Interleukin 7 Regulates the Survival and Generation of Memory CD4 Cells (original) (raw)
Abstract
Cytokines, particularly those of the common γ chain receptor family, provide extrinsic signals that regulate naive CD4 cell survival. Whether these cytokines are required for the maintenance of memory CD4 cells has not been rigorously assessed. In this paper, we examined the contribution of interleukin (IL) 7, a constitutively produced common γ chain receptor cytokine, to the survival of resting T cell receptor transgenic memory CD4 cells that were generated in vivo. IL-7 mediated the survival and up-regulation of Bcl-2 by resting memory CD4 cells in vitro in the absence of proliferation. Memory CD4 cells persisted for extended periods upon adoptive transfer into intact or lymphopenic recipients, but not in IL-7− mice or in recipients that were rendered deficient in IL-7 by antibody blocking. Both central (CD62L+) and effector (CD62L−) memory phenotype CD4 cells required IL-7 for survival and, in vivo, memory cells were comparable to naive CD4 cells in this regard. Although the generation of primary effector cells from naive CD4 cells and their dissemination to nonlymphoid tissues were not affected by IL-7 deficiency, memory cells failed to subsequently develop in either the lymphoid or nonlymphoid compartments. The results demonstrate that IL-7 can have previously unrecognized roles in the maintenance of memory in the CD4 cell population and in the survival of CD4 cells with a capacity to become memory cells.
Keywords: CD4 memory, cell subsets, T cell survival, homeostasis
Introduction
Prolonged protective immunity depends on the persistence of memory T cells that arise during the primary immune response. As with naive T cells, the survival and the overall size of the memory T cell pool is tightly regulated (1), even though naive and memory T cell populations are thought to be regulated in an independent manner (2, 3). For naive T cells, recent advances have shown that their homeostasis is largely controlled by signals from contact with self-MHC–peptide ligands and the cytokine IL-7 (4, 5). In contrast to naive T cells, homeostatic control of memory CD4 and CD8 cell populations appears to occur independently of TCR engagement (6, 7), although repeated exposures to the priming or cross-reactive epitopes can improve the longevity and functionality of memory T cells (1, 8). In terms of cytokines, homeostasis of memory CD8 cells appears to be largely controlled by the common γ chain (γc) receptor family cytokines, IL-15 and IL-7, whereas these cytokines have appeared to be dispensable for memory CD4 cells.
Clues to the cytokine requirements for homeostasis of memory T cells were initially gained from the profiles of receptor expression. The receptor for IL-15, as assessed by expression of the β chain (CD122), is expressed at significantly higher levels on memory phenotype CD8 cells than on memory phenotype CD4 cells (9). This difference presumably explains why IL-15 has a profound role on the homeostasis of memory CD8 cells, but not on memory CD4 cells (9–13). In contrast to the IL-15 receptor, the receptor for IL-7 is expressed at comparable high levels on all populations of T cells, including both memory CD4 and CD8 cells (14, 15). The role for IL-7 in memory CD8 cell homeostasis has been recently realized by the finding that IL-7 can synergize with, or act in place of IL-15 to support survival and homeostatic division of memory CD8 cells (11, 13, 16). In contrast to other subsets of T cells, the involvement of IL-7 and IL-15 on homeostasis of memory CD4 cells has yet to be fully explored, even though this is a distinct possibility as human memory CD4 cells were found to proliferate in vitro in response to IL-7 and IL-15 (17). In this regard, a recent analysis in the mouse showed that CD4 cells deficient in the γc chain, a crucial component of receptors for IL-7 and related cytokines, have a normal lifespan once they are activated and become memory cells, even though they have a short lifespan as naive cells (18). In addition, we have found previously that murine memory phenotype CD4 cells undergo efficient homeostatic proliferation in IL-7–deficient mice (13). Although these findings together suggest that agents other than IL-7 and IL-15 regulate homeostasis of memory CD4 cells, it is nevertheless possible that these cytokines may have roles, but that memory CD4 cells are more flexible in their homeostatic regulation than memory CD8 cells. Consistent with this hypothesis, a recent work suggests that TCR signals may mask a contribution of IL-7 to the homeostasis of memory CD4 cells (19). Thus, it is conceivable that multiple factors may be involved in controlling homeostasis of memory CD4 cells depending on the history of antigen exposure, stage of differentiation, or anatomic location (20).
In light of the increasing appreciation of the role of IL-7 in homeostasis of resting T cells, we sought to carefully address whether IL-7 can also contribute to the homeostasis of memory CD4 cells. To this end, we have studied the survival of defined populations of antigen-specific memory CD4 cells generated under in vivo conditions. This approach was taken to avoid potential problems associated with using CD4 cells that have developed in the absence of cytokine signaling (18, 21) and spontaneously generated memory phenotype cells of unknown history (13). We show that memory CD4 cells express high levels of IL-7R that are comparable to naive T cells and respond to IL-7 by prolonged in vitro survival. Strikingly, under in vivo conditions, the absence of IL-7 results in the dramatic disappearance of resting memory CD4 cells, and a failure to generate memory cells in both lymphoid and nonlymphoid tissues. We conclude that IL-7 regulates the survival of memory CD4 cells, playing critical roles in the initial development as well as long term maintenance of memory cells.
Materials and Methods
Mice.
C57BL/6, B10.BR, B6.PL Thy1a/Cy, C57BL/6.SCID, and IL-7− mice were purchased from The Jackson Laboratories. C57BL/6 Rag2− mice were obtained from Taconic. B10.BR.SCID mice were provided by P. Linton (Sidney Kimmel Cancer Center, San Diego, CA) and bred in our facility to C57BL/6.SCID mice. IL-7− mice (22) were bred at The Scripps Research Institute. OT-II TCR (vα2 and vβ5) CD4 transgenic mice that respond to peptide 323-339 of OVA were from W. Heath (Walter and Eliza Hall, Melbourne, Australia; reference 23). AND TCR (vα11 and vβ3) CD4 transgenic B10.BR mice that are specific for peptide 88-103 of pigeon cytochrome c (PCC; reference 24) were bred in our vivarium. The TCR transgenic mice were bred to B6.PL Thy1a/Cy mice to facilitate tracking by Thy 1.1 in syngeneic Thy 1.2 recipients.
Antibodies for Flow Cytometry and In Vivo Blocking.
mAbs specific for IL-7 (M25; reference 25) and IL-7R (A7R34 and CD127; reference 14) were generated from the hybridoma lines and purified by Protein G separation for blocking studies. Mouse IgG (mIgG) and rat IgG (rIgG) obtained from Jackson ImmunoResearch Laboratories were used as the respective controls. TCR-specific reagents (vα2 [B20.1], vα11 [RR8.1], vβ3 [KJ25], and vβ5 [MR9-4]), anti–IL-7R (SB/14/CD127), anti–L-selectin (Mel-14/CD62L), anti-CD44 (IM7), anti-CD4 (GK1.5), Thy 1.1 (CD90/OX7), anti–IFN-γ (R46A2 and XMG1.2), IL-4 (11B11 and BVD6–24G2), anti–Bcl-2 (3F11), and anti-BrdU (B44) were obtained from BD Biosciences. Reagents for cell permeabilization and intracellular staining (ICS) with mAbs specific for IFN-γ, Bcl-2, and bromodeoxyuridine (BrdU) were obtained from BD Biosciences, and staining was performed according to the manufacturer's instructions. Polyclonal anti–IL-13 (goat anti–mouse) antibodies were obtained from R&D Systems. Isotype control (rIgG1, rIgG2a, and rIgG2b) mAb and Thy 1.1 (CD90.1/HIS51) were obtained from eBioscience.
Memory T Cell Priming.
Naive TCR transgenic CD4 cells were primed in adoptive hosts. CD4 cells were isolated from the spleens and pooled LNs (inguinal, axillary, brachial, cervical, and mesenteric) of OT-II Thy 1.1 or AND Thy 1.1 mice by negative selection using magnetic sorting with an enrichment cocktail obtained from Stem Cell Technologies, Inc. according to the manufacturer's instructions. vα2, vβ5+ OT-II cells, or vα11, vβ3+ AND cells were quantitated by flow cytometry. 5 × 106 transgenic OT-II cells were injected i.v. into C57BL/6 or C57BL/6 Rag 2− recipients. These mice were immunized by i.p. injection of 100 μg alum-precipitated OVA (Sigma-Aldrich) with 109 Bordetella pertussis vaccine organisms (26). 5 × 106 AND (B10.BR × B6 PL Thy 1.1) F1 cells were injected i.v. into (C57BL/6 × B10.BR) F1 SCID recipients. These mice were immunized by s.c. injection of 50 μg PCC peptide in CFA (BD Diagnostic Systems) distributed on the back and at the base of the tail. At 4–8 wk after priming, donor CD4 cells were enriched by magnetic sorting using negative selection as described in this paragraph, and when isolated from intact mice, anti-Thy 1.2 magnetic beads (Miltenyi Biotec) were also used. In some experiments, resting CD4 cells were isolated by Percoll density separation as described previously (27). CD44hi CD4 cells were sorted by positive selection using a FACSVantage™ flow cytometer (Becton Dickinson). In some experiments, CD62L+ and CD62L− memory CD4 cells were sorted magnetically after labeling with biotinylated anti-CD62L and streptavidin beads (Imag; BD Biosciences). To evaluate the contribution of IL-7 to the maintenance of memory, 0.5–5 × 106 in vivo–primed transgenic CD4 cells were injected i.v. into a second set of recipients that included intact normal mice, SCID mice, IL-7− mice, or IL-7R− mice.
Analysis of Memory CD4 Cell Cytokine Responses.
In vivo–primed memory CD4 cells were tested for frequency of IFN-γ producers by ICS 12 h after restimulation of 2 × 106 cells plated in 24-well plates with 2 × 106 mitomycin-treated splenic APCs and 8 μg/ml OVA peptide in 2-ml cultures (28). For ELISPOT analysis, donor CD4 cells were isolated by positive selection with biotinylated-anti-Thy 1.1 and streptavidin magnetic beads (Imag; BD Biosciences), and 1.5 × 104 cultured together with 2 × 105 APCs and 8 μg/ml OVA peptide for 24 h in Multiscreen-IP plates (Millipore) that were coated with 2 μg/ml anti–IL-4 or 4 μg/ml anti–IL-13 capture antibodies. ELISPOTs were developed by sequential incubation of plates with the appropriate detecting antibodies (biotin-anti–IL-4 or -anti–IL-13, respectively), streptavidin-alkaline phosphatase, and nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate substrate (Kirkegaard & Perry Laboratories) as described previously (29). ELISPOTs were counted with an ImmunoSpot Analyzer (Cellular Technology Limited).
In Vitro Analysis of CD4 Cell Responses to IL-7.
Naive or memory CD4 cells were cultured at 106/ml in 2-ml volumes for 2 wk with or without 10 ng/ml murine rIL-7 (R&D Systems) in RPMI 1640 media (Irvine Scientific) containing 7% FCS (Irvine Scientific), 200 μg/ml penicillin, 200 U/ml streptomycin, 4 mM l-glutamine, 10 mM Hepes, and 5 × 10−5 M 2-ME. Viable cell recovery was determined by harvesting and counting cells by trypan blue exclusion at various times after incubation. Expression of IL-7R, CD44, CD62L, and Bcl-2 was assessed by staining with PE-conjugated mAb using a FACSCalibur™ flow cytometer with CELLQuest™ software (Becton Dickinson). For blocking studies, mAbs to IL-7 and IL-7R were each added at 10 μg/ml at the initiation of culture.
In Vivo Analysis of CD4 Cell Responses to IL-7.
Donor naive or memory CD4 cells were identified in the spleens, pooled peripheral LNs (cervical, inguinal, axillary, and brachial), mesenteric LNs, livers, and lungs of recipient mice at various times after transfer as indicated in the text for individual experiments by staining with PerCP (peridiunin chlorophyll-a protein)-labeled anti-Thy 1.1, allophycocyanin-labeled CD4, and FITC and PE-labeled anti-TCR α and β reagents specific for the OT-II (vα2 and vβ5) or AND (vα11 and vβ3) transgenic receptors. Each experimental design was repeated a minimum of twice using at least 2 recipients per group. PBS containing 10 U heparin was used to perfuse the lungs via the atrium of the heart, and to perfuse the liver through the hepatic vein before excision of these tissues. After disruption to obtain single cell suspensions, lymphocytes were isolated at the interface of 40 and 80% layers of Percoll density gradients.
For antibody blocking studies, anti–IL-7 was administered i.p. to adoptive recipients in 1-mg doses on the day of cell transfer, and every other day thereafter for the duration of the experiments, up to 2 wk. BrdU (Sigma-Aldrich) was administered by i.p. injection of 800 μg, followed by treatment in the drinking water at 800 μg/ml for 5–9 d before analysis by flow cytometry. To evaluate memory generated in vivo, recipients of naive OT-II cells were primed on the day of cell transfer by i.p. injection of 100 μg alum-precipitated OVA with 109 B. pertussis organisms and boosted 3 wk later by i.p. administration of the same dose of Ag and adjuvant.
Results
IL-7 Promotes Survival of Resting Memory Cells In Vitro.
Ag-specific memory CD4 cells were generated by transferring purified naive CD4 OT-II Thy 1.1 TCR transgenic cells into Rag-2− recipients and immunizing the hosts with alum-precipitated OVA protein plus B. pertussis vaccine organisms. Approximately 10% of the injected OT-II cells were recovered from the hosts at day 1, before immunization, and nearly all of these cells were found to have undergone vigorous proliferation when analyzed 5 d after immunization by BrdU uptake or by dilution of carboxy fluorescein diacetate succinimidyl ester (unpublished data). Typically, OT-II cells underwent four- to fivefold expansion during this time, and ∼50% of these cells survived as memory cells 1 mo later (unpublished data). At this time, OT-II cells in the spleen were resting and possessed an effector memory phenotype (Fig. 1 A, CD44hi and CD62Llo). The memory OT-II cells expressed similar levels of IL-7Rα as naive OT-II cells (Fig. 1 A), and 67% responded to OVA peptide–loaded APC by producing IFN-γ, as measured by ICS (Fig. 1 B). Although the frequency of IL-4 producers was too low to be reliably assessed by ICS, using ELISPOT analysis, we determined that 3% of the cells secreted this cytokine and that a similar number secreted the coordinately regulated cytokine, IL-13 (Fig. 1 B). Consistent with our previous studies, naive OT-II cells secreted only IL-2 under the same conditions (28). As described previously (4), spontaneous homeostatic proliferation of OT-II cells driven by lymphopenia was not a confounding problem as we found that both naive and memory OT-II cells did not undergo homeostatic proliferation in Rag-2− hosts (unpublished data).
Figure 1.
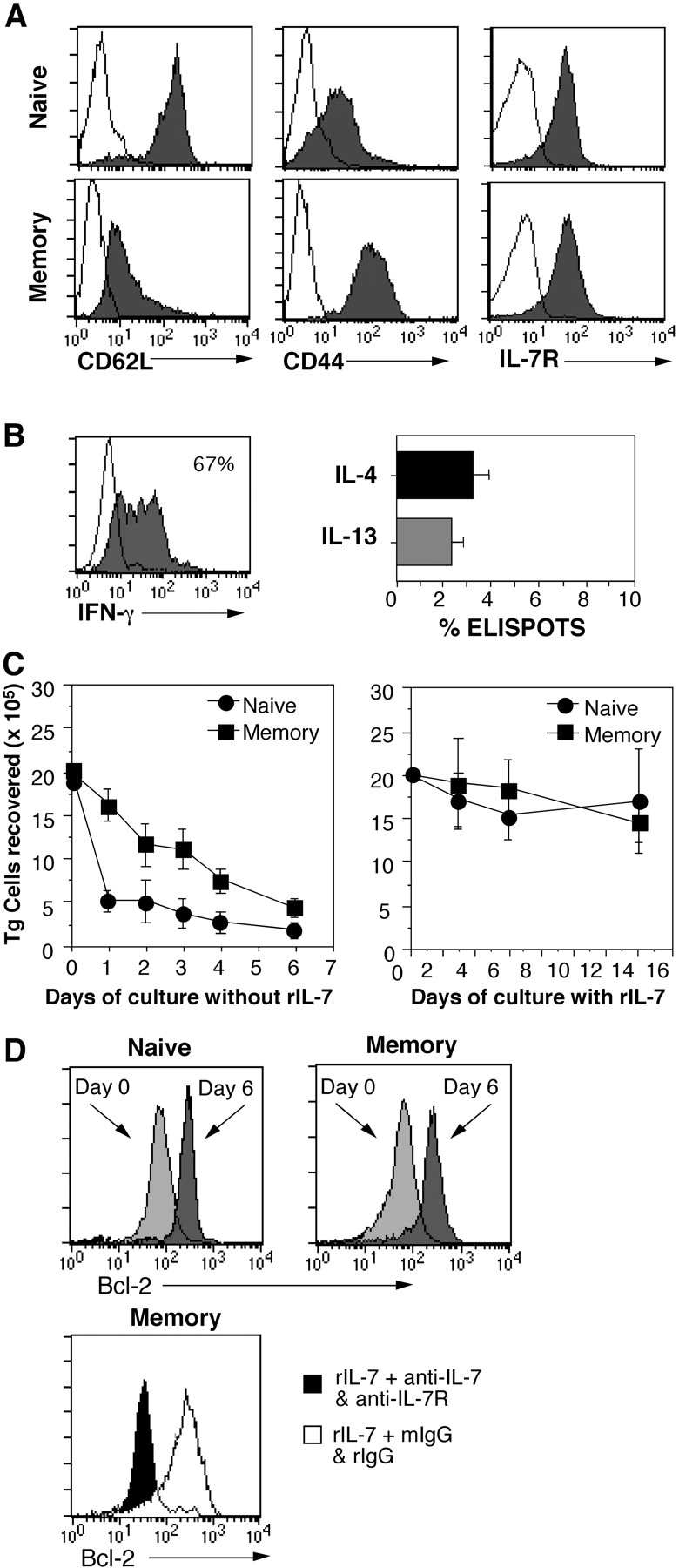
In vivo–primed TCR transgenic memory CD4 cells survive in response to rIL-7 in vitro. Purified naive OT-II Thy 1.1 CD4 cells were transferred into C57BL/6 Rag2− mice (5 × 106 cells/recipient) and immunized with OVA protein and adjuvant as indicated in Materials and Methods. 1 mo later, resting memory OT-II cells were isolated and compared with freshly isolated naive OT-II cells. (A) Phenotype of memory OT-II cells. Naive and memory OT-II cells were stained for expression of CD62L, CD44, and IL-7Rα and analyzed by flow cytometry (shaded histograms; unshaded histograms denote background staining). (B) Frequencies of effector cytokine producers among memory OT-II cells. Memory OT-II Thy 1.1 cells were restimulated with OVA peptide in the presence of splenic APC and tested for secretion of IFN-γ at 12 h by ICS, and after enrichment of Thy 1.1 cells, for production of IL-4 or IL-13 at 24 h by ELISPOT analysis. (C) IL-7 promotes survival of OT-II cells. Naive and memory OT-II Thy 1.1 cells were cultured at 106/ml for the indicated number of days without or with rIL-7 at 10 ng/ml (left and right, respectively). (D) Blocking IL-7 prevents up-regulation of Bcl-2. Naive (top left) and memory (top right) OT-II Thy 1.1 CD4 cells were stained for expression of Bcl-2 (light gray histograms). The cells were cultured as in C for 6 d with 10 ng/ml rIL-7 and stained for BcL-2 (dark gray histograms). Memory cell cultures were also treated with either 10 μg/ml each anti–IL-7 and anti–IL-7R mAb or with an equivalent amount of rat and mouse IgG (bottom, rIgG and mIgG, respectively). The cells were stained for Bcl-2 on day 6 after culture with blocking mAb (shaded histogram) or with control mAb (unshaded histogram).
To study the in vitro effects of IL-7 on CD4 cells, naive and resting memory OT-II cells were purified by magnetic sorting and Percoll density separation and cultured with murine rIL-7 (Fig. 1 C). Without rIL-7 (Fig. 1 C, left), naive OT-II cells died precipitously within 24 h. A more gradual decline was observed for memory OT-II cells, but by day 6, few cells survived. Notably, adding a single dose of rIL-7 (10 ng/ml) rescued survival of both naive and memory OT-II cells for 2 wk (Fig. 1 C, right). In other experiments, memory OT-II cells were kept alive as long as 4 wk without significant cell loss (unpublished data). These results are in agreement with previous data showing that resting murine T cells can use IL-7 for survival in vitro (30). Consistent with papers suggesting that IL-7 does not induce T cell division in vitro in the absence of TCR engagement or costimulation (31, 32), both naive and memory OT-II cells survived with no surface phenotypic change in a resting state with low forward scatter and without cycling. Comparable results were obtained using naive and memory CD4 AND TCR transgenic cells specific for PCC, with memory cells generated in vivo from naive cells in an analogous fashion by priming in SCID recipients with PCC peptide and CFA.
As expected, the efficacy of exogenous rIL-7 on memory OT-II cell survival was abolished in the presence of blocking antibodies to IL-7 and IL-7R (unpublished data). Because induction of the antiapoptotic protein, Bcl-2, is a hallmark of responses to IL-7 (32), we determined if IL-7 also has such an effect on memory CD4 cells. Naive and memory OT-II cells expressed similar basal levels of Bcl-2 before rIL-7 addition and up-regulated Bcl-2 to comparable levels in response to rIL-7 during the 6-d culture period (Fig. 1 D, top). Blocking IL-7 and its binding to the IL-7R inhibited Bcl-2 induction in the residual surviving CD4 memory cells (Fig. 1 D, bottom). These finding indicate that IL-7 can serve as a survival factor for memory CD4 cells under in vitro conditions.
IL-7 Promotes Survival of Resting Memory Cells In Vivo.
To assess the contribution of IL-7 to the survival of memory CD4 cells under in vivo conditions, resting memory OT-II Thy-1.1 cells were purified from OVA-primed Rag-2− recipients by sorting CD44hi, vα2+ CD4 cells and transferred into intact C57BL/6 or IL-7− recipients (both Thy-1.2+). Analysis of the recipients 1 wk later revealed that the recoveries of Thy-1.1+ CD4 donor OT-II cells were markedly lower in IL-7− mice compared with control C57BL/6 hosts (Fig. 2 A). One reason that memory OT-II cells failed to survive efficiently in IL-7− mice may be that these knockouts possess atrophied lymphoid tissues due to defective T and B cell lymphopoiesis. To rule out this possibility, two approaches were taken. First, survival of purified OT-II memory cells in IL-7− mice was compared with that in IL-7Rα− mice, which also possess small lymphoid tissues, but produce IL-7 (33). As shown in Fig. 2 B, although the recoveries of memory OT-II cells at 1 wk after transfer were somewhat lower in IL-7R− mice than in normal C57BL/6 hosts, a profound loss of OT-II memory cells was observed only in the IL-7− mice.
Figure 2.

Survival of memory CD4 cells in IL-7–deficient recipients. (A) Comparison with intact recipients. Resting OT-II Thy 1.1 memory CD4 cells were generated and isolated as for Fig. 1 and sorted by flow cytometry to obtain CD44hi cells. These cells were transferred into normal C57BL/6 or IL-7− mice (2 × 106 cells/recipient). 1 wk later, donor cell recoveries were quantitated in the host spleens and LNs from the total nucleated cell counts and fractions of transgenic donor (vα2, vβ5, and Thy 1.1+) CD4 cells. (B) Comparison with IL-7R− recipients. OT-II Thy 1.1 memory CD4 cells were isolated and transferred as in A into IL-7R− and IL-7− mice, and analyzed for transgenic donor cell recovery after 1 wk. (C) Susceptibility of CD62L+ memory cells to IL-7 deprivation. OT-II Thy 1.1 memory cells were isolated from the LNs of OVA-primed Rag2− that were primed as for Fig. 1. The cells were stained for CD62L before (top) and after magnetic selection for positively expressing cells (bottom, overlay of sorted CD62L− and CD62L+ subsets). CD62L+ cells were transferred into either IL-7R or IL-7 hosts (2 × 106 cells/recipient), and the recovery of donor memory CD4 cells was assessed after 10 d. (D) Naive OT-II Thy 1.1 CD4 cells were primed by transfer into normal C57BL/6 mice (5 × 106 each) and immunization of the recipients with OVA and adjuvant. 4 wk later, donor memory CD4 cells isolated from the spleens were transferred into IL-7R− and IL-7− mice (0.5 × 106 cells/recipient). These mice were evaluated 1 wk after cell transfer for the presence of transgenic donor cells in the LNs and spleens.
Second, dependence on IL-7 for survival of memory OT-II cells was measured under normal T-sufficient conditions by depleting circulating IL-7 using mAb specific for IL-7 (25). Thus, purified OT-II Thy 1.1 memory cells generated in Rag-2− recipients were transferred into normal C57BL/6 mice, and the hosts were injected with 1 mg anti–IL-7 or mIgG on an every other day basis. Analysis of spleen and of hosts 1, 7, and 14 d later revealed that a progressive loss of memory OT-II cells occurred in recipients treated with anti–IL-7 mAb, whereas the numbers of these cells stabilized in hosts injected with control Ab (Fig. 3 A). The difference in the recoveries was not due to variability in cell division as OT-II memory cells failed to undergo proliferation in both types of hosts (Fig. 3 B, top). In addition to reducing the lifespan of donor memory OT-II cells, anti–IL-7 treatment also decreased survivability of host polyclonal memory CD4 cells without altering their homeostatic turnover (Fig. 3 B, bottom). Thus, the recovery of host memory phenotype (CD44hi) CD4 cells at the day 14 time point was significantly lower in mice injected with anti–IL-7 mAb compared with control mice (Fig. 3 C). These data suggest that dependence on IL-7 for survival may be a general property of memory CD4 cells. However, memory T cells are heterogeneous and central memory cells have been distinguished on the basis of their expression of CD62L (L-selectin) that, together with CCR7, regulates the capacity to localize in LNs (34, 35). Therefore, it was of interest to determine if the survival of transgenic memory CD4 cells that reside in LNs was affected by withdrawal of IL-7.
Figure 3.

Effects blocking IL-7 on transgenic and polyclonal memory CD4 cells. (A) Blocking of transgenic memory cell survival in normal recipients. OT-II Thy 1.1 memory cells were primed in vivo and isolated as for Fig. 1. The cells were transferred into normal C57 BL/6 mice (2 × 106 cells/recipient). On the day of cell transfer, separate groups of recipients were injected with 1 mg of either anti–IL-7 or mIgG. Additional doses were given every other day through day 12. On days 1, 7, and 14, animals from each group were evaluated for transgenic donor cell recovery from the spleens and LNs. (B) Lack of division of donor transgenic memory cells but expansion of host polyclonal memory cells in anti–IL-7–treated recipients. Recipients were administered BrdU as described in Materials and Methods to assess proliferation of OT-II Thy 1.1 memory cells and in a separate experiment, division of host memory phenotype (CD44hi) CD4 cells. BrdU uptake was assessed by ICS of splenic lymphocytes. Histograms gated on donor Thy 1.1, vα2, and vβ5+ cells (top) and on host CD44hi and CD4 cells (bottom) are shown. (C) Diminished recovery of host naive and memory CD4 cells. The effects of anti–IL-7 treatment on the recovery of naive phenotype (CD44lo) and memory phenotype (CD44hi) CD4 cells were assessed by quantitating Thy 1.2+ host CD4 cells from the spleens and LNs of the recipients from A on day 14. Comparable results were obtained using high versus low expression of α4 integrin to distinguish memory versus naive phenotype host CD4 cells (not depicted). (D) Recovery of naive and resting memory CD4 cells after IL-7 treatment of immunodeficient mice. Naive CD4 cells were isolated from (AND B10.BR × B6PL.Thy)F1 mice and were transferred into (B10.BR × C57BL/6) F1 SCID mice (5 × 106 cells/recipient). One set of recipients was treated with anti–IL-7 or mIgG as for A without immunization. A second set of recipients was primed with PCC peptide as described in Materials and Methods. 1 mo later, when AND CD4 cells from the spleen were uniformly CD62L− and CD44hi, recipients were treated either with anti–IL-7 or with mIgG as for A. Recovery of transgenic donor (vα11, vβ3, and Thy 1.1+) from the spleens and LNs was determined on day 14 after treatment for naive and primed recipients (left and right, respectively).
Although memory OT-II cells induced by immunization of Rag-2− recipients were predominantly in the spleen (>90%) and were CD62L− (Fig. 1), transgenic memory cells were detected in LNs, where donor cells were exclusively CD44hi and comprised of 28% CD62L+ cells (Fig. 2 C, top left). To evaluate the IL-7 dependence of this population, CD62L+ cells were purified to >90% (Fig. 2 C, bottom left) from enriched CD4 cells using positive selection and transferred into either IL-7R or IL-7–deficient recipients. In the absence of IL-7, OT-II cells with a central memory phenotype survived poorly in either the spleen or LNs of recipients (Fig. 2 C, middle and right, respectively).
In light of our finding that CD4 cells with both central and effector memory phenotypes require IL-7 for persistence, it was of interest to compare the relative dependency of memory CD4 cells on IL-7 for survival with that of naive CD4 cells. A potentially similar requirement was suggested by our observation that anti–IL-7 treatment reduced the recovery of polyclonal naive phenotype (CD44lo) as well as memory phenotype (CD44hi) CD4 cells from otherwise intact recipients (Fig. 3 C). Thus, we measured the decay of naive and memory AND TCR transgenic cells in SCID hosts where CD4 cell survival could be assessed independently of the presence of peripheral T cells. As shown in Fig. 3 D, naive or memory AND cells residing in SCID hosts treated for 14 d with anti–IL-7 or control IgG decayed to similar extents when IL-7 was blocked. To measure survival of naive cells, purified AND cells from (B10.BR × B6 PL Thy 1.1) F1 mice were injected into (B10.BR × C57BL/6) F1 SCID mice and the recipients were treated immediately with anti–IL-7 or control IgG Abs (Fig. 3 D, left). For memory cell analysis, SCID mice were injected with naive AND cells and immunized with PCC peptide with CFA; these mice were rested for 1 mo and treated with anti–IL-7 or control antibody for 14 d (Fig. 3 D, right). Importantly, we found that naive AND cells failed to undergo homeostatic proliferation in the SCID hosts (unpublished data).
Despite the clarity of the data in Figs. 2 (A–C) and 3, a potential caveat is the fact that OT-II memory cells were generated in the presence of elevated basal levels of IL-7 in lymphopenic Rag-2− hosts. Therefore, it is possible that such conditions favored production of memory cells dependent on IL-7. To address this issue, OT-II memory cells were generated under conditions of normal IL-7 levels by transferring naive OT-II cells into intact T-sufficient C57BL/6 hosts and priming the mice with OVA protein and adjuvant. Although many fewer OT-II memory cells were recovered from normal mice than from Rag-2− mice 1 mo later, both types of memory OT-II cells displayed similar dependence for IL-7. Thus, memory OT-II cells from C57BL/6 hosts survived with significantly reduced efficiency in IL-7− hosts as compared with IL-7Rα− hosts at 1 wk (Fig. 2 D).
IL-7 Regulates the Generation of Memory CD4 Cells in Both Lymphoid and Nonlymphoid Tissues.
The aforementioned results support the conclusion that IL-7 can promote the survival of memory CD4 cells that reside in the lymphoid compartment. Because memory CD4 cells are thought to be generated in situ in multiple tissues throughout the body from the widely disseminated effector cells (36), next, we sought to determine whether IL-7 also affects the development of memory CD4 cells. To this end, purified naive OT-II Thy 1.1 cells were transferred into groups of IL-7− and IL-7Rα− mice immunized with OVA protein and adjuvant, and the fate of OT-II cells followed in both lymphoid and peripheral tissues. Examination of one group of recipients on day 5 revealed that similar numbers of activated OT-II cells were recovered in the spleens, LNs, lungs, and livers from both types of mice (Fig. 4 A). As seen with Rag-2− hosts, prominent proliferation of OT-II cells was observed in the draining mesenteric LNs at this time by BrdU incorporation (unpublished data). Strikingly, analysis of the recipients on day 21 showed that OT-II cells were no longer detectable in the lymphoid or nonlymphoid tissues of IL-7− recipients, whereas these cells were clearly present in all the tissues examined in IL-7Rα− hosts (Fig. 4 B).
Figure 4.

Depletion of primed CD4 cells in the absence of IL-7. (A) Cell recovery from lymphoid and nonlymphoid tissues after primary immunization. Naive OT-II Thy CD4 cells were transferred into IL-7− or IL-7R− mice (5 × 106 cells/recipient). The mice were immunized with OVA as described in Materials and Methods. On day 5, donor cell recovery was assessed in the spleen, LNs, lung, and liver as depicted in Fig. 2. (B) Recovery of primed CD4 cells after rest and challenge of IL-7− and IL-7R− recipients. Mice from the same experiment shown in A were evaluated for the presence of donor CD4 cells at 3 wk after OT-II Thy 1.1 CD4 cell transfer and immunization (left), and 5 d later after a secondary response was induced by challenge with OVA (right). Thy 1.1 and vβ5 staining of the total CD4 cells recovered from each tissue is shown. (C) Summary. Total donor OT-II cell recovery from each site before and after boosting with OVA, as determined from the cell counts as for Fig. 2.
To better ascertain whether memory OT-II cells generated in IL-7− hosts disappeared completely from various tissues, IL-7− and IL-7R− hosts from the aforementioned experiment that were rested for 3 wk after priming were challenged with OVA protein and adjuvant. As depicted in Fig. 4 (B and C), at 5 d after boosting, OT-II cells were very sparse in all the tissues we examined from IL-7− hosts, whereas control IL-7R− mice showed significant expansion of OT-II cells in both lymphoid and nonlymphoid tissues. We conclude that IL-7 deprivation can lead to impaired development of memory CD4 cells in both the lymphoid and nonlymphoid compartments.
Discussion
In this paper, we investigated the potential of IL-7 to function in the homeostasis of memory CD4 cells in vivo. Our results suggest that IL-7 is a key regulator of memory CD4 cell survival not only long term, but also during earlier stages when resting memory cells develop from primary cells that have undergone response to antigen. The notion that IL-7 might be a cytokine candidate for control of memory CD4 cell homeostasis was initially revealed by our in vitro assessment of the effects of rIL-7 on the survival of resting memory CD4 cells compared with naive CD4 cells (Fig. 1). By using highly purified resting TCR transgenic CD4 cells, we ensured greater uniformity of memory populations than in previous analyses of memory phenotype cells from normal animals. Importantly, we observed naive and memory CD4 cells express equivalent amounts of IL-7Rα and Bcl-2 and up-regulate Bcl-2 to comparable levels after exposure to IL-7, supporting the possibility that both populations have the potential to be similarly regulated by this cytokine. IL-7–mediated survival by memory CD4 cells was not accompanied by division, as shown previously for naive CD4 cells (32). The data suggest that survival and homeostatic division can be separately regulated in both naive and memory CD4 cells.
In vivo, IL-7 deprivation resulted in the disappearance of transferred memory CD4 cells in both intact and immunodeficient recipients (Fig. 2). Although deficiency of IL-7 also disrupts both T and B cell lymphopoeisis (37), it is striking that IL-7– and IL-7R–deficient animals, which display a similar phenotype with regard to a lack of mature lymphocytes, show a profound difference in their ability to maintain resting memory CD4 cells. Few memory CD4 cells were recoverable from either the lymphoid or nonlymphoid compartments in the absence of IL-7. The potential for IL-7 to regulate memory CD4 cell survival in vivo is further supported by the results of IL-7 blocking studies in normal recipients, where both donor and host memory CD4 cells decayed in recipients rendered deficient in IL-7 (Fig. 3). These data provide strong support for a conclusion that IL-7 is at least one factor that contributes to maintaining memory CD4 cells in vivo.
Although several previous studies have not detected a requirement for IL-7 in regulating the homeostasis of memory CD4 cells, it is important to bear in mind that a primary focus has been proliferative capacity in lymphopenic recipients (13). Our data suggest that IL-7 may not affect memory CD4 cell expansion. In addition, we find that under lymphopenic conditions where heterogeneous populations of normal memory CD4 cells undergo homeostatic division, reduced recovery is observed in the absence of IL-7 (unpublished data), in line with our current results from IL-7 blocking studies in normal recipients. The use of irradiated hosts for analysis of cytokine dependence in some studies may further complicate analysis of requirements for individual cytokines due to the rapid induction of multiple cytokines, including TGF-β (38) and IL-6 (39), that have the potential to mediate T cell survival.
Analyses of γc cytokine receptor–deficient mice indicate that some T cells can be generated in the absence of a capacity to respond to this cytokine family (21). In addition, when TCR transgenic mice are crossed to γc receptor knockout mice, CD4 cells with an activated phenotype that are highly susceptible to apoptosis arise. These findings suggest that, whereas other mediators can support expansion of the few naive CD4 cells that are generated in the absence of a γc cytokine response, most of γc-deficient naive CD4 cells fail to survive. However, TCR transgenic CD4 cells from γc receptor–deficient mice can survive upon activation and differentiation into memory CD4 cells in γc receptor–deficient recipients (18). Although this finding was interpreted to indicate that CD4 memory cell homeostasis is regulated independently of γc family cytokines, it is also possible that the finding either applies to only a minor subset of memory CD4 cells or is a reflection of a capacity to engage aberrant compensatory survival mechanisms that arise when responses to γc cytokines are genetically impaired (18). In either case, γc-deficient T cells might utilize other cytokines that become available, and these cytokines may promote homeostatic division under conditions of lymphopenia to sustain long-term memory.
Consistent with previous conclusions that CD4 cells do not depend solely on γc family cytokines for proliferation during an Ag-induced response (18), we found that naive TCR transgenic CD4 cells could be comparably expanded by cognate Ag and that the resulting effector cells distributed normally in both IL-7–sufficient and IL-7–deficient recipients. Our results suggest that IL-7 is not critical for survival of CD4 cells during Ag-induced activation or division, or for their survival during dissemination to nonlymphoid sites. However, the primed population disappeared from both lymphoid and nonlymphoid tissues only under conditions of IL-7 deprivation. Thus, we envision that IL-7 is necessary for the survival of developing memory cells. Because our previous analyses demonstrate that cytokines are not required for activated CD4 cells to return to rest and acquire properties of memory cells (28), it is less likely that IL-7 also regulates the differentiation of memory cells. IL-7 is not only produced by stromal cells in lymphoid tissues but also by epithelium, liver, and skin (14, 37, 40). Thus, it is possible that local availability of IL-7 can support the survival of memory cells in many different tissues, and that the decay we observed in nonlymphoid sites is not due to T cell migration/recirculation that results in IL-7 withdrawal only in the lymphoid compartment.
Although it could be argued that memory CD4 cells primed in lymphopenic animals might be atypical because of their induction and maintenance in an environment where survival factors may be elevated due to the lack of normal consumption, our finding that transgenic CD4 cells induced in normal recipients showed a similar dependence on IL-7 for survival suggests that this is an unfounded concern. In addition, when polarized Th1 and Th2 cells were induced under conditions where fully differentiated effectors are generated, and rested to induce a memory-like population (28), we observed similar susceptibility to IL-7 withdrawal in vivo (unpublished data). Thus, recently primed CD4 cells also appear to be highly susceptible to sudden withdrawal of IL-7. Consistent with the antiapoptotic effects of IL-7, our data suggest that IL-7 may play a key role in preventing the demise of effectors by activated T cell autonomous death (41), thereby promoting CD4 cell survival during the effector to memory transition. Thus, IL-7 may be an important survival factor for early memory CD4 cells as well as for maintaining long term memory CD4 cells.
Because we studied TCR transgenic memory CD4 cells generated by specific immunization protocols, it is possible that the findings may not be generally typical for memory CD4 cells. However, our observations that transgenic memory CD4 cells with either central or effector memory phenotypes can utilize IL-7 for persistence and that transgenic and polyclonal naive and memory CD4 cells have comparable IL-7 dependence are striking (Figs. 2 and 3). Together with previous papers on naive and memory CD8 cells (11, 13, 15, 16, 30, 42–44), our results support the hypothesis IL-7 is a key cytokine for the physiologic survival of resting peripheral T cells and in this capacity contributes to regulation of the pool sizes of naive and memory cells for both the CD4 and CD8 subsets. Nevertheless, alterations in cytokine receptor expression during an immune response and the relative abundance of various cytokines in the local milieu may determine changes in the preferential usage of growth and/or survival factors.
It remains unclear why IL-7 appears to function to mediate survival but not homeostatic proliferation of memory CD4 cells. However, this finding is reminiscent of the fact that IL-2 and IL-4, in addition to IL-7, transduce signals that promote survival of resting T cells without inducing division (45). Furthermore, a recent analysis of polyclonal memory phenotype CD4 cells suggests that TCR-mediated signals can support homeostatic cycling of memory CD4 cells in the absence of IL-7 but that IL-7 is required in addition for optimal survival (19). Our results also suggest that survival and homeostatic turnover of T cells need not be linked in vivo. Mediators that might exclusively promote turnover of memory CD4 cells have yet to be identified, and it is possible that multiple cytokines could exhibit redundancy in this regard. Importantly, our data suggest that an essential step in the maintenance of CD4 cell memory is the provision of survival signals from cytokines, and that IL-7 may play a prominent role in this process.
Acknowledgments
This work was supported by National Institutes of Health grants AI32978, AI46530, and DK59438 to L.M. Bradley and AI41079, AI45809, and AG20186 to C.D. Surh. C.D. Surh is a Scholar of the Leukemia and Lymphoma Society. J.T. Tan is supported by postdoctoral fellowship grant PF-02-121-01LIB from the American Cancer Society.
Abbreviations used in this paper: APC, allophycocyanin; BrdU, bromodeoxyuridine; γc, common γ chain; ICS, intracellular staining; mIgG, mouse IgG; PCC, pigeon cytochrome c; rIgG, rat IgG.
References
- 1.Welsh, R.M., and L.K. Selin. 2002. No one is naive: the significance of heterologous T-cell immunity. Nat. Rev. Immunol. 2:417–426. [DOI] [PubMed] [Google Scholar]
- 2.Homann, D., L. Teyton, and M.B.A. Oldstone. 2001. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T cell memory. Nat. Med. 7:913–919. [DOI] [PubMed] [Google Scholar]
- 3.Varga, S.M., L.K. Selin, and R.M. Welsh. 2001. Independent regulation of lymphocytic choriomeningitis virus-specific T cell memory pools: relative stability of CD4 memory under conditions of CD8 memory T cell loss. J. Immunol. 166:1554–1561. [DOI] [PubMed] [Google Scholar]
- 4.Ernst, B., D.-S. Lee, J.M. Chang, J. Sprent, and C.D. Surh. 1999. The peptide ligands mediating positive selection in the thymus control T cell survival and homeostatic proliferation in the periphery. Immunity. 11:173–181. [DOI] [PubMed] [Google Scholar]
- 5.Tanchot, C.F., A. Lemonnier, B. Perarnau, and A.A. Freitas. 1997. Differential requirements for survival and proliferation of CD8 naive or memory T cells. Science. 276:2057–2062. [DOI] [PubMed] [Google Scholar]
- 6.Murali-Krishna, K., L.L. Lau, S. Sambhara, F. Lemonnier, J. Altman, and R. Ahmed. 1999. Persistence of memory CD8 T cells in MHC class I-deficient mice. Science. 2886:1377–1381. [DOI] [PubMed] [Google Scholar]
- 7.Swain, S.L., H. Hui, and G. Huston. 1999. Class II-independent generation of CD4 memory T cells from effectors. Science. 286:1381–1383. [DOI] [PubMed] [Google Scholar]
- 8.Kassiotis, G., S. Garcia, E. Simpson, and B. Stockinger. 2002. Impairment of immunological memory in the absence of MHC despite survival of memory T cells. Nat. Immunol. 3:244–250. [DOI] [PubMed] [Google Scholar]
- 9.Zhang, X., S. Sun, I. Hwang, D.F. Tough, and J. Sprent. 1998. Potent and selective stimulation of memory phenotype CD8+ T cells in vivo by IL-15. Immunity. 8:591–599. [DOI] [PubMed] [Google Scholar]
- 10.Becker, T.C., E.J. Wherry, D. Boone, K. Murali-Krishna, R. Anita, A. Ma, and R. Ahmed. 2002. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J. Exp. Med. 195:1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldrath, A.W., P.V. Sivakumar, M. Glaccum, M.K. Kennedy, M.J. Bevan, C. Benoist, D. Mathis, and E.A. Butz. 2002. Cytokine requirements for acute and basal homeostatic proliferation of naive and memory CD8+ T cells. J. Exp. Med. 195:1515–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Judge, A.D., X. Zhang, H. Fuji, C.D. Surh, and J. Sprent. 2002. Interleukin 15 controls both proliferation and survival of a subset of memory phenotype CD8+ T cells. J. Exp. Med. 196:935–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan, J., B. Ernst, W.C. Kieper, E. LeRoy, J. Sprent, and C.D. Surh. 2002. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J. Exp. Med. 195:1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sudo, T., S. Nishikawa, N. Ohno, N. Akiyama, M. Takakoshi, H. Yoshida, and S.-I. Nishikawa. 1993. Expression and function of the interleukin 7 receptor in murine lymphocytes. Proc. Natl. Acad. Sci. USA. 90:9125–9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan, J.T., E. Dudler, E. LeRoy, R. Murray, J. Sprent, K.I. Weinberg, and C.D. Surh. 2001. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc. Natl. Acad. Sci. USA. 98:8732–8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schluns, K.S., W.C. Kieper, S.C. Jameson, and L. Lefrancois. 2000. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat. Immunol. 1:426–432. [DOI] [PubMed] [Google Scholar]
- 17.Geginat, J., F. Sallusto, and A. Lanzavecchia. 2001. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4+ T cells. J. Exp. Med. 194:1711–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lantz, O., I. Grandjean, P. Matzinger, and J.P. Di Santo. 2000. γ chain required for naive CD4+ T cell survival but not for antigen proliferation. Nat. Immunol. 1:54–58. [DOI] [PubMed] [Google Scholar]
- 19.Seddon, B., P. Tomlinson, and R. Zamoyska. 2003. Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat. Immunol. 4:680–686. [DOI] [PubMed] [Google Scholar]
- 20.Freitas, A.A., and B. Rocha. 1999. Peripheral T cell survival. Curr. Opin. Immunol. 11:152–156. [DOI] [PubMed] [Google Scholar]
- 21.Nakajima, H., E.W. Shores, M. Noguchi, and W. Leonard. 1997. The common cytokine receptor γ chain plays an essential role in regulating lymphoid homeostasis. J. Exp. Med. 186:189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Freeden-Jeffry, U., P. Vieira, L.A. Lucian, T. McNeil, S.E.G. Burdach, and R. Murray. 1995. Lymphopenia in interleukin (IL)-7 gene deleted mice identifies IL-7 as a nonredundant cytokine. J. Exp. Med. 181:1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barnden, M.J., J. Allison, W.R. Heath, and F.R. Carbone. 1998. Defective TCR expression in transgenic mice constructed using cDNA based α- and β-chain genes under the control of heterologous regulatory elements. Immunol. Cell. Biol. 76:34–40. [DOI] [PubMed] [Google Scholar]
- 24.Kaye, J., M.-L. Hsu, M.E. Sauron, S.C. Jameson, N.R.J. Gascoigne, and S. Hedrick. 1989. Selective development of CD4 T cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature. 341:746–749. [DOI] [PubMed] [Google Scholar]
- 25.Grabstein, K.H., T.J. Waldschmidt, F.D. Finkelman, B.W. Hess, A.R. Alpert, N.E. Boiani, A.E. Namen, and P.J. Morrissey. 1993. Inhibition of murine B and T lymphopoiesis in vivo by an anti-interleukin 7 monoclonal antibody. J. Exp. Med. 178:257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linton, P.J., B. Bautista, E. Biederman, E.S. Bradley, J. Harbertson, R.M. Kondrack, R.C. Padrick, and L.M. Bradley. 2003. Costimulation via OX40L expressed by B cells is sufficient to determine the extent of primary CD4 cell expansion and Th2 cytokine secretion in vivo. J. Exp. Med. 197:875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bradley, L.M., J. Harbertson, and S.R. Watson. 1999. Memory CD4 cells do not migrate into peripheral lymph nodes in the absence of antigen. Eur. J. Immunol. 29:3273–3284. [DOI] [PubMed] [Google Scholar]
- 28.Harbertson, J., E. Biederman, K.E. Bennett, R.M. Kondrack, and L.M. Bradley. 2002. Withdrawal of stimulation may initiate the transition of effector to memory CD4 cells. J. Immunol. 168:1095–1102. [DOI] [PubMed] [Google Scholar]
- 29.Targoni, O.S., J. Baus, H.H. Hofstetter, M.D. Hesse, A.Y. Karulin, B.O. Boehm, T.G. Forsthuber, and P.V. Lehmann. 2001. Frequencies of neuroantigen-specific T cells in the central nervous system versus the immune periphery during the course of experimental allergic encephalomyelitis. J. Immunol. 166:4757–4764. [DOI] [PubMed] [Google Scholar]
- 30.Vella, A.T., T.K. Teague, J. Ihle, J. Kappler, and P. Marrack. 1997. Interleukin 4 (IL-4) or IL-7 prevents the death of resting T cells: Stat6 is probably not required for the effect of IL-4. J. Exp. Med. 186:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grabstein, K.H., A.E. Namen, K. Shanebeck, R.F. Voice, S.G. Reed, and M. Widmer. 1990. Regulation of T cell proliferation by IL-7. J. Immunol. 144:938–941. [PubMed] [Google Scholar]
- 32.Rathmell, J.C., E.A. Farkash, W. Gao, and C.B. Thompson. 2001. IL-7 enhances the survival and maintains the size of naive T cells. J. Immunol. 167:6869–6876. [DOI] [PubMed] [Google Scholar]
- 33.Peschon, J.J., P.J. Morrissey, K.H. Grabstein, F.J. Ramsdell, E. Maraskovsky, B.C. Gliniak, L.S. Park, S.F. Ziegler, D.E. Williams, C.B. Ware, et al. 1994. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J. Exp. Med. 180:1955–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iezzi, G., D. Scheidegger, and A. Lanzavecchia. 2001. Migration and function of antigen-primed nonpolarized T lymphocytes in vivo. J. Exp. Med. 193:987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sallustro, F., D. Lenig, R. Forster, M. Lipp, and A. Lanzavecchia. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 140:708–712. [DOI] [PubMed] [Google Scholar]
- 36.Reinhardt, R.L., A. Khoruts, R. Merica, T. Zell, and M.K. Jenkins. 2001. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 410:101–105. [DOI] [PubMed] [Google Scholar]
- 37.Hofmeister, R., A.R. Khaled, N. Benbernou, E. Rajnavolgi, K. Muegge, and S.K. Durum. 1999. Interleukin-7: physiological roles and mechanisms of action. Cytokine Growth Factor Rev. 10:41–60. [DOI] [PubMed] [Google Scholar]
- 38.Ehrhart, E.J., P. Segarini, M.L. Tsang, A.G. Carroll, and M.H. Barcellos-Hoff. 1997. Latent transforming growth factor beta-1 activation in situ: quantitative and functional evidence after low dose gamma irradiation. FASEB J. 11:991–1002. [DOI] [PubMed] [Google Scholar]
- 39.Chang, C.M., A. Limanni, W.H. Baker, M.E. Dobson, J.F. Kalinich, and M.L. Patchen. 1997. Sublethal gamma irradiation increases IL-1α, IL-6, and TNF-α mRNA levels in murine hematopoietic tissues. J. Interferon Cytokine Res. 17:567–572. [DOI] [PubMed] [Google Scholar]
- 40.Wagner, L.A., T. Brown, S. Gil, I. Frank, W. Carter, R. Tamura, and E.A. Wayner. 1999. The keritinocyte-derived cytokine IL-7 increases adhesion of the epidermal T cell subset to the skin basement membrane protein laminin-5. Eur. J. Immunol. 29:2530–2538. [DOI] [PubMed] [Google Scholar]
- 41.Hildeman, D.A., Y. Zhu, T.C. Mitchell, J. Kappler, and P. Marrack. 2002. Molecular mechanisms of activated T cell death in vivo. Curr. Opin. Immunol. 14:354–359. [DOI] [PubMed] [Google Scholar]
- 42.Ku, C.C., M. Murakami, A. Sakamoto, J. Kappler, and P. Marrack. 2000. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science. 288:675–678. [DOI] [PubMed] [Google Scholar]
- 43.Vivien, L., C. Benoist, and D. Mathis. 2001. T lymphocytes need IL-7 but not IL-4 or IL-6 to survive in vivo. Int. Immunol. 13:763–768. [DOI] [PubMed] [Google Scholar]
- 44.Li, X., G. Demirci, S. Ferrari-Lacraz, C. Groves, A. Coyle, T.R. Malek, and T.B. Storm. 2001. IL-15 and IL-2: a matter of life and death for T cells in vivo. Nat. Immunol. 7:114–118. [DOI] [PubMed] [Google Scholar]
- 45.Boise, L.H., A.J. Minn, C.H. June, T. Lindsten, and C.B. Thompson. 1995. Growth factors can enhance lymphocyte survival without committing the cell to undergo division. Proc. Natl. Acad. Sci. USA. 92:5491–5495. [DOI] [PMC free article] [PubMed] [Google Scholar]