Characterization of the CD4+ T Cell Response to Epstein-Barr Virus during Primary and Persistent Infection (original) (raw)
Abstract
The CD8+ T cell response to Epstein-Barr virus (EBV) is well characterized. Much less is known about the evolution of the CD4+ T cell response. Here we show that EBV stimulates a primary burst of effector CD4+ T cells and this is followed by a period of down-regulation. A small population of EBV-specific effector CD4+ T cells survives during the lifelong persistent phase of infection. The EBV-specific effector CD4+ T cells accumulate within a CD27+ CD28+ differentiation compartment during primary infection and remain enriched within this compartment throughout the persistent phase of infection. Analysis of CD4+ T cell responses to individual epitopes from EBV latent and lytic cycle proteins confirms the observation that the majority of the effector cells express both CD27 and CD28, although CD4+ T cells specific for lytic cycle antigens have a greater tendency to express CD45RA than those specific for the latent antigens. In clear contrast, effector CD4+ T cells specific for cytomegalovirus (CMV) accumulate within the CD27− CD28+ and CD27− CD28− compartments. There are striking parallels in terms of the differentiation of CD8+ T cells specific for EBV and CMV. The results challenge current ideas on the definition of memory subsets.
Keywords: immunity, antigens CD27, antigens CD28, Epstein-Barr virus, cytomegalovirus
Introduction
Antigen-experienced T cells comprise a heterogeneous population with enormous diversity in terms of antigen specificity, expression of cell surface molecules, and effector capacity. In the context of CD8+ T cells an effort has been made to understand this complexity by considering cells as distributed between three differentiation compartments defined by expression of the costimulatory molecules CD27 and CD28 (1–7). A linear model of CD8+ T cell maturation has been proposed, with cells progressing from a CD45RA+ CD27+ CD28+ (naive) phenotype via a CD45RA− CD27+ CD28+ (early antigen–experienced) to a CD45RA−/+ CD27+ CD28− (intermediate antigen–experienced), and then to a CD45RA−/+ CD27− CD28− (late antigen–experienced) phenotype (6, 7).
Longitudinal studies show that EBV-specific CD8+ T cells accumulate within the CD27+ CD28+ and CD27+ CD28− compartments during primary EBV infection (8, 9) and remain enriched within these compartments during the lifelong persistent phase of EBV infection (6, 9, 10). CD8+ T cells specific for the latent proteins are more likely to be enriched within the CD27+ CD28+ compartment whereas those specific for lytic cycle proteins accumulate within both CD27+ CD28+ and CD27+ CD28− compartments. HIV-specific CD8+ T cells differentiate into the CD27+ CD28+ and CD27+ CD28− compartments during primary HIV infection but come to lie predominantly within the CD27+ CD28− compartment during the lifelong persistent phase of HIV infection (6). During persistent CMV infection, CMV-specific CD8+ T cells are enriched within the CD27− CD28− compartment (6). Thus, memory CD8+ T cells with different specificities accumulate in distinct differentiation compartments. In this context, “memory” is used as a term to describe the population of specific T cells that persists after resolution of primary infection regardless of their phenotypic and functional properties.
Investigation of the differentiation of virus-specific CD8+ T cells was facilitated by the use of multicolor flow cytometry and fluorescent-labeled HLA peptide tetrameric complexes, enabling the characterization of epitope-specific CD8+ T cells in samples of peripheral blood. Similar studies of the CD4+ T cell response to viruses have been hampered by paucity of reagents capable of identifying CD4+ T cells on the basis of specificity. Here we have analyzed CD4+ T cell differentiation in the context of the immune response to EBV. To detect virus-specific CD4+ T cells, we adapted an assay that has been developed to analyze the CD4+ T cell response to human CMV (11–13) and used a lysate of EBV-infected cells to stimulate samples of PBMCs in vitro. Small populations of CD4+ T cells from EBV-seropositive, but not EBV-seronegative, donors respond to stimulation by this lysate by expressing IFN-γ. We have used this assay to analyze the primary CD4+ T cell response to EBV and have gone on to examine the memory CD4+ T cell response to EBV in selected patients. We have then extended the study to include an analysis of memory EBV-specific CD4+ T cells in healthy EBV-seropositive individuals and have compared these cells with CMV-specific CD4+ T cells in the same individuals. CD4+ T cells characterized using these methods are, by definition, part of the effector subset, as they are identified on the basis of their capacity to express IFN-γ after short-term stimulation with antigen. The work has allowed us to characterize the effector CD4+ T cell response to EBV and compare the phenotype of effector CD4+ T cells specific for two different herpesviruses.
Materials and Methods
Patients and Samples.
36 patients were identified during the acute stage of infectious mononucleosis (IM) due to primary EBV infection, using an EBV-specific latex agglutination test. Samples of peripheral blood were taken immediately after diagnosis and, in seven patients, 4 mo later. Peripheral blood was also taken from 28 healthy EBV-seropositive individuals and 5 healthy EBV-seronegative individuals. EBV serological status was determined by testing first for the presence of EBV nuclear antigen (EBNA)1 IgG and then, in individuals negative for this antibody, for EBV IgG. Two samples of cord blood were also collected. PBMCs were separated by centrifugation on a lymphoprep gradient (Nycomed Pharam) and used in experiments immediately. The Oxfordshire Clinical Research Ethics Committee approved this study.
Stimulation of PBMCs.
Freshly separated PBMCs were incubated for 18 h in RPMI (GIBCO BRL) supplemented with 10% fetal calf serum and 10 U/ml recombinant IL-2, either alone or in the presence of an EBV-infected B cell lysate, a CMV-infected fibroblast cell lysate (both from East Coast Biologics, Inc.), or peptides from EBV latent or lytic cycle proteins, including PQCRLTPLSRLPFGM, RPFFHPVGEADYFEY, NPKFENIAEGLRALL, and TSLYNLRRGTALAI from EBNA1 (14), TVFYNIPPMPL and PRSPTVFYNIPPMPL from EBNA2 (15 and unpublished data), SDDELPYIDNMEPV (SDD) from EBNA3C (14), NFDVGGKKHQLDLDFGQLTP (NFD) from GP350 (unpublished data), and pools of overlapping 20 mer peptides covering the amino acid sequences of BMLF1, BZLF1, and GP350. All antigens were used at a final concentration of 10 μg/ml. Brefeldin A (Sigma-Aldrich) was added after 1 h at a final concentration of 5 μg/ml. The incubation was performed in V-bottomed tubes at 37°C in the presence of 5% CO2. The CMV-infected fibroblast cell lysate has been extensively used as a tool for investigating responses to CMV. The EBV-infected B cell lysate is derived from activated B cells infected with the P3HR1 strain of EBV and enriched for viral proteins. The manufacturers report that it contains proteins reactive with antibodies to viral capsid and early and nuclear antigens. As controls, PBMCs were also incubated with 10 μg/ml of a lysate from uninfected B cells, selected from PBMCs using dynabeads conjugated to anti-CD19 mAb, and with an uninfected fibroblast cell lysate (Virusys).
ELISA to Detect EBV Proteins.
MaxiSorp plates were coated with (a) the commercial EBV-infected B cell lysate, (b) a lysate preparation of uninfected B cells, positively selected from PBMCs using CD19-coated dynabeads, or (c) a lysate preparation of B cell blasts that had been infected overnight with a recombinant vaccinia virus expressing BMRF1 (r-VV BMRF1) at a multiplicity of infection (MOI) of 10:1 or (d) no protein. Plates were washed with PBS/0.1% Tween and blocked with buffer containing 1% BSA before being incubated with saturating concentrations of antibodies specific for BZLF1 (DakoCytomation), BMRF1 (Virusys), and LMP-1 (DakoCytomation), in blocking buffer. Antibody binding was detected using ALK anti-IgG1 followed by the addition of _p_-Nitrophenyl phosphate (Sigma-Aldrich).
Staining of PBMCs.
Cells were washed in PBS buffer containing 0.16% BSA and 0.1% sodium azide (PBA) and stained for 30 min on ice with saturating amounts of antibodies including anti–CD45RA-FITC (BD Biosciences), anti–CD45RO-FITC (DakoCytomation), anti–CD45RO-PE (BD Biosciences), anti–CD27-FITC (BD Biosciences), anti–CD27-PE (BD Biosciences), anti–CD62L-FITC (BD Biosciences), anti–CD57-FITC (BD Biosciences), anti–CD28-FITC (BD Biosciences), anti–CD28-PE (BD Biosciences), and anti–CD28-APC (BD Biosciences). Cells were stained for CCR7 by incubating with anti–human CCR7 mAb (BD Biosciences) and binding was detected by subsequent incubation with rabbit anti–mouse IgG-FITC (DakoCytomation). All antibodies were diluted in PBA supplemented by 1% human serum. Cells were then washed in PBA supplemented by 1% human serum and 1% mouse serum, fixed, and permeabilized with cytofix-cytoperm (BD Biosciences), as per the manufacturer's instructions. Aliquots of 106 cells were stained with 0.1 μg anti–IFN-γ–APC (BD Biosciences) or with mouse IgG2a-APC (BD Biosciences) as a negative control. All antibodies were diluted in cytoperm buffer and saturating amounts of anti–CD4-PerCP (BD Biosciences) and anti–CD3-PE (DakoCytomation) or anti–CD8-PE (BD Biosciences) and anti–CD3-PerCP (BD Biosciences) were also included in the intracellular staining mix. After 60 min, cells were washed in cold PBS and resuspended in PBS containing 1% formaldehyde and 1% fetal calf serum. Analysis was performed on a FACSCalibur™ flow cytometer (BD Biosciences) using CELLQuest™ software. Where possible, 500,000 events were collected for analysis. Fewer cells were available from some patients with IM and in these cases 200,000 events were collected. Gates were set on lymphocytes according to their forward and side scatter characteristics. Using these methods, we are able to detect populations of responding cells present at frequencies as low as 0.03% CD4+ T cells. In all but one experiment described here, the constitutive expression of IFN-γ by CD4+ T cells was <0.01%. In one patient with IM we found that 0.3% CD4+ T cells expressed IFN-γ directly ex vivo. For the purposes of analysis this frequency was subtracted from that of CD4+ T cells responding after stimulation with the viral lysate, although it is likely that the cells were EBV specific.
Results and Discussion
Detection of EBV-specific CD4+ T Cells within Preparations of PBMCs.
To characterize the CD4+ T cell response to EBV we developed an assay to detect EBV-specific T cells within samples of peripheral blood. We used a commercially available lysate prepared from EBV-infected B cells and enriched for viral antigens as a source of EBV antigens. By ELISA we confirmed the manufacturer's reports that the lysate contained both lytic cycle and latent proteins and were able to detect BZLF1, BMRF1, and LMP-1. These proteins were not detected in a sample of lysed uninfected B cells (Fig. 1 a and unpublished data). The concentration of BMRF1 within the commercial lysate was greater than that found within a detergent lysate of B cells infected overnight with r-VV BMRF1 at an MOI of 10:1 (Fig. 1 a).
Figure 1.
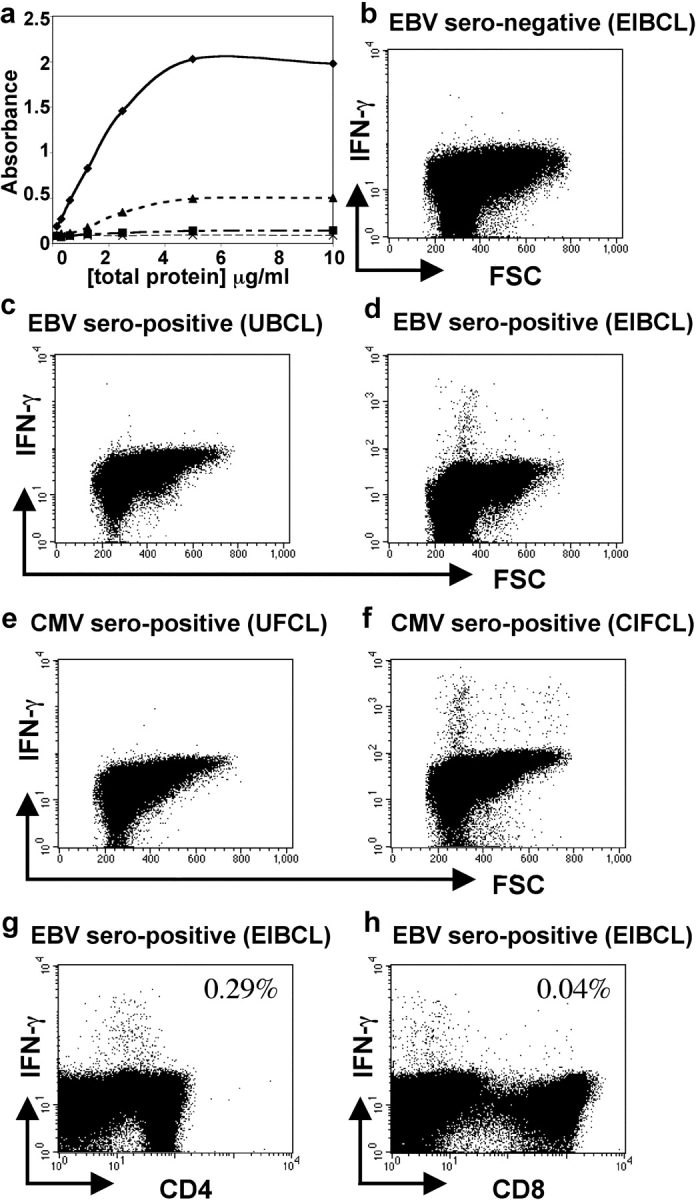
Detection of EBV-specific CD4+ T cells in samples of PBMCs. (a) BMRF1 was detected at higher concentrations in the EBV-infected cell lysate preparation (♦) than in a lysate from B cells infected overnight with r-VV-BMRF1 (MOI 10:1; ▴). BMRF1 was not detected in a lysate prepared from uninfected B cells (▪) or when plates were not coated with protein (X). PBMCs from (b) an EBV-seronegative and (c and d) an EBV-seropositive donor were cultured in the presence of (b and d) lysate from EBV-infected cells (EIBCL) or (c) lysate from uninfected B cells (UBCL). (e and f) PBMCs from a CMV-seropositive donor were cultured in presence of (e) lysate from uninfected fibroblasts (UFCL) or (f) lysate from CMV-infected fibroblasts (CIFCL). Cells were stained for expression of IFN-γ and CD3, CD4, and CD8. A population of cells within PBMCs from the EBV-seropositive donor responded to the EIBCL and a population from the CMV-seropositive donor responded to the CIFCL. (g and h) Responding EBV-specific IFN-γ–expressing cells were predominantly CD4+ T cells.
Incubation of PBMCs taken from EBV-seropositive donors, in R10 supplemented by the EBV-infected B cell lysate, in the presence of brefeldin A, stimulated detectable IFN-γ expression in a subset of lymphocytes (Fig. 1 d). In contrast, PBMCs from these donors, when incubated in R10 alone or with an uninfected B cell lysate, both in the presence of brefeldin A, did not express IFN-γ (Fig. 1 c and unpublished data). Furthermore, we found that the lysate did not stimulate lymphocytes, taken from any of five EBV-seronegative adults or two samples of cord blood, to express IFN-γ (Fig. 1 b and unpublished data). Analysis of responding lymphocytes in the EBV-seropositive donors showed that they were CD3+ T cells and that most expressed CD4 and not CD8 (Fig. 1, g and h). We observed some down-regulation of CD4 by responding T cells, particularly at 18 h. The assay was more sensitive at 18 h compared with 6 h, with higher numbers of CD4+ T cells expressing detectable IFN-γ at this time point. The type of antigen preparation used would be expected to preferentially stimulate a CD4+ T cell response in vitro. Engagement of CD8+ T cells would require cross-presentation to occur and this process was relatively inefficient under the assay conditions used.
In parallel experiments we showed that culture of PBMCs from a CMV-seropositive donor with a CMV-infected fibroblast cell lysate stimulated expression of IFN-γ in a subset of lymphocytes (Fig. 1 f). PBMCs from the same donor did not express IFN-γ after incubation with a lysate from an uninfected fibroblast cell line or with medium alone (Fig. 1 e and unpublished data).
The Magnitude of the CD4+ T Cell Response to EBV during Primary and Persistent Infection.
This experimental protocol allowed us to probe the primary CD4+ T cell response to EBV in patients with acute IM. We studied 36 patients and found that the frequency of responding CD4+ T cells ranged from 0.04 to 5.2% CD4+ T cells (mean 1.4%; Fig. 2 a). Although the overall magnitude of the response might be underestimated, the results show that primary infection with EBV stimulates a significant, early CD4+ T cell response in vivo. The magnitude of the response is comparable to the size of the CD4+ T cell response to primary lymphocytic choriomeningitis virus or Listeria monocytogenes infection in mice and to HIV in humans (16–19). The size of these responses is substantially lower than that of the primary CD8+ T cell response to the same pathogens (8, 19–22).
Figure 2.
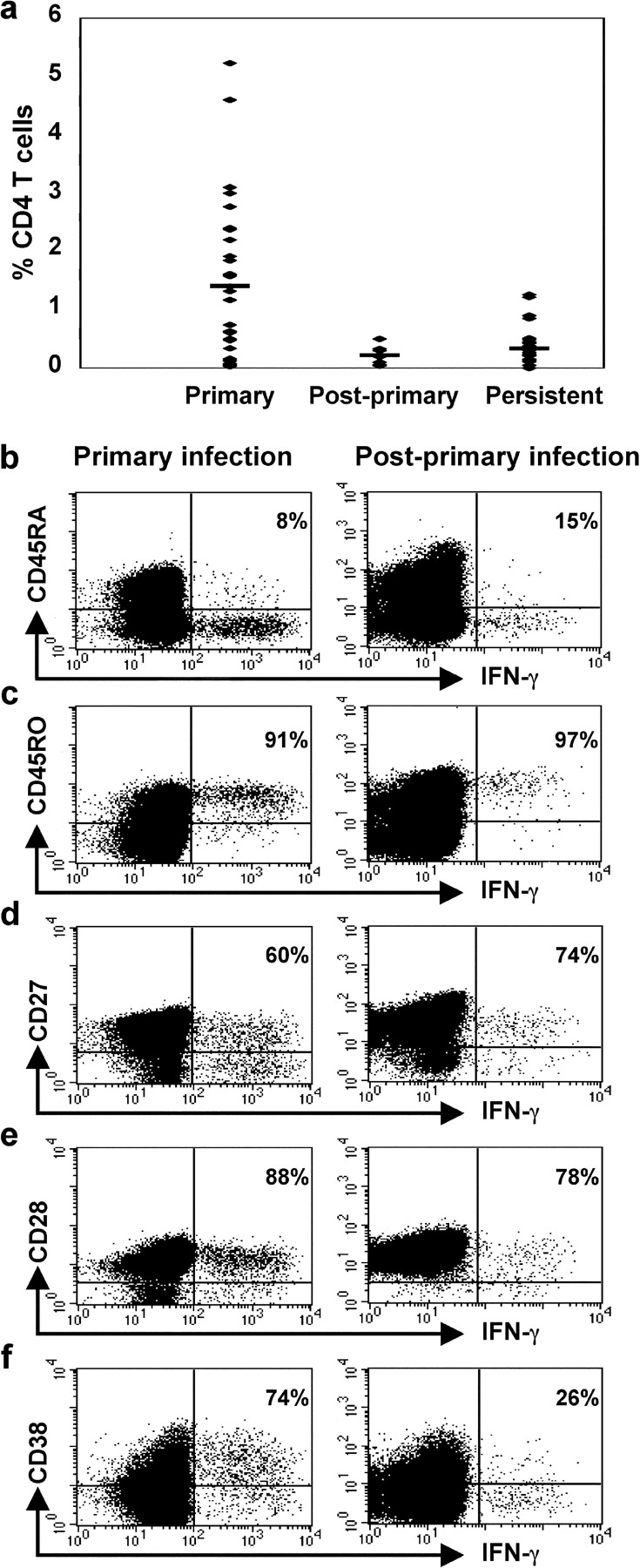
Frequency and phenotype of EBV-specific CD4+ T cells. (a) The frequency of CD4+ T cells that responded to the EBV-infected B cell lysate in 36 patients with IM (Primary), in 7 patients 4 mo after primary infection (Post-primary), and in 28 healthy EBV-seropositive individuals (Persistent). A mean of 1.4% CD4+ T cells responded during the primary phase of infection, a mean of 0.22% CD4+ T cells responded 4 mo later, and a mean of 0.34% CD4+ T cells responded during the persistent phase of infection. (b–f) PBMCs, taken from a patient with IM at the time of his acute illness (left column) and then again 4 mo later (right column), were cultured with lysate from EBV-infected cells. Cells were stained for expression of (b) CD45RA, (c) CD45RO, (d) CD27, (e) CD28, and (f) CD38, and then for expression of intracellular IFN-γ and CD4. Gates were set to include only CD4+ lymphocytes. The percentages of IFN-γ–expressing cells that stain with antibodies for each of the phenotypic markers are shown.
We obtained further specimens of PBMCs from seven patients 4 mo after primary infection. The magnitude of the response to the lysate was significantly lower at the second time point than during primary infection in this cohort (P < 0.005; mean reduction of 4.2-fold; Fig. 2 a).
We went on to determine the frequency of CD4+ T cells that responded to the EBV-infected cell lysate within PBMCs from healthy EBV carriers. In 5 out of 28 EBV-seropositive individuals we could not detect populations of responding cells. In the remaining 23 individuals we found that between 0.05 and 1.26% CD4+ T cells responded to the lysate (overall mean 0.34%; Fig. 2 a). The frequency of EBV-specific CD4+ T cells that we detected in these seropositive individuals was significantly lower than that detected in patients suffering from primary infection (P < 0.0001).
These results support the idea that EBV stimulates a primary CD4+ T cell burst and that many of the responding effector cells die or lose functional capacity, leaving a smaller population of effector cells to mediate protection during the persistent phase of infection. Our experiments do not address the fine specificity of the response, but a recently published study shows that CD4+ T cells specific for both lytic cycle proteins and latent proteins contribute to the primary response in patients with IM (23). It is possible that the dominant CD4+ T cell specificities change over time (22, 24).
Characterization of EBV-specific CD4+ T Cells during Primary and Persistent Infection.
We characterized the EBV-specific effector CD4+ T cell response in more detail by investigating the phenotype of the responding T cells. Experiments were performed on samples of PBMCs taken from 10 patients with IM due to primary EBV infection, 5 patients 4 mo later, and 10 healthy EBV-seropositive individuals.
Results from an experiment performed on patient NS128 while he was suffering from IM and then 4 mo later are shown in Fig. 2. During the primary phase of infection, 1.61% CD4+ T cells responded to culture with the EBV-infected cell lysate. The majority of these cells did not express CD45RA (Fig. 2 b) and did express CD45RO (Fig. 2 c). During the primary phase of infection most of the responding cells expressed CD27 (Fig. 2 d) and CD28 (Fig. 2 e) and had lost expression of CCR7 (unpublished data). 74% of the responding cells expressed CD38 (Fig. 2 f), suggesting recent activation in vivo.
4 mo later the frequency of responding CD4+ T cells had fallen to 0.33%. Most of these EBV-specific CD4+ T cells did not express CD38 (Fig. 2 f). In other respects the phenotype of the cells was similar to that seen during the primary phase of infection. The cells were predominantly, but not exclusively, CCR7− (unpublished data).
Although some inter-individual variation was observed, similar results were found when experiments were performed on paired samples of PBMCs taken from four other individuals during the primary and early persistent phases of EBV infection. Overall, we found no significant difference between frequency of expression of CD45 isoforms or of CD27 and CD28 at the two time points. Furthermore, the phenotype of EBV-specific CD4+ T cells identified in samples of PBMCs from 10 healthy EBV-seropositive individuals was not significantly different from that found in samples of PBMCs taken from the 5 patients shortly after recovery from acute EBV infection. An example of this is shown in the first column of Fig. 3 . The majority of the EBV-specific cells in healthy individuals with persistent EBV infection continue to express CD45RO (Fig. 3 b) rather than CD45RA (Fig. 3 a), and CD27 (Fig. 3 c) and CD28 (Fig. 3 d). One of the healthy EBV-seropositive individuals studied has been infected for at least 15 yr and it is likely that many others have also been persistently infected for long periods of time. Thus, the phenotype of the EBV-reactive effector CD4+ T cells is established early during the primary response and, apart from loss of expression of markers of recent activation, shows little change during the development or maintenance of the memory response. A summary of the results of all 10 analyses performed on blood taken during the primary and persistent phases of EBV infection are shown in Fig. 4 , a–d.
Figure 3.
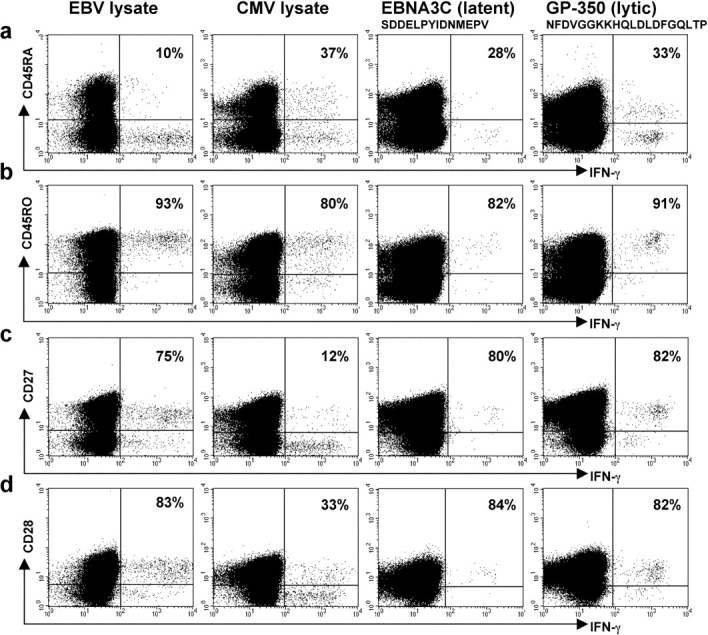
Phenotype of EBV- and CMV-specific CD4+ T cells during persistent infection. (a–d) PBMCs, taken from a healthy individual who was seropositive for both EBV and CMV, were cultured with lysate from EBV-infected cells (first column) or CMV-infected cells (second column). PBMCs from a healthy individual were cultured in the presence of the SDD (third column) or the NFD peptide (fourth column). Cells were stained for expression of (a) CD45RA, (b) CD45RO, (c) CD27, and (d) CD28, and then for expression of intracellular IFN-γ and CD4. Gates were set to include only CD4+ lymphocytes. The percentages of IFN-γ–expressing cells that stain with antibodies for each of the phenotypic markers are shown.
Figure 4.
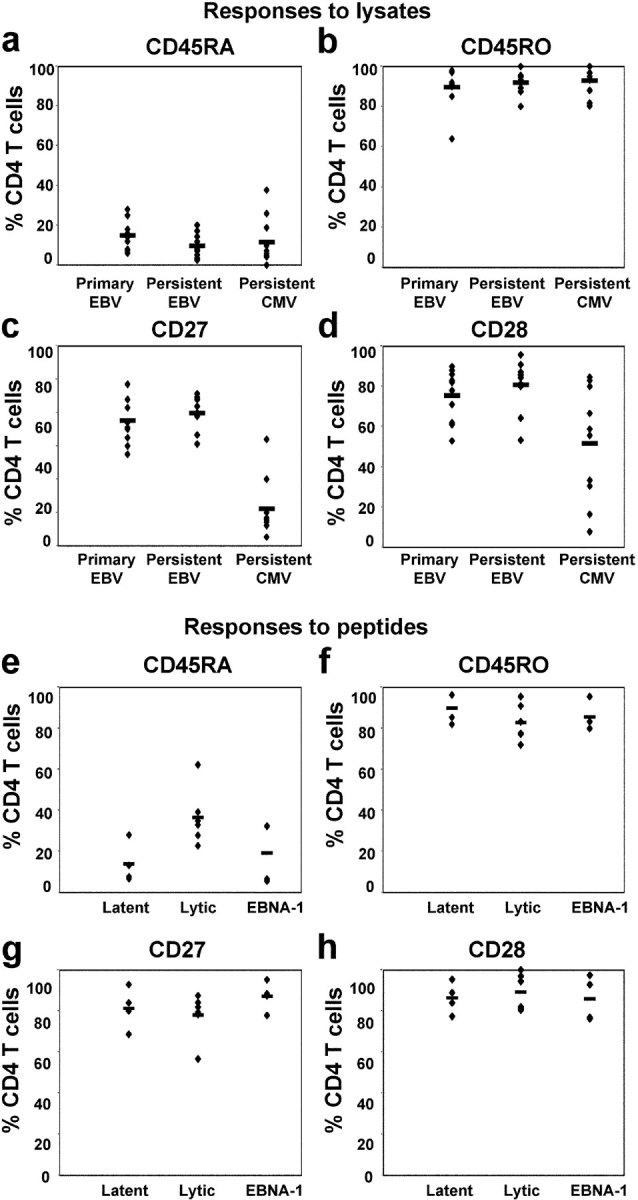
CD4+ T cell phenotype correlates with specificity. PBMCs from 10 individuals with primary EBV infection and 10 individuals with persistent EBV infection were cultured in vitro in R10 with lysate from EBV-infected cells. PBMCs from 10 individuals with persistent CMV infection were cultured with lysate from CMV-infected cells. Frequency of expression of (a) CD45RA, (b) CD45RO, (c) CD27, and (d) CD28 on CD4+ T cells that responded to the cell lysates by expressing IFN-γ is shown. PBMCs from healthy EBV-seropositive individuals were cultured in the presence of peptides from the EBV latent proteins (EBNA2 or EBNA3A), the EBV lytic cycle proteins (BZLF1, BMLF1, or GP350), or EBNA1. Frequency of expression of (e) CD45RA, (f) CD45RO, (g) CD27, and (h) CD28 on responding CD4+ T cells is shown.
Characterization of CD4+ T Cells Specific for Epitopes from EBV Latent and Lytic Cycle Proteins.
The frequency of CD4+ T cells specific for HLA class II–restricted epitopes from EBV is usually low, with fewer than 1 in 10,000 PBMCs showing a detectable response to a given peptide in ELISPOT assays. Furthermore, few class II–restricted epitopes have been mapped within the EBV lytic cycle proteins. These constraints limit ex vivo analysis of EBV epitope-specific CD4+ T cells. Nevertheless, we identified some EBV-seropositive individuals in whom we could detect populations of CD4+ T cells that responded to stimulation with peptides from EBV latent and lytic cycle proteins. We characterized eight responses to peptides from latent proteins, four of these being specific for EBNA1, three for EBNA2, and one for EBNA3C, and six responses to peptides from lytic cycle proteins, one specific for BZLF1, two for BMLF1, and three for GP350. A representative example of results from one donor, in whom 0.03% CD4+ T cells responded to a peptide epitope (SDD) from the latent protein EBNA3C and 0.16% CD4+ T cells responded to a peptide (NFD) from the lytic cycle protein GP350, is shown in Fig. 3 (third and fourth columns). The epitope-specific CD4+ T cells were predominantly CD45RO+, CD27+, and CD28+ (Fig. 3, b–d). The majority did not express CD45RA, although a clear minority of NFD-specific cells did express this CD45 isoform (Fig. 3 a). A summary of all results is shown in Fig. 4, e–h. Responses to EBNA1 are considered separately from responses to the other latent proteins studied (EBNA2 and EBNA3C) because EBNA1 might be expressed in lytic cycle as well as latent infection (25). Most importantly, overall, the phenotype of CD4+ T cells specific for individual epitopes from EBV proteins was similar to that of CD4+ T cells responding to the EBV-infected B cell lysate, with the majority of cells expressing CD45RO, CD27, and CD28. Interestingly, we found that mean CD45RA expression on CD4+ T cells specific for epitopes from the lytic cycle proteins was higher than on CD4+ T cells specific for epitopes from the latent proteins (P < 0.025 for lytic vs. EBNA2 and EBNA3 and P < 0.02 for lytic vs. EBNA1, EBNA2, and EBNA3). Identification of further, dominant class II–restricted epitopes from EBV lytic cycle proteins is now required to enable additional studies to be performed to confirm this observation. Notably, CD8+ T cells specific for EBV lytic cycle proteins are more likely to express CD45RA than CD8+ T cells specific for EBV latent proteins.
Comparison of the Phenotype of EBV-specific CD4+ T Cells with That of CMV-specific CD4+ T Cells in Healthy Seropositive Individuals.
The observation that EBV-specific effector cells are enriched within the CD27+ subset of CD4+ T cells contrasts with previously published work on CD4+ T cell responses, in which effector cells have been found to lack expression of CD27. To directly compare the phenotype of the CD4+ T cells specific for two different pathogens, we identified four individuals who were seropositive for both EBV and CMV and characterized the CD4+ T cells that responded to stimulation with the EBV and CMV lysate preparations. Results from a typical experiment are shown in Fig. 3. Despite the fact that the assays were performed in an identical manner, we found differences in the phenotype of the responding cells. The CMV-specific CD4+ T cells had usually lost expression of CD27 (Fig. 3 c) and many had also lost expression of CD28 (Fig. 3 d). Subpopulations of responding cells expressed CD45RA (Fig. 3 a). The response to CMV was studied in six additional CMV-seropositive donors and the responding CD4+ T cells were again found to be predominantly CD27− and to be heterogeneous with respect to expression of CD28. The differences between expression of CD27 and CD28 by EBV- and CMV-specific CD4+ T cells was significant (P < 0.00001 and P < 0.02, respectively). A summary of the results for all 10 donors studied are shown in Fig. 4, a–d.
Virus-specific Differentiation of CD4+ T Cells.
Multicolor staining of PBMCs suggests that antigen-experienced CD4+ T cells can be considered as being distributed between three major, phenotypically distinct differentiation compartments defined by expression of CD27 and CD28 (unpublished data). Cells within the CD27+ CD28+ compartment are more likely to express CCR7 whereas cells within the CD27− CD28− compartment frequently express CD57 and perforin (unpublished data). The patterns of expression of CD27 and CD28 on EBV- and CMV-specific CD4+ T cells (Fig. 4, c and d) suggest that CD4+ T cells specific for different viruses might be enriched within distinct differentiation compartments. To confirm this we performed a more detailed phenotypic analysis of EBV- and CMV-specific CD4+ T cells in four donors, looking at the phenotype of the CD28+ and CD28− subsets. The experiments were performed using EBV- and CMV-infected cell lysates as sources of antigen. A representative example of the results is shown in Fig. 5 . The vast majority of the EBV-specific CD4+ T cells expressed CD27, CD28, and CD45RO. Only a minority of cells had lost expression of CD27, and a proportion of these had also lost expression of CD28 (Fig. 5 a). Thus, these EBV-specific effector CD4+ T cells were highly enriched within the first (CD27+ CD28+) differentiation compartment (Fig. 5 e).
Figure 5.
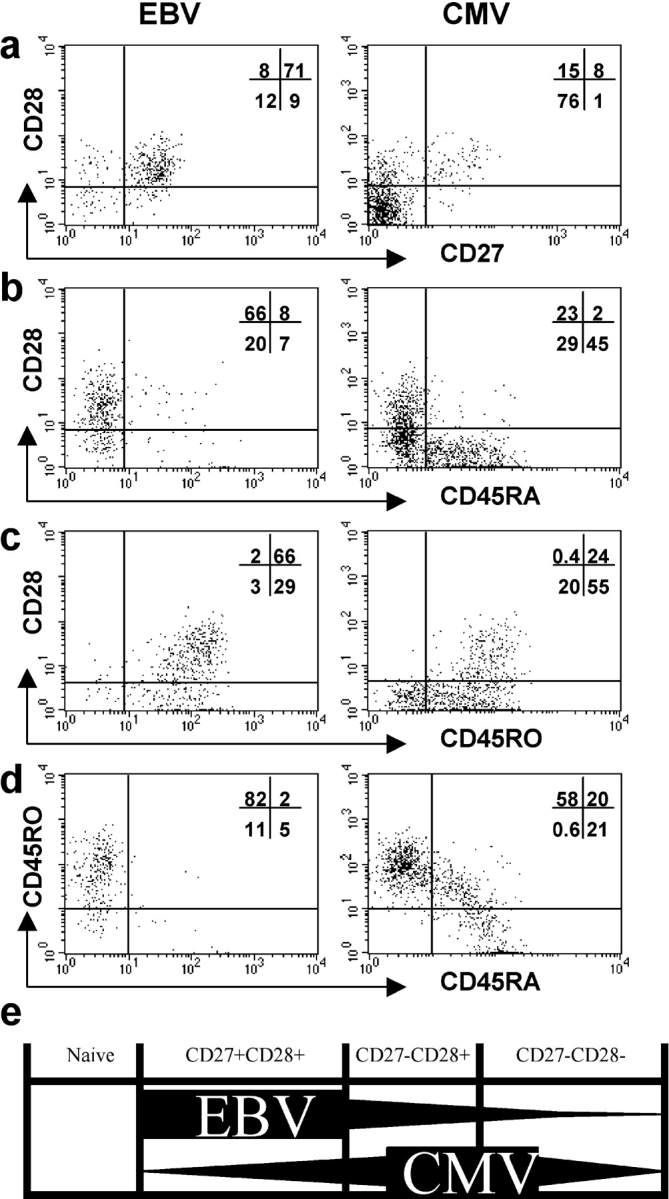
Phenotype of CD28+ and CD28− subsets of EBV- and CMV-specific CD4+ T cells during persistent infection. PBMCs from an EBV- and CMV-seropositive healthy individual were cultured with lysate from EBV-infected cells (left column) or from CMV-infected cells (right column). Cells were stained for expression of CD28 and (a) CD45RA, (b) CD45RO, (c) CD27, and then for expression of intracellular IFN-γ and CD4. Gates were set to include only CD4+ lymphocytes that expressed IFN-γ. The distribution of these cells between the quadrants is shown. (e) A cross-sectional representation of proposed CD4+ T cell differentiation compartments. Naive cells express CD27, CD28, and CD45RA. Antigen-experienced CD4+ T cells predominantly lie in one of three compartments, the first of which is characterized by expression of both CD27 and CD28, the second by expression of CD28 but not CD27, and the third by lack of expression of CD28 and CD27. EBV-specific CD4+ T cells are highly enriched in the first compartment. CMV-specific CD4+ T cells accumulate within the second and third compartments.
In clear contrast, the CMV-specific CD4+ T cells tended to lack expression of CD27 and be heterogeneous with respect to expression of CD28 (Fig. 5 a). In the example shown, subpopulations of the CD28− CD4+ T cells had reexpressed CD45RA and down-regulated CD45RO (Fig. 5, b and c). A small population (20%) of CMV-specific cells expressed low levels of both the CD45RO and RA isoforms (Fig. 5 d). Overall, these CMV-specific CD4+ effector T cells were enriched within the second (CD27− CD28+) and third (CD27− CD28−) differentiation compartments (Fig. 5 e).
Concluding Comments.
Different virus infections pose different challenges to the immune system and variations in initial antigen load, antigen persistence, antigen location, and pathway of antigen presentation may all affect T cell differentiation. Importantly, requirements for effective immune control may differ between viruses and the differences in dominant T cell phenotypes may represent an adaptation aimed at optimal control of virus infection.
In the context of virus-specific CD8+ T cells, it has been argued that sustained high antigen load may drive cells from the early CD27+ CD28+ compartment through to the late CD27− CD28− compartment. The observation that the characteristic virus-specific phenotypes develop shortly after primary infection and are stable thereafter, even where virus persists, argues against extent of antigen exposure as the only factor influencing responding cell phenotype. Moreover, recent evidence suggests that the emergence of CMV-specific CD27− CD8+ T cells within the circulation actually correlates with a fall in viral load (26). One interpretation of this is that the CD27− cells are generated during primary infection, at which time they are distributed at peripheral sites of CMV infection. As CMV infection becomes latent, these CD27− T cells emerge from the infected tissues and appear within the blood (26).
In contrast, the EBV latent proteins might be expressed intermittently throughout life in B cells, predominantly within lymphoid tissue. Switch into EBV lytic infection may also occur within B cells in the tonsillar environment. It is therefore interesting that EBV-specific T cells are enriched, both during the primary and persistent phases of infection, within the first differentiation compartment where many cells express CCR7. By staining for IL-2 instead of IFN-γ, we have been able to confirm the presence of populations of EBV-specific CCR7+ CD4+ T cells (unpublished data and 27). Such cells would be expected to recirculate not to peripheral tissue but to lymphoid tissues where EBV-infected B cells express EBV antigens. Thus, again the phenotype of the virus-specific CD4+ T cells may reflect the biology of virus infection.
CD27 and CD28 play an important role in modulating the function of CD4+ T cells. Expression of these molecules influences the capacity of T cells to proliferate and/or survive both in vitro and in vivo (3–5, 28–30). However, our data do not support the idea that expression of CD27 and CD28 might be used as a means of identifying effector T cells. We find that cells within each of the three proposed CD4+ T cell differentiation compartments are able to express IFN-γ after short-term stimulation in vitro. Thus, CD4+ T cells may have antigen-specific effector function regardless of expression of CD27 or CD28.
In conclusion, we have dissected the phenotypic diversity of the pool of memory CD4+ T cells and have provided evidence in support of the idea that the phenotype of antigen-experienced CD4+ T cells correlates with their specificity. Circulating EBV- and CMV-specific CD4+ T cells are enriched within different phenotypic compartments in a manner that is clearly analogous to that seen for their CD8+ counterparts. Importantly, CD4+ T cells with effector capacity are present within each of the proposed compartments. This work challenges the idea that the effector pool of CD4+ T cells is derived from a resting memory pool by a process of differentiation that involves changes in expression of CD27, CD28, or CD45RA. Rather, it suggests that the phenotypic heterogeneity of the memory pool reflects the different phenotypic characteristics of T cells specific for different pathogens.
Acknowledgments
We are grateful to General Practitioners for help in recruiting patients with acute IM and to the patients and healthy donors who participated in this study.
M. Callan is an MRC Senior Clinical Fellow and this work has been funded by a grant from the MRC.
Abbreviations used in this paper: EBNA, EBV nuclear antigen; IM, infectious mononucleosis; MOI, multiplicity of infection.
References
- 1.Hintzen, R.Q., R. de Jong, S.M.A. Lens, M. Brouwer, P. Baars, and R.A.W. van Lier. 1993. Regulation of CD27 expression on subsets of mature T lymphocytes. J. Immunol. 151:2426–2435. [PubMed] [Google Scholar]
- 2.Azuma, M., J.H. Phillips, and L.L. Lanier. 1993. CD28− T lymphocytes – antigenic and functional properties. J. Immunol. 150:1147–1159. [PubMed] [Google Scholar]
- 3.Hamann, D., P.A. Baars, M.H.G. Rep, B. Hooibrink, S.R. Kerkhof-Garde, M.R. Klein, and R.A.W. van Lier. 1997. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med. 186:1407–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamann, D., S. Kostense, K.C. Wolthers, S.A. Otto, P.A. Baars, F. Miedema, and R.A. van Lier. 1999. Evidence that human CD8+CD45RA+CD27− cells are induced by antigen and evolve through extensive rounds of division. Int. Immunol. 11:1027–1033. [DOI] [PubMed] [Google Scholar]
- 5.Posnett, D.N., J.W. Edinger, J.S. Manavalan, C. Irwin, and G. Marodon. 1999. Differentiation of human CD8+ T cells: implications for in vivo persistence of CD8+ CD28− cytotoxic effector clones. Int. Immunol. 11:229–241. [DOI] [PubMed] [Google Scholar]
- 6.Appay, V., P.R. Dunbar, M.F. Callan, P. Klenerman, G.M.A. Gillespie, L. Papagno, G.S. Ogg, A. King, F. Lechner, C.S. Spina, et al. 2002. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8:379–385. [DOI] [PubMed] [Google Scholar]
- 7.Tomiyama, H., T. Matsuda, and M. Takiguchi. 2002. Differentiation of human CD8+ T cells from a memory to memory/effector phenotype. J. Immunol. 168:5538–5550. [DOI] [PubMed] [Google Scholar]
- 8.Callan, M.F.C., L. Tan, N. Annels, G.S. Ogg, J. Wilson, C.A. O'Callaghan, N. Steven, A.J. McMichael, and A.B. Rickinson. 1998. Direct visualisation of antigen-specific CD8+ T cells during the primary immune response to Epstein-Barr Virus in vivo. J. Exp. Med. 187:1395–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hislop, A., N. Annels, N. Gudgeon, A.M. Leese, and A.B. Rickinson. 2002. Epitope-specific evolution of human CD8+ T cell responses from primary to persistent phases of Epstein-Barr virus infection. J. Exp. Med. 195:893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tan, L., N. Gudgeon, N.E Annels, P. Hansasuta, C.A. O'Callaghan, S. Rowland-Jones, A.J. McMichael, A.B. Rickinson, and M.F.C. Callan. 1999. A re-evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers. J. Immunol. 162:1827–1835. [PubMed] [Google Scholar]
- 11.Suni, M.A., L.J. Picker, and V.C. Maino. 1998. Detection of antigen-specific T cell cytokine expression in whole blood by flow cytometry. J. Immunol. Methods. 212:89–98. [DOI] [PubMed] [Google Scholar]
- 12.Rentenaar, R.J., L.E. Gamadia, N. van der Hoek, F.N.J. van Diepen, R. Boom, J.F.L. Well, P.M.E. Wertheim-van Dillen, R.A.W. van Lier, and I.J.M. ten Berge. 2000. Development of virus-specific CD4+ T cells during primary cytomegalovirus infection. J. Clin. Invest. 105:541–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harari, A., G.P. Rizzardi, K. Ellefsen, D. Ciuffreda, P. Champagne, P.A. Bart, D. Kaufmann, A. Telenti, R. Sahli, G. Tambussi, et al. 2002. Analysis of HIV-1 and CMV-specific memory CD4 T cell responses during primary and chronic infection. Blood. 100:1381–1387. [DOI] [PubMed] [Google Scholar]
- 14.Leen, A., P. Meij, I. Redchenko, J. Middleldorp, E. Bloemena, A. Rickinson, and N. Blake. 2001. Differential immunogenicity of Epstein-Barr virus latent-cycle proteins for human CD4+ T-helper 1 responses. J. Virol. 75:8649–8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khanna, R., S.R. Burrows, S.A. Thomas, D.J. Moss, P. Cresswell, L.M. Poulsen, and L. Cooper. 1997. Class I processing-defective Burkitt's lymphoma cells are recognized efficiently by CD4+ EBV-specific CTLs. J. Immunol. 158:3619–3625. [PubMed] [Google Scholar]
- 16.Whitmore, J.K., M.S. Asano, K. Murali-Krishna, M. Suresh, and R. Ahmed. 1998. Long-term CD4 Th1 and Th2 memory following acute lymphocytic chriomeningitis virus infection. J. Virol. 72:8281–8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varaga, S.M., and R.M. Welsh. 1998. Stability of virus-specific CD4+ T cell frequencies from acute infection into long-term memory. J. Immunol. 161:367–374. [PubMed] [Google Scholar]
- 18.Oxenius, A., S. Fidler, M. Brady, S.J. Dawson, K. Ruth, P.J. Easterbrook, J.N. Weber, R.E. Phillip, and D.A. Price. 2001. Variable fate of virus-specific CD4+ T cells during primary HIV-1 infection. Eur. J. Immunol. 31:3782–3788. [DOI] [PubMed] [Google Scholar]
- 19.Murali-Krishna, K., J.D. Altman, M. Suresh, D.J.D. Sourdive, A.J. Zajac, J.D. Miller, J. Slansky, and R. Ahmed. 1998. Counting antigen-specific CD8+ T cells: a re-evaluation of bystander activation during viral infection. Immunity. 8:177–187. [DOI] [PubMed] [Google Scholar]
- 20.Butz, E.A., and M.J. Bevan. 1998. Massive expansion of antigen-specific CD8+ T cells during an acute virus infection. Immunity. 8:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Callan, M.F.C., N. Steven, P. Krausa, J. Wilson, P.A. Moss, G. Gillespie, J.I. Bell, A.B. Rickinson, and A.J. McMichael. 1996. Large clonal expansions of CD8+ T cells in acute infectious mononucleosis. Nat. Med. 2:906–911. [DOI] [PubMed] [Google Scholar]
- 22.Callan, M.F.C., C. Fazou, H.B. Yang, T. Rostron, K. Poon, C. Hatton, and A.J. McMichael. 2000. CD8(+) T-cell selection, function, and death in the primary immune response in vivo. J. Clin. Invest. 106:1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Precopio, M.L., J.L. Sullivan, C. Willard, M. Somasundaran, and K. Luzuriaga. 2003. Differential kinetics and specificity of EBV-specific CD4+ and CD8+ T cells during primary infection. J. Immunol. 170:2590–2598. [DOI] [PubMed] [Google Scholar]
- 24.Catalina, M.D., J.L. Sullivan, K.R. Bak, and K. Luzuriaga. 2001. Differential evolution and stability of epitope-specific CD8+ T cell responses in EBV infection. J. Immunol. 167:4450–4457. [DOI] [PubMed] [Google Scholar]
- 25.Schaefer, B.C., J.L. Strominger, and S.H. Speck. 1996. A simple reverse transcriptase PCR assay to distinguish EBNA1 gene transcripts associated with type I and II latency from those arising during induction of the viral lytic cycle. J. Virol. 70:8204–8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gamadia, L., E.B.M. Remmerswaal, J.F. Weel, F. Bernelman, R.A.W. van Lier, and I.J.M. Ten Berge. 2003. Primary immune responses to CMV: a critical role for IFN-γ producing CD4+ T cells in protection against CMV disease. Blood. 101:2686–2692. [DOI] [PubMed] [Google Scholar]
- 27.Sallusto, F., D. Lenig, R. Forster, M. Lipp, and A. Lanzavecchia. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 401:708–712. [DOI] [PubMed] [Google Scholar]
- 28.Hendricks, J., L.S. Gravestein, K.I. Tesselaar, R.A.W. van Lier, T.N.M. Schumacher, and J. Borst. 2000. CD27 is required for generation and long term maintenance of T cell immunity. Nat. Immunol. 1:433–440. [DOI] [PubMed] [Google Scholar]
- 29.Arens, R., K. Tesselaar, P.A. Baars, G.M.W. van Schinjndel, J. Hendriks, S.T. Pals, P. Krimpenfort, J. Borst, M.H.J. van Oers, and R.A.W. van Lier. 2001. Constitutive CD27/CD70 interaction induces expansion of effector type T cells and results in IFN gamma-mediated B cell depletion. Immunity. 15:801–812. [DOI] [PubMed] [Google Scholar]
- 30.Suresh, M., J.K. Whitmire, L.E. Harrington, C.P. Larsen, T.C. Pearson, J.D. Altman, and R. Ahmed. 2001. Role of CD28-B7 interactions in generation and maintenance of CD8 T cell memory. J. Immunol. 167:5565–5573. [DOI] [PubMed] [Google Scholar]