Overexpression of the Dynamitin (p50) Subunit of the Dynactin Complex Disrupts Dynein-dependent Maintenance of Membrane Organelle Distribution (original) (raw)
Abstract
Dynactin is a multisubunit complex that plays an accessory role in cytoplasmic dynein function. Overexpression in mammalian cells of one dynactin subunit, dynamitin, disrupts the complex, resulting in dissociation of cytoplasmic dynein from prometaphase kinetochores, with consequent perturbation of mitosis (Echeverri, C.J., B.M. Paschal, K.T. Vaughan, and R.B. Vallee. 1996. J. Cell Biol. 132:617–634). Based on these results, dynactin was proposed to play a role in linking cytoplasmic dynein to kinetochores and, potentially, to membrane organelles. The current study reports on the dynamitin interphase phenotype. In dynamitin-overexpressing cells, early endosomes (labeled with antitransferrin receptor), as well as late endosomes and lysosomes (labeled with anti–lysosome-associated membrane protein-1 [LAMP-1]), were redistributed to the cell periphery. This redistribution was disrupted by nocodazole, implicating an underlying plus end–directed microtubule motor activity. The Golgi stack, monitored using sialyltransferase, galactosyltransferase, and _N_-acetylglucosaminyltransferase I, was dramatically disrupted into scattered structures that colocalized with components of the intermediate compartment (ERGIC-53 and ERD-2). The disrupted Golgi elements were revealed by EM to represent short stacks similar to those formed by microtubule-depolymerizing agents. Golgi-to-ER traffic of stack markers induced by brefeldin A was not inhibited by dynamitin overexpression. Time-lapse observations of dynamitin-overexpressing cells recovering from brefeldin A treatment revealed that the scattered Golgi elements do not undergo microtubule-based transport as seen in control cells, but rather, remain stationary at or near their ER exit sites. These results indicate that dynactin is specifically required for ongoing centripetal movement of endocytic organelles and components of the intermediate compartment. Results similar to those of dynamitin overexpression were obtained by microinjection with antidynein intermediate chain antibody, consistent with a role for dynactin in mediating interactions of cytoplasmic dynein with specific membrane organelles. These results suggest that dynamitin plays a pivotal role in regulating organelle movement at the level of motor–cargo binding.
Cytoplasmic dynein is a multisubunit, minus end– directed microtubule motor protein complex that has been implicated in a wide variety of cellular processes (Paschal and Vallee, 1987). Dynactin is a similar-sized complex found in cytoplasmic dynein preparations, which appears to serve as an accessory factor in cytoplasmic dynein function (reviewed in Allan, 1996; Schroer, 1996; Vallee and Sheetz, 1996). Structural analysis of the dynactin complex showed that it contains a 37-nm F-actin–like filament composed of the actin-related protein Arp-1, capped at one end by actin-capping protein and at the other end by a 62-kD subunit (Schafer et al., 1994). A large, multifunctional polypeptide, p150_Glued_, the mammalian homologue of the product of the Drosophila gene Glued (Holzbaur et al., 1991), protrudes as a filamentous projection from one end of the Arp1 filament. Dynamitin, a 50-kD subunit present at four to five moles per mole of dynactin, has not yet been localized within the complex, but may be important in regulating its assembly (Echeverri et al., 1996; see below).
Dynactin has been observed to stimulate cytoplasmic dynein-mediated vesicle movement in vitro (Schroer and Sheetz, 1991), suggesting a role in the regulation of dynein activity. Mutations in dynactin genes have also been found to produce dynein-like phenotypes in several organisms. In yeast, deletion of the Act5/Act3 locus, which encodes a homologue of Arp1, produces a defect in nuclear migration that results in chromosome missegregation (Clark and Meyer, 1994; Muhua et al., 1994), comparable to the phenotype produced by a dynein heavy chain knockout (Eshel et al., 1993). In Neurospora, mutations in ro-4 (Arp1) and ro-3 (p150_Glued_) block nuclear migration into the hyphae (Plamann et al., 1994), a phenotype also seen with mutations in the cytoplasmic dynein heavy chain in both Neurospora (ro-1; Plamann et al., 1994) and Aspergillus (NudA; Xiang et al., 1994). In Drosophila, the long-known defects in nervous system and eye development produced by mutations in the Glued gene (Plough and Ives, 1935; Meyerowitz and Kankel, 1978) have also been observed with mutations in the cytoplasmic dynein heavy chain (Dhc64C), some alleles of which also suppress mutations in Glued (McGrail et al., 1995). Together, these data suggest that dynein and dynactin function in a common pathway.
Evidence for a direct interaction between the intermediate chain subunits of cytoplasmic dynein and the p150_Glued_ component of dynactin has come from blot overlay analysis and affinity chromatography (Karki and Holzbaur, 1995; Vaughan and Vallee, 1995). Because the intermediate chains reside at the base of the dynein molecule (Steffen et al., 1996) and have been implicated in anchoring flagellar dynein within the axoneme (King and Witman, 1990; King et al., 1991), these data suggested a role for the dynactin complex in anchoring cytoplasmic dynein to its cargo organelles. Further evidence in support of this model has come from molecular analysis of the 50-kD dynamitin subunit of dynactin (Echeverri et al., 1996). Overexpression of dynamitin in COS-7 cells caused a pronounced increase in mitotic index. Mitotic cells were found to be delayed in prometaphase, and spindle organization was also perturbed. Sucrose density gradient analysis of extracts of transfected cell cultures revealed that the p150_Glued_ component of dynactin was dissociated from the rest of the complex, demonstrating a direct effect of dynamitin overexpression on dynactin itself. p150_Glued_ and Arp1 labeling of prometaphase kinetochores was clearly reduced, as was dynein labeling, supporting a role for dynactin in anchoring or targeting dynein to the kinetochore.
It is uncertain whether dynactin has a more general role in interphase dynein function. By extension of the mitotic results, it was proposed that dynactin serves to link cytoplasmic dynein to membranous organelles as well (Echeverri et al., 1996), perhaps as part of a spectrin-like lattice on the organelle surface (Vaughan and Vallee, 1995; Vallee et al., 1995). In support of the latter aspect of this hypothesis, spectrin and adducin have been found as components of dynactin immunoprecipitates, and spectrin was observed to colocalize with overexpressed Arp1 in cultured mammalian cells (Holleran et al., 1996). However, phenotypic evidence of a role for dynactin in dynein-mediated membrane dynamics has been limited. For example, although Arp1-overexpression affected Golgi morphology (Holleran et al., 1996), it is uncertain whether this represented physical distortion of the Golgi by the extensive array of Arp1 filaments rather than interference with dynactin activity. It also remains unclear whether dynactin is restricted in function to dynein-mediated processes, or whether it is involved in plus end motility as well. The latter prospect is of interest in view of the partial competition between kinesin and dynein for binding to microsomal membranes (Yu et al., 1992). Data have also been reported for a weak interaction between cytoplasmic dynein and kinectin (Kumar et al., 1995), a protein proposed to serve as a membrane surface receptor for kinesin (reviewed in Burkhardt, 1996).
Dynamitin overexpression offers the promise of a powerful new means of addressing a number of important outstanding issues regarding both dynein and dynactin function. Analysis of the mitotic phenotype of dynamitin overexpressors has already provided physiological evidence for a role for cytoplasmic dynein in prometaphase. In this study, we characterize the dynamitin overexpression phenotype in interphase mammalian cells. Our data provide strong support for a specific role for dynactin in dynein function, and they provide direct evidence for the involvement of dynactin and dynein in maintaining the dynamic distribution of the Golgi apparatus, intermediate compartment, early and late endosomes, and lysosomes. Evidence for a residual plus end–directed motor activity associated with late endosomes and lysosomes is presented, indicating that dynactin disruption specifically inhibits minus end–directed motor activity.
Materials and Methods
cDNAs and Antibodies
Cloning of pCMV-H50 and pCMVH50m was described previously (Echeverri et al., 1996). pCMVβ was obtained from Clontech Laboratories (Palo Alto, CA). pCMV-GFP-LpA, encoding the full-length green fluorescent protein (GFP)1 under the control of the cytomegalovirus promoter, was a gift from Dr. H. Stunnenberg (European Molecular Biology Laboratory [EMBL], Heidelberg, Germany). pCNG2, encoding the GFP-, β1,2_N_-acetylglucosaminyltransferase I (NAGT-I) fusion protein under control of the cytomegalovirus promoter, was a gift of Drs. D. Shima and G. Warren (Imperial Cancer Research Fund, London, U.K.).
Monoclonal anti–dynamitin 50-1 was described previously (Paschal et al., 1993). Rabbit antipeptide antisera against the 17–amino acid c-myc epitope and the 13–amino acid vesicular stomatitis virus (VSV-G) epitope were generated and affinity purified, and will be described elsewhere. The mAbs 9E10 and P5D4 recognizing the c-myc and VSV-G epitopes, respectively, have been described elsewhere (Evan et al., 1985). Rabbit anti–β-galactosidase was purchased from 5 Prime—3 Prime, Inc., (Boulder, CO). Mouse monoclonal anti–mitochondria AE-1 was purchased from Biodesign International (Kennebunk, ME). Rabbit anti–human lysosome-associated membrane protein-1 (LAMP)-1 and -2 were gifts from Dr. S. Carlsson (University of Umea, Umea, Sweden). Monoclonal anti– human LAMP-1 H4A3, developed by Dr. T. August (Johns Hopkins University, Baltimore, MD) was purchased from the Developmental Studies Hybridoma Bank, and was maintained by Johns Hopkins University and the University of Iowa (Iowa City, IA) under contract NO1-HD-2-3144 from the National Institute of Child Health and Human Development. Monoclonal anti–human transferrin receptor B3/25 was purchased from Boehringer Mannheim (Indianapolis, IN). Rabbit anticatalase antibody (Baumgart et al., 1989) was a gift from Dr. H.D. Fahimi (University of Heidelberg, Heidelberg, Germany). Rabbit polyclonal anti-TGN38 (Luzio et al., 1990) was a gift from Drs. P. Luzio (University of Cambridge, Cambridge, U.K.) and G. Banting (University of Bristol, Bristol, U.K.). Monoclonal anti-ERGIC53 G1/93 (Schweizer et al., 1990) was a gift from Dr. H.-P. Hauri (Biozentrum, Basel, Switzerland). Rabbit polyclonal antibody recognizing rat mannosidase II (Moremen and Robbins, 1991) was a gift from Dr. K. Moremen (University of Georgia, Athens, GA). Mouse monoclonal antidynein intermediate chain antibody 70.1 (Steuer et al., 1990) and isotype-matched mouse anti-IgG were purchased from Sigma Chemical Co. (St. Louis, MO). Mouse anti-myosin was a gift from Dr. Michael Glotzer (EMBL). Rabbit anti–protein disulfide isomerase was a gift from Dr. Steve Fuller (EMBL), and rabbit anti-ERD2 (Sönnichsen et al., 1994) was a gift from Drs. P. Nguyen Van and H.D. Söling (University of Göttingen, Göttingen, Germany).
Cell Culture and Transient Transfection
HeLa cell lines stably transfected with α-2,6-sialyltransferase (ST) epitope-tagged with a VSV-G epitope, murine mannosidase II, NAGT-I epitope-tagged with myc, and rat TGN38 were described previously (Nilsson et al., 1994; Rabouille et al., 1995). Cells were maintained in DME, 5% FCS, 200 μg/ml G418 (GIBCO BRL, Gaithersburg, MD), penicillin, and streptomycin. For transient transfection, cells were grown on coverslips to 40% confluence. CsCl-purified DNA was mixed with Lipofectamine (GIBCO BRL) according to the manufacturer's recommendations. Cells were washed once with serum-free medium and incubated with the DNA/liposome mixture in serum-free medium for 6 h, after which DNA was removed and replaced with complete medium. Cells were fixed for immunofluorescence 18–22 h after application of the DNA. COS-7 cells were cultured and transfected as described previously (Echeverri et al., 1996).
Drug Treatments
Cells were treated with nocodazole (Sigma Chemical Co.) at 20 μM for 30 min at 4°C, and then for an additional 2.5 h at 37°C. Alternatively, cells were treated with 2 μg/ml brefeldin A (BFA; Epicentre Technologies, Madison, WI) for 20 min. For recovery studies, cells were washed three times with drug-free medium and were incubated for the indicated times at 37°C in the absence of drug.
Fluorescence Microscopy
Processing of coverslips for immunofluorescence labeling of membrane proteins was performed as described previously (Burkhardt et al., 1993). Briefly, coverslips were fixed with 2% paraformaldehyde/PBS, quenched with ammonium chloride, and permeabilized with PBS/0.01% saponin/ 0.25% gelatin (PSG). Samples were incubated with primary and secondary antibodies, diluted in PSG for 30 min to 1 h, and washed four times with PSG after each antibody. For actin labeling, cells were fixed with 3% paraformaldehyde in CB (10 mM MES/150 mM NaCl/5 mM EGTA/5 mM MgCl2/5 mM glucose), permeabilized for 1 min with 0.3% Triton X-100, and incubated with Bodipy phallicidin or rhodamine phalloidin (Molecular Probes Inc., Eugene, OR). For microtubule labeling, cells were fixed and permeabilized simultaneously with 3% paraformaldehyde/0.1% glutaraldehyde/0.3% Triton X-100 in CB, quenched with NaBH4, and incubated with antitubulin antibody and appropriate secondary antibody. Cells were mounted in Mowiol (Calbiochem-Novabiochem Corp., La Jolla, CA) containing 2.5% 1,4-diazobicyclo-[2.2.2]-octane (DABCO; Sigma Chemical Co.). Unless otherwise specified, samples were observed with epifluorescence optics and photographed by conventional means using a Zeiss Axiophot (Carl Zeiss, Jena, Germany) and Tmax 400 film (Kodak, Rochester, NY). Negatives were scanned at 1,200 dpi using a Scanmaker III flatbed scanner (Microtek, Düsseldorf, Germany) and processed using Adobe Photoshop (Adobe Systems Inc., Mountain View, CA). Confocal fluorescence microscopy was performed with the confocal scanning laser beam microscope (CCM) developed at the EMBL using a 100X Plan-neofluor (Zeiss, NA 1.3) objective. Confocal series were collected at 0.5-μm steps. Images were stored on a computer and processed with Adobe Photoshop.
Scoring of Golgi disruption and lysosome peripheralization was performed in a blind study. Cells were transfected with dynamitin or β-galactosidase, and were double labeled for the transfected proteins and either ST (anti-G tag) or LAMP. Without knowing which transfected protein was being scored, cells that were positive for the transfected protein were first scored arbitrarily as either “bright” or “dim” (corresponding to “high” and “moderate” overexpressors respectively), and then the phenotype of the Golgi or lysosomes was assessed. Cells exhibiting either one or two elongated juxtanuclear Golgi structures were scored as “contiguous,” those with a small number (three to five) of large Golgi elements as “partially fragmented,” and cells with numerous ST positive structures scattered throughout the cytoplasm were scored as “fragmented.” According to this system, the two transfected cells in Fig. 1 B would be scored as “fragmented,” while all of the untransfected cells would be scored as unfragmented. For lysosomes, cells were scored on a scale of 1–5, with a score of 1 given to cells where nearly all LAMP-positive structures were clustered in the juxtanuclear region, 2 to cells where lysosomes were primarily concentrated in the juxtanuclear region, 3 where lysosomes were spread, evenly or unevenly, through all parts of the cell, 4 where lysosomes were primarily concentrated in the peripheral cell processes, and 5 where lysosomes were almost exclusively concentrated in the peripheral processes. The cells shown in Fig. 2 H would receive scores of 4 and 5 on this scale. In each case, 50–60 cells were scored in each of three independent experiments. Data represent an average ± 1 SD.
Figure 1.
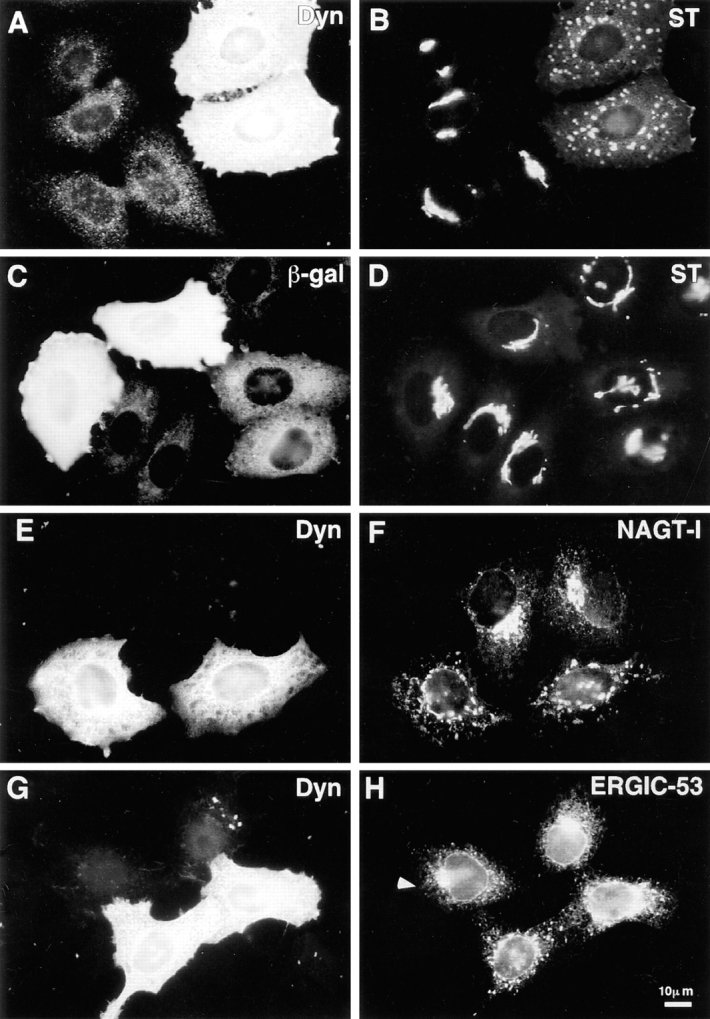
Disruption of exocytic organelles by dynamitin overexpression. HeLa cells were transiently transfected with dynamitin (A, B, E–H) or β-galactosidase (C and D), and the distribution of membrane markers in interphase cells was examined by double-label immunofluorescence microscopy. (A, C, E, and G) transfected cells labeled with antidynamitin (or anti-myc tag, E) or anti–β-galactosidase (C). (B and D) ST labeling of the _trans_-Golgi and TGN, showing that dynamitin overexpression disrupts the compact juxtanuclear Golgi complex. (F) NAGT-I labeling of the medial-Golgi shows disruption similar to ST. (H) ERGIC-53 labeling of the intermediate compartment between ER and _cis_-Golgi. Note that the juxtanuclear accumulation of ERGIC-53 is lost in the dynamitin-transfected cells, and that the normal punctate cytoplasmic fluorescence is somewhat exaggerated in dynamitin overexpressors.
Figure 2.
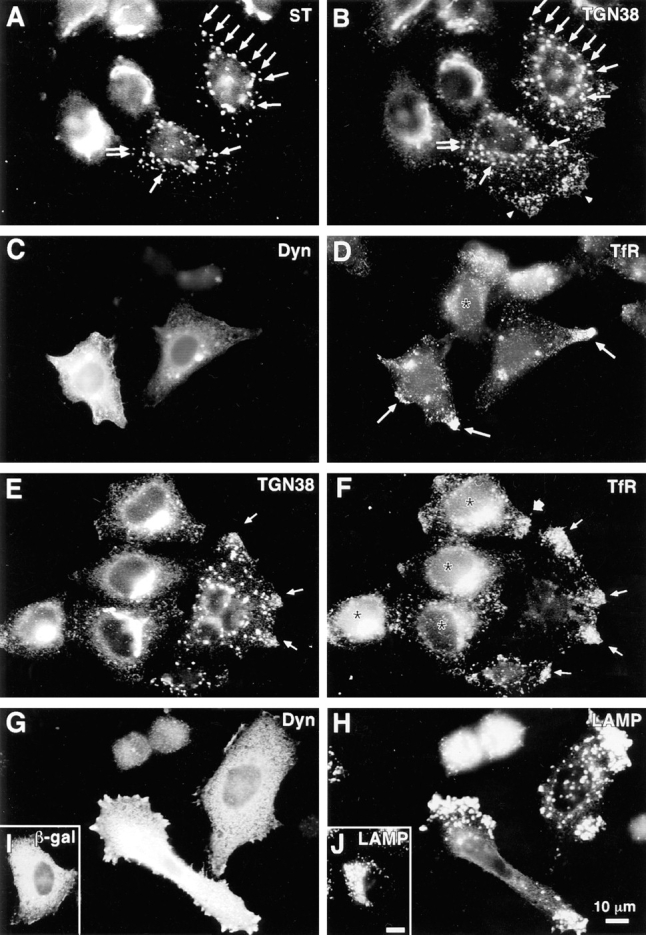
Disruption of endocytic organelles by dynamitin overexpression. HeLa cells were transfected with dynamitin (A–H) or β-galactosidase (I and J). Transfected cells are identified by labeling with antidynamitin (C and G), anti–β-galactosidase (I), or by the characteristic dynamitin-induced fragmentation of the Golgi (A and E). A and B show double labeling for ST and TGN38, respectively. In transfected cells, one pool of TGN38 colocalizes with ST (A and B, arrows), while another shifts to the periphery (B, arrowheads). (C–F) Effect of dynamitin on TfR distribution. TfR-labeled endosomes (D and F) are lost from the juxtanuclear recycling compartment, and in some cells, they accumulate at the tips of cell processes (arrows). This peripheral distribution is only infrequently observed in untransfected cells (broad arrow, F; see also Table I). Double labeling for TfR (F) and TGN38 (E) reveals that the two markers accumulate at overlapping peripheral sites (arrows). (H and J) The distribution of LAMP-1 in dynamitin overexpressors and control transfectants, respectively. Note the extreme shift of LAMP-positive late endosomes and lysosomes into the periphery. The cell elongation shown in H was sometimes observed in dynamitin-overexpressing cells, though in HeLa cells, this effect was infrequent and was not required for maximal organelle redistribution.
Microinjection
Antidynein intermediate chain antibody 70.1 ascites were dialyzed against 50 mM potassium glutamate, 0.5 mM MgCl2, pH 7.0, and concentrated using a centrifugal concentrator. Total protein concentration was determined using the BCA assay (Pierce Chemical Co., Rockford, IL) and was adjusted with dialysis buffer. Control experiments were performed using either isotype-matched mouse antimyosin or mouse anti-IgG ascites that were prepared similarly. 70-kD aldehyde-fixable FITC-dextran (Molecular Probes) was added to 1 mg/ml final concentration to enable identification of injected cells. Just before microinjection, antibody-dextran mixtures were cleared of aggregates by centrifugation for 10 min in an airfuge at 20 psi. Cells were grown overnight on coverslips and were microinjected using a Zeiss automated injection system. Cells were then returned to a CO2 incubator and incubated for 4 h at 37°C, after which they were fixed and prepared for immunofluorescence labeling of Golgi and lysosomes. Cells fluorescing in the FITC channel were selected at random, and the phenotype was scored as described above.
Cell Sorting and Golgi Length Analysis
To enrich transfected cells for analysis by EM, cells were cotransfected on 10-cm dishes with 5 μg each of pCMV-H50m and pCMV-GFP-LpA. Control cells were transfected with 10 μg of pCMV-GFP-LpA alone. Transfection was performed as described above, and at the end of the transfection period, cells were trypsinized and enrichment for GFP-positive cells was performed using a FACSTAR Plus® (Becton Dickinson & Co., San Jose, CA) cell sorter. Excitation was at 488 nm, the emitted fluorescence collected with a 530-nm bandpass filter, and gating was set to separate clear GFP expressors from negative cells, discarding low expressors. Sorted cells were then processed for electron microscopy. For epon sectioning, cells were fixed in 1% glutaraldehyde in cacodylate buffer, postfixed with osmium ferricyanide, stained en bloc with uranyl acetate, dehydrated through graded ethanols, and embedded in Epon 812. Thin sections of Golgi stacks showing three or more stacked cisternae were photographed at random at 31,000× using a Zeiss EM-10 electron microscope at 80 kV. For analysis of Golgi stack lengths, negatives were scanned using a flatbed scanner, opened with NIH Image software, and the length of a line drawn along the longest Golgi cisterna, following the contours of the stack, was determined using the curved line measurement tool. The proportion of measurements falling within various length ranges was then calculated to reveal population differences. To determine the percentage of cells expressing exogenous dynamitin within the sorted pool, parallel samples were prepared for ultrathin cryosectioning, and thawed cryosections were labeled with rabbit anti-myc antibody as described in Griffiths (1993). Randomly selected cells were scored as negative, weakly positive, or strongly positive for label. Control cells sorted into the GFP-negative pool were negative for dynamitin-myc labeling.
Live Cell Studies
COS-7 cells were seeded at 2 × 105 cells in growth medium (DME, 10% FBS, penicillin, streptomycin, l-glutamine) in 35-mm homemade observation chambers, and grown for 24 h before transfection. Transfections were carried out for 6 h in 2 ml serum- and antibiotic-free growth medium containing 2 μg total of plasmid DNA (pCMVH50m + pCNG2, or pCNG2 alone for control) and 8 μl Lipofectamine mixed according to supplier's instructions. In cotransfected samples, more than 90% of overexpressors expressed both constructs. Cells were then returned to normal growth medium and incubated at 37°C for 30–35 h before starting time-lapse observations.
Time-lapse observations were carried out using an inverted Leica DMIRB microscope (Leica, Wetzlar, Germany) equipped with temperature-, humidity- and CO2-controlled stage incubator, motor-driven z-axis stage control, and a 12-V/100-W halogen lamp (used at 7–9 V) for fluorescence illumination. All recordings (made at 3–7 s intervals) were obtained using a Leica 40×/1.00 PL Fluotar (Ph3) objective and a liquid-cooled CCD camera (CH250; Photometrics, Tucson, AZ). Camera settings, shutter, fine focusing, and epifluorescence filter wheels were all controlled through Metamorph software (version 2.5; Universal Imaging Corp., West Chester, PA).
Results
Dynamitin Overexpression Disrupts the Distribution of Specific Membrane Organelles
To define the role of dynactin in interphase cells, we used dynamitin overexpression to disrupt dynactin function, and evaluated the effects on membrane organelles. HeLa cell lines, which stably express epitope-tagged Golgi proteins (Rabouille et al., 1995), were transiently transfected with dynamitin under the control of the cytomegalovirus promoter to express high levels of exogenous dynamitin. In untransfected cells, ST was distributed in a compact juxtanuclear region (Fig. 1, A and B) corresponding to the trans portion of the Golgi stack and the _trans_-Golgi network (Rabouille et al., 1995). Overexpression of dynamitin induced a dramatic redistribution of ST into large punctate structures scattered throughout the cytoplasm (Fig. 1, A and B). This effect was specific for dynamitin, since a control plasmid expressing β-galactosidase had no effect (Fig. 1, C and D). Dynamitin overexpression led to a similar disruption of the medial Golgi markers NAGT-I (Fig. 1, E and F) and mannosidase II (not shown), as well as the _trans_-Golgi marker galactosyl-transferase (see Fig. 6 H). The severity of the Golgi disruption varied somewhat among transfected cells, usually as a result of variation in expression levels of exogenous dynamitin (see below), but the disrupted morphology did not differ among the stack markers tested.
Figure 6.
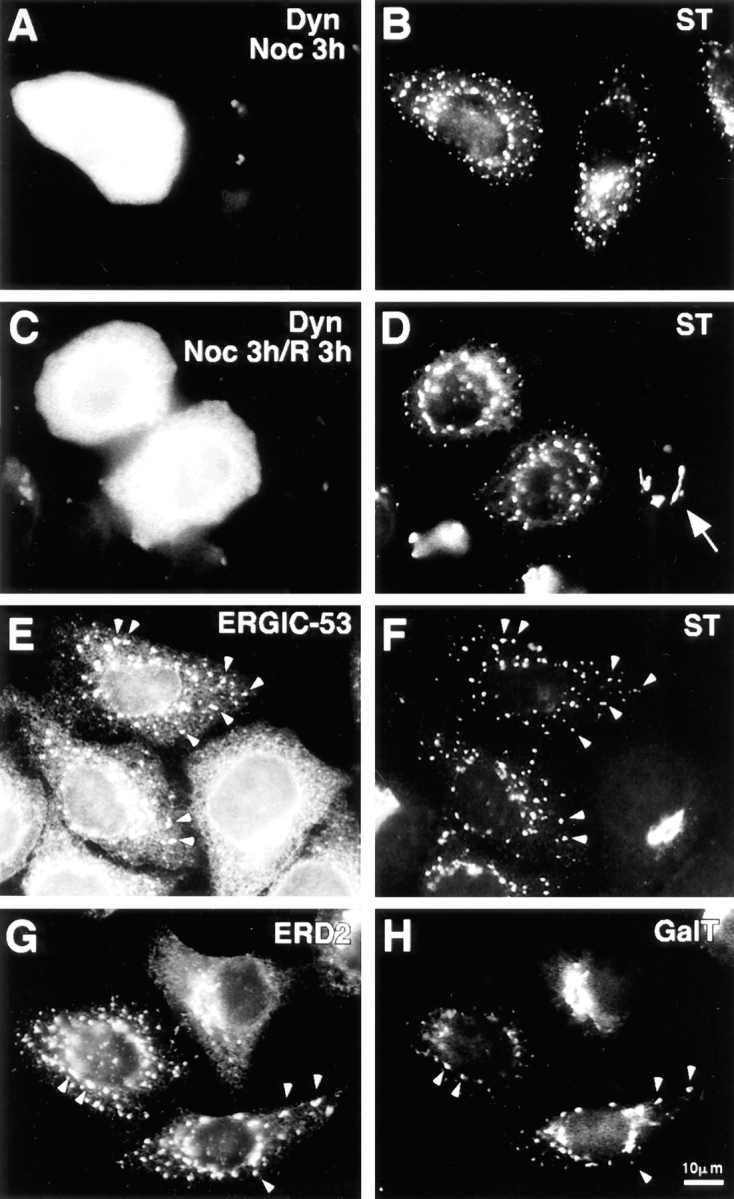
Dynamitin-mediated Golgi fragmentation resembles treatment with nocodazole. Cells transfected with dynamitin and incubated for ∼20 h to allow Golgi fragmentation were treated for 3 h with nocodazole (A and B), or treated and then allowed to recover in the absence of drug for an additional 3 h (C and D). Panels on the left show labeling for dynamitin, and those on the right for ST. After nocodazole treatment, the untransfected cell in B is indistinguishable from the dynamitin-transfected cell. Nocodazole has no effect on the dynamitin phenotype, but dynamitin overexpression prevents recovery from nocodazole. (D, arrow) An untransfected cell that has regenerated an intact Golgi complex upon nocodazole washout. (E–H) Golgi and IC/_cis_-Golgi network markers colocalize in dynamitin overexpressors. Cells transfected with dynamitin were labeled for the following pairs of marker proteins: ERGIC-53 (E) and ST (F); ERD-2 (G) and galactosyltransferase (H).
Two proteins that mark the dynamic cis and trans faces of the Golgi complex revealed dynamitin effects different from the stack markers. The distribution of ERGIC-53, a protein of the intermediate compartment that recycles between the ER and the _cis_-Golgi (Schweizer et al., 1990) was altered subtly (but reproducibly) by dynamitin overexpression. In untransfected HeLa cells, ERGIC-53 had a punctate cytoplasmic distribution superimposed on a reticular ER-like distribution, with accumulation in the Golgi region visible in some cells (Fig. 1, G and H). Dynamitin overexpression did not alter the ER-like ERGIC-53 staining, but the accumulation in the Golgi area was lost and the punctate cytoplasmic stain became more pronounced, often with larger, more irregularly shaped cytoplasmic aggregates (Fig. 1, G and H, see also Fig. 6 E for colocalization with ST). TGN38, a marker that recycles between the _trans_-Golgi network and the cell surface (Luzio et al., 1990; Reaves et al., 1993), had a predominantly juxtanuclear staining pattern in untransfected cells (Fig. 2 B, two left-most cells). Upon dynamitin overexpression, the TGN38 pattern was disrupted (Fig. 2 B, two right-most cells), but this marker appeared to divide into two separate pools. Some of the TGN38 was present in punctate structures similar to those seen for ST and the other Golgi markers; this pool of TGN38 colocalized with ST (Fig. 2, A and B, and arrows). Another pool of TGN38, however, was distributed in smaller vesicles at the periphery of the cell (Fig. 2 B, arrowheads). These structures were negative for ST and other stack markers.
The shift of a portion of TGN38 to the cell periphery suggested that dynamitin overexpression might also affect endocytic organelles. This possibility was assessed by labeling with antibodies to transferrin receptor (TfR), a marker for early and recycling endosomes (Huebers and Finch, 1987), and LAMP-1, a marker of late endosomes and lysosomes (Fukuda, 1991). In untransfected cells (Fig. 2, D and F, asterisks), TfR was detected in fine punctate organelles distributed throughout the cell. A distinct accumulation of TfR-positive vesicles was observed near the nucleus of most cells, most likely representing the recycling compartment (McGraw et al., 1993). In dynamitin-transfected cells (Fig. 2, C and D), the juxtanuclear accumulation of TfR was lost, and in about half of the cells analyzed, the TfR-positive organelles were found in the extreme cell processes (Fig. 2, D and F, thin arrows). As shown in Table I, these effects, though relatively subtle, were very consistent. When cells overexpressing dynamitin were double labeled with anti-TGN38 and anti-TfR, these markers were localized to overlapping peripheral sites (Fig. 2, E and F, thin arrows), consistent with the view that the peripheral pool of TGN38 is present in endosomes.
Table I.
Effect of Dynamitin Overexpression on the Distribution of TfR
| TfR phenotype | Dynamitin transfected (n = 4) | Untransfected (n = 4) | β-galactosidase transfected (n = 3) |
|---|---|---|---|
| Presence in juxtanuclear area | 5 ± 3 | 85 ± 3 | 80 ± 5 |
| Presence at tips of cell processes | 58 ± 8 | 14 ± 5 | 13 ± 4 |
When the dynamitin-overexpressing cells were labeled with anti-LAMP antibody, a dramatic redistribution of lysosomes and late endosomes was revealed. In control β-galactosidase–transfected cells (Fig. 2, I and J), late endocytic organelles were clustered in the juxtanuclear region. This pattern was also observed in untransfected controls (see Figs. 5 D and 10 D). After dynamitin transfection, however, late endocytic organelles were shifted away from the cell center, accumulating at the extreme peripheral processes of the cell (Fig. 2, G and H). In contrast to early endosomes, which became enriched in the cell periphery, the entire population of late endosomes and lysosomes was often found just under the cell surface. Although it was particularly noticeable in elongated cells (e.g., Fig. 2 H), this phenotype was observed in cells having a variety of different shapes (see Fig. 2 H, top cell, and Fig. 5 D). The observed redistribution of endocytic organelles resembled the effects of serum deprivation or cytoplasmic acidification (Heuser, 1989), but was much more dramatic. Indeed, when cells overexpressing dynamitin were incubated in acetate-Ringers solution to acidify the cytoplasm, the effect was the same as with dynamitin alone, and the net inward movement that normally occurs upon realkalinization was completely blocked (data not shown).
Figure 5.

An intact microtubule cytoskeleton is required for dynamitin-induced lysosome redistribution. Cells transfected with dynamitin and incubated for ∼20 h to allow accumulation of lysosomes in the periphery were incubated for 3 h in the presence of nocodazole to depolymerize microtubules (A and B). Nocodazole was then washed out, and the cells were allowed to recover for 3 h in the absence of drug (C and D). A and C show labeling with antidynamitin; B and D show anti-LAMP label. Note that in the presence of nocodazole, lysosomes are randomly distributed in both transfected and untransfected cells, whereas the juxtanuclear concentration in untransfected cells and the peripheral concentration in transfectants are regenerated when microtubules are allowed to repolymerize.
Figure 10.
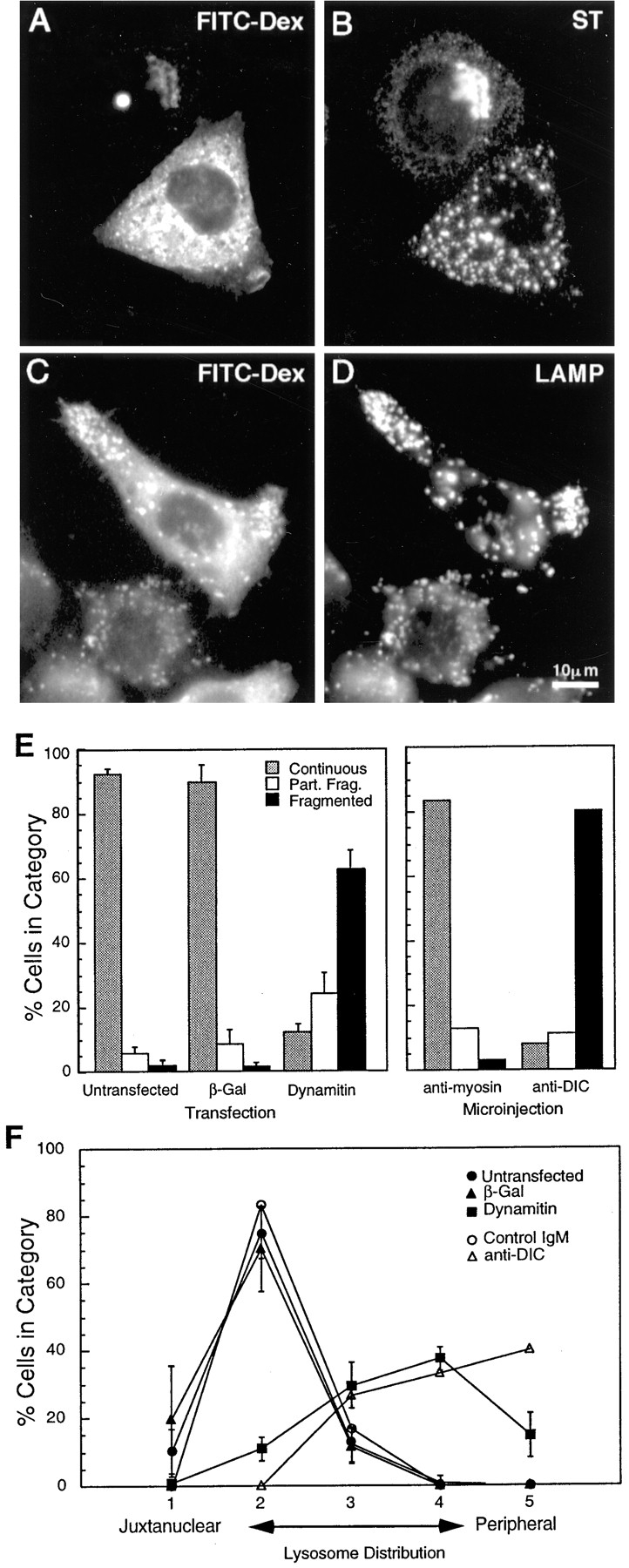
The effects of dynamitin overexpression are mimicked by microinjection of a function-blocking DIC antibody. HeLa cells were injected with a mixture of anti-DIC and fluorescent dextran, incubated for 4 h at 37°C, and then labeled for sialyltransferase (B) or LAMP (D). FITC-dextran-injected cells are shown in A and C (Note that the FITC signal was weak; the apparent punctate stain in A is an artifact of the strong LAMP fluorescence.) Anti-DIC disrupted the Golgi and peripheralized the lysosomes in a manner strikingly similar to dynamitin overexpression. (E and F) Quantitative comparison of the Golgi (E) and the lysosome (F) phenotypes resulting from microinjection and transfection. Cells were either injected with antibodies (E, right-hand portion; F, open symbols), or transfected with dynamitin or β-galactosidase (E, left-hand portion; F, closed symbols), and individual cells positive for the transfected proteins or for injected dextran were scored for the severity of the organelle redistribution, as described in Materials and Methods. For transfected cells, 50–60 cells were scored in each of three independent experiments. Data represent average scores ± 1 SD. For injected cells, data represent an average of 60–70 cells accumulated from two independent experiments. The effects of interfering with dynein and with dynactin were strikingly similar, and they were specific to these protein complexes.
The effects of dynamitin on the intermediate compartment, Golgi, endosomes and lysosomes were highly reproducible, and were the same regardless of whether or not the dynamitin construct encoded a COOH-terminal epitope tag from the c-myc protein (the cells shown in Fig. 1 B were transfected with myc-tagged dynamitin, while those in Fig. 1 F were transfected with wild-type dynamitin). Moreover, the severity of organelle redistribution correlated with the levels of dynamitin expression. In cells that expressed moderate levels of exogenous dynamitin (based on fluorescence intensity of staining), 48% showed complete Golgi disruption and another 36% showed partial disruption. In comparison, 77% of high expressors showed complete and 19% showed partial disruption. Similar dose dependence was seen for lysosome redistribution; in moderate expressors, 24% of cells showed lysosomes predominantly in the periphery and 2% had lysosomes exclusively in the periphery, whereas in the high expressors, these figures were 50 and 19%, respectively.
Whereas dynamitin overexpression had pronounced effects on some membrane organelles, effects on other organelles were not detectable. Labeling of cells with anti– protein disulfide isomerase revealed no obvious changes in the distribution of the ER (Fig. 3, A and B). Similarly, no clear perturbation was observed in the distribution of mitochondria (Fig. 3, C and D) or peroxisomes (not shown), even in the strongest dynamitin overexpressors where effects on other organelles were prominent. Taken together, these results show that the effects of dynamitin overexpression are specific for a subset of membrane organelles that includes the intermediate compartment, Golgi complex, early and late endosomes, and lysosomes. Moreover, the differences in the response among the affected organelles indicates organelle-specific differences in the way their positioning and movement are controlled.
Figure 3.
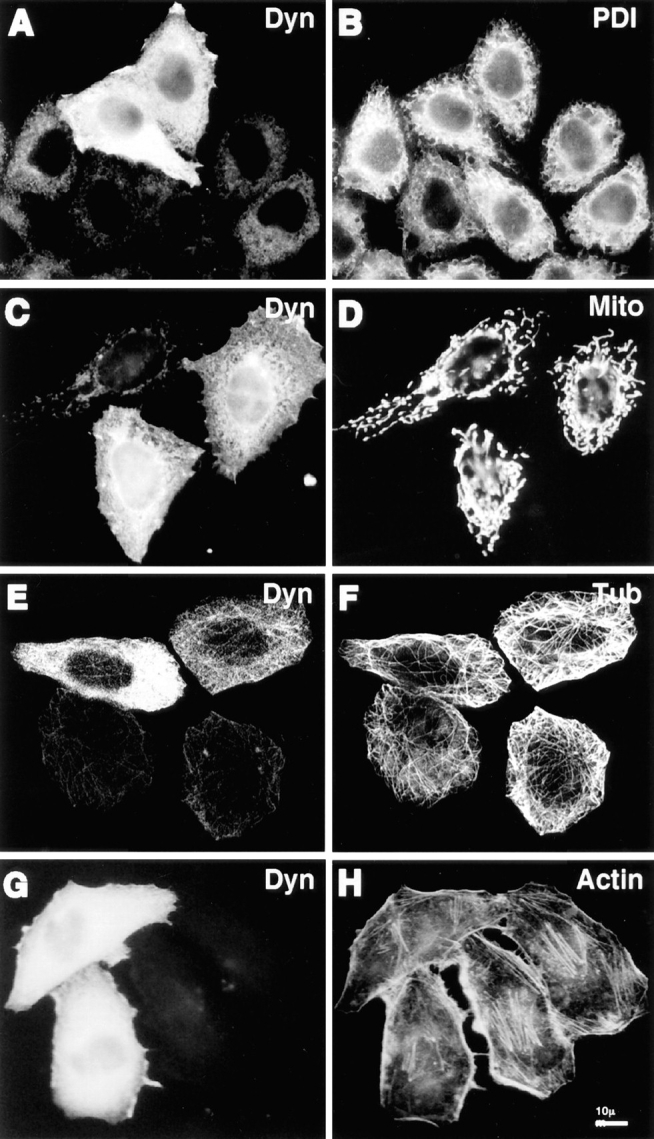
Cytoskeletal structures and certain organelles are not grossly affected by dynamitin overexpression in HeLa cells. HeLa cells transiently transfected with dynamitin were labeled with anti-dynamitin antibody (A, C, E, and G), and were double labeled with organelle markers. (B) Protein disulfide isomerase labeling the ER. (D) Antimitochondria antibody. (F) Anti–tubulin–labeling microtubules. (H) Bodipy phallicidin labeling filamentous actin. E and F show projections of confocal images to better show the microtubule arrays. The apparent microtubule staining pattern in E is an artifact resulting from the much stronger microtubule fluorescence.
In interphase HeLa cells, we observed no gross effects of dynamitin overexpression on the organization of major cytoskeletal filament systems. As shown in Fig. 3, E and F the overall distribution of microtubules in dynamitin-overexpressing HeLa cells was similar to control untransfected cells, and there was no evidence of microtubule bundling. We also examined this question in COS-7 cells, which have microtubule arrays more clearly focused at the centrosome. We found that 85–90% of COS-7 cells cotransfected with dynamitin and VSV-G–tagged ST exhibited specific Golgi disruptions similar to those seen in HeLa cells. Up to half of these cells also exhibited a less focused microtubule organizing center, though the microtubule array often still appeared to radiate from the perinuclear area (Fig. 4 D). Golgi fragmentation and dispersal was observed in dynamitin-overexpressing cells with clearly radial microtubule arrays (Fig. 4, A and B), as well as in those with less well-organized microtubules (Fig. 4 C). No changes were observed in the sensitivity to nocodazole- induced microtubule depolymerization, nor in the rate of centrosome-based microtubule regrowth after nocodazole washout in either cell type (not shown). Also, no change was detected in the population of stable microtubules containing acetylated α-tubulin in HeLa cells. Similarly, we observed no gross changes in the actin cytoskeleton in response to dynamitin transfection. This was true in HeLa cells (Fig. 3, G and H), COS-7 cells (Fig. 4, G and H), and BHK cells (not shown).
Figure 4.
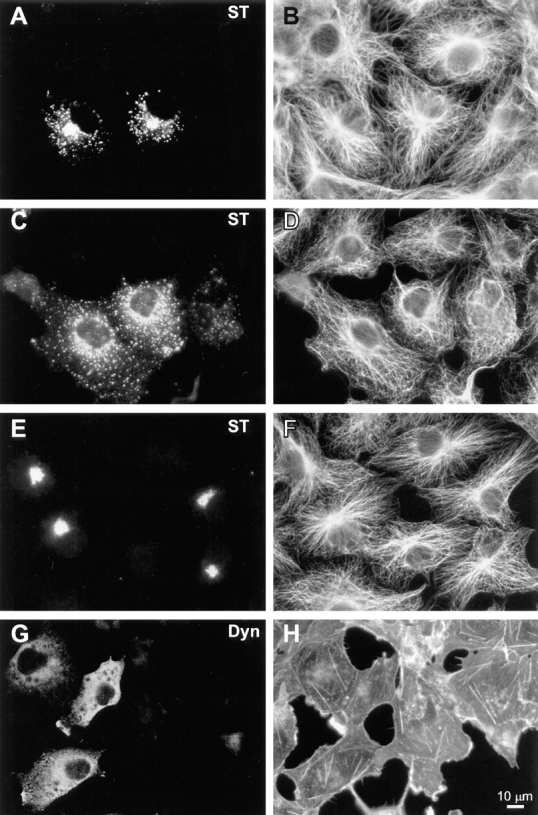
Effects of dynamitin overexpression on cytoskeletal structures in COS-7 cells do not correlate with Golgi disruption. (A–F) COS-7 cells were transiently transfected to coexpress VSV-G–tagged ST with either myc-tagged dynamitin (A–D), or β-galactosidase (E and F). More than 90% of overexpressing cells in cotransfected cultures were found to express both transfected constructs. Double- labeling of antitubulin (B, D, and F) with anti–VSV-G tag (A, C, and E) revealed clear Golgi disruptions in dynamitin-transfected cells with normal radial microtubule arrays (A and B), as well as in those showing less well- focused arrays (C and D). Golgi distribution and microtubule organization in β-galactosidase–transfected cells (E and F) were indistinguishable from those of control untransfected cells. As seen in HeLa cells, COS-7 cells overexpressing myc-tagged dynamitin alone and double-labeled with anti–myc tag (G) and rhodamine phalloidin (H) showed no apparent perturbations of the F-actin cytoskeleton.
Dynamitin-induced Redistribution of Lysosomes Requires Microtubules
Two mechanisms could explain the accumulation of endocytic organelles at the cell periphery in dynamitin-overexpressing cells: the balance of microtubule motor activities could be altered in favor of movement toward microtubule plus ends, or the organelles, once released from microtubules, could bind to the cortical actin cytoskeleton. To distinguish between these two possibilities, cells were first transfected with dynamitin to shift lysosomes to the periphery, and were then treated with nocodazole under conditions that completely depolymerize microtubules in these cells. As expected, nocodazole treatment of untransfected cells inhibited the accumulation of lysosomes in the cell center, leaving the lysosomes homogeneously distributed throughout the cytoplasm. In dynamitin-overexpressing cells, nocodazole abolished the accumulation of lysosomes in the periphery (Fig. 5, A and B). Upon removal of the drug (Fig. 5, C and D), lysosomes returned to the periphery in dynamitin-transfected cells and to the cell center in untransfected controls. These results show that ongoing interaction with microtubules is required to maintain the peripheral lysosomal distribution in dynamitin-transfected cells. Cytochalasin D had no effect on lysosome distribution in either normal or dynamitin-overexpressing cells (not shown), indicating that opposing microtubule motor proteins rather than actin-based interactions are primarily responsible for determining lysosome distribution in these cells.
Dynamitin Overexpression Results in the Generation of Peripheral Golgi “Stacklets”
The disruption of the Golgi complex by dynamitin overexpression is strikingly similar to the well-known effects of microtubule-depolymerizing agents (reviewed in Thyberg and Moskalewski, 1985). Indeed, we found that nocodazole-induced Golgi dispersion in untransfected cells was indistinguishable from the effects of dynamitin overexpression (Fig. 6, A and B). Cells overexpressing dynamitin showed no additional effects upon nocodazole treatment. In untransfected cells, removal of nocodazole allowed the reformation of a juxtanuclear Golgi complex within a few hours, while no change in the dispersed Golgi elements was seen in the dynamitin transfectants (Fig. 6, C and D), indicating that movement of the dispersed Golgi elements toward the cell center requires a functional dynactin complex.
In cells treated with microtubule depolymerizing agents, the dispersed Golgi elements have been shown to consist of short Golgi stacks (Pavelka and Ellinger, 1983; Thyberg and Moskalewski, 1985). These “stacklets” contain the full complement of Golgi marker proteins, and they colocalize at the immunofluorescence level with markers of the intermediate compartment (Cole et al., 1996). This was also found to be the case in dynamitin-overexpressing cells. As shown in Fig. 6, E and F, ST was present at the same peripheral sites where the intermediate compartment marker ERGIC-53 was concentrated. Similarly, galactosyltransferase, a _trans_-Golgi/TGN marker (Rabouille et al., 1995), colocalized with ERD-2, another ER-Golgi itinerant protein (Sönnichsen et al., 1994), which in these cells is normally concentrated in the juxtanuclear region (Fig. 6, G and H).
To determine whether a stacked Golgi structure was maintained after dynamitin overexpression, EM was performed. Cells were doubly transfected with dynamitin and GFP, the GFP-positive and negative cells were sorted by flow cytometry, and both pools were prepared for epon embedding and transmission EM. Immunolabeling of thawed cryosections prepared in parallel with epon specimens showed that 73% of cells sorted into the GFP-positive pool expressed detectable levels of exogenous (myc-tagged) dynamitin, with 58% expressing high levels. When this mixed population of cells were examined by EM, short Golgi stacks were observed in many cells (Fig. 7, A–C). In many cases, a single large multivesicular organelle that may represent part of the intermediate compartment was seen in close apposition to the short stacks (e.g., Fig. 7 B, arrow). When the lengths of the stacks were determined on randomly selected sections, the results showed that in control (GFP-negative) cells (shown in Fig. 7 D), the predominant stack length was 0.9–1.2 μm (Fig. 7 E). In the dynamitin/GFP transfectants, a second major maximum at 0.3–0.6 μm was observed. These short stacks resemble those observed after nocodazole treatment (data not shown).
Figure 7.
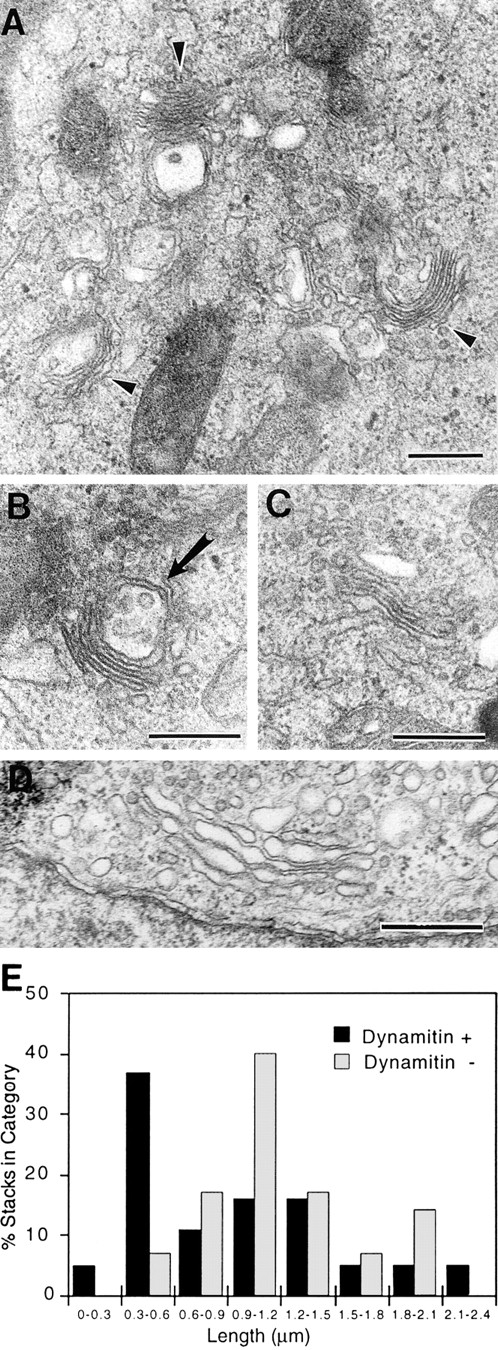
Dynamitin overexpression generates short Golgi stacks. Cells were cotransfected with dynamitin and GFP, and sorted into GFP-positive and GFP-negative pools. The GFP-positive pool was substantially enriched for dynamitin-positive cells, with 73% of cells expressing detectable dynamitin levels and 58% expressing high levels. The two cell populations were processed for EM, and the lengths of 20 randomly selected Golgi stacks in each sample were measured as described in Materials and Methods. (A–C) Representative short stacks found frequently in GFP/ dynamitin–positive cells. (D) Control Golgi stack. (E) Stack length distribution in the two cell populations shows a large increase in short stacks in the dynamitin/GFP–positive pool. The arrow in B denotes a multivesicular body seen in association with a short stack. Such structures were relatively common. Bars, 0.5 μm.
Golgi-to-ER Movement Is Not Blocked by Dynamitin
Since the Golgi normally sits near the microtubule-organizing center (MTOC), while the ER is distributed throughout the cell, “retrograde” protein traffic from the Golgi to the ER represents movement toward microtubule plus ends, and this movement has been shown to depend at least in part on kinesin (Lippincott-Schwartz et al., 1995). To ask what effect overexpression of dynamitin has on this process, dynamitin-transfected cells were treated with BFA, which leads to a dramatic redistribution of Golgi proteins into the ER (Klausner et al., 1992). After treatment with BFA, ST was observed in a reticular pattern characteristic of ER labeling, both in control cells and in dynamitin overexpressors (Fig. 8, A and B). No differences were observed in the duration of treatment or in the dose of BFA required to induce ST redistribution. Upon removal of BFA, ST in untransfected cells returned to a juxtanuclear distribution, while in the dynamitin overexpressors, the dispersed punctate pattern was regenerated (Fig. 8, C and D). The formation of dispersed elements in dynamitin-overexpressing cells occurred within a few minutes of BFA washout, well before the reformation of a juxtanuclear Golgi complex in untransfected cells (Fig. 8, E and F). Thus, the progression of Golgi proteins from the ER into the dispersed Golgi elements can occur under conditions where dynactin function is disrupted, and this process does not rely on passage through an intact juxtanuclear Golgi compartment.
Figure 8.

BFA treatment of dynamitin overexpressing cells. Transfected cells were treated with BFA for 20 min (A and B), or they were treated and allowed to recover in the absence of drug for 1 h (C and D) or for 10 min (E and F). Dynamitin overexpression did not prevent the redistribution of Golgi markers into the ER. The fragmented Golgi phenotype was generated at early times after BFA removal, without passage of marker proteins through a juxtanuclear Golgi complex.
Recovery from BFA treatment was also observed by time-lapse microscopy in living COS-7 cells transfected with a NAGT-I–GFP fusion protein, either alone or combined with dynamitin. Control cells in these experiments clearly showed the appearance of NAGT-I–GFP–labeled vesicles throughout the cytoplasm within a few minutes after BFA washout, followed by their rapid centripetal movements along linear tracks consistent with microtubules, leading to the reformation of a perinuclear Golgi complex after 20–30 min (Fig. 9, A–E). In cells overexpressing dynamitin, similar labeled vesicles also reappeared throughout the cytoplasm after BFA washout, but these exhibited no sustained linear movements in any direction (Fig. 9, F–J). It was noted that many of these vesicles did align to form short linear arrays suggestive of static interactions with microtubules (Fig. 9, F–J). These data directly demonstrate that dynamitin overexpression inhibits microtubule-based ER-to-Golgi transport, resulting in the accumulation of Golgi-destined components in the periphery, most likely at their sites of emergence from the ER.
Figure 9.
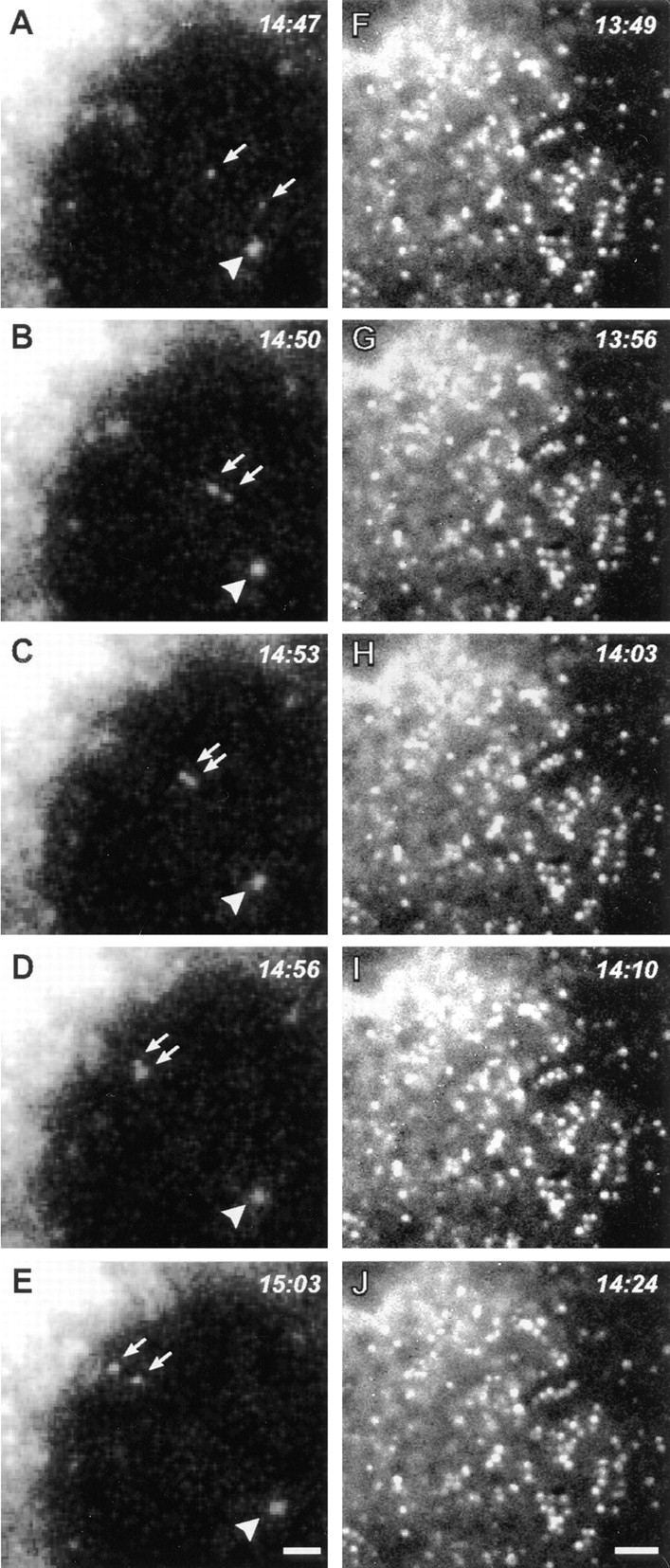
Time-lapse analysis of living COS-7 cells during recovery from BFA directly demonstrates dynamitin-induced inhibition of ER-to-Golgi transport. COS-7 cultures were transiently transfected with a GFP–NAGT-I fusion protein either alone (A–E) or with myc-tagged dynamitin (F–J). More than 90% of overexpressing cells in fixed, cotransfected cultures were found to express both transfected constructs. After a 20-min exposure to BFA, the drug was washed out and the GFP–NAGT-I fluorescence was recorded by time-lapse microscopy at 3- to 7-s intervals. (A–E) Cytoplasmic region beneath the nucleus with reforming Golgi complex at upper left corner. (F–J) Cytoplasmic region between nucleus at upper left corner and cell periphery at lower right corner. Control cells typically exhibited rapid centripetal movements of newly formed GFP–NAGT-I–positive vesicles (A–E, arrows; arrowhead indicates static vesicle for reference), leading to the reformation of a perinuclear Golgi complex (A–E, upper left corner). Similar labeled vesicles appeared throughout the cytoplasm of dynamitin-transfected cells, but no sustained movements were observed in any direction, as seen in F–J. Note partial alignments of vesicles in F–J, forming short linear arrays suggestive of interactions with microtubules. Bars, 1 μm.
Disruption of Cytoplasmic Dynein Activity Mimics the Effects of Dynamitin Overexpression
In view of earlier in vitro and genetic evidence implicating dynactin in cytoplasmic dynein function, the simplest explanation for the observed dynamitin-induced redistribution of membrane organelles is that some or all of the cytoplasmic dynein-dependent transport processes in the cell are being inhibited. Thus, a direct inhibition of dynein activity would be predicted to have similar effects. We therefore tested the effects of microinjecting an antibody to the 74-kD intermediate chain of cytoplasmic dynein (DIC), which appears to block dynein-driven movement in vitro (Heald et al., 1996). As shown in Fig. 10, A–D, microinjection of anti-DIC dispersed the Golgi complex and redistributed lysosomes and late endosomes to the cell periphery in a manner strikingly similar to the effects of dynamitin overexpression. The effects of anti-DIC were dose dependent (not shown), and control antibodies used at the same concentration had little or no effect. To facilitate comparisons, the phenotype of microinjected or transfected cells was randomly scored as described in Materials and Methods. As shown in Fig. 10, E and F, the results of interfering with either cytoplasmic dynein or dynactin were remarkably similar. Our results thus show that both dynein and dynactin probably function in a common pathway to maintain the intracellular localization of the Golgi complex, intermediate compartment, endosomes, and lysosomes.
Discussion
This study represents the first analysis of the effects of disrupting dynactin function in interphase mammalian cells. Our data show that dynactin plays an obligate role in maintaining the intracellular distribution of membrane organelles. In dynactin-disrupted cells, the Golgi complex is fragmented such that cis, medial, and trans stack marker proteins colocalize with markers of the intermediate compartment in large punctate structures scattered throughout the cytoplasm. Time-lapse analysis of living cells confirms that Golgi-destined components remain in the periphery and do not undergo the centripetal microtubule-based transport seen in control cells. EM shows that the arrested structures represent short Golgi stacks that closely resemble those resulting from treatment with microtubule-depolymerizing agents. Endocytic organelles ranging from early endosomes to lysosomes are redistributed to the cell periphery, and the recycling compartment loses its juxtanuclear localization. The phenotype of dynactin-disrupted cells is replicated in cells microinjected with a function-blocking antidynein antibody, providing further physiological evidence for a role for dynactin in dynein function.
The Role of Dynactin in Endosome/Lysosome Movement
Maintenance of lysosomes and recycling endosomes in the juxtanuclear region is known to require microtubules (Matteoni and Kreis, 1987; McGraw et al., 1993). Our results extend these findings by showing that cytoplasmic dynein and dynactin mediate this effect. We find that the redistribution of endocytic organelles to the cell periphery in dynamitin overexpressors is itself an active process requiring the continued presence of intact microtubules. The simplest interpretation of these results is that the normal distribution of these organelles represents a steady-state equilibrium between opposing transport processes. Centripetal, minus end–directed movement requires ongoing dynein and dynactin activity, and this is opposed by a plus end microtubule motor. The opposing plus end motor is almost certainly kinesin, since perturbation of kinesin activity by several independent means has been shown to affect outward lysosome movement (Hollenbeck and Swanson, 1990; Feiguin et al., 1994; Nakata and Hirokawa, 1995). In support of this conclusion, a recent study of phagolysosome motility has suggested that at least these specialized endocytic organelles can use dynein, dynactin, and kinesin to move along microtubules in vitro (Blocker et al., 1997). We therefore suggest that the observed bidirectional movement of endocytic organelles (Matteoni and Kreis, 1987) is controlled by a simple dynamic equilibrium between dynein and kinesin activity. At steady state, this movement would facilitate multiple rounds of fusion and fission, mixing endocytosed material with lysosomal hydrolases (Bomsel et al., 1990). In response to certain environmental stimuli, the net distribution of endocytic organelles could be rapidly altered by regulating the balance between the motor activities. This could mediate, for example, the collapse of lysosomes to the cell center, which occurs when macrophages ingest a large phagocytic load (Knapp and Swanson, 1990), or the release of “secretory lysosomes” from fibroblasts and leukocytes (Rodriguez et al., 1997). Experimental manipulations, such as acidification of the cytosol (Heuser, 1989), serum deprivation, and treatment with phosphatase inhibitors (Lin and Collins, 1993; Lin et al., 1994), apparently affect the balance of motor activity and suggest that phosphorylation plays a role in the regulatory mechanism. Based on our current results, it appears that a change in dynein activity alone could account for these phenomena.
Dynactin, Dyneins, and the Golgi Complex
In dynamitin overexpressors, the Golgi complex becomes “fragmented,” with short stacks distributed throughout the cytoplasm, and similar results are obtained by microinjection of antidynein antibody. These effects closely resemble Golgi fragmentation resulting from microtubule depolymerization, suggesting that dynein–dynactin–mediated interactions represent the dominant form of Golgi– microtubule interactions in these cells. Golgi elements have been reported to migrate along microtubules to the cell center during the recovery from nocodazole treatment (Ho et al., 1989), a process that resembles the reorganization of scattered Golgi elements after mitosis, and cytoplasmic dynein has been implicated in a similar process in permeabilized cells (Corthesy Theulaz et al., 1992). We show that dynactin is required for this bulk movement of Golgi stacklets in vivo, since dynamitin overexpression blocks the recovery from nocodazole. In interphase cells, the primary biosynthetic organelles transported to the cell center are thought to be elements of the intermediate compartment, carrying newly synthesized cargo proteins. Our results indicate that movement of the intermediate compartment is also dependent on dynactin (and, by extension, cytoplasmic dynein), since the distribution of intermediate compartment markers, but not of ER markers, is perturbed, and the juxtanuclear pool of ERGIC-53 is lost. Indeed, we have directly observed the inhibition of ER–Golgi motility in cells recovering from BFA treatment. As described below, the resulting blockage of biosynthetic protein transport is likely to induce the observed Golgi fragmentation.
Kinesin has been localized to Golgi complex (Fath et al., 1994; Lippincott-Schwartz et al., 1995), and, in astrocytes, kinesin antisense treatment resulted in collapse of the Golgi toward the nucleus (Feiguin et al., 1994). We have occasionally observed threadlike Golgi projections in cells overexpressing moderate levels of dynamitin, suggesting that kinesin-mediated extension of Golgi membranes may occur; however, dynamitin overexpression does not result in redistribution of Golgi membranes to the cell periphery. This can be explained if Golgi membranes relieved of minus end motor activity fuse with the ER before reaching the periphery. As in cells treated with nocodazole (Cole et al., 1996), dynactin disruption results in the accumulation of Golgi proteins at the same sites with intermediate compartment markers and newly synthesized proteins exiting the ER (Burkhardt, J.K., unpublished data). It is therefore likely that, as proposed by Cole et al. (1996) for nocodazole-treated cells, fragmentation of the Golgi complex results from the disruption of forward protein transport, but continued retrograde transport, so that short Golgi stacks form de novo at ER exit sites. Our results showing that these short stacks can be generated upon release from BFA treatment directly supports this model.
Golgi disruption has been reported in cells microinjected with antibodies to DHC2, a dynein heavy chain isoform that has been localized to the Golgi complex (Vaisberg et al., 1996). Antibodies to DHC-1, the conventional dynein heavy chain, did not have this effect (Vaisberg, et al., 1996). Microinjection with anti-DHC2 antibody disrupted Golgi morphology in 40–45% of cells, far fewer than we have observed with anti-DIC microinjections or with dynamitin overexpression. It is not known whether this is caused by technical differences, or if it indicates that DHC2 is only partially responsible for Golgi localization. From this, however, it does seem likely that dynactin interacts with multiple dynein isoforms. In this context, it is important to note that the 70.1 anti-dynein intermediate chain antibody used for these studies reacts with at least two DIC isoforms (Vaughan, K.T., K.K Pfister, and R.B. Vallee, unpublished observations). Clearly, ongoing movement toward the cell center, driven by one or more dynein isoforms together with the dynactin complex, is required to maintain the structural integrity and juxtanuclear localization of the Golgi stack.
Organelle Specificity of the Dynamitin Overexpression Phenotype
We could detect no effect of dynamitin overexpression on the distribution of the ER, mitochondria, or peroxisomes, though each of these organelles has been proposed to interact with cytoplasmic dynein. Mitochondria move bidirectionally along microtubules, and their anterograde movement is supported in vitro by one or more kinesin family members (Nangaku et al., 1994; Elluru et al., 1995). The retrograde motor has not been identified, and there is currently no direct evidence for the involvement of either cytoplasmic dynein or dynactin. Indeed, there is evidence that mitochondrial movement is controlled differently than that of other organelles, since it includes long periods of apparent static attachment to cytoskeletal elements (reviewed in Hollenbeck, 1996). Thus, even if dynactin is involved in mitochondrial movement, an additional tethering mechanism might mask any effects of dynamitin overexpression. Similar issues arise regarding peroxisome motility, since analysis in living cells showed that only 5–15% of these organelles move in a microtubule-dependent manner (Rapp et al., 1996; Wiemer et al., 1997). Since peroxisomes have been shown to bind avidly to microtubules in vitro (Schrader et al., 1996), the localization of these organelles may also involve a tethering mechanism that would not be disrupted by perturbing dynactin activity.
Unlike the situation with mitochondria and peroxisomes, there is evidence that the ER does interact with cytoplasmic dynein. Cytoplasmic dynein can drive the formation of ER networks in vitro in a phosphorylation-dependent manner (Allan, 1995). It may be, however, that the distribution of the ER in vivo depends primarily on the activity of plus end motors. Kinesin, which is thought to be responsible for actively generating the spread distribution of the ER (Houliston and Elinson, 1991; Toyoshima et al., 1992; Feiguin et al., 1994), may dominate in determining ER distribution in the cell types we have examined, much as we find that dynein dominates in determining the distribution of Golgi elements. Once established, the reticular structure itself may tend to maintain its distribution in the cell, given that depolymerization of microtubules with nocodazole only slowly results in retraction of the microtubule network (Terasaki et al., 1986). As with mitochondria and peroxisomes, our finding that the ER is not altered by dynactin disruption does not rule out a role for dynactin. In each case, however, it appears that steady-state distribution is controlled by a mechanism that does not rely on ongoing dynactin activity. In this regard, our results clearly show that the distribution of these organelles is controlled by a mechanism different from that used by the Golgi and endocytic organelles.
The Mechanism of Dynactin Function and the Role of Dynamitin
Echeverri et al. (1996) reported that dynamitin overexpression induces the disruption of the dynactin complex, separating p150_Glued_, the subunit that interacts with cytoplasmic dynein, from the Arp1 filament, the portion of the complex thought to be responsible for cargo attachment. In mitotic cells, this disruption results in the release of cytoplasmic dynein from prometaphase kinetochores. Our current findings indicate that dissociation of the dynactin complex similarly interferes with vesicular motility and is consistent with a model in which dynactin serves to link cytoplasmic dynein to the surface of membranous organelles. However, the fine vesicular dynein and dynactin staining pattern observed during interphase makes it difficult to evaluate the dissociation of dynein from membrane organelles. It will be of considerable interest to determine whether both p150_Glued_ and Arp1 are dissociated from membranes as they were observed to be from kinetochores, or whether attachment occurs via a different mechanism.
It does seem clear from our studies that the effects of dynamitin overexpression on the distribution of membrane organelles is unlikely to result from indirect effects on the organization of the cytoskeleton. We could find no perturbation of actin filament organization, and although we did observe a decrease in the number of cells that exhibit a highly organized MTOC, it is difficult to see how this could account for the dramatic shift of endocytic organelles toward the cell surface. Moreover, the MTOC effects were much less pronounced than the organelle effects, and many cells could be identified where membrane organelle redistribution was observed in the absence of any detectable change in microtubule organization. We therefore conclude that dynamitin overexpression affects the distribution of membrane organelles by directly disrupting their transport along microtubules.
In contrast with the relatively clear effects of dynamitin overexpression, the interphase phenotype produced by overexpression of other dynactin subunits has been more difficult to interpret. Full-length p150_Glued_ or truncation mutant forms of the polypeptide exhibited continuous microtubule decoration, loss of centrosomal association, and microtubule bundling (Waterman-Storer et al., 1995). Effects on membranous organelles would therefore be difficult to interpret and have not been reported. Similarly, although changes in Golgi morphology have been observed in cells overexpressing Arp1, extensive cytoplasmic Arp1 filament arrays were formed in the transfected cells, raising the possibility that membrane redistribution could be a secondary effect of these changes (Holleran et al., 1996).
One of the outstanding questions about dynamitin has been the range of its interactions with other motor proteins. Our findings show that an intact dynactin complex is an obligate component for dynein-mediated motility of membrane organelles. On the other hand, dynactin appears not to be required for plus end–directed movements, some of which have been shown previously to be kinesin driven. Dynamitin overexpression or microinjection with antidynein antibody blocks dynein activity without affecting kinesin activity, just as disrupting kinesin activity allows ongoing movement that we now show to be dynein driven (Feiguin et al., 1994; Hollenbeck and Swanson, 1990). These results have clear implications for the mechanism by which organelle motility is regulated. They are inconsistent with a model in which the two motors compete for a common receptor. Instead, it is more likely that the balance of power is regulated by regulating the activity of one or both motors. In view of the potent effects of dynamitin overexpression on dynein activity, it is worth considering that dynamitin represents a key regulatory subunit of the dynactin complex. Regulation of dynamitin activity could modulate the attachment of cytoplasmic dynein to kinetochores and vesicular organelles, and possibly also to other structures, such as microtubules (Heald et al., 1996) and NuMA (Merdes et al., 1996). Whatever proves to be the case, dynamitin overexpression appears to be a general, effective tool for blocking both dynactin and dynein function in the cell, and should be of considerable value in future physiological and mechanistic studies.
Acknowledgments
The authors are grateful to the numerous colleagues who provided antibodies, cell lines, and cDNA constructs, and particularly to Drs. Shima and Warren for the NAGT-GFP construct. We thank Liz Jusino and Jesse Argon for technical assistance; Anja Habermann at EMBL and Ed Williamson at the University of Chicago Cancer Center EM facility for advice and assistance with EM; Drs. Gareth Griffiths, Kevin Vaughan, Marty Jacobson, Carol Murphy, and Sigrid Reinsch for many helpful discussions; and Jamie White, Dr. Brian Storrie, and Dr. Yair Argon for critical reading of the manuscript.
This work was supported by grants from the Human Frontier Science Program to J.K. Burkhardt, and the National Institutes of Health GM43474 to R.B. Vallee.
Abbreviations used in this paper
BFA
brefeldin A
DIC
dynein intermediate chain
GFP
green fluorescent protein
LAMP
lysosome-associated membrane protein
MTOC
microtubule-organizing center
NAGT-I
β1,2 _N_-acetylglucosaminyltransferase I
ST
α2,6 sialyltransferase
TfR; transferrin receptor; VSV-G
vesicular stomatitis virus G protein
Footnotes
Address all correspondence to Janis K. Burkhardt, Department of Pathology, The University of Chicago, 5841 S. Maryland Ave., MC1089, Chicago, IL 60637. Tel.: (773) 834-0869. Fax: (773) 702-3701. e-mail: jburkhar@flowcity.bsd.uchicago.edu
References
- Allan V. Protein phosphatase 1 regulates the cytoplasmic dynein-driven formation of endoplasmic reticulum networks in vitro. J Cell Biol. 1995;128:879–891. doi: 10.1083/jcb.128.5.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan V. Motor proteins: a dynamic duo. Curr Biology. 1996;6:630–633. doi: 10.1016/s0960-9822(09)00434-5. [DOI] [PubMed] [Google Scholar]
- Baumgart E, Völkl A, Hashimoto T, Fahimi HD. Biogenesis of peroxisomes: immunocytochemical investigation of peroxisomal membrane proteins in proliferating rat liver peroxisomes and in catalase-negative loops. J Cell Biol. 1989;108:2221–2231. doi: 10.1083/jcb.108.6.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blocker A, Severin FF, Burkhardt JK, Bingham JB, Yu H, Olivio J-C, Schroer TA, Hyman AA, Griffiths G. Molecular requirements for bi-directional movement of phagosomes along microtubules. J Cell Biol. 1997;137:113–129. doi: 10.1083/jcb.137.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomsel M, Parton R, Kuznetsov SA, Schroer TA, Gruenberg J. Microtubule- and motor-dependent fusion in vitro between apical and basolateral endocytic vesicles from MDCK cells. Cell. 1990;62:719–731. doi: 10.1016/0092-8674(90)90117-w. [DOI] [PubMed] [Google Scholar]
- Burkhardt JK. In search of membrane receptors for microtubule-based motors. Is kinectin a kinesin receptor? . Trends Cell Biol. 1996;6:127–131. doi: 10.1016/0962-8924(96)20002-9. [DOI] [PubMed] [Google Scholar]
- Burkhardt JK, Wiebel FA, Hester S, Argon Y. The giant organelles in beige and Chediak-Higashi fibroblasts are derived from late endosomes and mature lysosomes. J Exp Med. 1993;178:1845–1856. doi: 10.1084/jem.178.6.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SW, Meyer DI. ACT3: a putative centractin homologue in S. cerevisiaeis required for proper orientation of the mitotic spindle. J Cell Biol. 1994;127:129–138. doi: 10.1083/jcb.127.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole NB, Sciaky N, Marotta A, Song J, Lippincott-Schwartz J. Golgi dispersal during microtubule disruption: regeneration of Golgi stacks at peripheral endoplasmic reticulum exit sites. Mol Biol Cell. 1996;7:631–650. doi: 10.1091/mbc.7.4.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corthesy Theulaz, I., A. Pauloin, and S.R. Pfeffer. Cytoplasmic dynein participates in the centrosomal localization of the Golgi complex. J Cell Biol. 1992;118:1333–1345. doi: 10.1083/jcb.118.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverri CJ, Paschal BM, Vaughan KT, Vallee RB. Molecular characterization of the 50-kD subunit of dynactin reveals function for the complex in chromosome alignment and spindle organization during mitosis. J Cell Biol. 1996;132:617–633. doi: 10.1083/jcb.132.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elluru RG, Bloom GS, Brady ST. Fast axonal transport of kinesin in the rat visual system: functionality of kinesin heavy chain isoforms. Mol Biol Cell. 1995;6:21–40. doi: 10.1091/mbc.6.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshel D, Urrestarazu LA, Vissers S, Jauniaux JC, van Vliet-Reedijk JC, Planta RJ, Gibbons IR. Cytoplasmic dynein is required for normal nuclear segregation in yeast. Proc Nat Acad Sci USA. 1993;90:11172–11176. doi: 10.1073/pnas.90.23.11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Lewis GK, Ramsay G, Bishop JM. Isolation of monoclonal antibodies specific for the human c-myc proto-oncogene product. Mol Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fath KR, Trimbur GM, Burgess DR. Molecular motors are differentially distributed on Golgi membranes from polarized epithelial cells. J Cell Biol. 1994;126:661–675. doi: 10.1083/jcb.126.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiguin F, Ferreira A, Kosik KS, Caceres A. Kinesin-mediated organelle translocation revealed by specific cellular manipulations. J Cell Biol. 1994;127:1021–1039. doi: 10.1083/jcb.127.4.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M. Lysosomal membrane glycoproteins. Structure, biosynthesis, and intracellular trafficking. J Biol Chem. 1991;266:21327–21330. [PubMed] [Google Scholar]
- Griffiths, G. 1993. Fine Structure Immunocytochemistry. Springer Verlag, Berlin. 459.
- Heald R, Tournebize R, Blank T, Sandaltzopoulos R, Becker P, Hyman A, Karsenti E. Self-organization of microtubules into bipolar spindles around artificial chromosomes in Xenopusegg extracts. Nature (Lond) 1996;382:420–425. doi: 10.1038/382420a0. [DOI] [PubMed] [Google Scholar]
- Heuser J. Changes in lysosome shape and distribution correlated with changes in cytoplasmic pH. J Cell Biol. 1989;108:855–864. doi: 10.1083/jcb.108.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho WC, Allan VJ, van Meer G, Berger EG, Kreis TE. Reclustering of scattered Golgi elements occurs along microtubules. Eur J Cell Biol. 1989;48:250–263. [PubMed] [Google Scholar]
- Hollenbeck P. The pattern and mechanism of mitochondrial transport in axons. Frontiers Biosci. 1996;1:91–102. doi: 10.2741/a118. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, Swanson JA. Radial extension of macrophage tubular lysosomes supported by kinesin. Nature (Lond) 1990;346:864–866. doi: 10.1038/346864a0. [DOI] [PubMed] [Google Scholar]
- Holleran E, Tokito M, Karki S, Holzbaur E. Centractin (Arp1) associates with spectrin revealing a potential mechanism to link dynactin to intracellular organelles. J Cell Biol. 1996;135:1815–1829. doi: 10.1083/jcb.135.6.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzbaur EL, Hammarback JA, Paschal BM, Kravit NG, Pfister KK, Vallee RB. Homology of a 150K cytoplasmic dynein-associated polypeptide with the Drosophilagene Glued. Nature (Lond) 1991;351:579–583. doi: 10.1038/351579a0. [DOI] [PubMed] [Google Scholar]
- Houliston E, Elinson RP. Evidence for the involvement of microtubules, ER, and kinesin in the cortical rotation of fertilized frog eggs. J Cell Biol. 1991;114:1017–1028. doi: 10.1083/jcb.114.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebers HA, Finch CA. The physiology of transferrin and transferrin receptors. Physiol Rev. 1987;67:520–582. doi: 10.1152/physrev.1987.67.2.520. [DOI] [PubMed] [Google Scholar]
- Karki S, Holzbaur EL. Affinity chromatography demonstrates a direct binding between cytoplasmic dynein and the dynactin complex. J Biol Chem. 1995;270:28806–28811. doi: 10.1074/jbc.270.48.28806. [DOI] [PubMed] [Google Scholar]
- King SM, Wilkerson CG, Witman GB. The M r 78,000 intermediate chain of Chlamydomonasouter arm dynein interacts with alpha-tubulin in situ. J Biol Chem. 1991;266:8401–8407. [PubMed] [Google Scholar]
- King SM, Witman GB. Localization of an intermediate chain of outer arm dynein by immunoelectron microscopy. J Biol Chem. 1990;265:19807–19811. [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp PE, Swanson JA. Plasticity of the tubular lysosomal compartment in macrophages. J Cell Sci. 1990;95:433–439. doi: 10.1242/jcs.95.3.433. [DOI] [PubMed] [Google Scholar]
- Kumar J, Yu H, Sheetz MP. Kinectin, an essential anchor for kinesin-driven vesicle motility. Science (Wash DC) 1995;267:1834–1837. doi: 10.1126/science.7892610. [DOI] [PubMed] [Google Scholar]
- Lin SX, Collins CA. Regulation of the intracellular distribution of cytoplasmic dynein by serum factors and calcium. J Cell Science. 1993;105:579–588. doi: 10.1242/jcs.105.2.579. [DOI] [PubMed] [Google Scholar]
- Lin SX, Ferro KL, Collins CA. Cytoplasmic dynein undergoes intracellular redistribution concomitant with phosphorylation of the heavy chain in response to serum starvation and okadaic acid. J Cell Biol. 1994;127:1009–1019. doi: 10.1083/jcb.127.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Cole NB, Marotta A, Conrad PA, Bloom GS. Kinesin is the motor for microtubule-mediated Golgi-to-ER membrane traffic. J Cell Biol. 1995;128:293–306. doi: 10.1083/jcb.128.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzio JP, Brake B, Banting G, Howell KE, Braghetta P, Stanley KK. Identification, sequencing and expression of an integral membrane protein of the trans-Golgi network (TGN38) Biochem J. 1990;270:97–102. doi: 10.1042/bj2700097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteoni R, Kreis TE. Translocation and clustering of endosomes and lysosomes depends on microtubules. J Cell Biol. 1987;105:1253–1265. doi: 10.1083/jcb.105.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrail M, Gepner J, Silvanovich A, Ludmann S, Serr M, Hays TS. Regulation of cytoplasmic dynein function in vivo by the Drosophila gluedcomplex. J Cell Biol. 1995;131:411–425. doi: 10.1083/jcb.131.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw TE, Dunn KW, Maxfield FR. Isolation of a temperature-sensitive variant Chinese hamster ovary cell line with a morphologically altered endocytic recycling compartment. J Cell Physiol. 1993;155:579–594. doi: 10.1002/jcp.1041550316. [DOI] [PubMed] [Google Scholar]
- Merdes A, Ramyar K, Vechio JD, Cleveland DW. A complex of NuMA and cytoplasmic dynein is essential for mitotic spindle assembly. Cell. 1996;87:447–458. doi: 10.1016/s0092-8674(00)81365-3. [DOI] [PubMed] [Google Scholar]
- Meyerowitz E, Kankel D. A genetic analysis of visual system development in Drosophila melanogaster. . Dev Biol. 1978;62:112–142. doi: 10.1016/0012-1606(78)90096-9. [DOI] [PubMed] [Google Scholar]
- Moremen KW, Robbins PW. Isolation, characterization, and expression of cDNAs encoding murine α-mannosidase II, a Golgi enzyme that controls conversion of high mannose to complex N-glycans. J Cell Biol. 1991;115:1521–1534. doi: 10.1083/jcb.115.6.1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhua L, Karpova TS, Cooper JA. A yeast actin-related protein homologous to that in vertebrate dynactin complex is important for spindle orientation and nuclear migration. Cell. 1994;78:669–679. doi: 10.1016/0092-8674(94)90531-2. [DOI] [PubMed] [Google Scholar]
- Nakata T, Hirokawa N. Point mutation of adenosine triphosphate-binding motif generated rigor kinesin that selectively blocks anterograde lysosome membrane transport. J Cell Biol. 1995;131:1039–1053. doi: 10.1083/jcb.131.4.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, Hirokawa N. KIF1B, a novel microtubule plus end- directed monomeric motor protein for transport of mitochondria. Cell. 1994;79:1209–1220. doi: 10.1016/0092-8674(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Nilsson T, Hoe MH, Slusarewicz P, Rabouille C, Watson R, Hunte F, Watzele G, Berger EG, Warren G. Kin recognition between medial Golgi enzymes in HeLa cells. EMBO J. 1994;13:562–574. doi: 10.1002/j.1460-2075.1994.tb06294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschal BM, Holzbaur EL, Pfister KK, Clark S, Meyer DI, Vallee RB. Characterization of a 50-kDa polypeptide in cytoplasmic dynein preparations reveals a complex with p150GLUED and a novel actin. J Biol Chem. 1993;268:15318–15323. [PubMed] [Google Scholar]
- Paschal BM, Vallee RB. Retrograde transport by the microtubule-associated protein MAP 1C. Nature (Lond) 1987;330:181–183. doi: 10.1038/330181a0. [DOI] [PubMed] [Google Scholar]
- Pavelka M, Ellinger A. Effect of colchicine on the Golgi complex of rat pancreatic acinar cells. J Cell Biol. 1983;97:737–748. doi: 10.1083/jcb.97.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plamann M, Minke PF, Tinsley JH, Bruno KS. Cytoplasmic dynein and actin-related protein Arp1 are required for normal nuclear distribution in filamentous fungi. J Cell Biol. 1994;127:139–149. doi: 10.1083/jcb.127.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plough H, Ives P. Induction of mutations by high temperature in Drosophila. . Genetics. 1935;20:42–69. doi: 10.1093/genetics/20.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille C, Hui N, Hunte F, Kieckbusch R, Berger EG, Warren G, Nilsson T. Mapping the distribution of Golgi enzymes involved in the construction of complex oligosaccharides. J Cell Sci. 1995;108:1617–1627. doi: 10.1242/jcs.108.4.1617. [DOI] [PubMed] [Google Scholar]
- Rapp S, Saffrich R, Anton M, Jakle U, Ansorge W, Gorgas K, Just WW. Microtubule-based peroxisome movement. J Cell Sci. 1996;109:837–849. doi: 10.1242/jcs.109.4.837. [DOI] [PubMed] [Google Scholar]
- Reaves B, Horn M, Banting G. TGN38/41 recycles between the cell surface and the TGN: brefeldin A affects its rate of return to the TGN. Mol Biol Cell. 1993;4:93–105. doi: 10.1091/mbc.4.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol. 1997;137:1–12. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DA, Gill SR, Cooper JA, Heuser JE, Schroer TA. Ultrastructural analysis of the dynactin complex: an actin-related protein is a component of a filament that resembles F-actin. J Cell Biol. 1994;126:403–412. doi: 10.1083/jcb.126.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader M, Burkhardt J, Baumgart E, Lüers G, Spring H, Völkl A, Fahimi H. Interaction of peroxisomes with microtubules. Tubular and spherical peroxisomes in HepG2 cells and their alterations induced by microtubule-active drugs. Eur J Cell Biol. 1996;69:24–35. [PubMed] [Google Scholar]
- Schroer TA. Structure and function of dynactin. Sem Cell Devel Biol. 1996;7:321–328. [Google Scholar]
- Schroer TA, Sheetz MP. Two activators of microtubule-based vesicle transport. J Cell Biol. 1991;115:1309–1318. doi: 10.1083/jcb.115.5.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer A, Fransen JA, Matter K, Kreis TE, Ginsel L, Hauri HP. Identification of an intermediate compartment involved in protein transport from endoplasmic reticulum to Golgi apparatus. Eur J Cell Biol. 1990;53:185–196. [PubMed] [Google Scholar]
- Sönnichsen B, Füllekrug J, Nguyen P, Van, Diekmann P, Robinson DG, Mieskes G. Retention and retrieval: two mechanisms cooperate to maintain calreticulin in the endoplasmic reticulum. J Cell Sci. 1994;107:2705–2717. doi: 10.1242/jcs.107.10.2705. [DOI] [PubMed] [Google Scholar]
- Steffen W, Hodgkinson JL, Wiche G. Immunogold localization of the intermediate chain localization within the complex of cytoplasmic dynein. J Struct Biol. 1996;117:227–235. doi: 10.1006/jsbi.1996.0087. [DOI] [PubMed] [Google Scholar]
- Steuer ER, Wordeman L, Schroer TA, Sheetz MP. Localization of cytoplasmic dynein to mitotic spindles and kinetochores [see comments] Nature (Lond) 1990;345:266–268. doi: 10.1038/345266a0. [DOI] [PubMed] [Google Scholar]
- Terasaki M, Chen LB, Fujiwara K. Microtubules and the endoplasmic reticulum are highly interdependent structures. J Cell Biol. 1986;103:1557–1568. doi: 10.1083/jcb.103.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyberg J, Moskalewski S. Microtubules and the organization of the Golgi complex. Exp Cell Res. 1985;159:1–16. doi: 10.1016/s0014-4827(85)80032-x. [DOI] [PubMed] [Google Scholar]
- Toyoshima I, Yu H, Steuer ER, Sheetz MP. Kinectin, a major kinesin-binding protein on ER. J Cell Biol. 1992;118:1121–1131. doi: 10.1083/jcb.118.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisberg EA, Grissom PM, McIntosh JR. Mammalian cells express three distinct dynein heavy chains that are localized to different cytoplasmic organelles. J Cell Biol. 1996;133:831–842. doi: 10.1083/jcb.133.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee RB, Vaughan KT, Echeverri CJ. Targeting of cytoplasmic dynein to membranous organelles and kinetochores. Cold Spring Harbor Symp Quant Biol. 1995;60:803–811. doi: 10.1101/sqb.1995.060.01.086. [DOI] [PubMed] [Google Scholar]
- Vallee R, Sheetz MP. Targeting of motor proteins. Science (Wash DC) 1996;271:1539–1544. doi: 10.1126/science.271.5255.1539. [DOI] [PubMed] [Google Scholar]
- Vaughan KT, Vallee RB. Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chains and p150Glued. J Cell Biol. 1995;131:1507–1516. doi: 10.1083/jcb.131.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Karki S, Holzbaur EL. The p150Glued component of the dynactin complex binds to both microtubules and the actin- related protein centractin (Arp-1) Proc Natl Acad Sci USA. 1995;92:1634–1638. doi: 10.1073/pnas.92.5.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemer EA, Wentzel T, Deerinck TJ, Ellisman MH, Subramani S. Visualization of the peroxisomal compartment in living mammalian cells: dynamic behavior and association with microtubules. J Cell Biol. 1997;136:71–80. doi: 10.1083/jcb.136.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang X, Beckwith SM, Morris NR. Cytoplasmic dynein is involved in nuclear migration in Aspergillus nidulans. . Proc Natl Acad Sci USA. 1994;91:2100–2104. doi: 10.1073/pnas.91.6.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Toyoshima I, Steuer ER, Sheetz MP. Kinesin and cytoplasmic dynein binding to brain microsomes. J Biol Chem. 1992;267:20457–20464. [PubMed] [Google Scholar]