C-Reactive Protein Binds to Apoptotic Cells, Protects the Cells from Assembly of the Terminal Complement Components, and Sustains an Antiinflammatory Innate Immune Response: Implications for Systemic Autoimmunity (original) (raw)
Abstract
C-reactive protein (CRP) is a serum protein that is massively induced as part of the innate immune response to infection and tissue injury. As CRP has been detected in damaged tissues and is known to activate complement, we assessed whether apoptotic lymphocytes bound CRP and determined the effect of binding on innate immunity. CRP bound to apoptotic cells in a Ca2+-dependent manner and augmented the classical pathway of complement activation but protected the cells from assembly of the terminal complement components. Furthermore, CRP enhanced opsonization and phagocytosis of apoptotic cells by macrophages associated with the expression of the antiinflammatory cytokine transforming growth factor β. The antiinflammatory effects of CRP required C1q and factor H and were not effective once cells had become necrotic. These observations demonstrate that CRP and the classical complement components act in concert to promote noninflammatory clearance of apoptotic cells and may help to explain how deficiencies of the classical pathway and certain pentraxins lead to impaired handling of apoptotic cells and increased necrosis with the likelihood of immune response to self.
Keywords: apoptosis, C-reactive protein, complement, macrophages, autoimmunity
Introduction
The vast majority of the cells of the immune system are short lived, requiring that billions of dying cells be eliminated per day. Exactly how these cells are removed is poorly understood, but exposure of different protein, carbohydrate, and lipid moieties on cells dying through programmed pathways (apoptosis) provides signals to phagocytes for their rapid ingestion (for review see references 1 and 2). In inflammatory states, serum proteins such as pentraxins have been implicated in the removal of damaged cells 3. Pentraxins are a family of highly conserved cyclic pentameric proteins that include C-reactive protein (CRP) and serum amyloid P component (SAP; for review see references 4 and 5). Whereas SAP is constitutively expressed at low levels in human serum, release of proinflammatory cytokines such as IL-1 and TNF-α results in up to a 1,000-fold elevation of CRP 4 5.
We previously reported that serum complement facilitates the uptake of apoptotic cells by phagocytes in vitro 6, and others have shown that the acute phase pentraxin in mice, SAP, facilitates the clearance of chromatin 7. As deficiencies in the early complement components and acute phase pentraxins predispose humans and mice to lupus-like diseases 8 9 10 and an increased number of apoptotic cells are observed in the kidneys of C1q-deficient (C1qd) mice 10, we attempted to define the relationship between these serum proteins, apoptotic cells, and phagocytosis. Here we report that human CRP amplifies early classical pathway activation, inhibits the assembly of the terminal complement components, and promotes opsonization of apoptotic cells associated with the expression of TGF-β, an antiinflammatory cytokine, by macrophages. Based on these observations, we propose that a key function of CRP is to promote the clearance of dying cells.
Materials and Methods
Antibodies and Reagents.
Purified human C1q, factor H (FH), C1qd human serum, mAbs to C3bi, C1q, and a neoantigen created by the formation of the membrane attack complex (MAC; C5-9), anti–Sc5-9, were purchased from Quidel Corp. C1qd serum had the following properties (normal range in parenthesis): CH50 179 U/ml when reconstituted with 100 μg/ml C1q (≥40 U/ml) and APH50 348 U/ml (≥50 U/ml). Two sera with low or absent FH were provided by Dr. C.D. West (Children's Hospital Research Foundation, Cincinnati, OH). Serum 1 had a partial FH deficiency (FHlo) [3.7 (37–68 mg/dl)], with C3 = 23.7 (83–177); factor B = 9.0 (13.3–31.5); C5 = 3.1 (3.8–7.6); C6 = 5.9 (4.0–6.9); C7 = 4.5 (3.5–7.9); C8 = 3.4 (3.0–5.8); and 9 = 3.4 (2.5–7.5). Serum 2 had a complete (no protein detected) FH deficiency with C3 = 8.3; factor B = 4.5; C5 = 1.1; C6 = 3.3; C7 = 5.1; C8 = 2.0; and C9 = 0.7. mAbs to Fas (CH11), CD14, and a polyclonal sheep antiserum to FH were purchased from Upstate Biotechnology, Jackson ImmunoResearch Laboratories, and The Binding Site, respectively. Human CRP and rabbit polyclonal anti–human CRP were purchased from Calbiochem. Annexin V and propidium iodide (PI) were obtained from Trevigen, Inc. The following secondary reagents were used: biotinylated or FITC-, PE-, or Cy5-conjugated sheep anti–mouse IgG, goat anti–human IgG, donkey anti–rabbit IgG, goat anti–sheep IgG, and streptavidin (Jackson ImmunoResearch Laboratories or Santa Cruz Biotechnology, Inc.). Appropriate isotype controls were purchased from Sigma-Aldrich. Human autoantibodies relatively monospecific for Sm RNP or antiribosomal P proteins were selected from well characterized patient sera 11 12.
The anti-CRP and anti-FH antisera were verified to be monospecific when tested against normal serum and cell lysates by Western blotting, whereas the anti-C3bi antibody bound to C3b as well as C3bi (not shown). For that reason, the specificity will be referred to as C3b/bi. SDS-PAGE and Coomassie blue staining confirmed purity of the isolated CRP and FH proteins (not shown).
Cell Culture and Apoptosis Induction.
Peripheral blood–derived T cells from normal donors or Jurkat T cells were cultured in RPMI 1640 medium supplemented with 10% FBS, l-glutamine, and penicillin–streptomycin as described 6. Normal T cells were activated by incubation with 2 μg/ml PHA for 24 h. Nonadherent cells were collected and washed and then cultured at 106 cells/ml for 5 d in medium containing 50 U/ml of human recombinant IL-2 (Chemicon International, Inc.).
Apoptosis of Jurkat T cells or activated peripheral blood–derived T cells was induced by incubation with 50 ng/ml of an agonist anti-Fas mAb, CH11, or 0.1–0.5 μg/ml staurosporine (Sigma-Aldrich) for 4–6 h. The percentage of early and late apoptotic cells was quantified by flow cytometry analysis using annexin V and PI according to the manufacturer's instructions and was routinely 70–80%. Distinction between apoptotic and necrotic cells was based on exclusion of trypan blue as determined by light microscopy. Necrosis was induced by heating cells to 65°C for 1 h or by repeat freeze–thaw cycles 13.
Complement and CRP Binding Studies.
Normal human sera (NHS) and other sera used for complement binding and activation were stored at −70°C in small aliquots. Where appropriate, complement was inactivated by heating to 56°C for 30 min (heat-inactivated sera [HIS]). Complement binding studies were performed by incubation of 106 cells with 100 μl of medium containing 20% human serum in TC buffer (140 mM NaCl, 2 mM CaCl2, 10 mM Tris, pH 8.0 or 7.4, supplemented with 1 mM Mg2+ and 1% BSA) for 30 min at 37°C. CRP binding studies were performed by incubation of 106 cells with CRP in TC buffer for 30 min at room temperature. All subsequent wash steps were performed in the presence of TC buffer, as CRP requires Ca2+ for binding 14.
Flow Cytometry, Immunofluorescence, and Confocal and Western Blot Analysis.
Antibody staining was performed by incubation of the cells with antibodies for 30 min at 4°C. Flow cytometry was performed on a FACScan™ instrument operating with CELLQuest™ software (Becton Dickinson) as described previously 6. Dead cells and debris were excluded from analysis. Data were displayed on a logarithmic scale of increasing fluorescence intensity, and each histogram plot was generated from at least 104 events.
Live and apoptotic cells were cytocentrifuged onto glass slides. The glass-adherent cells were fixed with 2% paraformaldehyde, permeabilized with 99% methanol, and, in some cases, counterstained with 5 μg/ml of PI. Incubation with CRP, annexin V, or antibodies was performed as described above, either in the fluid phase before cytocentrifugation or after fixation and permeabilization. Cells were stained with the appropriate antibodies or ligands and viewed either by immunofluorescence (Nikon Eclipse E600) or confocal (Zeiss LSM 510 laser scanning microscope) microscopy. Western blot analysis was performed as described previously 15 and developed using an ECL kit (Amersham Pharmacia Biotech) according to the manufacturer's instructions.
Macrophage Phagocytosis.
Human macrophages were isolated from peripheral blood monocytes of normal donors and cultured on 12-well plastic dishes for 7 d as described previously 6. To quantify macrophage phagocytosis of apoptotic cells, a flow cytometric assay modified from reference 16 was developed. In brief, Jurkat cells were labeled with the dye TAMRA (5- (and 6)-carboxy tetramethyl rhodamine succinimidyl ester; Molecular Probes) according to the manufacturer's instructions. Apoptosis was induced by staurosporine as above and incubated with serum factors for 30 min at 37°C. 106 labeled cells were then incubated with ∼3.5 × 106 adherent macrophages for 1 h at 37°C in the absence of serum. After extensive washing, monolayers were released with trypsin–EDTA, and the macrophages were stained with FITC–anti-CD14 mAb and analyzed by two-color flow cytometry. Macrophages were gated by forward and side scatter and CD14 positivity. Apoptotic cells that were not bound or internalized were excluded by this gating. The percentage of macrophages that bound or ingested TAMRA-labeled target cells was calculated. In some experiments, phagocytosis of apoptotic cells was verified by counting the number of surface-bound and internalized cells by light microscopy as described 6.
Cytokine Assays.
2 × 106 target cells were preincubated with CRP and/or serum as above, washed, and then incubated with adherent macrophages at a 2:1 ratio for 16 h at 37°C in 10% heat-inactivated FCS. The culture medium was collected, briefly centrifuged, and tested for the presence of TNF-α or TGF-β using OptEIA™ kits (PharMingen) according to the manufacturer's instructions. Purified standards were obtained from PharMingen.
Results
Complement Assembles on the Surfaces of Apoptotic Cells and Leads to Enhanced Cell Lysis.
We previously reported that the C3 cleavage product, C3bi, is bound to the surfaces of apoptotic cells and that both the classical and alternative complement pathways were required for maximal C3bi fixation 6. Activation of the early components of complement promote opsonization and clearance of particles, whereas activation of the terminal complement components are proinflammatory and result in cell lysis and necrosis 17. To determine the potential consequences of the interaction between complement and apoptotic cells, we first quantified the binding of the early (C1q) and late (C5-9; the MAC) complement components to apoptotic cells by flow cytometry.
C1q has previously been detected on apoptotic keratinocytes, although these cells had been fixed before analysis 18. To determine if C1q bound to unfixed apoptotic cells, apoptotic Jurkat cells were incubated with 20% NHS. As shown by representative examples in Fig. 1 A and by kinetic studies in Fig. 1 B, C1q bound to the cell surface within minutes, and evidence of C3 activation (C3b/bi) and MAC assembly was also detected by 30 min postincubation. Despite the fact that nucleated cells are relatively resistant to the effects of lysis by the MAC 19, assembly of the MAC was associated with increasing permeability to PI over time (Fig. 1 B). Binding of C1q, C3b/bi, and the MAC and increased cell permeability were not confined to apoptotic Jurkat T cells, as similar observations were observed with apoptotic peripheral blood–derived human T cells, although lower C1q binding suggests that C3b/bi deposition was due to activation of the alternative 6 and/or lectin pathway (Fig. 1 C).
Figure 1.

Complement assembles on the surfaces of apoptotic cells and leads to cell lysis. Apoptosis was induced in Jurkat (A and B) and peripheral blood–derived (C) T cells with staurosporin and then analyzed by flow cytometry for binding of complement components as well as cell permeability to PI as described in Materials and Methods. In this and subsequent figures, “apoptotic cells” (Apo) include 20–30% live cells (annexin V negative). In A and B, live or apoptotic cells were incubated with 20% NHS, HIS, or C1qd serum for 30 min before analysis. Gates were set with isotype control antibodies. In A, cells were analyzed after 20 min for C1q and MAC binding and after 30 min for C3b/bi and MAC binding. In B, cells were analyzed at different time points as indicated. The mean ± SE of three experiments is shown. In C, apoptotic cells were incubated with HIS (thin line) or NHS (thick line) and then analyzed at 30 min except for PI staining, which was evaluated at 2 h. Results are expressed as the change in mean channel fluorescence (ΔMCF). The results shown in A and C are representative of three to five experiments.
To determine whether C1q binding was absolutely required for complement activation, C3b/bi deposition and the assembly of the MAC were assessed on apoptotic cells incubated with NHS or C1qd serum. As shown in Fig. 1 A, right two panels, the MAC was detected on apoptotic cells incubated with either source of serum, although MAC assembly was lower in C1qd serum. These findings indicate that C1q is fixed to the surfaces of apoptotic cells and is associated with complement activation and assembly of the MAC but that C1q binding is not absolutely required for downstream complement activation. In the absence of C1q, complement is presumably activated by the lectin and/or alternative pathways, as also occurs to some extent in normal serum.
CRP Binds to the Surfaces of Apoptotic Cells.
CRP is an acute phase protein that binds to damaged cells and has been proposed to have a scavenging function, promoting the removal of cell debris 4 5. We postulated that it may play a similar role in the clearance of apoptotic cells. To examine this question, live, apoptotic, and necrotic cells were incubated with 0.5 μg/ml (equivalent to the normal serum concentration; Fig. 2 A, d) or 50 μg/ml (equivalent to a moderate acute phase response, Fig. 2 A, a–c and e–g) of purified CRP, washed, and then examined for CRP binding by flow cytometry. CRP bound not only to necrotic cells as expected (b) but also to apoptotic Jurkat cells that had been induced to die by either anti-Fas antibody (e) or by staurosporine (f ) as well as to apoptotic human peripheral blood T cells (g). Binding was specific, because EDTA, which chelates Ca2+ and disrupts CRP–phosphocholine interactions 14, totally abrogated binding to apoptotic cells (c).
Figure 2.
CRP binds to the surfaces of apoptotic cells. (A) Live (a), necrotic (b), or apoptotic (c–g) T cells were incubated with purified CRP, 0.5 (d) or 50 μg/ml (a–c, e–g), for 20 min at room temperature. Jurkat T cells were used for all experiments except for g, where normal human T cells were used. Apoptosis was induced by staurosporin, except for in e, where anti-Fas was used. Necrosis was induced by heat (65°C for 1 h) as described in Materials and Methods. CRP binding was assessed by flow cytometry using either isotype control (thin line) or anti-CRP antibody (thick line). In c, CRP binding studies were performed in the presence of 10 mM EDTA. The results are expressed as the percentage of cells positive for binding and are representative of five individual experiments. (B) Apoptotic Jurkat cells were incubated with CRP (50 μg/ml) as in A and analyzed by two-color flow cytometry with anti-CRP and annexin V. The kinetics of binding are shown in C (mean ± SD of three experiments). In C, time 0 is taken from the exposure of cells to the apoptotic stimulus (whereas the kinetics of complement binding in Fig. 1 B and 3 A were performed with cells that were already apoptotic). (D) Live (a) or apoptotic (b–f) Jurkat cells were incubated with 50 μg/ml CRP. The cells were washed and stained with anti-CRP, followed by Cy-5–conjugated (blue, a–c) or PE-conjugated (red, d–f) secondary antibodies. In a–c, the cells were then cytocentrifuged onto glass slides, fixed, and counterstained with PI (red) and analyzed by confocal microscopy. In d–f, the cells were not counterstained and were analyzed by immunofluorescence. In c, cells were coincubated with CRP and annexin V–FITC (green) to compare the patterns of staining between CRP and annexin V. In d, apoptotic cells were permeabilized before incubation with CRP and anti-CRP so as to compare the patterns of binding to apoptotic cells that were not permeabilized (b and e) before exposure to CRP. In e and f, nonpermeabilized (e) or permeabilized (f) apoptotic cells were incubated with 50 μg/ml CRP and an anti-Sm/RNP–containing antiserum (1:200 dilution). Sm/RNP antibodies were detected using FITC-conjugated goat anti–human IgG (green). Colocalization of snRNP and CRP appears yellow. Confocal images a and c are projections of a stack of 12 slices, and b is a single optical slice at ×64 magnification. Magnification for immunofluorescence is ×60. In c, inset = ×224.
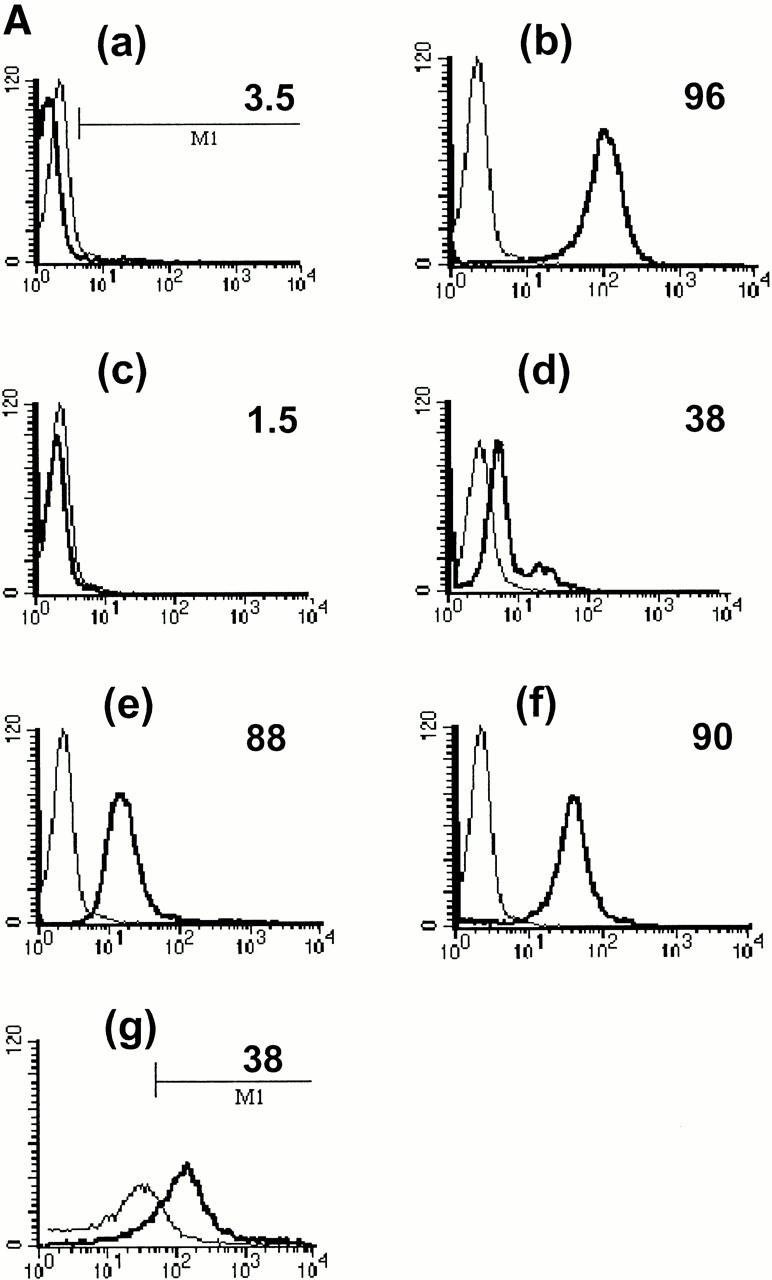








Annexin V binds to phosphatidylserine that “flips” to the outer surface of the cell membrane 20 21. Despite the fact that 87% of apoptotic Jurkat T cells stained positive for both annexin V and CRP (Fig. 2 B), the kinetics of binding of CRP and annexin V were different (Fig. 2 C), and excess CRP failed to inhibit binding of annexin V (not shown), suggesting that CRP bound to a molecule other than phosphatidylserine on the cell surface. Different patterns of binding were also detected by confocal microscopy (Fig. 2 D). As shown in Fig. 2 D c, although colocalization (white) was present on each cell, annexin V (green) and CRP (blue) staining were, for the most part, distinct, with CRP staining showing a more particulate pattern (b, c, and e).
In permeabilized cells, CRP has previously been shown to bind to small nuclear ribonucleoproteins (snRNPs; reference 22) that are detected by anti-Sm/RNP autoantibodies and, under certain conditions, to chromatin (reference 23; for review see reference 24). To determine whether CRP bound to ligands on the surface or within the cell, immunofluorescence and confocal microscopy were performed. As shown in Fig. 2 D, CRP failed to stain the surfaces of live cells (a) but intensely stained the peripheries of apoptotic cells, particularly around apoptotic blebs (b, c, and e). In contrast to permeabilized cells where nuclear staining was consistent with binding to chromatin and/or snRNPs (d and f), staining of the nuclei was not observed in apoptotic cells (b), even when multiple images at different planes were examined.
As both snRNPs and chromatin redistribute to apoptotic blebs 25, we tested whether the blebs “leaked” these potential ligands, thereby accounting for the peripheral staining pattern of CRP. Both immunofluorescence (e) and flow cytometry (not shown) revealed that very little snRNP was present on the surfaces of the blebs. In permeabilized cells (f ), colocalization of CRP and snRNP was observed in the nucleus and also in apoptotic blebs. Finally, incubation of apoptotic cells with a soluble cell extract failed to enhance CRP binding to apoptotic cells as assessed by flow cytometry (not shown). Together, these findings indicate that CRP likely binds to ligand(s) other than chromatin or snRNP on the surfaces of apoptotic cells.
CRP Enhances and Sustains Early Classical Pathway Activation but Attenuates MAC Formation on Apoptotic Cells.
CRP has previously been shown to activate complement by binding to the collagenous tail of C1q 26. We therefore examined the role of CRP in binding and activation of complement on apoptotic cells. When apoptotic cells were preincubated with serum containing CRP, C1q binding to the apoptotic cell surface was amplified and sustained over time compared with controls without the addition of CRP (Fig. 3 A). These findings are consistent with other models demonstrating that CRP amplifies the classical complement pathway on the surface of the activating particle 27 28.
Figure 3.
CRP enhances the deposition of early components of complement yet protects apoptotic cells against the formation of the MAC. Apoptotic Jurkat cells were incubated with 20% NHS or HIS for the times shown in the presence (+CRP) or absence of 50 μg/ml CRP. The percentage of cells positive for C1q staining is shown in A and reflects the mean ± SE of three experiments. An IgM anti–β2-microglobulin antibody (+Ab) was used as a positive control for C1q binding to live Jurkat cells in NHS. Representative flow cytometry histograms of changes in the binding of C1q, C3b/bi, and MAC to apoptotic cells in the presence or absence of 50 μg/ml CRP are shown in B, and the results of 6–10 experiments are summarized in C. The results in C (mean ± SD) are presented as the percentage of cells that bound the complement factors in the presence of CRP as compared with binding in the control (20% NHS without added CRP). *P = 0.03; **P < 0.05; ***P < 0.0001 compared with control without added CRP.



To determine the effect of CRP on other key downstream complement components that induce lysis, such as C3b/bi, ligands for complement receptors, and assembly of the MAC, we quantified the levels of these proteins on apoptotic cells at intervals after incubation with serum containing normal or elevated concentrations of CRP. As shown in Fig. 3 B, CRP enhanced C3b/bi deposition on the surfaces of apoptotic cells as expected from increased classical pathway activation and the role of FH as a cofactor for the generation of C3bi but, paradoxically, markedly reduced MAC assembly. The somewhat lower increment in C3b/bi, as compared with C1q, recruitment may be explained by the role of FH in dissociation of the C3 convertase.
CRP Reduces MAC Assembly on Apoptotic Cells by Recruiting FH.
CRP has been shown to activate the classical pathway of complement on nucleated cells without activating the MAC or causing cytolysis 29. As recent studies have shown that CRP binds to FH 30 31, a complement regulatory protein that accelerates the decay of the C3 and C5 convertases 32, we tested whether CRP recruitment of FH was responsible for the reduction in MAC formation.
Western blot (Fig. 4 A) and flow cytometry analysis (Fig. 4 B) confirmed that CRP added to normal but not FHlo serum increased FH binding to apoptotic cells. The greater difference in FH binding observed by Western as compared with flow cytometric analysis is most likely explained by the superior binding of the polyclonal antiserum to denatured protein on Western blots. Interestingly, FH recruitment was reduced in C1qd serum compared with NHS, with or without added CRP (Fig. 4A and Fig. B). As FH recruitment in NHS could be attributed to enhanced CRP and/or C3b deposition on apoptotic cells, we sought to determine by flow cytometry whether cell bound CRP could recruit FH in the absence of other serum factors. As shown in Fig. 4 C, increased binding of CRP to apoptotic cells was associated with increased FH detection, thus demonstrating specific recruitment by CRP.
Figure 4.

Protection from complement-mediated lysis requires FH recruitment by CRP. Apoptotic Jurkat cells were incubated with 20% NHS, C1qd serum, or FH-deficient (FHlo) serum for 30 min in the presence or absence of 50 μg/ml CRP. In A, FH binding (155 kD) was detected by Western blotting using anti-FH antiserum, biotinylated anti–sheep IgG, and ECL reagents. Protein loading between lanes was compared by Western blotting the same membrane with antiribosomal P antiserum (P0 is 38 kD). The roles of CRP, C1q, and FH on the percentage of apoptotic cells binding FH (B) and the MAC (E) were determined by flow cytometric analysis. The mean ± SE of three experiments is presented. Where indicated by +, CRP was added to a concentration of 50 μg/ml or FH was added back to FHlo serum to a concentration of 50 μg/ml. (C) To determine whether CRP could recruit FH in the absence of C3b or other serum factors, apoptotic Jurkat cells were incubated with purified CRP (0.5 μg/ml, thin line, or 50 μg/ml, thick line), washed, and then incubated with purified FH (50 μg/ml). The cells were washed and stained with isotype control (dotted lines), anti-CRP (left panel), or anti-FH (right panel) and analyzed by flow cytometry. (D) Flow cytometric analysis of apoptotic Jurkat cells incubated with FHlo serum demonstrating C3b/bi binding (thick line) as compared with normal serum (thin line) and isotype control (dotted line). In F, apoptotic Jurkat T cells were incubated with PI in the presence of NHS (thick line) or FHlo serum (thin line). The experiments in the left panel were performed without added CRP and those on the right with 50 μg/ml CRP. PI staining was quantified by flow cytometry.
To determine whether FH was required for the attenuation of MAC formation, we incubated apoptotic cells with FHlo serum that contained residual functional C3 (Fig. 4 D) and terminal complement components rather than the total FH-deficient serum, in which the complement components were almost completely consumed. As FHlo serum failed to attenuate MAC deposition (Fig. 4 E) or lysis (Fig. 4 F) on CRP-coated apoptotic cells but this inhibitory activity could be restored by the addback of FH (Fig. 4 E), we conclude that the recruitment of FH by CRP is required for the attenuation of MAC assembly. Interestingly, CRP failed to inhibit MAC formation in C1qd serum (Fig. 4 E). This may be explained by reduced C3b/bi deposition on these cells (reference 6; results not shown), leading to an overall reduction in FH binding.
CRP Enhances Phagocytosis of Apoptotic Cells.
We previously reported that complement activation on the surfaces of apoptotic cells facilitates uptake of these cells by macrophage CR3 and CR4 receptors 6. In this study, we have shown that CRP amplified classical pathway complement activation on the surface of the apoptotic cell. These findings lead to the prediction that CRP would enhance phagocytosis of these cells. As shown in Fig. 5, although isolated CRP that had bound to apoptotic cells failed to increase phagocytosis in the absence of serum, CRP in the presence of serum markedly increased phagocytosis. This effect was complement dependent, as no increase was observed when CRP was added to heat-inactivated (not shown) or C1qd serum. The reason purified C1q did not fully restore phagocytosis equivalent to that of normal serum is uncertain, but it could be due to partial denaturation of the commercial preparation, as a higher concentration of C1q (250 μg/ml) restored phagocytosis 6. These findings indicate that CRP promotes classical pathway activation, which, in turn, enhances the phagocytosis of apoptotic cells. The similar uptake of apoptotic cells in FHlo serum and NHS is most likely explained by the high level of binding of C3b and/or C3bi to apoptotic cells in both sources of serum (Fig. 4 D).
Figure 5.
CRP enhances phagocytosis of apoptotic cells. TAMRA-labeled apoptotic Jurkat cells were incubated with 50 μg/ml CRP and/or 20% HIS, NHS, C1qd, or FHlo human serum for 30 min. The cells were washed in serum-free medium and then incubated with human macrophages at a ratio of 5:1 for 1 h in the absence of serum. The macrophages were disadhered and then labeled with an anti-CD14 mAb and analyzed by flow cytometry. The results are expressed as the percentage of macrophages that bound or internalized TAMRA-positive cells. A representative experiment is shown in A, and a summary (mean ± SE of four experiments) is shown in B. *C1q reconstituted.


CRP Maintains the Expression of Macrophage TGF-β in the Presence of Complement.
It has been shown that phagocytosis of apoptotic or necrotic cells leads to the synthesis of anti- or proinflammatory cytokines such as TGF-β or TNF-α, respectively 33 34. To determine the effects of CRP and complement on macrophage cytokine production, TGF-β and TNF-α protein concentrations were quantified by ELISA in culture supernates after the ingestion of apoptotic or necrotic cells. As shown in Fig. 6, phagocytosis of apoptotic cells induced high expression of TGF-β in the presence of high CRP and normal serum. Induction of TGF-β was not, however, observed in HIS or C1qd sera, indicating an obligate requirement for complement in the antiinflammatory effect of interaction between apoptotic cells and macrophages. As expected, necrotic cells were potent inducers of TNF-α, an effect that was not affected by the presence of high CRP. Surprisingly, the addition of high levels of CRP with apoptotic cells also induced TNF-α in the absence of serum or in C1qd serum (Fig. 6).
Figure 6.

Phagocytosis of CRP-adherent apoptotic cells maintains TGF-β production in the presence of early complement components. Apoptotic or necrotic Jurkat cells were preincubated with the components indicated for 30 min and then incubated with macrophages in medium containing 10% FBS overnight. The supernatants were collected, and the concentrations of TNF-α and TGF-β were quantified by ELISA. The results are presented as the mean ± SE of triplicates and are representative of three independent experiments.
Apoptotic cells incubated in FH-deficient serum had high levels of bound C3b/bi and the MAC and increased permeability to PI (Fig. 4) and were efficiently phagocytosed by macrophages (Fig. 5 A). Despite efficient phagocytosis of apoptotic cells, the cytokine profile (low TGF-β, high TNF-α) was similar to that induced by necrotic cells and was not affected by the presence of high CRP.
Discussion
There is currently considerable interest in the immunological consequences of phagocytosis and processing of dead and dying cells (for review see reference 35). Most evidence would suggest that apoptotic cells dying in their normal microenvironments are phagocytosed by macrophages and that this process induces the expression of antiinflammatory cytokines such as TGF-β 13 33 34 36. In contrast, necrotic cells provoke proinflammatory cytokines and may promote maturation of dendritic cells, leading to efficient antigen presentation and an immune response 13 33 34. We have recently shown that apoptotic cells fix the C3 component of complement to their surfaces, resulting in the facilitation of receptor-mediated macrophage phagocytosis 6. In this report, we demonstrate that the acute phase protein, CRP, also binds to the surfaces of apoptotic cells, resulting in amplification of classical pathway activation, reduced terminal complement component assembly, increased phagocytosis by macrophages, and sustained production of TGF-β. The classical pathway of complement and CRP appear to work in concert to promote the clearance of apoptotic cells in an antiinflammatory context.
CRP was initially characterized by its binding to the C-polysaccharide of Streptococcus pneumonia 37, suggesting a primary function in clearance of encapsulated bacteria. However, CRP in certain species does not bind to C-polysaccharide 38. Furthermore, the striking degree of sequence homology between species and lack of polymorphisms within species 39 suggests that the selection pressures to maintain homeostatic function are more important than pressures from infectious organisms. Since the demonstration of CRP deposition predominantly along the membranes of damaged cells in noninfectious tissue injury 40, increasing attention has focused on the role of CRP binding to self-molecules. CRP specifically binds to phosphocholine in membrane bilayers 14 as well as to H1-containing chromatin 23 and snRNPs 22.
In addition to binding to subcellular particles from lysed or permeabilized cells, our results indicate that CRP binds to the cell surface membranes of intact apoptotic cells. This binding was specific, as evidenced by the requirement for Ca2+ and could readily be distinguished from the speckled nuclear localization detected in permeabilized cells. The intense staining of CRP on nonpermeabilized apoptotic cells compared with the minimal surface binding of snRNP and failure to augment CRP binding after the exposure of apoptotic cells to cell lysates suggests that CRP bound to a ligand(s) other than snRNPs or chromatin on the apoptotic cell surface. Translocation of phosphatidylserine from the inside to the outside of the cell is an early event in apoptosis and can be detected by binding of annexin V 41. In damaged cells, it has also been proposed that the lipid bilayer becomes unpacked, facilitating hydrolysis of phospholipids by cytoplasmic or secretory phospholipase A2 42. Kinetic studies revealed that CRP binding to apoptotic cells occurred later than that of annexin V. In addition, confocal microscopy showed an even distribution of annexin V along the membranes of early apoptotic cells compared with CRP aggregates observed on apoptotic cells that had started to bleb. Taken together with the evidence that CRP binds to lysophosphocholines in a Ca2+-dependent manner only in bilayers that have undergone an alteration of normal organization 14, it seems likely that CRP bound to lysophophospholipids on apoptotic cells.
One of the best known effects of solid phase binding of CRP is activation of complement 27. Whereas activation of the classical pathway facilitates opsonization of particles for phagocytosis, a process that is noninflammatory unless other receptors such as FcγRs or CD14 are engaged 43 44 45 46 47, assembly of the MAC results in necrotic cell lysis 17 48. Furthermore, assembly of late complement components may induce sublethal cell injury and the release of proinflammatory cytokines 49. As activation of the early and late complement components would be predicted to have opposing effects on the innate immune response, we compared the outcome of CRP binding to apoptotic cells on early and late complement pathways. Levels of CRP equivalent to that found in an acute phase response (50 μg/ml) enhanced and sustained the deposition of both C1q and, to a lesser extent, C3b/bi on the apoptotic cells. The relatively lower recruitment of C3b/bi may be explained by the balance between the increased deposition of C3bi on the activator surface and destabilization of the amplification loop, both promoted by FH. Despite amplification of the classical pathway, CRP paradoxically attenuated the formation of the MAC on the surfaces of apoptotic cells, thereby protecting the cells from lysis. This effect was achieved by the recruitment of FH, a complement regulatory protein that accelerates the decay of the C3 and C5 convertases 32. Although a similar, FH-dependent 30 inhibitory effect of CRP on alternative pathway activation on Streptococcus pneumonia and artificially sensitized erythrocytes has been observed 50 51, we propose that the contrasting effects of CRP on the early and late complement pathways that act in concert to opsonize and protect apoptotic cells from lysis is the primary function of this acute phase protein in early tissue injury.
Elevated levels of CRP in serum also had striking effects on the phagocytosis of apoptotic cells and quality of cytokines expressed by macrophages that, most likely, dictate the nature of the immune response to the ingested particle 13 33 34. Elevated serum CRP markedly increased phagocytosis of apoptotic cells and maintained TGF-β expression. Failure of elevated CRP to augment TGF-β beyond that seen in normal serum is, most likely, explained by near maximal expression of TGF-β under these experimental conditions. In contrast, as shown previously 13 33 34, phagocytosis of necrotic cells, including those produced by exposure to FHlo serum (Fig. 4 F), induced expression of the proinflammatory cytokine TNF-α. Elevated TNF-α expression was observed whether or not the necrotic cells were preincubated with CRP, consistent with studies demonstrating that, once tissue necrosis has already occurred, CRP does not exert an antiinflammatory effect 52. Although CRP can engage FcγRs in serum-free medium 53, the failure of isolated CRP or elevated CRP in HIS to enhance phagocytosis suggests that increased uptake was mediated predominantly through complement rather than via Fcγ receptors.
The above observations indicate that CRP promotes the clearance of apoptotic cells, helps to protect the cells from necrotic lysis through recruitment of FH, and maintains the cytokine profile antagonistic to an immune response. It should be emphasized that all of these effects required the presence of C1q, indicating a pivotal role for the early complement component(s) in the antiinflammatory effects of CRP. The cytokine response in C1qd serum was particularly informative with respect to the known relationship between C1q deficiency and autoimmunity. The minimal expression of TGF-β by macrophages incubated with apoptotic cells in C1qd serum is either explained by failure of ligation of C1q receptors or failure of uptake of the apoptotic cells. In contrast, when CRP levels were elevated in C1qd serum, TNF-α expression was markedly increased. These results suggest that, in an acute phase response, macrophage receptor engagement by CRP, and possibly other ligands, in the absence of coengagement of the C1q receptors exerts a proinflammatory effect. As CRP has recently been shown to bind to FcγRIIa 53 54, a receptor associated with activation of the respiratory burst and a proinflammatory cytokine production by macrophages 47, we propose that it is the engagement of this receptor by CRP in the absence of serum or in C1qd serum that promotes TNF-α production but that complement receptors have a dominant inhibitory effect in normal serum. Experiments to study this further are underway.
The results of this study also suggest a common mechanism to help explain how early complement pathway and serum pentraxin deficiencies predispose individuals to systemic autoimmune disorders such as systemic lupus erythematosus (SLE). Whereas apoptotic cells in central lymphoid organs would be expected to be rapidly cleared by local phagocytes, most likely by noncomplement receptors 1 2, dying cells in the circulation or at sites of tissue injury/inflammation may not be immediately phagocytosed 55 56 57 58. It is at these sites that the dying cells would bind serum proteins, including pentraxins, and activate the early components of complement. In normal individuals, phagocytosis would be efficiently achieved through complement 6 as well as other receptors, and the cytokine response is antiinflammatory. In the absence of C1q and other early complement components, opsonization of apoptotic cells would be less efficient, providing increased opportunity for necrotic cell lysis, uptake would be reduced, and the cytokines expressed by macrophages would become proinflammatory, promoting an immune response to self.
SLE is a relapsing disease, with exacerbations possibly induced by minor infections. Whereas serum complement components and CRP normally increase during acute infections, patients with SLE paradoxically demonstrate low levels of early complement 59 and CRP 60. The results of this study indicate that deficiency of C1q, as might arise from inherited deficiency, hypermetabolism, or antibodies to C1q 61 not only fails to protect apoptotic cells from lysis but also permits the normally protective effect of CRP to perpetuate the inflammatory response. This may explain how tolerance to self-antigens is broken.
Acknowledgments
We thank Dr. C.D. West (Cincinnati, OH) for providing FH-deficient and FHlo sera with their respective complement profiles and Dr. Dalit Ashany (Hospital for Special Surgery) for helpful discussion.
This work was supported in part by the National Institutes of Health (grant AR-45482).
Footnotes
D. Gershov and S. Kim contributed equally to this work.
Abbreviations used in this paper: C1qd, C1q-deficient; CRP, C-reactive protein; FH, factor H; HIS, heat-inactivated sera; MAC, membrane attack complex; NHS, normal human sera; PI, propidium iodide; SAP, serum amyloid P component; SLE, systemic lupus erythematosus; snRNPs, small nuclear ribonucleoproteins.
References
- Ren Y., Savill J. Apoptosisthe importance of being eaten. Cell. Death. Differ. 1998;5:563–568. doi: 10.1038/sj.cdd.4400407. [DOI] [PubMed] [Google Scholar]
- Gregory C.D. CD14-dependent clearance of apoptotic cellsrelevance to the immune system. Curr. Opin. Immunol. 2000;12:27–34. doi: 10.1016/s0952-7915(99)00047-3. [DOI] [PubMed] [Google Scholar]
- Gewurz H., Mold C., Siegel J., Fiedel B. C-reactive protein and the acute phase response. Adv. Int. Med. 1982;27:345–372. [PubMed] [Google Scholar]
- Steel D.M., Whitehead A.S. The major acute phase reactantsC-reactive protein, serum amyloid P component and serum amyloid A protein. Immunol. Today. 1994;15:81–88. doi: 10.1016/0167-5699(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Szalai A.J., Agrawal A., Greenhough T.J., Volanakis J.E. C-reactive protein. Structural biology, gene expression and host defense function. Immunol. Res. 1997;16:127–136. doi: 10.1007/BF02786357. [DOI] [PubMed] [Google Scholar]
- Mevorach D., Mascarenhas J., Gershov D.A., Elkon K.B. Complement-dependent clearance of apoptotic cells by human macrophages. J. Exp. Med. 1998;188:2313–2320. doi: 10.1084/jem.188.12.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickerstaff M.C.M., Botto M., Hutchinson W.L., Herbert J., Tennent G.A., Bybee A., Mitchell D.A., Cook H.T., Butler P.J.G., Walport M.J. Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity. Nat. Med. 1999;5:694–697. doi: 10.1038/9544. [DOI] [PubMed] [Google Scholar]
- Rordorf C., Schnebli H.P., Baltz M., Tennent G.A., Pepys M.B. The acute phase response in (NZB × NZW)F1 and MRL/1 mice. J. Exp. Med. 1982;156:1268–1273. doi: 10.1084/jem.156.4.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan B.P., Walport M.J. Complement deficiency and disease. Immunol. Today. 1991;12:301–306. doi: 10.1016/0167-5699(91)90003-C. [DOI] [PubMed] [Google Scholar]
- Botto M., Dell'Agnola C., Bygrave A.E., Thompson E.M., Cook T., Petry F., Loos M., Pandolfi P.P., Walport M.J. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- Elkon K.B., Hines J.J., Chu J.L., Parnassa A.P. Epitope mapping of recombinant HeLa SmB and B′ peptides obtained by the polymerase chain reaction. J. Immunol. 1990;145:636–643. [PubMed] [Google Scholar]
- Elkon K.B., Parnassa A.P., Foster C.L. Lupus autoantibodies target the ribosomal P proteins. J. Exp. Med. 1985;162:459–471. doi: 10.1084/jem.162.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallucci S., Lolkema M., Matzinger P. Natural adjuvantsendogenous activators of dendritic cells. Nat. Med. 1999;5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- Volanakis J.E., Wirtz K.W.A. Interaction of C-reactive protein with artificial phosphatidylcholine bilayers. Nature. 1979;281:155–157. doi: 10.1038/281155a0. [DOI] [PubMed] [Google Scholar]
- Vaishnaw A.K., Orlinick J.R., Chu J.L., Krammer P.H., Chao M.V., Elkon K.B. Molecular basis for the apoptotic defects in patients with CD95 (Fas/Apo-1) mutations. J. Clin. Invest. 1999;103:355–363. doi: 10.1172/JCI5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess K.L., Babcock G.F., Askew D.S., Cook-Mills J.M. A novel flow cytometric method for quantifying phagocytosis of apoptotic cells. Cytometry. 1997;27:145–152. [PMC free article] [PubMed] [Google Scholar]
- Liszewski, M.K., and J.P. Atkinson. 1993. The complement system. In Fundamental Immunology, 3rd ed. Raven Press, New York.
- Korb L.C., Ahearn J.M. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes. J. Immunol. 1997;158:4525–4528. [PubMed] [Google Scholar]
- Morgan P. Complement membrane attack on nucleated cellsresistance, recovery and non-lethal effects. Biochem. J. 1989;264:1–14. doi: 10.1042/bj2640001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.J., Reutelingsperger C.P.M., McGahon A.J., Rader J.A., van Schie R.C.A.A., LaFace D.M., Green D.R. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulusInhibition by overexpression of Bcl-2 and Ab1. J. Exp. Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoven B., Schlegel R.A., Williamson P. Mechanisms of phosphatidylserine exposure, a phagocyte recognition signal, on apoptotic T lymphocytes. J. Exp. Med. 1995;182:1597–1601. doi: 10.1084/jem.182.5.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuClos T.W. C-reactive protein reacts with the U1 small nuclear ribonucleoprotein. J. Immunol. 1989;143:2553–2559. [PubMed] [Google Scholar]
- Robey F.A., Jones K.D., Tanaka T., Liu T.-Y. Binding of C-reactive protein to chromatin and nucleosome core particles. J. Biol. Chem. 1984;259:7311–7316. [PubMed] [Google Scholar]
- Du Clos T.W. The interaction of C-reactive protein and serum amyloid P component with nuclear antigens. Mol. Biol. Rep. 1996;23:253–260. doi: 10.1007/BF00351177. [DOI] [PubMed] [Google Scholar]
- Casciola-Rosen L.A., Anhalt G., Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med. 1994;179:1317–1330. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H.X., Siegel J.N., Gewurz H. Binding and complement activation by C-reactive protein via the collagen-like region of C1q and inhibition of these reactions by monoclonal antibodies to C-reactive protein and C1q. J. Immunol. 1991;146:2324–2330. [PubMed] [Google Scholar]
- Kaplan M.H., Volanakis J.E. Interaction of C-reactive protein complexes with the complement system. I. Consumption of human complement associated with the reaction of C-reactive protein with pneumococcal C-polysaccharide and with the choline phosphatides, lecithin and sphingomyelin. J. Immunol. 1974;112:2135–2147. [PubMed] [Google Scholar]
- Siegel J., Rent R., Gewurz H. Interactions of C-reactive protein with the complement system. I. Protamine-induced consumption of complement in acute phase sera. J. Exp. Med. 1974;140:631–647. doi: 10.1084/jem.140.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman S., Gewurz H., Mold C. Binding of C-reactive protein to nucleated cells leads to complement activation without cytolysis. J. Immunol. 1986;136:1354–1359. [PubMed] [Google Scholar]
- Aronen M., Lehto T., Meri S. Regulation of the alternative pathway complement activation by an interaction of C-reactive protein with factor H. Immunol. Infect. Dis. 1993;3:83–88. [Google Scholar]
- Mold C., Stein M.P., DuClos T.W. Regulation of complement by direct binding of factor H to C-reactive protein. Mol. Immunol. 1998;35:346–350. [Google Scholar]
- Vik D.P., Munoz-Canoves P., Chaplin D.D., Tack B.F. Factor H. Curr. Top. Microbiol. Immunol. 1990;153:147–162. doi: 10.1007/978-3-642-74977-3_8. [DOI] [PubMed] [Google Scholar]
- Fadok V.A., Bratton D.L., Konowal A., Freed P.W., Westcott J.Y., Henson P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voll R.E., Herrmann M., Roth E.A., Stach C., Kalden J.R., Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- Steinman R.M., Turley S., Mellman I., Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J. Exp. Med. 2000;191:411–416. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern M., Savill J., Haslett C. Human monocyte derived macrophage phagocytosis of senescent eosinophils undergoing apoptosis. Am. J. Pathol. 1996;149:911–921. [PMC free article] [PubMed] [Google Scholar]
- Tillett W.S., Francis T. Serological reactions in pneumonia with a non-protein fraction of pneumococcus. J. Exp. Med. 1930;52:561–571. doi: 10.1084/jem.52.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltz M.L., De Beer F.C., Feinstein A., Munn E.A., Milstein C.P., Fletcher T.C., March J.F., Taylor J., Bruton C., Clamp J.R. Phylogenetic aspects of C-reactive protein and related proteins. Ann. NY Acad. Sci. 1982;389:49–75. doi: 10.1111/j.1749-6632.1982.tb22125.x. [DOI] [PubMed] [Google Scholar]
- Thompson D., Pepys M.B., Wood S.P. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. 1999;7:169–177. doi: 10.1016/S0969-2126(99)80023-9. [DOI] [PubMed] [Google Scholar]
- Kushner I., Kaplan M.H. Studies of acute phase protein. J. Exp. Med. 1961;114:961–974. doi: 10.1084/jem.114.6.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermes I., Haanen C., Steffens-Nakken H., Reutelingsperger C. A novel assay for apoptosisflow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein-labeled annexin V. J. Immunol. Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Hack C.E., Wolbink G.J., Schalkwijk C., Speijer H., Hermens W.T., van den Bosch H. A role for secretory phospholipase A2 and C-reactive protein in the removal of injured cells. Immunol. Today. 1997;18:111–115. doi: 10.1016/s0167-5699(97)01002-5. [DOI] [PubMed] [Google Scholar]
- Aderem A.A., Wright S.D., Silverstein S.C., Cohn Z.A. Ligated complement receptors do not activate the arachidonic acid cascade in resident peritoneal macrophages. J. Exp. Med. 1985;161:617–622. doi: 10.1084/jem.161.3.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright S.D., Silverstein S.C. Receptors for C3b and C3bi promote phagocytosis but not the release of toxic oxygen from human phagocytes. J. Exp. Med. 1983;158:2016–2023. doi: 10.1084/jem.158.6.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K., Johnston R.B., Jr. Dissociation of phagocytosis from stimulation of the oxidative metabolic burst in macrophages. J. Exp. Med. 1984;159:405–416. doi: 10.1084/jem.159.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petty H.R., Todd R.F. Integrins as promiscuous signal transduction devices. Immunol. Today. 1996;17:209–212. doi: 10.1016/0167-5699(96)30013-3. [DOI] [PubMed] [Google Scholar]
- Caron E., Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–1721. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- Reference deleted in proof.
- Morgan B.P. Regulation of the complement membrane attack pathway. Crit. Rev. Immunol. 1999;19:173–198. [PubMed] [Google Scholar]
- Mold C., Gewurz H. Inhibitory effect of C-reactive protein on alternative C pathway activation by liposomes and streptococcus pneumoniae. J. Immunol. 1981;127:2089–2092. [PubMed] [Google Scholar]
- Mold C., Gewurz H., Du Clos T.W. Regulation of complement activation by C-reactive protein. Immunopharmacology. 1999;42:23–30. doi: 10.1016/s0162-3109(99)00007-7. [DOI] [PubMed] [Google Scholar]
- Griselli M., Herbert J., Hutchinson W.L., Taylor K.M., Sohail M., Krausz T., Pepys M.B. C-reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J. Exp. Med. 1999;190:1733–1739. doi: 10.1084/jem.190.12.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj D., Stein M.P., Volzer M., Mold C., DuClos T.W. The major receptor for C-reactive protein on leukocytes is Fcγ receptor II. J. Exp. Med. 1999;190:585–590. doi: 10.1084/jem.190.4.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein M.P., Edberg J.C., Kimberly R.P., Mangan E.K., Bharadwaj D., Mold C., Du C.T.W. C-reactive protein binding to FcγRIIa on human monocytes and neutrophils is allele-specific. J. Clin. Invest. 2000;105:369–376. doi: 10.1172/JCI7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein G.S., Yeo M., Zvaifler N.J. Apoptosis in rheumatoid arthritis synovium. J. Clin. Invest. 1995;96:1631–1638. doi: 10.1172/JCI118202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T., Aono H., Hasunuma T., Yamamoto K., Shirai T., Hirohata K., Nishioka K. Apoptosis and functional Fas antigen in rheumatoid arthritis synoviocytes. Arthritis. Rheum. 1995;38:485–491. doi: 10.1002/art.1780380405. [DOI] [PubMed] [Google Scholar]
- James T.N. The variable morphological coexistence of apoptosis and necrosis in human myocardial infarctionsignificance for understanding its pathogenesis, clinical course, diagnosis and prognosis. Coron. Artery. Dis. 1998;9:291–307. doi: 10.1097/00019501-199809050-00007. [DOI] [PubMed] [Google Scholar]
- Zhu C., Wang X., Hagberg H., Blomgren K. Correlation between caspase-3 activation and three different markers of DNA damage in neonatal cerebral hypoxia-ischemia. J. Neurochem. 2000;75:819–829. doi: 10.1046/j.1471-4159.2000.0750819.x. [DOI] [PubMed] [Google Scholar]
- Lloyd W., Schur P.H. Immune complexes, complement, and anti-DNA in exacerbations of systemic lupus erythematosus. Medicine. 1981;60:208–217. doi: 10.1097/00005792-198105000-00004. [DOI] [PubMed] [Google Scholar]
- De Beer F.C., Fagan E.A., Hughes G.R.V., Mallya R.A., Lanham J.G., Pepys M.B. Serum amyloid-A concentration in inflammatory diseases and its relationship to the incidence of reactive systemic amyloidosis. Lancet. 1982;2:231–233. doi: 10.1016/s0140-6736(82)90321-x. [DOI] [PubMed] [Google Scholar]
- Walport M.J., Davies K.A., Botto M. C1q deficiencies and C1q in autoimmunity. Immunobiology. 1998;199:265–285. doi: 10.1016/S0171-2985(98)80032-6. [DOI] [PubMed] [Google Scholar]