Interaction of fascin and protein kinase Cα: a novel intersection in cell adhesion and motility (original) (raw)
Abstract
Coordination of protrusive and contractile cell–matrix contacts is important for cell adhesion and migration, but the mechanisms involved are not well understood. We report an unexpected direct association between fascin, an actin-bundling component of filopodia, microspikes and lamellipodial ribs, and protein kinase Cα (PKCα), a regulator of focal adhesions. The association is detectable by protein–protein binding in vitro, by coimmunoprecipitation from cell extracts, and in live cells as fluorescence resonance energy transfer detected by fluorescence imaging lifetime microscopy. The interaction is physiologically regulated by the extracellular matrix context of cells, depends on activation of PKCα and is mediated by the C1B domain of PKCα. Strikingly, a fascin mutant, fascin S39D, associates constitutively with PKCα. Through use of a newly developed set of membrane-permeable peptides that separately inhibit either fascin/PKCα or fascin/actin binding, we have uncovered that specific blockade of the fascin/PKCα interaction increases cell migration on fibronectin in conjunction with increased fascin protrusions and remodeling of focal adhesions. These results identify the fascin–PKCα interaction as an important novel intersection in the regulation and networking of cell–matrix contacts.
Keywords: cell adhesion/cell–matrix contacts/cytoskeleton/extracellular matrix/signaling
Introduction
Cell adhesion and motility are important biological processes in normal tissue organization and homeostasis that contribute to the causation of many human diseases when dysregulated (reviewed by Gumbiner, 1996; Danen and Yamada, 2001). Cell adhesion to extracellular matrix (ECM) is mediated by transmembrane multiprotein structures termed cell–matrix contacts, which are built up by the activation and clustering of ECM adhesion receptors and their linkage through intracellular protein complexes to the cytoskeleton (reviewed by Adams, 2001; Geiger et al., 2001). Precise coordination of matrix contacts is needed to achieve cell migration and other cell behaviors. Adhesion receptors and signaling molecules have jointly important roles in these processes (Lauffenburger and Horwitz, 1996; Giancotti and Ruoslahti, 1999; Schwartz and Ginsberg, 2002). Because the survival of normal cells depends on maintenance of adhesion to ECM (Meredith et al., 1993; Frisch and Francis, 1994), strategies that would regulate cell morphology or motility, without complete loss of cell–matrix attachment, are attractive and interesting propositions for tissue engineering, the regulation of stem cells and other biotherapeutic applications (reviewed by Sipe, 2002).
Whereas contractile adhesions are widely studied, the coordination of protrusive contacts (microspikes, filopodia and lamellipodia) is not well-understood (Adams, 2001). The assembly of protrusive spikes and lamellipodia that contain the actin-bundling protein, fascin, is a distinct response of cells to the ECM component, thrombospondin-1 (TSP-1), and certain other extracellular cues (Adams, 1995; reviewed by Kureishy et al., 2002). The correct organization of fascin and actin bundles is necessary for cell adhesion and migration on TSP-1 and contributes to cell migration on fibronectin (FN) (Adams, 1997; Adams and Schwartz, 2000). Fascin is concentrated in leading edge protrusions of migratory cells, and fascin-containing protrusions function in the motility and cell–cell interactions of many differentiated cells (reviewed by Kureishy et al, 2002).
Although fascin bundles F-actin slowly in vitro, the rapidity and specificity with which fascin-containing protrusions are formed by cells in response to distinct extracellular cues indicates that its actin-crosslinking activity is highly regulated in living cells (Kureishy et al., 2002). Whereas TSP-1 induces cross-linking of fascin and F-actin in protrusions, cell adhesion to FN triggers phosphorylation of fascin at S39, leading to rapid loss of fascin from actin-based structures during initial cell spreading, in a protein kinase Cα (PKCα)-dependent process (Adams, 1995; Adams et al., 1999). PKCα is activated by integrin-mediated adhesion and has significant regulatory effects on cell migration through its participation in β1 integrin-associated protein complexes (Vuori and Ruoslahti, 1993; Miranti et al., 1999; Ng et al., 1999a). However, it is not known whether fascin is a direct substrate of PKCα in whole cells, or whether the phosphorylation of fascin has biological significance beyond nullifying its actin-crosslinking activity (Yamakita et al., 1996; Ono et al., 1997; Adams et al., 1999). We hypothesized that a link between fascin and PKCα might represent a novel point of integration in the signaling of cytoskeletal organization between cell protrusions and other cell–ECM contacts. We report here a dynamically regulated direct association between fascin and PKCα that is of unexpected functional importance for the balance between fascin protrusions and focal adhesions and for cell migration on FN.
Results
Activation of PKCα is differentially regulated by ECM context
We hypothesized that the regulation of fascin phosphorylation by the ECM environment might be due to differential activation of PKCα. Phosphorylation of T250 of PKCα is an early event in PKCα activation (Ng et al., 1999b). C2C12 cells express PKCα as the major conventional PKC isoform: PKCβ is not expressed and PKCγ is present at very low levels (Adams et al., 1999). Using an antibody specifically reactive with phosphorylated T250, we found that 4-fold more active PKCα was present in C2C12 cells adherent to FN for 1 h, compared with cells adherent to TSP-1 or suspended over BSA (Figure 1A). The amount of active PKCα in FN-adherent cells increased from basal levels at 10 min to maximum levels at 1 h (Figure 1B). This time-course correlates with the phosphorylation of fascin in cells adherent to FN (Adams et al., 1999).
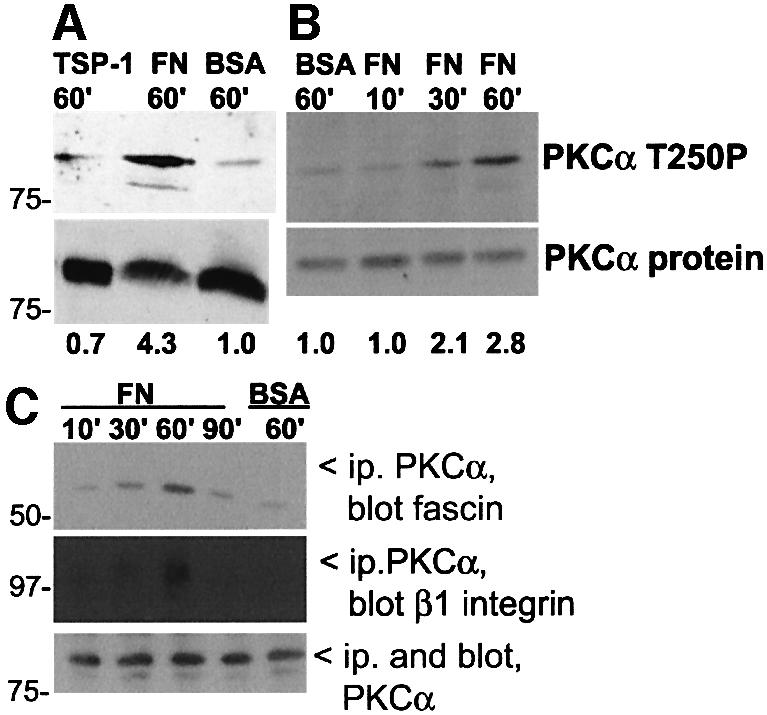
Fig. 1. Activation of PKCα by ECM adhesion. (A) C2C12 cells were plated for 1 h on surfaces coated with BSA, 50 nM FN or 50 nM TSP-1. Western blots of whole-cell lysates were probed with antibodies to activated PKCα or PKCα total protein. The mean relative signal intensity, normalized against protein load by densitometric scanning, is shown below. (B) Time-course of PKCα activation in FN-adherent cells, determined by the same methods as in (A). (C) Time-course of association of fascin and β1 integrin with PKCα. Detergent-soluble fractions of C2C12 cells were immunoprecipitated for PKCα and associated fascin or β1 integrin detected by immunoblot. All data are representative of three independent experiments.
Regulated and dynamic association of fascin and PKCα in living cells
To examine whether PKCα associates with fascin during cell–ECM adhesion, coimmunoprecipitation experiments were carried out. Association of fascin with PKCα was detected after 30 min adhesion to FN and was maximal at 60 min, whereas only minor association was detected in cells suspended over BSA (Figure 1C). Because activated PKCα has been reported to associate with integrin β1 subunit (Ng et al., 1999a) and β1-containing integrins mediate cell adhesion to FN, we tested whether β1 integrin was also present in the immunoprecipitates. β1 integrin was detected at the 1 h time-point (Figure 1C). Thus, association of fascin with PKCα was detected temporally earlier than β1 integrin.
To view directly the association between fascin and PKCα and its regulation by PKCα activity, the molecular proximity of the two proteins was analyzed by real-time fluorescence lifetime imaging microscopy (FLIM), carried out as described previously (Ng et al., 1999a,b, 2001; Legg et al., 2002). The extent of fluorescence resonance energy transfer (FRET) between EGFP-tagged fascin, as donor, and myc epitope-tagged PKCα detected with Cy3-conjugated anti-myc antibody, as acceptor, was examined in live MCF-7 cells stimulated with tetradecanoyl phorbol 13-acetate (TPA) to directly activate PKCα. These cells have very low endogenous levels of PKCα and thus any association of donor with the PKC acceptor can be measured with high accuracy. The FRET efficiency (Eff) time-course showed that there was little FRET between EGFP–fascin and Cy3-PKCα in untreated cells (Figure 2A; n = 6 cells). FRET was detected 4 min after addition of 400 nM TPA (indicated by red pixels on the Eff pseudocolour cell map, Figure 2A; n = 6 cells) and persisted until 30 min, when there was a partial reversal of <τ> and an overall reduction in FRET efficiencies. The pixel counts plotted against Eff profiles at different timepoints (hereafter referred to as Eff histograms) showed that the mean FRET efficiency detected in TPA-stimulated, EGFP–fascin/PKCα-coexpressing cells changed from 1.0% at time 0, to 4.2, 4.5, 5.1 and 3.4% at times 4, 12, 20 and 30 min, respectively (Figure 2A; n = 6 cells). These results demonstrate that fascin undergoes a TPA-responsive association with PKCα that involves a spatial separation of no more than 9 nm between donor and acceptor (Ng et al., 1999a).

Fig. 2. Association of fascin and PKCα detected in live cells by FLIM/FRET techniques. (A) Real-time FLIM between EGFP–WT fascin and Myc-tagged PKCα. In the +mAb 9E10-Cy3 field of GFP–fascin-expressing cells, only one of the two was injected with a visible amount of Cy3-conjugated mAb 9E10 (arrowed). Changes in donor fluorescence lifetimes, <τ> (the average of τp and τm), were monitored with time in both control and antibody-injected cells following treatment with 400 nM TPA. The FRET efficiency (Eff) pseudocolour cell map (Eff = 1 – τda/τd, where τda is the lifetime map of the donor in the presence of acceptor and τd is the average lifetime <τ> of the donor in the corresponding control, uninjected cell at each time point) is shown for each individual sampling point. The cumulative lifetimes (of all the pixels) of GFP–fascin alone (green) and that measured in the presence of the acceptor fluorophore (red) are plotted on the τp versus τm 2D histograms for each time point. At t = 0, the donor lifetimes in antibody-injected cells were the same as those of control cells, giving rise to the yellow overlap area. After TPA treatment, there was a time-dependent shortening of GFP fluorescence lifetime according to both τp and τm in the antibody-injected cell. The bottom left-hand panel shows the pixel counts versus Eff (%) profiles (Eff histograms) for the full time-course. (B) A similar experiment to that in (A), performed using MCF-7 cells microinjected with GFP-tagged S39D fascin and Myc-tagged PKCα constructs. In the +mAb 9E10-Cy3 field of GFP–fascin-expressing cells, only one of the two was injected with a sufficient amount of Cy3-conjugated mAb 9E10 (arrowed). Eff histogram analysis (lower right-hand panel) was performed using lifetimes obtained from this cell.
Phosphorylation of fascin at S39 modulates association kinetics with PKCα
The major site in fascin that is phosphorylated by PKCα is S39 (Yamakita et al., 1996; Ono et al., 1997). The phosphorylation motif is highly conserved in all species homologs of fascin (Kureishy et al., 2002). Over expression of S39D or S39A mutant fascins result in ECM context-specific impairments of cell spreading and cytoskeletal organization due to dysregulation of actin bundling by fascin (Adams et al., 1999; Kureishy et al., 2002). To investigate the importance of S39 phosphorylation of fascin for the association with PKCα, time-course FLIM experiments were performed on live cells with GFP-tagged fascin S39D as the donor, to mimic sustained phosphorylation of S39. In contrast to the results with wild-type (WT) fascin, the presence of anti-myc-Cy3 acceptor in unstimulated cells resulted in a significant decrease in the GFP fluorescence lifetimes (Figure 2B, see τp versus τm 2D histogram at time = 0). The mean FRET efficiency detected in EGFP–fascin S39D/PKCα-coexpressing cells was 5.0% at time 0 and increased moderately over treatment with TPA to 6.4 and 7.8% at 25 and 40 min, respectively (n = 6 cells). These results show that non-activated PKCα associates significantly with fascin S39D, and the interaction is at most 1.5-fold enhanced by PKCα activity, compared with the 5-fold enhancement of association with WT fascin (Figure 2A).
To corroborate these observations, MCF-7 cells were cotransfected with EGFP–fascin WT, EGFP–fascin S39D, or EGFP–fascin S39A (a mutant in which actin-bundling activity is not regulated by phosphorylation) (Yamakita et al., 1996), and PKCα-myc for 36 h, and stimulated with 400 nM TPA for 15 min to achieve the maximal interaction of fascin and PKCα, as determined from the time-course experiments with WT fascin (see Figure 2A). Cells were then fixed and stained for FRET/FLIM analyses (Figure 3). A cumulative analysis of all the EGFP–fascin/PKCα-coexpressing cells revealed that the mean FRET efficiency was 3.6% for fascin S39A, 5.7% for WT and 8.1% for fascin S39D. The mean FRET efficiency for unstained EGFP–fascin S39A/PKCα-coexpressing cells in these experiments was 3.6%, thus the donor lifetimes measured in the EGFP–fascin S39A-expressing cells in the presence of the anti-myc-Cy3 antibody were undistinguishable from those of the control unstained cells (n = 6; Figure 3, τp versus τm 2D histograms). Significant heterogeneity of the TPA-induced association was observed in the WT fascin-expressing cells, which probably reflected the transient association documented by the time-courses (Figure 2A). Thus, WT fascin and fascin S39D both associate closely with PKCα and, over the same time-course of TPA treatment, the non-phosphorylatable fascin S39A has a poor capacity to associate with PKCα, irrespective of PKCα activity status.

Fig. 3. Influence of phosphoregulation of fascin on its association with PKCα. MCF-7 cells transiently coexpressing either GFP–WT fascin, GFP–fascin S39A or GFP–fascin S39D, and myc-tagged PKCα, for 36 h were left untreated or stimulated with 400 nM TPA for 15 min. Cells were fixed in 4% paraformaldehyde and either left as controls (–anti-myc-Cy3) or stained with Cy3-conjugated anti-myc antibody (+anti-myc-Cy3). The pixel counts versus Eff (%) profiles (bottom right-hand panel) summarize all the FRET efficiency data for each construct (measured at each pixel of the cell from at least five donor and acceptor-positive cells), as well as for unstained, cotranfected cells as a negative control.
Direct binding of fascin and PKCα
Having discovered that fascin and PKCα associate physically, and that this association is regulated both by PKCα activation and by the phosphorylation/charge status of S39 of fascin, we wished to establish whether the binding was direct and, if so, the physiological importance of this process. To answer these questions, we developed a set of chemically synthesized, membrane-permeable TAT-fusion peptides containing the polybasic sequence from HIV TAT protein and WT or mutant versions of amino acids (aa) 33–47 from fascin (TAT-FAS peptides; Figure 4A). This very highly conserved region of fascin is surface-exposed in an α-helical loop and, in addition to containing the S39 regulatory site, is indirectly implicated in actin binding (reviewed by Kureishy et al., 2002).

Fig. 4. Biochemical properties of TAT-FAS peptides. (A) Design and amino acid sequences of TAT-FAS peptides. (B) Binding specificity of FAS-TAT peptides for actin. Pull-downs were performed with the different biotinylated TAT-FAS peptides bound to streptavidin–agarose as indicated. Candidate partner proteins were detected by immunoblot of SDS–PAGE gels. For each sample in (i) and (ii), the whole of the bound fraction and one-fifth of the supernatant (SN) was loaded per lane. (i) Binding to purified F-actin; (ii) binding specificity for actin in cell lysates; and (iii) competition of actin binding to purified fascin-His by free peptides. For each peptide: lane 1, 100 nM; lane 2, 300 nM; lane 3, 600 nM free peptide. Beads, Ni-NTA without fascin; actin +ve, cell extract. (C) Binding specificities of TAT-FAS peptides for PKCα. (i) Pull-down assays with TAT-FAS peptides bound to streptavidin–agarose as indicated were carried out on lysates of MCF-7 cells expressing GFP–PKCα-myc and immunoblotted for PKCα. Samples were reprobed for α-tubulin to demonstrate binding specificity. (ii) Cross-competition for PKCα binding to TAT–fascin S39D peptide–beads by free peptides. Cell extracts were preincubated with free TAT–fascin peptides as indicated for 1 h at 4°C before addition of TAT-FAS S39D–beads. For each sample, the whole of the bound fraction (B) and one-fifth of the supernatant (SN) was loaded per lane. (D) (i) Direct binding of fascin to active PKCα. Ni-NTA beads were left unloaded or loaded with 400 ng of purified recombinant fascin, washed, and incubated with 100 ng recombinant PKCα in the absence or presence of activating lipids and free TAT-FAS peptides as indicated. For each peptide: lane 1, 100 nM; lane 2, 300 nM; lane 3, 600 nM free peptide. (ii) Competition of fascin coimmunoprecipitation with PKCα by TAT-FAS S39D. C2C12 cells were preloaded with 300 nM TAT-FAS peptides as indicated, plated on 50 nM FN for 1 h, then immunoprecipitated for PKCα. Associated fascin was detected by immunoblot.
To establish the binding-properties of the peptides, biotinylated versions were used for in vitro pull-downs on streptavidin–agarose. The TAT-FAS WT and TAT-FAS S39A peptides bound to purified F-actin, whereas TAT-FAS S39D, a scrambled version of the WT peptide, or unfused TAT peptide did not bind (Figure 4Bi; data not shown). The specificity of interaction of TAT-FAS WT and TAT-FAS S39A with actin was established by pull-downs from cell extracts, in which only TAT-FAS WT and TAT-FAS S39A bound actin, and none of the peptides bound the abundant protein tubulin (Figure 4Bii). To establish that the actin-binding peptides can compete for binding of full-length fascin to actin, a recombinant His-tagged form of fascin was prepared and purified on nickel beads. The association between cellular actin and purified fascin-His was then measured in the presence or absence of the TAT-FAS peptides. TAT-FAS WT and, to a larger extent, TAT-FAS S39A inhibited the coprecipitation of actin with fascin in a dose-dependent manner, whereas the other peptides were not inhibitory (Figure 4Biii). Actin binding was not detected on beads lacking fascin (Figure 4Biii, last lane).
Pull-downs from cell extracts were then used to test which of the peptides bound to PKCα. TAT-FAS WT and TAT-FAS S39D bound PKCα, whereas TAT-FAS S39A and scrambled peptides did not. Consistent with the FLIM measurements from unstimulated cells, TAT-FAS S39D reproducibly bound more PKCα than TAT-FAS WT (Figure 4Ci). TAT-FAS WT or TAT-FAS S39D competed for the binding of GFP–PKCα to FAS-TAT S39D–agarose beads, whereas TAT-FAS S39A and scrambled control peptides were inactive (Figure 4Cii). To establish that full-length fascin indeed binds directly to PKCα, purified recombinant fascin was incubated with recombinant PKCα, in the absence or presence of activating lipids, and the association of the two proteins monitered by pull-down. Binding of PKCα was dependent on the presence of fascin on the beads and was strongly dependent on PKCα activation (Figure 4Di). The interaction was inhibited in a dose-dependent manner by TAT-FAS S39D and TAT-FAS WT, but not by TAT-FAS S39A or the scrambled control peptide, even at the highest concentration (Figure 4Di; data not shown). We also found that TAT-FAS S39D specifically blocked the coimmunoprecipitation of fascin with PKCα from cells adherent to FN for 1 h (Figure 4Dii). Thus, TAT-FAS S39D and S39A have distinct properties in perturbing either PKCα- or actin-binding by fascin.
The fascin-binding site maps to the C1B region of PKCα
To map the domain of PKCα required for fascin binding, TAT-FAS S39D peptide beads were used to pull-down WT or truncated, GFP-tagged versions of PKCα from MCF-7 cells (diagrammed in Figure 5Ai). All the proteins were expressed at similar levels, yet only WT GFP–PKCα and GFP–PKCαRDV3 or GFP–PKCαRD bound to TAT-FAS S39D. The GFP–PKCαC2V3 deletion mutant did not bind and no binding was detected on beads without TAT-FAS S39D peptide (Figure 5Aii). Thus, aa 1–289 of PKCα are sufficient for the interaction with fascin.

Fig. 5. Mapping the fascin-binding site on PKCα. (A) (i) Schematic diagram of the GFP-tagged or GST-tagged PKCα constructs. (ii) Pull-down of GFP–PKCα proteins, expressed in MCF-7 cells, by TAT-FAS–S39D peptide–beads. Western blots of bound proteins were probed with antibody to GFP. Only forms of PKCα containing the V1-PS-C1-V2 region (aa 1–289) bound to the peptide. (iii) Pull-down of fascin from C2C12 cells by GST–PKCα affinity matrices. The indicated purified GST–PKCα fusion proteins were bound to glutathione–agarose and incubated with C2C12 extracts. Western blots of bound proteins were probed with antibodies to fascin or to GST. Only proteins containing the C1B domain bound to fascin. (B) TAT-FAS peptides do not alter kinase activity of PKCα. In vitro kinase reactions were run for 30 min at 30°C with ezrin as exogenous substrate, in the absence or presence of activating lipids or TAT-FAS peptides as indicated. The reactions were resolved on 10% polyacrylamide gels and incorporation of [32P]ATP detected by autoradiography. Band intensities for ezrin (E) and PKCα (P) were quantified by scanning densitometry and were taken as 100% in the presence of lipids and the absence of peptides for each band. Band intensities in the presence of the peptides were expressed as a percentage of the control value and are shown under each lane. All results are representative of at least three independent experiments.
To confirm this result on intact fascin and to map the fascin-binding site, a set of purified glutathione _S_-transferase (GST) fusion proteins covering aa 1–337 of PKCα were tested for fascin binding in C2C12 extracts (diagrammed in Figure 5Ai). The proteins RDV3 (aa 1–337), RD (aa 1–289) and C1 (aa 32–155) bound to fascin, whereas C2V3 (aa 175–337), or unfused GST did not (Figure 5Aii). The C1 region contains two structural domains, C1A and C1B, which are 37% homologous in sequence (reviewed by Jaken and Parker, 2000). Fascin-binding activity was retained by C1B (aa 102–151) but not by C1A (aa 37–86) (Figure 5Aiii). Thus, fascin binding maps to aa 102–151 of PKCα.
In vitro kinase assays with ezrin as substrate were used to establish whether the TAT-FAS peptides altered the activity of PKCα. Under conditions previously determined to be optimal for PKCα autophosphorylation and ezrin phosphorylation (Ng et al., 2001), no alterations to kinase activity were detected in the presence of any of the peptides, each tested over a range of concentrations (Figure 5B).
TAT-FAS peptides selectively modulate cell spreading without general effects on cell signaling
We next established the conditions for loading the TAT-FAS peptides into live cells. In pilot experiments, 95% of C2C12 cells treated with 100–500 nM of FITC-conjugated TAT-FAS WT were loaded by 45 min, and no toxicity was detected over 3 days. The peptide localized to the cytoplasm and the nucleus (data not shown). In agreement with the results of the in vitro kinase assays, cells treated with any of the peptides at 200 nM for 1 h did activate PKCα, as measured by the abundance of the activation-specific T250P phosphoepitope (Figure 6A; data not shown) (Ng et al., 1999b). Furthermore, none of the peptides affected the activation state of the known target of PKCα, Raf-1, upon TPA-induced activation of PKCα, as measured by immunoblotting of cell extracts with antibodies specific to the S338P or S259P major phosphorylation sites of Raf-1 (Figure 6B; data not shown) (Morrison and Cutler, 1997; Cheng et al., 2001). In contrast, the TPA-dependent phosphorylation of fascin at S39 was blocked by TAT-FAS S39D and was partially inhibited by TAT-FAS WT (Figure 6C). We also documented that the profile of phosphotyrosine-containing proteins in cells spreading on FN was not altered, although a modest increase in paxillin phosphorylation was apparent in cells treated with TAT-FAS S39D (Figure 6D). We conclude that the peptides have appropriate properties to enable investigation of the functional consequences of disrupting the fascin/PKCα interaction in living cells.

Fig. 6. Effects of TAT-FAS peptides on cell signaling pathways. (A) Thirty micrograms of extracts from adherent C2C12 cells, preloaded for 2 h with 200 nM peptides as indicated, were probed by immunoblot for the content of active (T250 phosphorylated) PKCα. Five hundred nanograms of purified, autophosphorylated PKCα was used as a positive control for T250P antibody reactivity. (B) C2C12 cells were left untreated (UT), treated with 50 nM bisindolylmaleimide (BIM), or stimulated with 50 nM TPA (TPA) for 15 min as indicated. Cells pretreated for 1 h with 200 nM TAT-FAS peptides as indicated were also stimulated with TPA. Thirty micrograms of cell extracts were probed by immunoblot for Raf S338P and reprobed for Raf-1 protein. Peptide loading without TPA did not affect Raf-1 phosphorylation (not shown). (C) Thirty micrograms of extracts from adherent C2C12 cells, untreated, or stimulated with 50 nM TPA for 1 h, with or without 200 nM TAT-FAS peptides as indicated, were probed by immunoblot for S39-phosphorylated fascin and reprobed for fascin protein. Cells treated with peptides in the absence of TPA resembled untreated controls (not shown). (D) C2C12 cells were placed in suspension for 2 h (Sus), left untreated (UT) or preloaded for 1 h with 200 nM peptide as indicated. Untreated and peptide-treated cells were plated on FN for 20 min in the continued presence of the peptides. Thirty micrograms of cell extracts were probed by immunoblot for phosphotyrosine or for vinculin as a loading control. Focal adhesion kinase (FAK) and paxillin (pax) were identified in separate experiments by immunoprecipitation with anti-phosphotyrosine followed by immunoblot with antibodies specific for FAK or paxillin. Relative signal intensities for FAK-ptyr, normalized against protein load, are listed below. All data are representative of three independent experiments.
We first explored the role of the fascin–PKCα interaction in cell adhesion to FN. The parameters selected were based on our previous studies of the concentration and time dependence of C2C12 cell attachment, spreading and cytoskeletal organization in response to TSP-1 and FN under serum-free conditions. On both matrices, maximal quantitative cell attachment is achieved by 1 h of adhesion with a 50 nM coating concentration (Adams and Lawler, 1994; Fischer et al., 1997). Spreading and assembly of the appropriate matrix contacts are also complete by 1 h for cells on 50 nM coatings (Adams, 1995). On low concentrations of FN, cells remain round or incompletely spread at 1 h (Fischer et al., 1997). The peptides were first tested over a range of concentrations for their effects on cell–matrix attachment under optimal conditions. None of the peptides has statistically significant effects on initial cell attachment to FN or TSP-1 (Figure 7A; data not shown).

Fig. 7. Effects of TAT-FAS peptides on FN-induced cell adhesion and migration. (A) Introduction of TAT-FAS peptides into C2C12 cells does not affect initial attachment to FN. Cells were preloaded with peptides at the indicated concentrations for 1 h and cell attachment quantitated after 1 h at 37°C on surfaces coated with 50 nM FN in the continued presence of peptides. Each point is the mean of three independent experiments; bars represent the SEM. (B) Effects of TAT-FAS peptides on cell area. C2C12 cells were preloaded with 200 nM peptides for 1 h and plated for 1 h at 37°C on surfaces coated with the indicated concentrations of FN in the continued presence of peptides, then fixed and imaged by phase-contrast microscopy. Cell perimeters were outlined morphometrically and areas calculated. Each point represents the mean of triplicate experiments (60–80 cells in total); bars indicate the SEM. (C) Effect of TAT-FAS peptides on cell spreading on FN. C2C12 cells were preloaded with 200 nM peptides for 1 h, plated onto 50 nM FN at 37°C for the indicated times in the continued presence of peptides, then fixed and stained for F-actin. Cell perimeters were outlined morphometrically and areas calculated. Each point represents the mean of triplicate experiments (80–100 cells in total); bars indicate the SEM. (D) Effects of TAT-FAS peptides on FN-induced cell migration. C2C12 cells were preloaded with 200 nM peptides for 1 h, plated for 1 h at 37°C on surfaces coated with the indicated concentrations of FN and imaged for 2 h by time-lapse phase-contrast microscopy in the continuing presence of peptides. Migration speeds were calculated by measuring the change in position of cell nuclei over time, with exclusion of mitotic cells. Each bar represents the mean of triplicate experiments (60–80 cells in total); bars indicate the SEM. Differences significant to at least P = 0.05 are indicated by an asterisk. (E) Effects of TAT-FAS peptides on lamellipodial persistence. Directional persistence of cell migration was calculated as the ratio of the number of changes in direction of lamellipodial extensions, divided by the total distance moved in the 2-h filming period. Each bar represents the mean of triplicate experiments (60–80 cells in total); bars indicate the SEM. Cells treated with TAT-FAS S39A had significantly decreased lamellipodial persistence (P <0.001).
Next, we examined effects of the peptides on cell spreading on a range of FN concentrations at the 1 h timepoint. TAT-FAS WT and TAT-FAS S39D decreased spread cell area by 30 and 40%, respectively, at the 25 and 50 nM coating concentrations, whereas the other peptides had no significant effect (Figure 7B). At the 6 nM coating concentration, cell spreading is minimal and no significant effect was detected with any of the peptides (Figure 7B; Fischer et al., 1997). These studies were extended by measuring the time dependence of the TAT-FAS S39D (which specifically inhibits the fascin–PKCα interaction) effect on cell spreading on 50 nM FN. Control cells or cells treated with scrambled or TAT-FAS S39A peptides had similar timecourses of spreading over 90 min (Figure 7C). In contrast, FAS-TAT S39D-treated cells had significantly reduced areas at times after 30 min (Figure 7C). These results all indicate that TAT-FAS S39D inhibits post-attachment events that are involved in cell spreading.
Role of the fascin–PKCα interaction in cytoskeletal organization
To investigate the role of the fascin–PKCα interaction in cytoskeletal organization, conditions for optimal spreading and cytoskeletal organization of control cells were selected (i.e. cells plated on 50 nM TSP-1 or FN for 1 h), so that any effects of the peptides could be clearly distinguished. On TSP-1, the TAT-FAS WT peptide partially inhibited cell spreading and fascin protrusions, whereas cells loaded with TAT-FAS S39A were completely rounded and without fascin spikes (Figure 8A). TAT-FAS S39D-treated cells retained arrays of fascin spikes and spread similar to control cells (Figure 8A). The results demonstrate that the fascin–actin interaction (susceptible to inhibition by TAT-FAS WT and TAT-FAS S39A; Figure 5) is important for cell spreading on TSP-1, and are in agreement with previous data on the necessity of the fascin–actin interaction for cell adhesion and migration on TSP-1 (Adams et al., 1999; Adams and Schwartz, 2000). In the context of the current study, the results provide a control demonstration that the peptides have appropriate properties in a physiological assay.

Fig. 8. Effects of TAT-FAS peptides on F-actin organization and matrix contact structures. C2C12 cells were preloaded for 1 h with 200 nM peptides as indicated, plated onto 50 nM TSP-1 (A), or 50 nM FN (B and C), for 1 h in the continued presence of peptides and fixed and stained for (A) fascin, or (B) F-actin, fascin or phosphotyrosine. Bars, 10 µm. (C) Higher magnification views of cell edges. Bar, 5 µm. Images are representative of six independent experiments.
Further experiments focused on cell adhesion to FN, where PKCα activity is necessary for full cell spreading (Adams et al., 1999). Treatment of cells with TAT-FAS WT or TAT-FAS S39D correlated with major reductions in stress fiber organization and a striking increase in phosphotyrosine staining in focal adhesions in cells plated on FN (Figure 8B). Adhesions tended to be localized at cell margins rather than distributed across the ventral surfaces of cells (Figure 8B, compare scr. control panel with TAT-FAS WT- and TAT-FAS S39D-treated panels). Alterations to focal adhesion size and distribution were also apparent by staining for vinculin (Figure 8C; data not shown). Cells treated with TAT-FAS S39A were more angular in morphology than control cells. However, stress fibers were maintained in the cell bodies and the distribution and phosphotyrosine content of focal adhesions was similar to that of control cells (Figure 8B). In time-course experiments, we found that the effects of TAT-FAS S39D on F-actin and focal adhesions were first apparent after 30 min, and became more pronounced compared with control cells over the next hour, in agreement with the time-course of effects on cell area (Figure 7C; data not shown).
Under normal conditions, fascin-containing spikes and ruffles are formed transiently during initial attachment to FN and spread cells have diffusely distributed fascin (Adams, 1995). Fascin distribution was not altered in cells treated with the scrambled peptide or TAT-FAS S39A (Figure 8B). In striking contrast, cells treated with TAT-FAS WT or TAT-FAS S39D were enriched for fascin in extended protrusions, ruffles or bunches of short spikes at cell margins (Figure 8B and C). These results identify a requirement for the fascin–PKCα interaction for full cell spreading and cytoskeletal organization on fibronectin.
Role of the fascin–PKCα interaction in FN-induced cell migration
To relate the documented changes in cell spreading and cytoskeletal organization to other cell behaviors, we examined the effects of the peptides on haptotactic cell migration on FN. Because cell migration speeds depend on the concentration of the substratum ligand (Palecek et al., 1997), these assays were carried out on three coating concentrations of FN to enable proper comparison of dose–response behavior under each experimental condition. As expected, control cells produced a clear bell-shaped dose–response curve, with maximum migration on the intermediate concentration of FN (Figure 7D, white bars). Cells containing FAS-TAT WT or S39D were significantly more migratory at all FN concentrations (Figure 7D, gray bars). Cells treated with FAS-TAT S39A had the same overall speed as control cells (Figure 7D, cross-hatched bars). However, these cells did not maintain a clear leading edge, and moved non-directionally with a significant decrease in the persistence of lamellipodia (Figure 7E). Thus, peptide pertubation of the interaction of fascin and PKCα resulted in increased cell migration on FN, whereas perturbation of fascin binding to actin altered lamellipodial characteristics, resulting in aberrant motility.
Discussion
We report evidence for a regulated interaction of fascin with PKCα that is functionally necessary in cell-adhesive behavior on FN. This interaction is direct, because it occurs in vitro on the purified proteins, is detectable by FRET/FLIM measurements, and can be recapitulated on recombinant domains and short peptides. Importantly, as demonstrated by the biochemical and functional activities of the TAT-FAS S39D peptide, the functionality of the interaction does not depend on actin binding by fascin. For full-length WT fascin and PKCα, the extent of association depends on PKCα activity and is triggered during cell spreading on FN. Strikingly, the interaction is rendered largely independent of PKCα activation by substitution of a phosphomimetic aspartic acid for serine at aa 39 of fascin. We postulate the existence of a positive feedback loop in cells, whereby after the extension of fascin-containing protrusions during initial cell spreading on FN, activation of PKCα drives phosphorylation of fascin. Phosphorylated fascin, by retaining PKCα, then accelerates phosphorylation of other fascin molecules. This process would be important for effective down-regulation of the actin-crosslinking activity of fascin that is necessary for cells on FN to progress to full spreading and focal adhesion assembly (Adams et al., 1999). It could also be important to maintain active PKCα at appropriate subcellular locations to participate in focal adhesion assembly and maturation.
The fascin-binding site on PKCα is located in the C1B domain of the regulatory domain. The comparable regions of PKCβI and βII have 90% sequence identity; thus, it is possible that fascin may also interact with PKCβ in cells which express this isoform. Other protein interactions mapped to the regulatory domain of conventional PKCs include the interaction of RACK1 with the C2 region of PKCβ (Mochly-Rosen et al., 1995) and the binding of integrin β1 subunit to the V3 region of PKCα (Ng et al., 1999a). The C2 region has also been implicated in the binding of vinculin tail domain to PKCα (Ziegler et al., 2002). These interactions anchor or translocate PKCs to appropriate cellular sites and may serve to place PKCs within alternate signaling complexes that couple to distinct effectors (reviewed by Jaken and Parker, 2000). During cell spreading on FN, integrin β1 subunit coassociates with PKCα and fascin. In cells on FN, fascin-bound PKCα was found to be hyperphosphorylated at the T250 residue (data not shown). We hypothesize that the subpopulation of PKCα that is activated as a result of integrin engagement by FN may adopt a more favorable (i.e. open, autophosphorylated) conformation for binding to fascin in situ. It will be of future interest to characterize the extent of protein complex formation on active PKCα.
The novel TAT-FAS peptides we have developed provided a sensitive means to probe the functional significance of the fascin–PKCα interaction, because the monomeric peptides lack the actin cross-linking activity of intact fascin, do not themselves activate PKCα and can be employed to distinguish definitively between the actin binding and PKCα binding activities of fascin aa 33–47. Although cells treated with TAT-FAS WT or S39D peptides attached normally to FN, decreased cell spreading and changes in cytoskeletal organization that included enhancement of fascin-containing protrusions and alterations to focal adhesion assembly occurred subsequently. Elevated phosphotyrosine content is well known as a marker of actively remodeling focal adhesions at cell margins (reviewed by Parsons et al., 2000), and our results demonstrated that the combination of increased fascin protrusions and active, cortical focal adhesions is favorable to cell migration. The spatial concentration of fascin with actin in protrusions is clearly necessary in this process, because TAT-FAS S39A-treated cells, which had lamellipodia that were not enriched for fascin, had reduced directional persistence.
In other experimental systems, the extension of cell protrusions has been correlated with development of tractional force through focal adhesions (Pelham and Wang, 1999; Parker et al., 2002). Our studies uncover an unexpected molecular mechanism of functional significance for crosstalk between distinct forms of cell–ECM contacts. There are numerous in vivo contexts where stimulation of cell protrusions and accelerated cell migration could be of therapeutic benefit, for example in wound repair or spinal cord injury. Conversely, enhancement of the fascin–PKCα interaction could be valuable to decrease cell migration in FN-rich matrices, for example to block metastatic spread of tumor cells. We suggest that regulation of the fascin–PKCα interaction could be advantageous as a potential therapeutic approach, because, as demonstrated here, the balance of cell–matrix contacts and subsequent cell behavior can be modulated without general alterations to cell signaling pathways or loss of cell–ECM attachment.
Materials and methods
Materials and transient expression procedures
C2C12 mouse skeletal myoblastic cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 20% fetal calf serum (FCS). MCF-7 human breast cancer cells were grown in DMEM containing 10% FCS. For FRET measurements, cells were microinjected in the nucleus with EGFP–fascin (Adams and Schwartz, 2000) and PKCα-myc (Ng et al., 1999a) at a plasmid ratio of 1:2 and incubated at 37°C for 3–4 h, until EGFP expression was detectable. EGFP–fascin-expressing cells on the same coverslip were either left untreated (–mAb 9E10-Cy3) or injected in the cytoplasm with a Cy3-conjugated mAb 9E10 (32) (+mAb 9E10-Cy3) and incubated for 30 min. The GFP- and GST-tagged PKCα constructs were as described previously (Parsons et al., 2002). Transfections, cell extractions, immunoprecipitations and preparation of recombinant proteins were carried out by standard procedures, as described in Supplementary data, available at The EMBO Journal Online.
FRET determination by FLIM measurements
The FLIM apparatus used for FRET determination was as described previously (Bastiaens and Squire, 1999). All images were taken using a Zeiss Plan-APOCHROMAT 100×/1.4NA phase 3 oil objective, and the homodyne phase sensitive images were recorded at a modulation frequency of 80.224 MHz. The donor (EGFP–fascin) was excited using the 488 nm line of an argon/krypton laser and the resultant fluorescence separated using a combination of dichroic beamsplitter (Q 505 LP; Chroma Technology Corp.) and narrow band emitter filter (BP514/10; Lys & Optik). Acceptor images (Cy3-conjugated anti-MYC antibody) were recorded using a 100 W mercury arc lamp (Zeiss Attoarc) as a source of sample illumination combined with a high Q Cy3 filter set (exciter: HQ 535/50; dichroic: Q 565 LP; emitter: HQ 610/75 LP; Chroma Technology Corp.).
PKCα kinase assays
PKCα kinase activity was measured by a standard in vitro 32P incorporation assay for 30 min at 30°C as described in Supplementary data. This timepoint was previously determined to be optimal for ezrin phosphorylation (Ng et al., 2001). Assays were run in the presence or absence of the TAT-FAS peptides at 0.1, 0.5 or 1 mg/ml, and were stopped by addition of an equal volume of Laemmli sample buffer, resolved on 8% polyacrylamide gels under reducing conditions, and phosphorylated proteins detected by autoradiography. Band intensities for ezrin and PKCα were quantified by scanning densitometry and were taken as 100% in the presence of lipids and the absence of peptides for each band. Band intensities obtained in the presence of peptides were expressed as a percentage of control. All measurements were made in triplicate.
Protein and peptide pull-downs and immunoblotting
Synthetic peptides corresponding to the HIV TAT protein membrane permeablization sequence (Becker-Hapak et al., 2001) fused to the sequence of aa 33–47 of fascin (identical in human and mouse fascin) were prepared in unconjugated, N-terminal biotinylated, or N-terminal FITC-conjugated forms, by Genemed Synthesis Inc. For pull-downs, biotinylated peptides were bound to streptavidin–agarose (Sigma) for 16 h at 4°C. After washing three times in 1% Triton X-100 in Tris-buffered saline, beads were incubated with 20–200 µg aliquots of cell lysates, or 10 µg of purified rabbit skeletal muscle actin (Sigma) in F-actin buffer (10 mM Tris pH 7.5, 130 mM KCl, 20 mM NaCl, 2 mM MgCl2, 1 mM β-mercaptoethanol, 0.1 mM EGTA, 0.1 mM ATP), for 4 h at 4°C. To examine direct binding between fascin and PKCα, 400 ng of the purified fascin was bound to Ni-NTA beads and mixed with 100 ng recombinant PKCα (PanVera), or with cell extracts, in buffer containing 20 mM HEPES, pH 7.4, 5 mM MgCl2, 1.25 mM CaCl2, 1 mM dithiothreitol, 1 mM ATP, 0.03% Triton X-100 in the presence or absence of activating lipid mixture, with or without TAT-FAS peptides, for 30 min at 30°C. Beads were collected by centrifugation, the supernatants stored separately, and the beads washed four times in buffer containing 0.1% Triton X-100. Bound proteins were collected into Laemmli sample buffer, resolved on 10% or 12.5% polyacrylamide gels, transferred to nitrocellulose (Amersham Hybond C-super, 0.22 µM pore size), and immunoblotted for fascin (Adams et al., 1999) or PKCα (Sigma monoclone MC-5 or rabbit polyclonal). In other experiments, blots were probed as appropriate with antibodies to actin (Sigma clone AC-40), tubulin (Sigma clone DM1A) or Raf-1 (either S338P rat monoclonal from Upstate Biotechnology, S259P rabbit polyclonal from Cell Signaling or c-Raf-1 protein mouse monoclonal antibody from BD Transduction Labs). Rabbit antiserum reactive with FAS-S39PO4 was raised against the synthetic peptide Ac-VNASAS(pS)LKKKQIWC-amide at New England Peptide. Immunoglobulins were preabsorbed on the equivalent non-phosphorylated peptide coupled to Sepharose CL-6B, followed by binding to phosphorylated peptide–Sepharose and elution with 0.2 M glycine at pH 2.8 and characterized for specific reactivity with phosphorylated fascin (J.C.Adams and M.A.Schwartz, in revision). Rabbit anti-phospho-T250 PKCα (Ng et al., 1999b) was preincubated for 30 min at room temperature with 1 µg/ml cognate dephosphorylated peptide (WDRTTRND) to block any reactivity with the non-phosphorylated form of PKCα.
Cell adhesion and cytoskeletal organization
Cell attachment was quantified as described previously (Anilkumar et al., 2002). Cytoskeletal organization assays were carried out as described, and are detailed in Supplementary data (Adams, 1995).
Measurement of cell spreading and haptotactic migration
The time-lapse setup was as described previously (Adams, 1997) and is detailed in Supplementary data. Images were analysed in Improvision Openlab 3.0.9. Cell perimeter and cell area were measured at the start of filming and cell movement was measured as displacement of cell nuclei over time. Lamellipodial persistence was determined by calculating the number of changes in direction of lamellipodial extensions (either extension of a new lamellipodium or redirection and regrowth of the existing lamellipodium) as a fraction of the total time period. At least 15 cells were scored from each sample and data from four replicate experiments were pooled (in total ∼60–80 cells for each experimental condition) in Microsoft Excel worksheets for statistical analysis. The significance of peptide effects was calculated by two-tailed _t_-test assuming equal variance in comparison to scrambled peptide-treated cells, or by pairwise _t_-tests between the TAT-FAS WT, S39A and S39D peptide treatments.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
This research was supported by the Wellcome Trust (SFBBR 038284 and ITF 057379), NIGMS RO1 068073 to J.C.A. and the UK Medical Research Council (Clinician Scientist Grant award to T.N.).
References
- Adams J.C. (1995) Formation of stable microspikes containing actin and the 55kDa actin-bundling protein, fascin, is a consequence of cell adhesion to thrombospondin-1: implications for the antiadhesive activities of thrombospondin-1. J. Cell Sci., 108, 1977–1990. [DOI] [PubMed] [Google Scholar]
- Adams J.C. (1997) Characterisation of cell–matrix adhesion requirements for the formation of fascin microspikes. Mol. Biol. Cell, 8, 2345–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J.C. (2001) Cell–matrix contact structures. Cell. Mol. Life Sci., 58, 371–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J.C. and Lawler,J. (1994) Cell-type specific adhesive interactions of skeletal myoblasts with thrombospondin-1. Mol. Biol. Cell, 5, 423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J.C. and Schwartz,M.A. (2000) Stimulation of fascin spikes by thrombospondin-1 is mediated by the GTPases Rac and Cdc42. J. Cell Biol., 150, 807–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J.C, Clelland,J.D., Collett,G.D.M., Matsumura,F., Yamashiro,S. and Zhang,L. (1999) Cell–matrix adhesions differentially regulate fascin phosphorylation. Mol. Biol. Cell, 10, 4177–4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anilkumar N., Annis,D.S., Mosher,D.F. and Adams,J.C. (2002) Trimeric assembly of the C-terminal region of thrombospondin-1 or thrombospondin-2 is necessary for cell spreading and fascin spike organisation. J. Cell Sci., 115, 2357–2366. [DOI] [PubMed] [Google Scholar]
- Bastiaens P.I. and Squire,A. (1999) Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell. Trends Cell Biol., 9, 48–52. [DOI] [PubMed] [Google Scholar]
- Becker-Hapak M., McAllister,S.S. and Dowdy,S.F. (2001) TAT-mediated protein transduction into mammalian cells. Methods, 24, 247–256. [DOI] [PubMed] [Google Scholar]
- Cheng J.-J., Wung,B.-S., Chao,Y.-J. and Wang,D.L. (2001) Sequential activation of protein kinase C (PKC)-α and PKC-ε contributes to sustained Raf/ERK1/2 activation in endothelial cells under mechanical strain. J. Biol. Chem., 276, 31368–31375. [DOI] [PubMed] [Google Scholar]
- Danen E.H. and Yamada,K.M. (2001) Fibronectin, integrins and growth control. J. Cell. Physiol., 189, 1–13. [DOI] [PubMed] [Google Scholar]
- Evan G.I., Lewis,G.K., Ramsay,G. and Bishop,J.M. (1985) Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol., 5, 3610–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D., Tucker,R.P., Chiquet-Ehrismann,R. and Adams,J.C. (1997) Cell-adhesive responses to tenascin-C splice variants involve formation of fascin microspikes. Mol. Biol. Cell, 8, 2055–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch S.M. and Francis,H. (1994) Disruption of epithelial cell–matrix interactions induces apoptosis. J. Cell Biol., 124, 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B., Bershadsky,A., Pankov,R. and Yamada,K.M. (2001) Transmembrane crosstalk between the extracellular matrix–cytoskeleton. Nat. Rev. Mol. Cell Biol., 2, 793–805. [DOI] [PubMed] [Google Scholar]
- Giancotti F.G. and Ruoslahti,E. (1999) Integrin signaling. Science, 285, 1028–1032. [DOI] [PubMed] [Google Scholar]
- Gumbiner B.M. (1996) Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell, 84, 345–357. [DOI] [PubMed] [Google Scholar]
- Jaken S. and Parker,P.J. (2000) Protein kinase C binding partners. BioEssays, 22, 245–254. [DOI] [PubMed] [Google Scholar]
- Kureishy N., Sapountzi,V., Prag,S., Anilkumar,N. and Adams,J.C. (2002) Fascins and their roles in cell structure and function. BioEssays, 24, 350–361. [DOI] [PubMed] [Google Scholar]
- Lauffenburger D.A. and Horwitz,A.F. (1996) Cell migration: a physically integrated molecular process. Cell, 84, 359–369. [DOI] [PubMed] [Google Scholar]
- Legg J.W., Lewis,C.A., Parsons,M., Ng,T. and Isacke,C.M. (2002) A novel PKC-regulated mechanism controls CD44–ezrin association and directional cell motility. Nat. Cell Biol., 4, 399–407. [DOI] [PubMed] [Google Scholar]
- Meredith J.E., Fazeli,B. and Schwartz,M.A. (1993) The extracellular matrix as a cell survival factor. Mol. Biol. Cell, 4, 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranti C.K., Ohno,S. and Brugge,J.S. (1999) Protein kinase C regulates integrin-induced activation of the extracellular regulated kinase pathway upstream of Shc. J. Biol. Chem., 274, 10571–10581. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D., Smith,B.L., Chen,C.H., Disatnik,M.H. and Ron,D. (1995) Interaction of protein kinase C with RACK1, a receptor for activated C-kinase: a role in beta protein kinase C mediated signal transduction. Biochem. Soc. Trans, 23, 596–600. [DOI] [PubMed] [Google Scholar]
- Morrison D.K. and Cutler,R.E. (1997) The complexity of Raf-1 regulation. Curr. Opin. Cell Biol., 9, 174–179. [DOI] [PubMed] [Google Scholar]
- Ng T., Shima,D., Squire,A., Bastiaens,P.I., Gschmeissner,S., Humphries,M.J. and Parker,P.J. (1999a) PKCα regulates beta1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J., 18, 3909–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T. et al. (1999b) Imaging protein kinase Cα activation in cells. Science, 283, 2085–2089. [DOI] [PubMed] [Google Scholar]
- Ng T. et al. (2001) Ezrin is a downstream effector of trafficking PKC–integrin complexes involved in the control of cell motility. EMBO J., 20, 2723–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S., Yamakita,Y., Yamashiro,S., Matsudaira,P.T., Gnarra,J.R., Obinata,T. and Matsumura,F. (1997) Identification of an actin-binding region and a protein kinase C phosphorylation site on human fascin. J. Biol. Chem., 272, 2527–2533. [DOI] [PubMed] [Google Scholar]
- Palecek S.P., Loftus,J.C., Ginsberg,M.H., Lauffenburger,D.A. and Horwitz,A.F. (1997) Integrin–ligand binding properties govern cell migration speed through cell–substratum adhesiveness. Nature, 385, 537–540. [DOI] [PubMed] [Google Scholar]
- Parker K.P. et al. (2002) Directional control of lamellipodial extension by constraining cell shape and orienting cell tractional forces. FASEB J., 16, 1195–1204. [DOI] [PubMed] [Google Scholar]
- Parsons J.T., Martin,K.H., Slack,J.K., Taylor,J.M. and Weed,S.A. (2000) Focal adhesion kinase: a regulator of focal adhesion dynamics and cell movement. Oncogene, 19, 5606–5613. [DOI] [PubMed] [Google Scholar]
- Parsons M. et al. (2002) Site-directed perturbation of protein kinase C–integrin interaction blocks carcinoma cell chemotaxis. Mol. Cell. Biol., 22, 5897–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham R.J. and Wang,Y.-L. (1999) High resolution detection of mechanical forces exerted by locomoting fibroblasts on the substrate. Mol. Biol. Cell, 10, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M.A. and Ginsberg,M.H. (2002) Networks and crosstalk: integrin signalling spreads. Nat. Cell Biol., 4, E65–E68. [DOI] [PubMed] [Google Scholar]
- Sipe J.D. (2002) Tissue engineering and reparative medicine. Ann. N. Y. Acad. Sci., 961, 1–9. [DOI] [PubMed] [Google Scholar]
- Vuori K. and Ruoslahti,E. (1993) Activation of protein kinase C precedes alpha 5 beta 1 integrin-mediated cell spreading on fibronectin. J. Biol. Chem., 268, 21459–21462. [PubMed] [Google Scholar]
- Yamakita Y., Ono,S., Matsumura,F. and Yamashiro,S. (1996) Phosphorylation of human fascin inhibits its actin binding and bundling activities. J. Biol. Chem., 271, 12632–12638. [DOI] [PubMed] [Google Scholar]
- Ziegler W.H., Tigges,U., Zieseniss,A. and Jockusch,B.M. (2002) A lipid-regulated docking site on vinculin for protein kinase C. J. Biol. Chem., 277, 7396–7404. [DOI] [PubMed] [Google Scholar]