contract it — contain a consensus furin site in their envelope glycoprotein (GP)145. But, by contrast, GP of the relatively milder Ebola Reston strain lacks a consensus furin site. This isolate is not pathogenic to humans141. Surprisingly, however, despite the apparent underlying structural similarities between HIV-1 gp160, influenza virus HA and Ebola virus GP, furin-catalysed cleavage of GP is not required for membrane fusion in cell-culture models146. The lack of a requirement for GP processing for membrane fusion is consistent with the presence of an internal fusion sequence in Ebola GP and related flaviviruses147. What, then, might account for the furin-dependent tropism of Ebola virus? One clue lies in the severe cytotoxicity and marked increase in vascular permeability of GP from the highly pathogenic Ebola Zaire, but not from the apathogenic Reston isolate148,149. Interestingly, the crucial region of GP required for this toxicity is adjacent to the furin cleavage site, indicating that proteolysis might unveil the cytotoxic domain.
INFECTIVITY.
The ability of a pathogen to invade a host and replicate, irrespective of its ability to cause disease.
Conclusions and perspectives
In summary, the proteolytic reaction catalysed by furin, which once seemed so ordinary, now clearly has enormous ramifications in biological research. Furin's ‘measure once, cut twice’ method of autoactivation has yielded new understanding of the interplay between protein folding and the distinct microenvironments of early and late secretory pathway compartments. Moreover, this activation method seems to extend beyond the PCs — it is used to control the formation of morphogen gradients during embryogenesis, as well as the correct folding of some viral envelope glycoproteins. The intracellular trafficking of furin is decidedly complex and has provided new insights into the regulation of protein traffic, including new roles for protein phosphorylation, the actin cytoskeleton and sorting adaptors. Surprisingly, the analysis of furin trafficking has also provided new insights into diverse processes ranging from secretory-granule formation to HIV immunoevasion.
Ongoing studies of furin should further clarify its activity in vivo, as well as its relationships with other PCs. Analyses of neuronal innervation, cell fate and juxtacrine- versus paracrine-signalling indicate that furin activity might be dynamically controlled, yet we do not know how. One possibility is that cellular furin inhibitors might be temporally expressed, or that the complex and diverse machinery that directs the highly regulated furin trafficking itinerary might control whether furin interacts with its various substrates. Furthermore, our models of furin processing must also account for the roles of the other broadly expressed PC family members that traffic similarly to furin — namely PC7, PC5/6B and PACE4. The extent of their roles in the furin pathway remains unclear.
Not only will the analysis of furin trafficking continue to lead to a better understanding of the basic processes that are involved both in controlling membrane traffic and in integrating these dynamic sorting steps with signal-transduction cascades, cell fate, other PCs and protease systems, but it will also clarify furin's role in a broad spectrum of human diseases. The finding that deadly bacterial and viral pathogens usurp the furin pathway to exert their virulence strongly argues for a strategy that targets furin for therapeutic intervention, even though such a strategy would have to consider that the very prevalence of furin might also create toxicity in the drugs that target it. Nevertheless, furin holds great promise as a key to solving both theoretical and practical questions in cell biology.
Acknowledgements
My apologies to colleagues whose work I did not cite because of space limitations. Special thanks go to A. Zhou, D. Steiner, members of my lab and collaborators for insightful discussions and review of the manuscript, and to R. Dresbeck for editing. G.T. is supported by grants from the National Institutes of Health.
Footnotes
Online links
DATABASES
The following terms in this article are linked online to:
InterPro: http://www.ebi.ac.uk/interpro/ P domain
LocusLink: http://www.ncbi.nlm.nih.gov/LocusLink/ AP-1 | AP-2 | AP-4 | BRI | CK2 | filamin | IGF1| MAPK | PP2A | protein kinase C | TGF-β | TNF
OMIM: http://www.ncbi.nlm.nih.gov/Omim/ Alzheimer's disease | familial British dementia | familial Danish dementia | rheumatoid arthritis
Swiss-Prot: http://www.expasy.ch/ ADAM10 | ADAM17 | ADAMTS-4 | APP | APRIL | ARF1 | ATR | BAFF | BMP-4 | CI-MPR | CPD | EDAR | EF | Furin | gelsolin | GGA3 | HA | IGF1R | KIF13A | Lefty-2 | LF | MMP2 | MT1-MMP | β-NGF | Nodal | PA | PACE4 | PACS-1 | PC5/6A | PC5/6B | PC7 | RAB6 | β-secretase | SMAD2 | SNX-15 | Sortilin | TGF-β1 | TNF-α
References
- 1.Seidah NG, Day R, Marcinkiewicz M, Chretien M. Precursor convertases: an evolutionary ancient, cell-specific, combinatorial mechanism yielding diverse bioactive peptides and proteins. Ann. N. Y. Acad. Sci. 1998;839:9–24. doi: 10.1111/j.1749-6632.1998.tb10727.x. [DOI] [PubMed] [Google Scholar]
- 2.Thacker C, Rose AM. A look at the Caenorhabditis elegans Kex2/Subtilisin-like proprotein convertase family. Bioessays. 2000;22:545–553. doi: 10.1002/(SICI)1521-1878(200006)22:6<545::AID-BIES7>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 3.Zhou A, Martin S, Lipkind G, LaMendola J, Steiner DF. Regulatory roles of the P domain of the subtilisin-like prohormone convertases. J. Biol. Chem. 1998;273:11107–11114. doi: 10.1074/jbc.273.18.11107. [DOI] [PubMed] [Google Scholar]
- 4.Siezen RJ, Creemers JW, Van de Ven WJ. Homology modelling of the catalytic domain of human furin. A model for the eukaryotic subtilisin-like proprotein convertases. Eur. J. Biochem. 1994;222:255–266. doi: 10.1111/j.1432-1033.1994.tb18864.x. [DOI] [PubMed] [Google Scholar]
- 5.Rockwell NC, Fuller RS. Specific modulation of Kex2/Furin family proteases by potassium. J. Biol. Chem. 2002;277:17531–17537. doi: 10.1074/jbc.M111909200. [DOI] [PubMed] [Google Scholar]
- 6.Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J. Biol. Chem. 1992;267:16396–16402. [PubMed] [Google Scholar]
- 7.Walker JA, et al. Sequence specificity of furin, a proprotein-processing endoprotease, for the hemagglutinin of a virulent avian influenza virus. J. Virol. 1994;68:1213–1218. doi: 10.1128/jvi.68.2.1213-1218.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krysan DJ, Rockwell NC, Fuller RS. Quantitative characterization of furin specificity. Energetics of substrate discrimination using an internally consistent set of hexapeptidyl methylcoumarinamides. J. Biol. Chem. 1999;274:23229–23234. doi: 10.1074/jbc.274.33.23229. [DOI] [PubMed] [Google Scholar]
- 9.Siezen RJ. Modelling and engineering of enzyme/substrate interactions in subtilisin-like enzymes of unknown 3-dimensional structure. Adv. Exp. Med. Biol. 1996;379:63–73. doi: 10.1007/978-1-4613-0319-0_8. [DOI] [PubMed] [Google Scholar]
- 10.Molloy SS, Thomas G. The Enzymes. Academic Press; San Diego, CA: 2001. pp. 199–235. [Google Scholar]
- 11.Komiyama T, Fuller RS. Engineered eglin c variants inhibit yeast and human proprotein processing proteases, Kex2 and furin. Biochemistry. 2000;39:15156–15165. doi: 10.1021/bi001907c. [DOI] [PubMed] [Google Scholar]
- 12.Cameron A, Appel J, Houghten RA, Lindberg I. Polyarginines are potent furin inhibitors. J. Biol. Chem. 2000;275:36741–36749. doi: 10.1074/jbc.M003848200. [DOI] [PubMed] [Google Scholar]
- 13.Jean F, et al. α1-Antitrypsin Portland, a bioengineered serpin highly selective for furin: application as an antipathogenic agent. Proc. Natl. Acad. Sci. U. S. A. 1998;95:7293–7298. doi: 10.1073/pnas.95.13.7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson ED, Thomas L, Hayflick JS, Thomas G. Inhibition of HIV-1 gp160-dependent membrane fusion by a furin-directed α1-antitrypsin variant. J. Biol. Chem. 1993;268:24887–24891. [PubMed] [Google Scholar]
- 15.Anderson ED, et al. The ordered and compartment-specific autoproteolytic removal of the furin intramolecular chaperone is required for enzyme activation. J. Biol. Chem. 2002;277:12879–12890. doi: 10.1074/jbc.M108740200. Illustrates furin's ’measure once, cut twice’ method of pro-enzyme activation and shows how the sequential autoproteolytic cleavages of the propeptide are controlled in a secretory pathway compartment-specific manner. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker D. Metastable states and folding free energy barriers. Nature Struct. Biol. 1998;5:1021–1024. doi: 10.1038/4130. [DOI] [PubMed] [Google Scholar]
- 17.Peters RJ, et al. Pro region C-terminus:protease active site interactions are critical in catalyzing the folding of α-lytic protease. Biochemistry. 1998;37:12058–12067. doi: 10.1021/bi980883v. [DOI] [PubMed] [Google Scholar]
- 18.Shinde U, Inouye M. Folding pathway mediated by an intramolecular chaperone: characterization of the structural changes in pro-subtilisin E coincident with autoprocessing. J. Mol. Biol. 1995;252:25–30. doi: 10.1006/jmbi.1995.0472. [DOI] [PubMed] [Google Scholar]
- 19.Yabuta Y, Takagi H, Inouye M, Shinde U. Folding pathway mediated by an intramolecular chaperone: propeptide release modulates activation precision of prosubtilisin. J. Biol. Chem. 2001;276:44427–44434. doi: 10.1074/jbc.M107573200. [DOI] [PubMed] [Google Scholar]
- 20.Griffiths G, Simons K. The trans Golgi network: sorting at the exit site of the Golgi complex. Science. 1986;234:438–443. doi: 10.1126/science.2945253. [DOI] [PubMed] [Google Scholar]
- 21.Gu F, Crump CM, Thomas G. Trans-Golgi network sorting. Cell Mol. Life Sci. 2001;58:1067–1084. doi: 10.1007/PL00000922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molloy SS, Anderson ED, Jean F, Thomas G. Bi-cycling the furin pathway: from TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 1999;9:28–35. doi: 10.1016/s0962-8924(98)01382-8. [DOI] [PubMed] [Google Scholar]
- 23.Molloy SS, Thomas L, VanSlyke JK, Stenberg PE, Thomas G. Intracellular trafficking and activation of the furin proprotein convertase: localization to the TGN and recycling from the cell surface. EMBO J. 1994;13:18–33. doi: 10.1002/j.1460-2075.1994.tb06231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bosshart H, et al. The cytoplasmic domain mediates localization of furin to the trans-Golgi network en route to the endosomal/lysosomal system. J. Cell Biol. 1994;126:1157–1172. doi: 10.1083/jcb.126.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schafer W, et al. Two independent targeting signals in the cytoplasmic domain determine trans-Golgi network localization and endosomal trafficking of the proprotein convertase furin. EMBO J. 1995;14:2424–2435. doi: 10.1002/j.1460-2075.1995.tb07240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jones BG, et al. Intracellular trafficking of furin is modulated by the phosphorylation state of a casein kinase II site in its cytoplasmic tail. EMBO J. 1995;14:5869–5883. doi: 10.1002/j.1460-2075.1995.tb00275.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takahashi S, et al. Localization of furin to the trans-Golgi network and recycling from the cell surface involves Ser and Tyr residues within the cytoplasmic domain. J. Biol. Chem. 1995;270:28397–28401. doi: 10.1074/jbc.270.47.28397. [DOI] [PubMed] [Google Scholar]
- 28.Wan L, et al. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell. 1998;94:205–216. doi: 10.1016/s0092-8674(00)81420-8. Identifies PACS-1 as a sorting protein that localizes phosphorylated furin to the TGN. Subsequent studies, as summarized in the text, revealed additional roles for PACS-1 in protein traffic and viral pathogenesis. [DOI] [PubMed] [Google Scholar]
- 29.Jung LJ, Kreiner T, Scheller RH. Expression of mutant ELH prohormones in AtT-20 cells: the relationship between prohormone processing and sorting. J. Cell Biol. 1993;121:11–21. doi: 10.1083/jcb.121.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vischer UM, Wagner DD. von Willebrand factor proteolytic processing and multimerization precede the formation of Weibel-Palade bodies. Blood. 1994;83:3536–3544. [PubMed] [Google Scholar]
- 31.Chen CD, et al. Furin initiates gelsolin familial amyloidosis in the Golgi through a defect in Ca(2+) stabilization. EMBO J. 2001;20:6277–6287. doi: 10.1093/emboj/20.22.6277. Explains how mutations in secreted plasma gelsolin disrupt its folding and render it susceptible to furin cleavage and subsequently to amyloid formation. Moreover, this study and the work in reference 32 provide evidence that furin-mediated processing in the biosynthetic pathway is a post-TGN event. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Band AM, Maatta J, Kaariainen L, Kuismanen E. Inhibition of the membrane fusion machinery prevents exit from the TGN and proteolytic processing by furin. FEBS Lett. 2001;505:118–124. doi: 10.1016/s0014-5793(01)02798-3. [DOI] [PubMed] [Google Scholar]
- 33.Simmen T, Nobile M, Bonifacino JS, Hunziker W. Basolateral sorting of furin in MDCK cells requires a phenylalanine-isoleucine motif together with an acidic amino acid cluster. Mol. Cell. Biol. 1999;19:3136–3144. doi: 10.1128/mcb.19.4.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simmen T, Honing S, Icking A, Tikkanen R, Hunziker W. AP-4 binds basolateral signals and participates in basolateral sorting in epithelial MDCK cells. Nature Cell Biol. 2002;4:154–159. doi: 10.1038/ncb745. [DOI] [PubMed] [Google Scholar]
- 35.Folsch H, Ohno H, Bonifacino JS, Mellman I. A novel clathrin adaptor complex mediates basolateral targeting in polarized epithelial cells. Cell. 1999;99:189–198. doi: 10.1016/s0092-8674(00)81650-5. [DOI] [PubMed] [Google Scholar]
- 36.Folsch H, Pypaert M, Schu P, Mellman I. Distribution and function of AP-1 clathrin adaptor complexes in polarized epithelial cells. J. Cell Biol. 2001;152:595–606. doi: 10.1083/jcb.152.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teuchert M, et al. Sorting of furin at the trans-Golgi network. Interaction of the cytoplasmic tail sorting signals with AP-1 Golgi-specific assembly proteins. J. Biol. Chem. 1999;274:8199–8207. doi: 10.1074/jbc.274.12.8199. [DOI] [PubMed] [Google Scholar]
- 38.Jacobsen L, et al. The sorLA cytoplasmic domain interacts with GGA1 and -2 and defines minimum requirements for GGA binding. FEBS Lett. 2002;511:155–158. doi: 10.1016/s0014-5793(01)03299-9. [DOI] [PubMed] [Google Scholar]
- 39.Misra S, Puertollano R, Kato Y, Bonifacino JS, Hurley JH. Structural basis for acidic-cluster-dileucine sorting-signal recognition by VHS domains. Nature. 2002;415:933–937. doi: 10.1038/415933a. [DOI] [PubMed] [Google Scholar]
- 40.Zhu Y, Doray B, Poussu A, Lehto VP, Kornfeld S. Binding of GGA2 to the lysosomal enzyme sorting motif of the mannose 6-phosphate receptor. Science. 2001;292:1716–1718. doi: 10.1126/science.1060896. [DOI] [PubMed] [Google Scholar]
- 41.Crump CM, et al. PACS-1 binding to adaptors is required for acidic cluster motif-mediated protein traffic. EMBO J. 2001;20:2191–2201. doi: 10.1093/emboj/20.9.2191. This paper and the work reported in reference 42 show the importance of PACS-1 and AP–1 in the retrieval of furin and other proteins from endosomes to the TGN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyer C, et al. mu1A-adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J. 2000;19:2193–2203. doi: 10.1093/emboj/19.10.2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiang Y, Molloy SS, Thomas L, Thomas G. The PC6B cytoplasmic domain contains two acidic clusters that direct sorting to distinct trans-Golgi network/endosomal compartments. Mol. Biol. Cell. 2000;11:1257–1273. doi: 10.1091/mbc.11.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piguet V, et al. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nature Cell Biol. 2000;2:163–167. doi: 10.1038/35004038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen MS, et al. The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 2001;20:2180–2190. doi: 10.1093/emboj/20.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakagawa T, et al. A novel motor, KIF13A, transports mannose-6-phosphate receptor to plasma membrane through direct interaction with AP-1 complex. Cell. 2000;103:569–581. doi: 10.1016/s0092-8674(00)00161-6. [DOI] [PubMed] [Google Scholar]
- 47.Liu G, et al. Cytoskeletal protein ABP-280 directs the intracellular trafficking of furin and modulates proprotein processing in the endocytic pathway. J. Cell Biol. 1997;139:1719–1733. doi: 10.1083/jcb.139.7.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stossel TP, et al. Filamins as integrators of cell mechanics and signalling. Nature Rev. Mol. Cell Biol. 2001;2:138–145. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 49.Barr VA, Phillips SA, Taylor SI, Haft CR. Overexpression of a novel sorting nexin, SNX15, affects endosome morphology and protein trafficking. Traffic. 2000;1:904–916. doi: 10.1034/j.1600-0854.2000.011109.x. [DOI] [PubMed] [Google Scholar]
- 50.Molloy SS, Thomas L, Kamibayashi C, Mumby MC, Thomas G. Regulation of endosome sorting by a specific PP2A isoform. J. Cell Biol. 1998;142:1399–1411. doi: 10.1083/jcb.142.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mallet WG, Maxfield FR. Chimeric forms of furin and TGN38 are transported with the plasma membrane in the trans-Golgi network via distinct endosomal pathways. J. Cell Biol. 1999;146:345–359. doi: 10.1083/jcb.146.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varlamov O, Kalinina E, Che FY, Fricker LD. Protein phosphatase 2A binds to the cytoplasmic tail of carboxypeptidase D and regulates post-trans-Golgi network trafficking. J. Cell Sci. 2001;114:311–322. doi: 10.1242/jcs.114.2.311. Uses proteomics and cell biology to explain how PP2A regulates the trafficking of an itinerant membrane protein in the TGN/endosomal system. This work extends earlier studies, reported in reference 50, of the importance of specific PP2A isoforms in regulating protein traffic. [DOI] [PubMed] [Google Scholar]
- 53.Urbe S, Page LJ, Tooze SA. Homotypic fusion of immature secretory granules during maturation in a cell-free assay. J. Cell Biol. 1998;143:1831–1844. doi: 10.1083/jcb.143.7.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tooze SA. Biogenesis of secretory granules in the trans-Golgi network of neuroendocrine and endocrine cells. Biochim. Biophys. Acta. 1998;1404:231–244. doi: 10.1016/S0167-4889(98)00059-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rudolf R, Salm T, Rustom A, Gerdes HH. Dynamics of immature secretory granules: role of cytoskeletal elements during transport, cortical restriction, and F-actin-dependent tethering. Mol. Biol. Cell. 2001;12:1353–1365. doi: 10.1091/mbc.12.5.1353. Provides the first real-time imaging that morphologically shows the maturation of MSGs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eaton BA, Haugwitz M, Lau D, Moore HP. Biogenesis of regulated exocytotic carriers in neuroendocrine cells. J. Neurosci. 2000;20:7334–7344. doi: 10.1523/JNEUROSCI.20-19-07334.2000. An insightful study that identifies the molecular machinery that controls the transition of ISGs to calcium-responsive MSGs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Littleton JT, Serano TL, Rubin GM, Ganetzky B, Chapman ER. Synaptic function modulated by changes in the ratio of synaptotagmin I and IV. Nature. 1999;400:757–760. doi: 10.1038/23462. [DOI] [PubMed] [Google Scholar]
- 58.Dittie AS, Thomas L, Thomas G, Tooze SA. Interaction of furin in immature secretory granules from neuroendocrine cells with the AP-1 adaptor complex is modulated by casein kinase II phosphorylation. EMBO J. 1997;16:4859–4870. doi: 10.1093/emboj/16.16.4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Varlamov O, Fricker LD. Intracellular trafficking of metallocarboxypeptidase D in AtT-20 cells: localization to the trans-Golgi network and recycling from the cell surface. J. Cell Sci. 1998;111:877–885. doi: 10.1242/jcs.111.7.877. [DOI] [PubMed] [Google Scholar]
- 60.Varlamov O, Eng FJ, Novikova EG, Fricker LD. Localization of metallocarboxypeptidase D in AtT-20 cells. Potential role in prohormone processing. J. Biol. Chem. 1999;274:14759–14767. doi: 10.1074/jbc.274.21.14759. [DOI] [PubMed] [Google Scholar]
- 61.Waites CL, et al. An acidic motif retains vesicular monoamine transporter 2 on large dense core vesicles. J. Cell Biol. 2001;152:1159–1168. doi: 10.1083/jcb.152.6.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paquet L, et al. The neuroendocrine precursor 7B2 is a sulfated protein proteolytically processed by a ubiquitous furin-like convertase. J. Biol. Chem. 1994;269:19279–19285. [PubMed] [Google Scholar]
- 63.Canaff L, Bennett HP, Hou Y, Seidah NG, Hendy GN. Proparathyroid hormone processing by the proprotein convertase-7: comparison with furin and assessment of modulation of parathyroid convertase messenger ribonucleic acid levels by calcium and 1,25- dihydroxyvitamin D3. Endocrinology. 1999;140:3633–3642. doi: 10.1210/endo.140.8.6882. [DOI] [PubMed] [Google Scholar]
- 64.Eskeland NL, et al. Chromogranin A processing and secretion: specific role of endogenous and exogenous prohormone convertases in the regulated secretory pathway. J. Clin. Invest. 1996;98:148–156. doi: 10.1172/JCI118760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klumperman J, et al. Cell type-specific sorting of neuropeptides: a mechanism to modulate peptide composition of large dense-core vesicles. J. Neurosci. 1996;16:7930–7940. doi: 10.1523/JNEUROSCI.16-24-07930.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee R, Kermani P, Teng KK. Hempstead, B. L. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. provocative study that explains the opposing roles of processed NGF and pro-β–NGF in controlling synaptic innervation and apoptosis. [DOI] [PubMed] [Google Scholar]
- 67.Mumm JS, et al. A ligand-induced extracellular cleavage regulates γ-secretase-like proteolytic activation of Notch1. Mol. Cell. 2000;5:197–206. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- 68.Bush G, et al. Ligand-induced signaling in the absence of furin processing of Notch1. Dev. Biol. 2001;229:494–502. doi: 10.1006/dbio.2000.9992. [DOI] [PubMed] [Google Scholar]
- 69.Isacson O, Seo H, Lin L, Albeck D, Granholm AC. Alzheimer's disease and Down's syndrome: roles of APP, trophic factors and ACh. Trends Neurosci. 2002;25:79–84. doi: 10.1016/s0166-2236(02)02037-4. [DOI] [PubMed] [Google Scholar]
- 70.Walter J, Kaether C, Steiner H, Haass C. The cell biology of Alzheimer's disease: uncovering the secrets of secretases. Curr. Opin. Neurobiol. 2001;11:585–590. doi: 10.1016/s0959-4388(00)00253-1. [DOI] [PubMed] [Google Scholar]
- 71.Weidemann A, et al. Identification, biogenesis, and localization of precursors of Alzheimer's disease A4 amyloid protein. Cell. 1989;57:115–126. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- 72.Luo JJ, Wallace MS, Hawver DB, Kusiak JW, Wallace WC. Characterization of the neurotrophic interaction between nerve growth factor and secreted α-amyloid precursor protein. J. Neurosci. Res. 2001;63:410–420. doi: 10.1002/1097-4547(20010301)63:5<410::AID-JNR1036>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 73.Wang HY, et al. β-Amyloid(1-42) binds to α7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J. Biol. Chem. 2000;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- 74.Lammich S, et al. Constitutive and regulated α-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl Acad. Sci. U.S.A. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Buxbaum JD, et al. Evidence that tumor necrosis factor α converting enzyme is involved in regulated α-secretase cleavage of the Alzheimer amyloid protein precursor. J. Biol. Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 76.Anders A, Gilbert S, Garten W, Postina R, Fahrenholz F. Regulation of the α-secretase ADAM10 by its prodomain and proprotein convertases. FASEB J. 2001;15:1837–1839. doi: 10.1096/fj.01-0007fje. Reports the regulated use of either furin or PC7 to cleave a proprotein substrate and might lead to a better understanding of the apparent redundancy of PCs in many systems. [DOI] [PubMed] [Google Scholar]
- 77.Lopez-Perez E, et al. Constitutive α-secretase cleavage of the β-amyloid precursor protein in the furin-deficient LoVo cell line: involvement of the pro-hormone convertase 7 and the disintegrin metalloprotease ADAM10. J. Neurochem. 2001;76:1532–1539. doi: 10.1046/j.1471-4159.2001.00180.x. [DOI] [PubMed] [Google Scholar]
- 78.Bennett BD, et al. A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer's β-secretase. J. Biol. Chem. 2000;275:37712–37717. doi: 10.1074/jbc.M005339200. [DOI] [PubMed] [Google Scholar]
- 79.Benjannet S, et al. Post-translational processing of β-secretase (β-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-β production. J. Biol. Chem. 2001;276:10879–10887. doi: 10.1074/jbc.M009899200. [DOI] [PubMed] [Google Scholar]
- 80.Creemers JW, et al. Processing of β-secretase by furin and other members of the proprotein convertase family. J. Biol. Chem. 2001;276:4211–4217. doi: 10.1074/jbc.M006947200. [DOI] [PubMed] [Google Scholar]
- 81.Walter J, et al. Phosphorylation regulates intracellular trafficking of β-secretase. J. Biol. Chem. 2001;276:14634–14641. doi: 10.1074/jbc.M011116200. [DOI] [PubMed] [Google Scholar]
- 82.Pastorino L, Ikin AF, Nairn AC, Pursnani A, Buxbaum JD. The carboxyl-terminus of BACE contains a sorting signal that regulates BACE trafficking but not the formation of total Aβ. Mol. Cell. Neurosci. 2002;19:175–185. doi: 10.1006/mcne.2001.1065. [DOI] [PubMed] [Google Scholar]
- 83.Kim SH, et al. Furin mediates enhanced production of fibrillogenic ABri peptides in familial British dementia. Nature Neurosci. 1999;2:984–988. doi: 10.1038/14783. [DOI] [PubMed] [Google Scholar]
- 84.Kim SH, Creemers JW, Chu S, Thinakaran G, Sisodia SS. Proteolytic processing of familial British dementia-associated BRI variants: evidence for enhanced intracellular accumulation of amyloidogenic peptides. J. Biol. Chem. 2002;277:1872–1877. doi: 10.1074/jbc.M108739200. [DOI] [PubMed] [Google Scholar]
- 85.Vidal R, et al. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature. 1999;399:776–781. doi: 10.1038/21637. [DOI] [PubMed] [Google Scholar]
- 86.Lee WM, Galbraith RM. The extracellular actin-scavenger system and actin toxicity. N. Engl. J. Med. 1992;326:1335–1341. doi: 10.1056/NEJM199205143262006. [DOI] [PubMed] [Google Scholar]
- 87.Maury CP, et al. Danish type gelsolin related amyloidosis: 654G-T mutation is associated with a disease pathogenetically and clinically similar to that caused by the 654G-A mutation (familial amyloidosis of the Finnish type) J. Clin. Pathol. 2000;53:95–99. doi: 10.1136/jcp.53.2.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grell M, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 89.Chen Y, et al. Mutations within a furin consensus sequence block proteolytic release of ectodysplasin-A and cause X-linked hypohidrotic ectodermal dysplasia. Proc. Natl Acad. Sci. U.S.A. 2001;98:7218–7223. doi: 10.1073/pnas.131076098. This work, together with the work reported in reference 91, shows a disease caused by mutation of a furin site in a signalling molecule and raises the possibility that furin cleavage might regulate juxtacrine versus paracrine signalling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Schneider P, et al. Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J. Biol. Chem. 2001;276:18819–18827. doi: 10.1074/jbc.M101280200. [DOI] [PubMed] [Google Scholar]
- 91.Tucker AS, et al. Edar/Eda interactions regulate enamel knot formation in tooth morphogenesis. Development. 2000;127:4691–4700. doi: 10.1242/dev.127.21.4691. [DOI] [PubMed] [Google Scholar]
- 92.Schneider P, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J. Exp. Med. 1999;189:1747–1756. doi: 10.1084/jem.189.11.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lopez-Fraga M, Fernandez R, Albar JP, Hahne M. Biologically active APRIL is secreted following intracellular processing in the Golgi apparatus by furin convertase. EMBO Rep. 2001;2:945–951. doi: 10.1093/embo-reports/kve198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Vaux DL. The buzz about BAFF. J. Clin. Invest. 2002;109:17–18. doi: 10.1172/JCI14780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roebroek AJ, et al. Failure of ventral closure and axial rotation in embryos lacking the proprotein convertase Furin. Development. 1998;125:4863–4876. doi: 10.1242/dev.125.24.4863. [DOI] [PubMed] [Google Scholar]
- 96.Constam DB, Robertson EJ. Tissue-specific requirements for the proprotein convertase furin/SPC1 during embryonic turning and heart looping. Development. 2000;127:245–254. doi: 10.1242/dev.127.2.245. [DOI] [PubMed] [Google Scholar]
- 97.Dubois CM, et al. Evidence that furin is an authentic transforming growth factor-β1-converting enzyme. Am. J. Pathol. 2001;158:305–316. doi: 10.1016/s0002-9440(10)63970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cui Y, Jean F, Thomas G, Christian JL. BMP-4 is proteolytically activated by furin and/or PC6 during vertebrate embryonic development. EMBO J. 1998;17:4735–4743. doi: 10.1093/emboj/17.16.4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cui Y, et al. The activity and signaling range of mature BMP-4 is regulated by sequential cleavage at two sites within the prodomain of the precursor. Genes Dev. 2001;15:2797–2802. doi: 10.1101/gad.940001. This work shows that furin's ‘measure once, cut twice’ method of activation extends to morphogen molecules to control embryogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gonzalez-Reyes L, et al. Cleavage of the human respiratory syncytial virus fusion protein at two distinct sites is required for activation of membrane fusion. Proc. Natl Acad. Sci. U.S.A. 2001;98:9859–9864. doi: 10.1073/pnas.151098198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zimmer G, Budz L, Herrler G. Proteolytic activation of respiratory syncytial virus fusion protein. Cleavage at two furin consensus sequences. J. Biol. Chem. 2001;276:31642–31650. doi: 10.1074/jbc.M102633200. [DOI] [PubMed] [Google Scholar]
- 102.Blanchette F, et al. Cross-talk between the p42/p44 MAP kinase and Smad pathways in transforming growth factor β 1-induced furin gene transactivation. J. Biol. Chem. 2001;276:33986–33994. doi: 10.1074/jbc.M100093200. [DOI] [PubMed] [Google Scholar]
- 103.Blanchette F, Rudd P, Grondin F, Attisano L, Dubois CM. Involvement of Smads in TGFβ1-induced furin (fur) transcription. J. Cell Physiol. 2001;188:264–273. doi: 10.1002/jcp.1116. [DOI] [PubMed] [Google Scholar]
- 104.Blanchette F, Day R, Dong W, Laprise MH, Dubois CM. TGFβ1 regulates gene expression of its own converting enzyme furin. J. Clin. Invest. 1997;99:1974–1983. doi: 10.1172/JCI119365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yamanishi Y, et al. Expression and regulation of aggrecanase in arthritis: the role of TGF-β. J. Immunol. 2002;168:1405–1412. doi: 10.4049/jimmunol.168.3.1405. These data, together with the work in reference 104, indicate a decidedly dark side of furin that initiates a signalling cascade that results in rheumatoid arthritis. [DOI] [PubMed] [Google Scholar]
- 106.Tang BL. ADAMTS: a novel family of extracellular matrix proteases. Int. J. Biochem. Cell. Biol. 2001;33:33–44. doi: 10.1016/s1357-2725(00)00061-3. [DOI] [PubMed] [Google Scholar]
- 107.Mbikay M, Sirois F, Yao J, Seidah NG, Chretien M. Comparative analysis of expression of the proprotein convertases furin, PACE4, PC1 and PC2 in human lung tumours. Br. J. Cancer. 1997;75:1509–1514. doi: 10.1038/bjc.1997.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bassi DE, et al. Elevated furin expression in aggressive human head and neck tumors and tumor cell lines. Mol. Carcinog. 2001;31:224–232. doi: 10.1002/mc.1057. [DOI] [PubMed] [Google Scholar]
- 109.Bassi DE, et al. Furin inhibition results in absent or decreased invasiveness and tumorigenicity of human cancer cells. Proc. Natl Acad. Sci. U.S.A. 2001;98:10326–10331. doi: 10.1073/pnas.191199198. This study, and the work in reference 115, shows how blocking furin can inhibit tumour metastasis in simple animal models. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sounni NE, et al. Expression of membrane type 1 matrix metalloproteinase (MT1-MMP) in A2058 melanoma cells is associated with MMP-2 activation and increased tumor growth and vascularization. Int. J. Cancer. 2002;98:23–28. doi: 10.1002/ijc.10134. [DOI] [PubMed] [Google Scholar]
- 111.Yana I, Weiss SJ. Regulation of membrane type-1 matrix metalloproteinase activation by proprotein convertases. Mol. Biol. Cell. 2000;11:2387–2401. doi: 10.1091/mbc.11.7.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kang T, Nagase H, Pei D. Activation of membrane-type matrix metalloproteinase 3 zymogen by the proprotein convertase furin in the trans-Golgi network. Cancer Res. 2002;62:675–681. [PubMed] [Google Scholar]
- 113.Aznavoorian S, et al. Membrane type I-matrix metalloproteinase-mediated degradation of type I collagen by oral squamous cell carcinoma cells. Cancer Res. 2001;61:6264–6275. [PubMed] [Google Scholar]
- 114.Kheradmand F, Rishi K, Werb Z. Signaling through the EGF receptor controls lung morphogenesis in part by regulating MT1-MMP-mediated activation of gelatinase A/MMP2. J. Cell Sci. 2002;115:839–848. doi: 10.1242/jcs.115.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu Y, Yakar S, Zhao L, Hennighausen L, LeRoith D. Circulating insulin-like growth factor-I levels regulate colon cancer growth and metastasis. Cancer Res. 2002;62:1030–1035. [PubMed] [Google Scholar]
- 116.Khatib AM, et al. Inhibition of proprotein convertases is associated with loss of growth and tumorigenicity of HT-29 human colon carcinoma cells: importance of insulin-like growth factor-1 (IGF-1) receptor processing in IGF-1-mediated functions. J. Biol. Chem. 2001;276:30686–30693. doi: 10.1074/jbc.M101725200. [DOI] [PubMed] [Google Scholar]
- 117.Cheng M, et al. Pro-protein convertase gene expression in human breast cancer. Int. J. Cancer. 1997;71:966–971. doi: 10.1002/(sici)1097-0215(19970611)71:6<966::aid-ijc10>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 118.Mahloogi H, Bassi DE, Klein-Szanto AJ. Malignant conversion of non–tumorogenic murine skin keritinocytes overexpressing PACE4. Carcinogenesis. 2002;23:565–572. doi: 10.1093/carcin/23.4.565. [DOI] [PubMed] [Google Scholar]
- 119.Klimpel KR, Molloy SS, Thomas G, Leppla S. H. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl Acad. Sci. U.S.A. 1992;89:10277–10281. doi: 10.1073/pnas.89.21.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jernigan JA, et al. Bioterrorism-related inhalational anthrax: the first 10 cases reported in the United States. Emerg. Infect. Dis. 2001;7:933–944. doi: 10.3201/eid0706.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Abrami L, et al. The pore-forming toxin proaerolysin is activated by furin. J. Biol. Chem. 1998;273:32656–32661. doi: 10.1074/jbc.273.49.32656. [DOI] [PubMed] [Google Scholar]
- 122.Gordon VM, Benz R, Fujii K, Leppla SH, Tweten RK. Clostridium septicum α-toxin is proteolytically activated by furin. Infect. Immun. 1997;65:4130–4134. doi: 10.1128/iai.65.10.4130-4134.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mock M, Fouet A. Anthrax. Annu. Rev. Microbiol. 2001;55:647–671. doi: 10.1146/annurev.micro.55.1.647. [DOI] [PubMed] [Google Scholar]
- 124.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 125.Beauregard KE, Collier RJ, Swanson JA. Proteolytic activation of receptor-bound anthrax protective antigen on macrophages promotes its internalization. Cell. Microbiol. 2000;2:251–258. doi: 10.1046/j.1462-5822.2000.00052.x. [DOI] [PubMed] [Google Scholar]
- 126.Gordon VM, Klimpel KR, Arora N, Henderson MA, Leppla SH. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect. Immun. 1995;63:82–87. doi: 10.1128/iai.63.1.82-87.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sellman BR, Mourez M, Collier RJ. Dominant-negative mutants of a toxin subunit: an approach to therapy of anthrax. Science. 2001;292:695–697. doi: 10.1126/science.109563. [DOI] [PubMed] [Google Scholar]
- 128.Ballard J, Sokolov Y, Yuan WL, Kagan BL, Tweten RK. Activation and mechanism of Clostridium septicum α toxin. Mol. Microbiol. 1993;10:627–634. doi: 10.1111/j.1365-2958.1993.tb00934.x. [DOI] [PubMed] [Google Scholar]
- 129.Schiavo G, van der Goot FG. The bacterial toxin toolkit. Nature Rev. Mol. Cell Biol. 2001;2:530–537. doi: 10.1038/35080089. [DOI] [PubMed] [Google Scholar]
- 130.Wedekind JE, et al. Refined crystallographic structure of Pseudomonas aeruginosa exotoxin A and its implications for the molecular mechanism of toxicity. J. Mol. Biol. 2001;314:823–837. doi: 10.1006/jmbi.2001.5195. [DOI] [PubMed] [Google Scholar]
- 131.Senzel L, Huynh PD, Jakes KS, Collier RJ, Finkelstein A. The diphtheria toxin channel-forming T domain translocates its own NH2-terminal region across planar bilayers. J. Gen. Physiol. 1998;112:317–324. doi: 10.1085/jgp.112.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Girod A, et al. Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nature Cell Biol. 1999;1:423–430. doi: 10.1038/15658. [DOI] [PubMed] [Google Scholar]
- 133.Jackson ME, et al. The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. J. Cell Sci. 1999;112:467–475. doi: 10.1242/jcs.112.4.467. [DOI] [PubMed] [Google Scholar]
- 134.Storrie B, et al. Recycling of Golgi-resident glycosyltransferases through the ER reveals a novel pathway and provides an explanation for nocodazole-induced Golgi scattering. J. Cell Biol. 1998;143:1505–1521. doi: 10.1083/jcb.143.6.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bass J, Turck C, Rouard M, Steiner DF. Furin-mediated processing in the early secretory pathway: sequential cleavage and degradation of misfolded insulin receptors. Proc. Natl Acad. Sci. U.S.A. 2000;97:11905–11909. doi: 10.1073/pnas.97.22.11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Moulard M, Decroly E. Maturation of HIV envelope glycoprotein precursors by cellular endoproteases. Biochim. Biophys. Acta. 2000;1469:121–132. doi: 10.1016/s0304-4157(00)00014-9. [DOI] [PubMed] [Google Scholar]
- 137.Ohnishi Y, et al. A furin-defective cell line is able to process correctly the gp160 of human immunodeficiency virus type 1. J. Virol. 1994;68:4075–4079. doi: 10.1128/jvi.68.6.4075-4079.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Binley JM, et al. Enhancing the proteolytic maturation of human immunodeficiency virus type 1 envelope glycoproteins. J. Virol. 2002;76:2606–2616. doi: 10.1128/JVI.76.6.2606-2616.2002. Solves a much-debated mystery as to why HIV-1 maintains an inefficient furin cleavage site in its envelope glycoprotein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zambon MC. The pathogenesis of influenza in humans. Rev. Med. Virol. 2001;11:227–241. doi: 10.1002/rmv.319. [DOI] [PubMed] [Google Scholar]
- 140.Takada A, Kawaoka Y. The pathogenesis of Ebola hemorrhagic fever. Trends Microbiol. 2001;9:506–511. doi: 10.1016/s0966-842x(01)02201-6. [DOI] [PubMed] [Google Scholar]
- 141.Feldmann H, Volchkov VE, Volchkova VA, Klenk HD. The glycoproteins of Marburg and Ebola virus and their potential roles in pathogenesis. Arch. Virol. Suppl. 1999;15:159–169. doi: 10.1007/978-3-7091-6425-9_11. [DOI] [PubMed] [Google Scholar]
- 142.Rott R, Klenk HD, Nagai Y, Tashiro M. Influenza viruses, cell enzymes, and pathogenicity. Am. J. Respir. Crit. Care Med. 1995;152:S16–19. doi: 10.1164/ajrccm/152.4_Pt_2.S16. [DOI] [PubMed] [Google Scholar]
- 143.Hatta M, Gao P, Halfmann P, Kawaoka Y. Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science. 2001;293:1840–1842. doi: 10.1126/science.1062882. [DOI] [PubMed] [Google Scholar]
- 144.Horimoto T, Kawaoka Y. Pandemic threat posed by avian influenza A viruses. Clin. Microbiol. Rev. 2001;14:129–149. doi: 10.1128/CMR.14.1.129-149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Volchkov VE, Feldmann H, Volchkova VA, Klenk HD. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl Acad. Sci. U.S.A. 1998;95:5762–5767. doi: 10.1073/pnas.95.10.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Neumann G, Feldmann H, Watanabe S, Lukashevich I, Kawaoka Y. Reverse genetics demonstrates that proteolytic processing of the Ebola virus glycoprotein is not essential for replication in cell culture. J. Virol. 2002;76:406–410. doi: 10.1128/JVI.76.1.406-410.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Heinz FX, Allison SL. The machinery for flavivirus fusion with host cell membranes. Curr. Opin. Microbiol. 2001;4:450–455. doi: 10.1016/s1369-5274(00)00234-4. [DOI] [PubMed] [Google Scholar]
- 148.Simmons G, Wool-Lewis RJ, Baribaud F, Netter RC, Bates P. Ebola virus glycoproteins induce global surface protein down-modulation and loss of cell adherence. J. Virol. 2002;76:2518–2528. doi: 10.1128/jvi.76.5.2518-2528.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Yang ZY, et al. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nature Med. 2000;6:886–889. doi: 10.1038/78645. [DOI] [PubMed] [Google Scholar]
- 150.Zhou A, Webb G, Zhu X, Steiner DF. Proteolytic processing in the secretory pathway. J. Biol. Chem. 1999;274:20745–20748. doi: 10.1074/jbc.274.30.20745. [DOI] [PubMed] [Google Scholar]
- 151.Seidah NG, Chretien M. Proprotein and prohormone convertases: a family of subtilases generating diverse bioactive polypeptides. Brain Res. 1999;848:45–62. doi: 10.1016/s0006-8993(99)01909-5. [DOI] [PubMed] [Google Scholar]
- 152.Steiner DF, Cunningham D, Spigelman L, Aten B. Insulin biosynthesis: evidence for a precursor. Science. 1967;157:697–700. doi: 10.1126/science.157.3789.697. [DOI] [PubMed] [Google Scholar]
- 153.Steiner DF, et al. Proinsulin and the biosynthesis of insulin. Recent Prog. Horm. Res. 1969;25:207–282. doi: 10.1016/b978-0-12-571125-8.50008-9. [DOI] [PubMed] [Google Scholar]
- 154.Chretien M, Li CH. Isolation, purification, and characterization of γ-lipotropic hormone from sheep pituitary glands. Can. J. Biochem. 1967;45:1163–1174. doi: 10.1139/o67-133. [DOI] [PubMed] [Google Scholar]
- 155.Julius D, Brake A, Blair L, Kunisawa R, Thorner J. Isolation of the putative structural gene for the lysinearginine-cleaving endopeptidase required for processing of yeast prepro-α-factor. Cell. 1984;37:1075–1089. doi: 10.1016/0092-8674(84)90442-2. [DOI] [PubMed] [Google Scholar]
- 156.Thomas G, et al. Yeast KEX2 endopeptidase correctly cleaves a neuroendocrine prohormone in mammalian cells. Science. 1988;241:226–230. doi: 10.1126/science.3291117. [DOI] [PubMed] [Google Scholar]
- 157.Roebroek AJ, et al. Characterization of human c-fes/fps reveals a new transcription unit (fur) in the immediately upstream region of the proto-oncogene. Mol. Biol. Rep. 1986;11:117–125. doi: 10.1007/BF00364823. [DOI] [PubMed] [Google Scholar]
- 158.Fuller RS, Brake AJ, Thorner J. Intracellular targeting and structural conservation of a prohormone-processing endoprotease. Science. 1989;246:482–486. doi: 10.1126/science.2683070. [DOI] [PubMed] [Google Scholar]
- 159.Bresnahan PA, et al. Human fur gene encodes a yeast KEX2-like endoprotease that cleaves pro-β-NGF in vivo. J. Cell Biol. 1990;111:2851–2859. doi: 10.1083/jcb.111.6.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Wise RJ, et al. Expression of a human proprotein processing enzyme: correct cleavage of the von Willebrand factor precursor at a paired basic amino acid site. Proc. Natl Acad. Sci. U.S.A. 1990;87:9378–9382. doi: 10.1073/pnas.87.23.9378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.From the Centers for Disease Control and Prevention Update: investigation of bioterrorism-related Anthrax–Connecticut, 2001. JAMA. 2002;287:34–35. [PubMed] [Google Scholar]
- 162.Update: Investigation of bioterrorism-related anthrax–Connecticut, 2001. M. M. W. R. Morb. Mortal. Wkl. Rep. 2001;50:1077–1079. [PubMed] [Google Scholar]
- 163.Duesbery NS, Vande Woude GF. Anthrax toxins. Cell. Mol. Life Sci. 1999;55:1599–1609. doi: 10.1007/s000180050399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Constam DB, Robertson EJ. SPC4/PACE4 regulates a TGFβ signaling network during axis formation. Genes Dev. 2000;14:1146–1155. [PMC free article] [PubMed] [Google Scholar]
- 165.Furuta M, et al. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proc. Natl Acad. Sci. U.S.A. 1997;94:6646–6651. doi: 10.1073/pnas.94.13.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mbikay M, et al. Impaired fertility in mice deficient for the testicular germ-cell protease PC4. Proc. Natl Acad. Sci. U.S.A. 1997;94:6842–6846. doi: 10.1073/pnas.94.13.6842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Constam DB, Robertson EJ. Regulation of bone morphogenetic protein activity by pro domains and proprotein convertases. J. Cell Biol. 1999;144:139–149. doi: 10.1083/jcb.144.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhu X, et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc. Natl Acad. Sci. U.S.A. 2002;99:10293–10298. doi: 10.1073/pnas.162352599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Jackson RS, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nature Genet. 1997;16:303–306. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- 170.Steiner DF. The proprotein convertases. Curr. Opin. Chem. Biol. 1998;2:31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]