Specific Migratory Dendritic Cells Rapidly Transport Antigen from the Airways to the Thoracic Lymph Nodes (original) (raw)
Abstract
Antigen transport from the airway mucosa to the thoracic lymph nodes (TLNs) was studied in vivo by intratracheal instillation of fluorescein isothiocyanate (FITC)-conjugated macromolecules. After instillation, FITC+ cells with stellate morphology were found deep in the TLN T cell area. Using flow cytometry, an FITC signal was exclusively detected in CD11cmed-hi/major histocompatibility complex class II (MHCII)hi cells, representing migratory airway-derived lymph node dendritic cells (AW-LNDCs). No FITC signal accumulated in lymphocytes and in a CD11chiMHCIImed DC group containing a CD8αhi subset (non–airway-derived [NAW]-LNDCs). Sorted AW-LNDCs showed long MHCIIbright cytoplasmic processes and intracytoplasmatic FITC+ granules. The fraction of FITC+ AW-LNDCs peaked after 24 h and had reached baseline by day 7. AW-LNDCs were depleted by 7 d of ganciclovir treatment in thymidine kinase transgenic mice, resulting in a strong reduction of FITC-macromolecule transport into the TLNs. Compared with intrapulmonary DCs, AW-LNDCs had a mature phenotype and upregulated levels of MHCII, B7-2, CD40, and intracellular adhesion molecule (ICAM)-1. In addition, sorted AW-LNDCs from FITC-ovalbumin (OVA)–instilled animals strongly presented OVA to OVA-TCR transgenic T cells. These results validate the unique sentinel role of airway DCs, picking up antigen in the airways and delivering it in an immunogenic form to the T cells in the TLNs.
Keywords: antigen-presenting cells, endocytosis, fluorescein isothiocyanate, respiratory mucosa, lymph nodes
Introduction
The lungs are extensively exposed to the outside world. On a daily basis, human lungs process >10,000 liters of ambient air. This represents a considerable amount of airborne foreign particles which, depending on their size, deposit at different levels within the airways. Consequently, the respiratory system is one of the most immunologically challenged organs of the body. Therefore, its immunological homeostasis must be kept under tight control to provide adequate defenses while avoiding inappropriate, potentially life-threatening inflammatory damage.
Dendritic cells (DCs) are now widely recognized as controlling the immune response 1 and a dense network of DCs has been described in the airway mucosa 2 3 4. DCs are the most efficient professional APCs to date and the only ones capable of eliciting a primary immune response. They perform their task through a tightly controlled sequence of events. DC precursors arise in the bone marrow and home to their target organ. At this stage, they are in an “immature” state characterized by high Ag uptake capacity and low expression of surface MHC and T cell costimulatory molecules. Upon contact with Ags, and in the presence of so-called danger signals 5, DCs evolve towards their mature state; the Ag uptake and processing machinery is shut down while Ag-loaded MHC and T cell costimulatory molecules are strongly upregulated on the cell surface. This is paralleled by a migration of the DCs out of the Ag-exposed site into interstitial afferent lymphatics, towards the T cell area of regional LNs 6. Once there, DCs attract, select, and activate the right Ag-specific naive T lymphocyte for proliferation and differentiation 7.
Migration is therefore a crucial component of the DC's physiology, and hence, of the initiation of an immune response. Many cellular functions involved in migration are differentially regulated in immature versus mature DCs. These include the response to chemokines 6 8, the expression of integrins 9 10, the downregulation of cadherins 11, and the production of tissue-degrading metalloproteinases 12. This knowledge is mostly derived from ex vivo studies involving Langerhans cells (LCs [epidermal DCs]) and the skin explant model, and may not be relevant for the airways 13. Our group and others have studied the migration of DCs in the airways by performing adoptive transfer of labeled, freshly isolated spleen or bone marrow–derived DCs into the trachea 14 15 16. Because of in vitro enrichment procedures and/or culture in growth factors, these data may not be representative for the migration of endogenous DCs residing in the airway mucosa. Conversely, functional studies using Ag delivery straight into the airways did not disclose enough information regarding the exact phenotype of the migratory, Ag-carrying DCs and did not visualize the migrating cell types 17.
We addressed these issues by studying the migration of endogenous airway DCs in vivo based on their ability to take up and transport fluorescently labeled macromolecules. We simultaneously obtained a better insight into the phenotype and function of these cells.
Materials and Methods
Animals.
Male C57BL/6, DBA/2, and BALB/c mice, 6–8-wk old, were purchased from Harlan. Heterozygous OVA-TCR transgenic mice (DO11.10 × BALB/c) were obtained from Dr. M. Moser (University of Brussels, Brussels, Belgium; 18). Thymidine kinase transgenic (TK-TG) mice were a donation from Prof. D. Klatzmann (Hôpital de la Pitié-Salpêtrière, Paris, France; 19). For experiments using TK-TG mice, bone marrow chimeric mice were generated by injecting TK-TG bone marrow cells into γ-irradiated DBA/2 mice, as described previously 19 20. All in vivo manipulations were approved by the local Ethics Committee.
Instillation of Macromolecule Solutions into the Trachea.
Fluorescein-conjugated macromolecules were diluted in sterile PBS to a final concentration of 10 mg/ml: FITC-dextran (FITC-DX; mol wt 40,000, anionic); FITC-OVA (mol wt 45,000; both from Molecular Probes); mannosylated FITC-BSA; and galactosylated FITC-BSA (both from Sigma-Aldrich).
For instillation, mice were anesthetized by intraperitoneal injection of 2.5% avertin, as described previously 15. 70 μl of macromolecule solution was deposited just above the glottis under sterile conditions using disposable and pyrogen-free 18 GA polyurethane catheters (Insyte-W; Becton Dickinson), followed by complete recovery of the animal.
Cryostat Sections of Thoracic LNs.
Paratracheal and parathymic LNs were extracted 48 h after intratracheal instillation of fluorescein-conjugated Ag. 5-μm cryostat sections were fixed in acetone and treated with mouse FcR-block followed by RPE-conjugated anti-B220 mAb. The sections were subsequently examined under fluorescence microscopy.
Buffers and Media for Preparation of Single Cell Suspensions and Immunofluorescent Labeling.
Digestion medium consisted of RPMI 1640, 5% FCS (both from GIBCO BRL), 1 mg/ml collagenase type 2 (Worthington Biochemical Corp.), and 0.02 mg/ml DNase I (grade II from bovine pancreas; Boehringer). EDTA-treated FCS was prepared by passing FCS through a 0.2-μm filter and mixing 1 ml of a 0.1 M disodium EDTA solution through every 10 ml of FCS. FACS-EDTA buffer contained PBS without Ca2+ or Mg2+, 0.1% azide, 5% EDTA-treated FCS, and 5 mM EDTA. Tissue culture medium (TCM) was prepared using RPMI 1640 supplemented with 5% FCS, penicillin/streptomycin, l-glutamine, and 2-mercaptoethanol (all from GIBCO BRL).
Preparation of Lung and LN Single Cell Suspensions.
Animals were killed and the pulmonary and systemic circulation was perfused with saline/EDTA to remove the intravascular pool of cells. Paratracheal and parathymic intrathoracic LNs were collected. Lungs were carefully separated from thymic and cardiovascular remnants and removed in toto, including the main bronchi and trachea. Due to the photosensitivity of the FITC material, organs from FITC-macromolecule–instilled animals were protected from direct light throughout the manipulation. Organs were thoroughly minced using iridectomy scissors and incubated for 30 min in digestion medium in a humidified incubator at 37°C and 5% CO2, according to a modified protocol 21. Organ fragments were resuspended, fresh digestion medium was added, and incubation was extended for another 15 min. After a final resuspension, very few organ debris were left. Samples were centrifuged and resuspended in calcium and magnesium–free PBS containing 10 mM EDTA for 5 min at room temperature on a shaker. Finally, the cells were subjected to RBC lysis, washed in FACS-EDTA, passed through a 50-μm cell strainer, and kept on ice until labeling. Cell viability after this procedure was consistently >95%.
Labeling of Single Cell Suspensions for Flow Cytometry.
mAbs used to identify mouse DC populations were: biotinylated anti-CD11c (N418), FITC- and PE-conjugated anti-IAb (AF6-120.1), and FITC-IEk (14-4-4S) (both from BD PharMingen). Additional markers used for phenotyping were (PE-conjugated or uncoupled): anti-CD8α (53-6.7), CD11b/Mac-1α (M1/70), CD40 (3/23), CD24/HSA (M1/69), LFA1α/CD11a (2D7), CD86/B7-2 (GL-1), and hamster anti–CD80/B7-1 (16-10-A1) (all from BD PharMingen); and rat anti–mouse DEC-205 (NLDC-145) and F4/80 (A3-1) (both from Serotec) and rat anti–mouse CD54/intracellular adhesion molecule (ICAM)-1 (KAT-1; Caltag). Isotype controls were RPE-conjugated or uncoupled rat IgG2a, rat IgG2b, and polyclonal hamster IgG (BD PharMingen). PE-conjugated goat anti–rat F(ab′)2 fragments (Caltag) were used as second step reagents.
All staining reactions were performed on ice in FACS-EDTA buffer. If possible, cells were preincubated with anti-CD16/CD32 (2.4G2) to reduce nonspecific binding of mAbs. Biotinylated anti-CD11c was revealed by incubation with streptavidin-PECy5 conjugate (Streptavidin Quantum Red™; Sigma-Aldrich). Unconjugated rat mAbs were revealed by PE-conjugated goat anti–rat F(ab′)2 fragments, followed by blocking with normal rat serum and staining with directly conjugated antibodies. Flow cytometry data acquisition was performed on a FACS Vantage™ flow cytometer running CELLQuest™ software (Becton Dickinson). FlowJo software (Treestar) was used for data analysis.
Microscopic Examination of Sorted LNDCs.
Animals were instilled with FITC-OVA and after 48 h the two clusters of pulmonary LN cells appearing in the CD11c+MHC class II (MHCII)+ quadrant were sorted on a FACS Vantage™ with a Sort Enhancement Module (SEM; Becton Dickinson). Cytospin slides were prepared from each subpopulation and examined under a fluorescence microscope.
In Vitro Pulsing of DCs with Fluorescein-conjugated Ag.
Single cell suspensions from thoracic LN (TLN) digests of several C57BL/6 mice were pooled. CD11c+ cells were enriched by layering the cells on a 14.5% metrizamide gradient (Sigma-Aldrich), as described 15. Cells in the interface were collected, washed three times, and transferred in triplicate to 96-well plates. Subsequently, they were pulsed with graded concentrations of FITC-OVA in TCM and incubated for 30 min on ice or in a humidified incubator at 37°C and 5% CO2. Background fluorescence was determined using PBS-pulsed LN cells. After incubation, cells were labeled with anti-CD11c and anti-MHCII (as described above). All labeling steps were performed on ice, in the presence of azide and 5 mM EDTA. FITC mean fluorescence intensity within the CD11c+MHCII+ subpopulations was assessed by flow cytometry.
Ag-specific Proliferation Assay.
BALB/c mice were instilled intratracheally with either FITC-OVA or FITC-DX and killed 48 h later. TLNs were extracted and pooled, and single cell suspensions were stained with a combination of anti-CD11c and anti-MHCII (IEk). The two distinct clusters of interest in the CD11c+MHCII+ quadrant were sorted on a FACS Vantage™ with SEM. OVA-specific T cells were isolated from the spleen and LNs of DO11.10 × BALB/c mice. After RBC lysis, T lymphocytes were enriched by magnetic negative depletion of MHCII+, B220+, and GR-1(RB6)+ cells (Dynabeads; Dynal). Graded concentrations of CD11c+MHCII+ DCs were added to 2 × 105 OVA-TCR transgenic T cells per well, in triplicate. After 72 h of culture in TCM, [3H]thymidine (1 μCi/well) was added to the cultures for the final 12 h. Cell proliferation was measured on an automated liquid scintillation counter (Microbeta Workstation).
Conditional Depletion of DCs.
After bone marrow reconstitution, TK-TG chimeric mice were treated by continuous infusion of ganciclovir (GCV, Cymevene®; Roche) or vehicle (PBS) using subcutaneously implanted miniosmotic pumps (ALZET model 2001; Alza Pharmaceuticals). To selectively eliminate dividing myeloid DC precursors, GCV concentration was adjusted to reach a dose delivery rate of 50–55 mg GCV/kg body wt/d, as described previously 19 20. After 7 d, the pumps were explanted and 1 d was allowed for any residual drug to wash out. Subsequently, mice were instilled with FITC-OVA and 48 h later TLNs were processed and labeled as described above.
Results and Discussion
Ag Transport from the Airways to the TLNs.
TLN cryostat sections obtained 48 h after intratracheal instillation of FITC-DX or FITC-OVA showed numerous FITC+ cells that were strictly confined to B220− T cell zones (Fig. 1 A). A closer look revealed stellar-shaped collections of FITC material reaching deep into the LN paracortex (Fig. 1 B). Based on the stellate morphology and localization within the T cell area, we hypothesized that DCs within the draining LNs contained FITC+ material 48 h after instillation of FITC-labeled macromolecules into the trachea.
Figure 1.

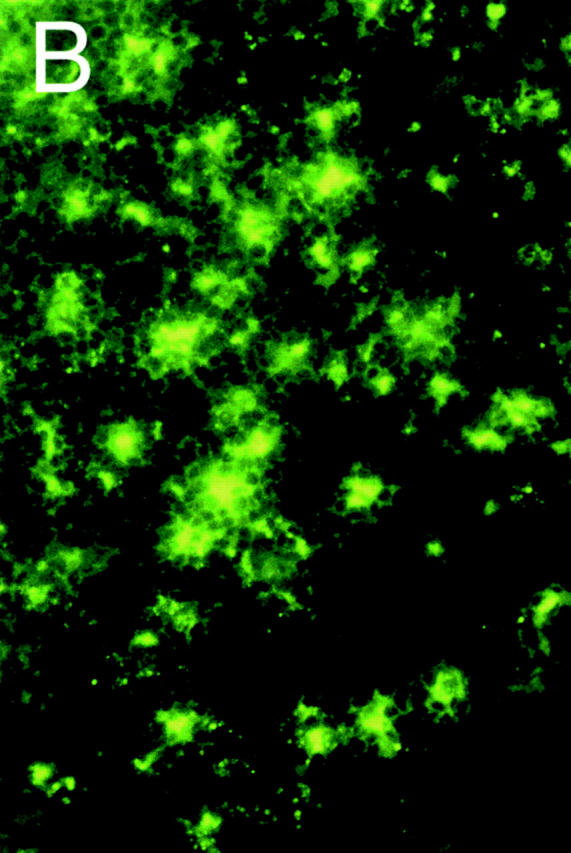
TLN cryostat section from a DX-FITC–instilled mouse, as seen under fluorescence microscopy. (A) Staining with the B cell marker B220 (red): FITC+ cells are confined to the B cell–negative, paracortical T cell–dependent zones. (B) High power magnification of the paracortical zone showing numerous infiltrating FITC+ cells with a stellate morphology.
To address this further, TLNs of FITC-OVA–instilled mice were subjected to collagenase/DNase/EDTA treatment. The use of enzymatic stromal digestion, Ca2+-free media, and EDTA has been documented to favor the isolation of as many DCs from lymphoid organs as possible by extracting tightly tissue-bound DC subsets and disrupting DC–lymphocyte rosettes 22. TLN cell suspensions were stained using the DC marker combination MHCII plus CD11c (αx chain of the p150,95 integrin abundant on murine DCs) and examined by flow cytometry, as described in earlier studies 23. The cells were analyzed on bivariate plots of MHCII versus CD11c and examined for FITC positivity (Fig. 2 A). Dead cells and debris were excluded based on light scatter properties. Two main clusters of cells could be distinguished in the MHCII+CD11c+ quadrant. One group of cells (group 1) had a very strong expression of MHCII and intermediate to high levels of CD11c (MHCIIhiCD11cmed-hi), while group 2 had intermediate expression of MHCII and high level expression of CD11c (MHCIImedCD11chi). As seen in Fig. 2 B, 24 h after instillation of FITC-OVA into the airways of mice, FITC positivity was exclusively detected in group 1 LNDCs. At that particular time point, the FITC signal was absent in group 2 LNDCs as well as in cells in the CD11c− lymphocyte gate (not shown on this density plot as these cells comprise ∼95% of the whole LN cell population).
Figure 2.

(A) Flow cytometric staining pattern of TLN single cell suspensions, labeled with rat anti–mouse MHCII (PE) and hamster anti–mouse CD11c (PE-Cy5). Two clusters were invariably distinguished in the CD11c+MHCII+ quadrant. (B) The animals were given an intratracheal instillation of a 10 mg/ml FITC-OVA solution (gray histogram) or PBS as a control (white histogram). After 24 h, only group 1 LNDCs acquired a strong FITC signal.
To further confirm that MHCIIhiCD11cmed-hi cells were indeed DCs containing FITC, cytospin preparations of sorted group 1 LNDCs were examined using fluorescence microscopy (Fig. 3a and Fig. b). FITC material appears in yellow-green, whereas MHCII stains red (anti-IAb–RPE). Group 1 LNDCs showed a typical DC morphology with long MHCII bright cytoplasmic extensions (Fig. 3 A) and FITC material concentrated in intracytoplasmic granules (none on the cell surface; Fig. 3 B). No FITC signal was visible in group 2 LNDCs (data not shown).
Figure 3.

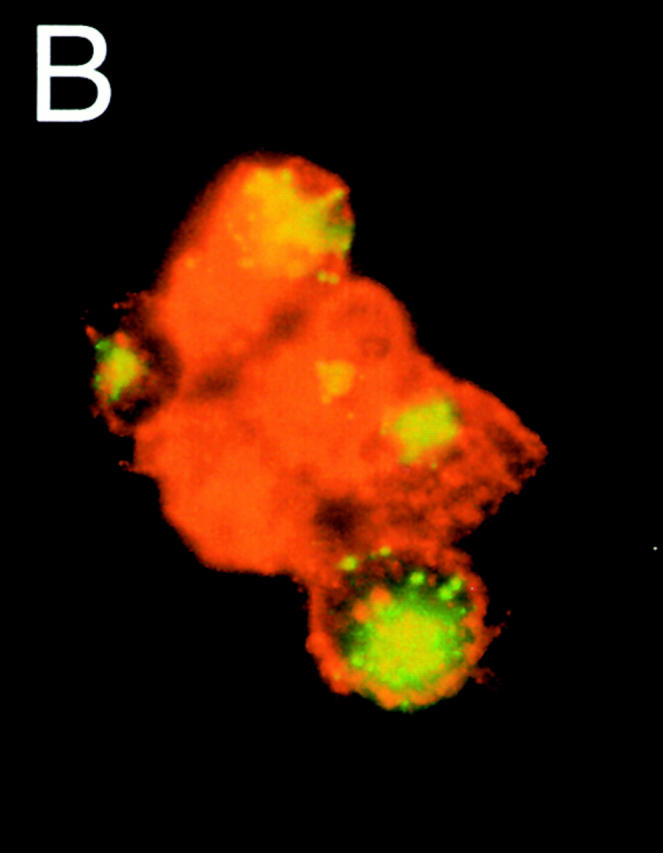
Morphology of sorted group 1 (MHCIIhi CD11cmed-hi) LNDCs 24 h after intratracheal instillation of DX-FITC, as seen under fluorescence microscopy. The cells, sometimes appearing in clusters (B), have numerous MHCII+ (bright red) cytoplasmic processes (A), kidney-shaped nuclei, and granular concentrations of FITC-bright material inside the cytoplasm.
To check whether FITC positivity in the LNs may be due to passive leakage of fluorescein-conjugated macromolecules from the airway mucosa to the draining TLNs, we assessed the capacity of both LNDC populations described above to take up FITC-OVA in vitro (Fig. 4). Both groups were equally capable of taking up FITC-OVA in a dose-dependent manner. For instance, pulsing the cells with 0.1 mg/ml FITC-OVA resulted in 86.5 ± 0.1% and 84.0 ± 1.9% FITC+ cells in group 1 versus group 2 LNDCs. This uptake was partially inhibited by cold, which hints to an active endocytic process. The capacity of group 2 LNDCs to take up Ags in vitro was in sharp contrast to the in vivo situation, in which virtually no FITC signal was seen in these cells after intratracheal instillation of FITC-OVA (Fig. 2 B, bottom). We could therefore exclude the possibility of an important passive leakage of free FITC-OVA from the airway mucosa to the draining LNs, at least in baseline conditions and with healthy animals. In fact, macromolecules such as albumins or dextrans are commonly used in physiological studies to test the integrity of endothelial and epithelial barriers, including the respiratory epithelium 24 25.
Figure 4.
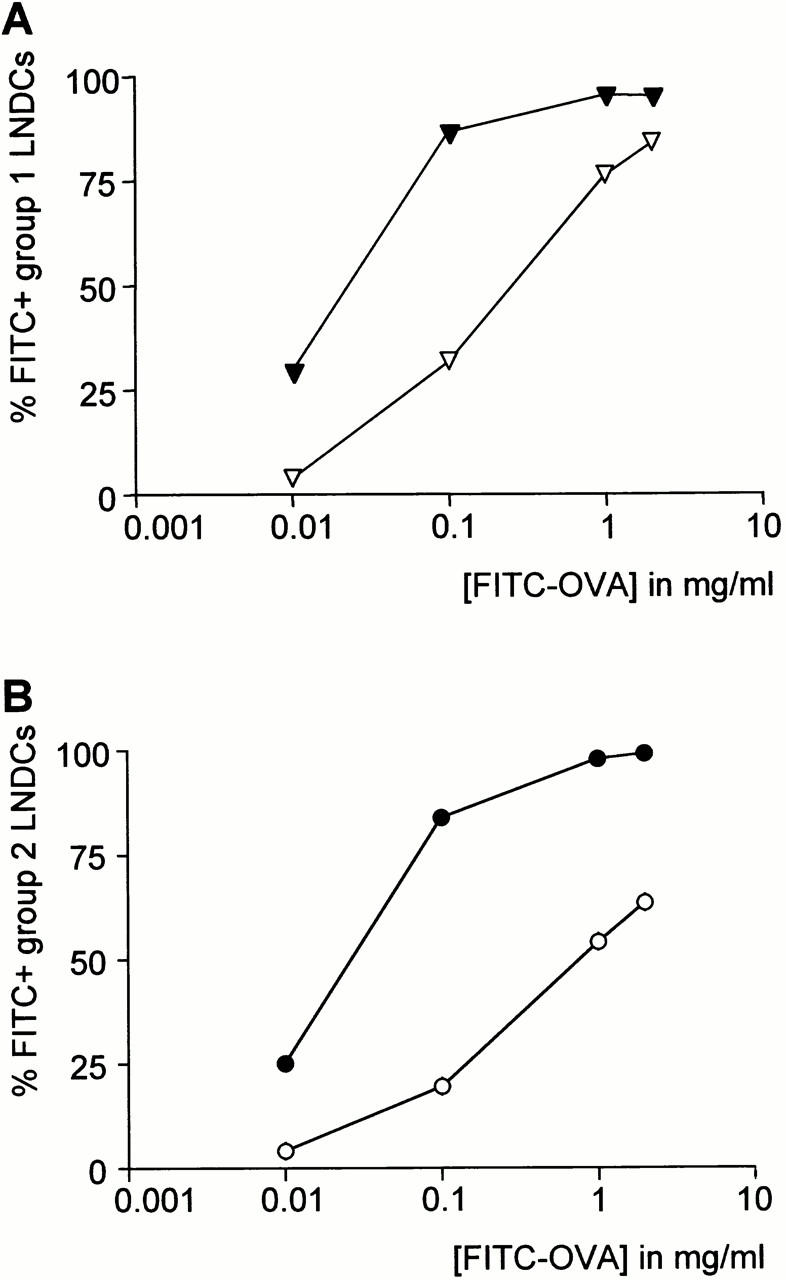
In vitro uptake of FITC-OVA by group 1 (A) and group 2 (B) LNDCs. Filled and open symbols represent uptake at 37°C and 4°C, respectively. Values are mean fractions of FITC+ cells ±SE derived from triplicate experiments. Both LNDC subgroups are equally capable of taking up FITC-OVA in vitro.
Based on the presence of FITC+ granular material within group 1 (MHCIIhiCD11cmed-hi) LNDCs and the exclusion of passive leakage of FITC+ material into the draining TLNs, we believe that these cells have taken up Ags and migrated from the airways, thus representing airway-derived (AW)-LNDCs, whereas group 2 (MHCIImedCD11chi) cells are non–airway-derived (NAW)-LNDCs.
Interestingly, transport of macromolecular Ags by AW-LNDCs appears to be selective. FITC-OVA and FITC-DX were comparable in their ability to be carried to the TLNs (Fig. 5). To verify the possible involvement of the mannose receptor (MR) in this process, we instilled mannosylated FITC-BSA and used galactosylated FITC-BSA as a control neoglycoconjugate which does not bind to the MR. Surprisingly, in vivo Ag uptake was low in both cases. This is in sharp contrast to human cultured DCs 26, but in accordance with recent reports demonstrating the absence of the MR on murine DCs in situ 27. Therefore, other MR-like receptors could be involved in the macromolecule uptake we observed. Although DEC-205, a lectin-like receptor shown to be involved in Ag processing by DCs 28, is expressed on murine pulmonary DCs (29; and our observation below), no glycosylated ligands for DEC-205 have yet been uncovered. Another possible candidate is Langerin, a recently described DC-specific C-type lectin receptor expressed on human DCs (including those within bronchial epithelium [30, 31]) and murine DCs (data not shown).
Figure 5.

Comparison of different fluorescein-conjugated macromolecules in their ability to be transported within AW-LNDCs. Bar heights represent mean percentage of FITC+ AW-LNDCs ±SE. Notice low uptake of mannosylated FITC-BSA compared with FITC-DX on FITC-OVA.
Kinetics of Ag Transport by AW-LNDCs.
Having confirmed the nature of migratory AW-LNDCs, a kinetics experiment was conducted (Fig. 6) and showed FITC-carrying AW-LNDCs appearing in the TLNs as soon as 6 h after the instillation of FITC-OVA, with a peak influx reached at 24 h. Similar migration rates were found in our previous studies using intratracheal instillation of carboxy fluorescein succinimidyl ester (CFSE)-labeled exogenous DCs in mice 15, in which we found CFSE+ DCs appearing in the LNs as soon as 12 h after injection. In addition, experiments using intratracheal delivery of soluble protein Ag describe an appearance of strong Ag-presenting activity in DCs of draining LNs 24 h after instillation 17. The kinetics of migration are also in agreement with the early occurrence of T cell activation and proliferation in Ag-specific TCR transgenic T cells when peptide- or Ag-pulsed DCs are deposited in the airways. In these models, some T cells have already proliferated three times 48 h after injection of Ags into the trachea 7. This rapid migration of DCs seems to occur in the absence of airway inflammation and is a fundamental feature of DCs at mucosal interfaces, not observed in skin LCs 32.
Figure 6.

Kinetics of DC entry into TLNs. Groups of mice were killed at several time intervals after intratracheal instillation of a 1% FITC-OVA solution. The fraction of FITC+ cells was determined within AW- (•) and NAW- (○) LNDCs (formerly group 1 and group 2 LNDCs, respectively). Each time point is derived from four to seven mice and represents the mean percentage of fluorescein-positive LNDCs ±SE, subtracting background autofluorescence obtained from PBS-instilled mice (dotted line).
A considerable fraction of cells in the AW-LNDC cluster did not acquire Ags after instillation. This could indicate that macromolecules instilled intratracheally do not reach DCs in all lung compartments. Alternatively, FITC− AW-LNDCs could either not be derived from peripheral immigrating DCs (an origin among myeloid resident LNDCs populations has been suggested in the case of skin LC–derived LNDCs 23) or they could be derived from a subset of lung DCs with a low trafficking rate. Indeed, considerable heterogeneity exists in the turnover rate (and functions) of different pulmonary DC subsets 32 33 34. Similarly, using TK-TG mice, 7 d of selective myeloid DC precursor depletion resulted in a partial elimination of AW-LNDCs from the TLNs (Table ). This indicates that a substantial fraction of AW-LNDCs has a transit time (from dividing precursor through lung to LNs) longer than 1 wk. In contrast, a similar 1-wk conditional DC depletion in a previous study resulted in >95% disappearance of MHCII+ DCs from the tracheal epithelium 20. In the present paper, the partial (37%) depletion of AW-LNDCs is accompanied by a drastic (86%) reduction in FITC-OVA transport (Fig. 7). Taken together, these data suggest that the majority of FITC-OVA–carrying DCs were derived from airway intraepithelial DCs. The remaining unaffected FITC+ AW-LNDCs could represent Ag-carrying DCs with a lower turnover rate.
Table 1.
Reduction in the Absolute Number of AW-LNDCs and NAW-LNDCs after a 1-wk Systemic GCV Treatment in HSV1-TK Transgenic Bone Marrow Chimeras
| PBS-sys | GCV-sys | Reduction | |
|---|---|---|---|
| % | |||
| AW-LNDC | 45,887 ± 4,985 | 28,854 ± 10,920 | 37 |
| NAW-LNDC | 3,322 ± 646 | 2,314 ± 613 | 30 |
Figure 7.
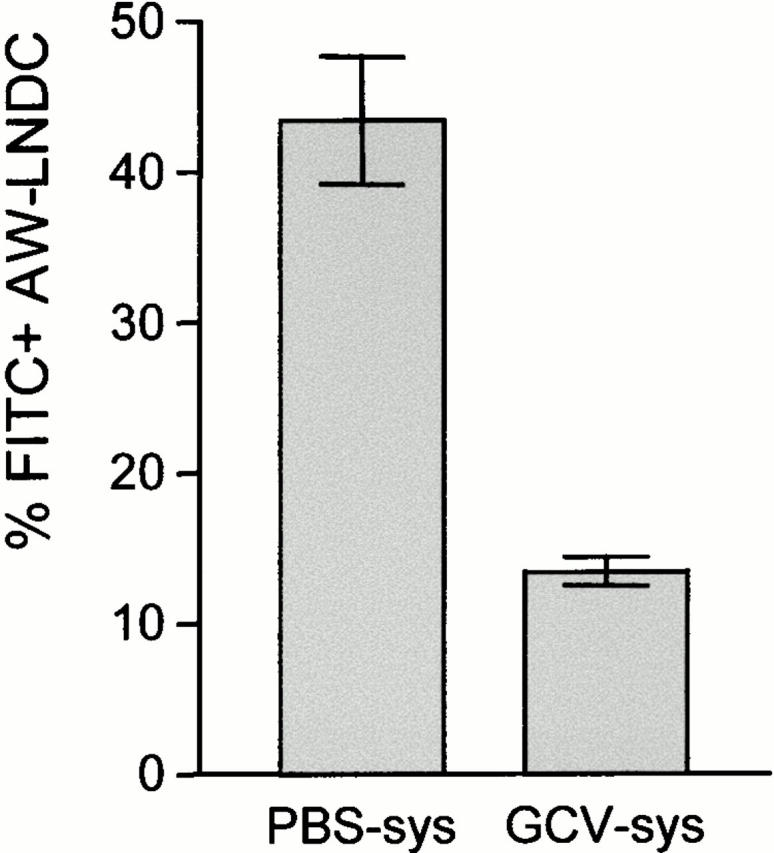
Treatment of TK-TG bone marrow chimeras with systemic (sys) GCV: impact on FITC-OVA transport within AW-LNDCs. Bars represent mean fraction of FITC+ AW-LNDCs ±SE. The depletion in absolute number of AW-LNDCs was taken into account when calculating the percentage of FITC+ cells in the GCV-treated group.
In our migration kinetics studies, the aim was to come as near as possible to the description of the steady state flux. DCs are exquisitely sensitive to specific environmental stimuli and maturation/migration can be triggered by very low dose endotoxin, mechanical stress, or damage in surrounding tissue 5. The following precautions were taken to avoid these triggers as much as possible: (a) disposable sterile and pyrogen-free instillation catheters were used; (b) there was no actual penetration of the catheter tip inside the trachea; (c) random testing for endotoxin contamination was performed on the instillates and found to be negative; and (d) low toxicity/high stability of succinimidyl esters of FITC-macromolecules were chosen as opposed to isothiocyanate esters, and free dye was removed through dialysis. Nevertheless, it can be argued that the aspiration of the macromolecule solution, however inert, could represent a local stress signal in itself.
Still, the trafficking rate we observe is of similar magnitude as previously described estimates of DC turnover after irradiation 32. This supports the idea that the FITC-conjugates instilled in our model are relatively inert “tell-tales” for tracking the movement of airway DCs.
Very early after instillation, some FITC signal was observed within resident LNDCs. However, at most 6% of these LNDCs became FITC+ (12 h after instillation) and, in contrast to FITC+ AW-LNDCs, these cells did not further accumulate beyond that early time point, reinforcing the notion that NAW-LNDCs do not import Ag deposited in the periphery.
As far as the disappearance of FITC+ AW-LNDCs from the TLNs is concerned, studies will be necessary to determine which DC apoptosis/survival-related factors are mainly involved. This is a significant issue, as preliminary data point towards an altered clearance of Ag-carrying AW-LNDCs in a model of allergic airway inflammation.
Phenotype of Lung and Mediastinal LNDCs.
We compared the phenotype of lung and TLN DCs after collagenase digestion and EDTA treatment of whole lungs and corresponding TLNs. We chose to phenotype cells right after organ digestion in order to avoid artefacts originating from additional DC enrichment and culture procedures. AW and NAW DC subsets in the LNs were outlined using the MHCII/CD11c double staining as described above. To pinpoint DCs in the lungs, we used a strategy based on findings by Havenith et al. 35 in which DCs and monocytic DC precursors were identified within the low autofluorescent cell fraction of rat bronchoalveolar lavage fluid. Additionally, in this study the phenotype of the low autofluorescent cell fraction was found to shift further towards typical mature DC morphology after overnight incubation. We applied this approach to murine lung cells and added the expression of CD11c to the immunofluorescent identification criteria (Fig. 8 A). Two peaks of autofluorescence could be distinguished within the CD11c+ gate. When sorted, CD11c+/low autofluorescence cells showed a predominant monocyte and immature DC morphology. After overnight incubation in cytokine-free, serum-supplemented medium this appearance shifted clearly towards typical DCs with numerous cytoplasmic extensions (data not shown). We did not consider high levels of surface MHCII as a DC identification criterion a priori in order to avoid a bias towards more mature forms. Nevertheless, CD11c+/low autofluorescence cells were >90% MHCII+. Therefore, these cells are hereafter referred to as “intrapulmonary DCs.” The mean fluorescence intensity of MHCII was even higher for CD11c+ TLN cells (Fig. 8 B).
Figure 8.

(A) Identification of DCs in collagenase digests of whole lungs (parenchymal tissue and large airways including trachea, no bronchoalveolar lavage) after collagenase/DNase/EDTA treatment. Pulmonary DCs, defined as CD11c+/low autofluorescence cells, were found to be MHCII+, in contrast to CD11c+/high autofluorescence cells. (B) CD11c+ TLN cells obtained using the same organ digestion protocol show even higher MHCII expression.
Fig. 9 shows a surface phenotypic comparison between pulmonary DCs and corresponding TLN DC subsets. Compared with their intrapulmonary counterpart, AW-LNDCs were clearly more mature as illustrated by an upregulation of MHCII (Fig. 8) and the T cell costimulatory molecules CD40, B7-2, and ICAM-1 (Fig. 9 B). This specific pattern of phenotypical shift is also seen after in vitro maturation induction of cultured DCs by such various factors as mechanical stress, LPS, necrotic cells, and a variety of other danger signals 5. Our observations suggest that even in the absence of specific danger signals or Ag, lung DCs undergo phenotypical maturation as they migrate to the TLNs.
Figure 9.


Phenotypic comparison between DCs identified in whole lung digests (pulm-DC defined as CD11c+/low autofluorescence; see Fig. 8 A) and DC subsets within TLNs (see Fig. 2 A). Cells were phenotyped right after organ digest, without any additional enrichment procedure. Semitransparent histograms, isotype controls; gray histograms, Ag-specific antibodies. (A) Lineage markers. (B) T cell costimulatory molecules/maturation markers. Compared with pulm-DCs, AW-LNDCs have upregulated levels of CD40, B7-2, and ICAM-1 (framed markers) and upregulate low levels of CD8α as well (arrow, A). Note the strong expression of CD8α on NAW-LNDCs.
Interestingly, we were unable to detect any significant levels of B7-1 on either murine pulm-DCs or AW-LNDCs. In contrast, this costimulatory molecule was clearly detected in a previous work performed on murine pulmonary DCs, albeit at weaker levels than B7-2 36. In this study however, the DCs were further enriched by an overnight adherence step, which could alter the phenotype. Using the same antibodies, we have previously reported that both B7-1 and -2 are very strongly expressed on cultured bone marrow DCs, illustrating that the absence of B7-1 might be typical of freshly isolated lung DCs 16. Another noteworthy difference between intrapulmonary DCs and AW-LNDCs was the consistent low level expression of CD8α on the latter cells (Fig. 9 A). In contrast, both cell types expressed the Mac-1 myelomonocytic marker as well as DEC-205 (traditionally observed on LCs, DCs derived from bone marrow culture 37 and thymic DCs 38). Although DCs seeding nonlymphoid peripheral organs are generally considered to be of myeloid origin, there have been reports of CD8α expression on LCs as they arrive into cutaneous LNs 39 40. It was also shown that in vitro CD40 ligation induces expression of CD8α on purified LCs 41. The fact that CD8α was absent from lung DCs, but clearly expressed on AW-LNDCs, also suggests a similar mechanism.
In contrast to the CD8αlowDEC-205high migratory LNDCs, the main feature of NAW- LNDCs was a high level expression of both DEC-205 and CD8α. The NAW-LNDC cluster was heterogeneous, however, as it also contained cells expressing the myeloid marker Mac-1. This is analogous to findings reported by Salomon et al. 23 in which three subpopulations of DCs were described in cutaneous LNs, only one of which (MHCIIhiCD11cmed-hi) became FITC+ after skin painting with FITC. The exact origin of NAW-LNDC subsets and their possible relationship to AW-LNDCs need further investigation.
AW-LNDCs Efficiently Present Ag Deposited in the Airways.
It was necessary to verify the functional impact of the findings outlined above, namely that AW-LNDC transport Ag from the airways to the TLNs and phenotypically mature while doing so. Therefore, we conducted an in vitro antigen-specific proliferation assay using AW-LNDCs from FITC-OVA–instilled BALB/c mice as APCs, and DO11.10 T cells as OVA-responsive cells. As can be seen in Fig. 10, after FITC-OVA instillation AW-LNDCs induced a very strong OVA-specific T cell proliferation, again stressing the unique role of these cells in the uptake and processing of Ag deposited in the airways. In this setting, FITC− NAW-LNDCs also induced some T cell proliferation, albeit several orders of magnitude weaker than when AW-LNDCs were used as APCs. The possibility of nonspecific T cell stimulation could be ruled out, as AW-LNDCs from FITC-DX–instilled animals induced no T cell proliferation in the same culture conditions. Several explanations exist for the low level induction of T cell proliferation by NAW-LNDCs. (a) Some contamination of NAW-LNDCs by AW-LNDCs may have occurred during fluorescence-activated cell sorting. (b) Alternatively, the minor early uptake of FITC+ material in NAW-LNDCs (see kinetics) could suffice to load these cells with very low levels of Ags, generating a low ligand density and a suboptimal T cell response. (c) Another possibility is the transfer of Ag between AW- and NAW-LNDCs. In previous work by Inaba et al. 42 43, it was shown that Ags from incoming migratory DCs and apoptotic cells can be efficiently presented on (possibly CD8α+) DCs residing in the T cell areas. It is possible in our model that NAW-LNDCs (containing a CD8a+DEC-205+ subset) have phagocytosed and processed apoptotic AW-LNDCs, including their OVA cargo. Alternatively, recent experiments also suggest that preformed MHC–peptide fragments can be transferred from one DC to another 44 45.
Figure 10.

OVA-TCR transgenic T cell proliferation induced by different subsets of TLN DCs. Values represent tritiated thymidine uptake by T cells, expressed as mean counts per minute ±SE derived from triplicate assays. ▾, AW-LNDCs from FITC-OVA–instilled mice; •, NAW-LNDCs from the same mice; ⋄, AW-LNDCs from FITC-DX–instilled mice.
In summary, we have described the kinetics of pulmonary DC migration from the airways to the T cell zones of the TLNs by relying on their Ag-carrying properties. This migration is accompanied by phenotypic maturation and very efficient presentation of the peripherally administered soluble Ag. The novel approach we used allows the study of the kinetics of DC-mediated Ag transport in pathological states such as allergic airway inflammation. In addition, it offers the possibility of investigating the relative contribution of chemokines, cytokines, metalloproteinases, and integrins and cadherins to the migration of airway DCs, by means of pharmacological blockade and the use of knockout or transgenic animals.
Acknowledgments
We would like to thank Prof. Magda De Smet (Ghent University Hospital, Ghent, Belgium) for allowing access to the FACS Vantage SE™ system used in our cell sorting experiments, and Dr. Tom Boterberg (Ghent University Hospital, Ghent, Belgium) for his help during the irradiation experiments. We are grateful to Prof. David Klatzmann (Hôspital de la Pitié-Salpêtriére, Paris, France) for providing the TK-TG mice. Many thanks also to Greet Barbier for her technical assistance.
This work was supported by the Fund for Scientific Research in Flanders (FWO Vlaanderen, Research Project G.0393.99) and by the Concerted Research Initiative of the University of Ghent (GOA Project 98-6). K. Vermaelen was supported by a grant from the Fund for Scientific Research in Flanders (FWO Vlaanderen). I. Carro-Muino was supported by a grant from the Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT). B. Lambrecht was supported by a grant from the Fund for Scientific Research in Flanders (FWO Vlaanderen) and the Dutch Asthma Fund.
Footnotes
Abbreviations used in this paper: AW, airway-derived; DC, dendritic cell; DX, dextran; GCV, ganciclovir; ICAM, intracellular adhesion molecule; LC, Langerhans cell; MR, mannose receptor; NAW, non–airway-derived; TCM, tissue culture medium; TK-TG, thymidine kinase transgenic; TLN, thoracic LN.
References
- Banchereau J., Steinman R.M. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Holt P.G., Schon-Hegrad M.A., Oliver J. MHC class II antigen–bearing dendritic cells in pulmonary tissues of the rat. Regulation of antigen presentation activity by endogenous macrophage populations. J. Exp. Med. 1988;167:262–274. doi: 10.1084/jem.167.2.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brokaw J.J., White G.W., Baluk P., Anderson G.P., Umemoto E.Y., McDonald D.M. Glucocorticoid-induced apoptosis of dendritic cells in the rat tracheal mucosa. Am. J. Respir. Cell Mol. Biol. 1998;19:598–605. doi: 10.1165/ajrcmb.19.4.2870. [DOI] [PubMed] [Google Scholar]
- Schon-Hegrad M.A., Oliver J., McMenamin P.G., Holt P.G. Studies on the density, distribution, and surface phenotype of intraepithelial class II major histocompatibility complex antigen (Ia)–bearing dendritic cells (DC) in the conducting airways. J. Exp. Med. 1991;173:1345–1356. doi: 10.1084/jem.173.6.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallucci S., Lolkema M., Matzinger P. Natural adjuvantsendogenous activators of dendritic cells. Nat. Med. 1999;5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- Cyster J.G. Chemokines and cell migration in secondary lymphoid organs. Science. 1999;286:2098–2102. doi: 10.1126/science.286.5447.2098. [DOI] [PubMed] [Google Scholar]
- Lambrecht B.N., Pauwels R.A., De St. Groth B. Fazekas. Induction of rapid T cell activation, division, and recirculation by intratracheal injection of dendritic cells in a TCR transgenic model. J. Immunol. 2000;164:2937–2946. doi: 10.4049/jimmunol.164.6.2937. [DOI] [PubMed] [Google Scholar]
- Sozzani S., Allavena P., D'Amico G., Luini W., Bianchi G., Kataura M., Imai T., Yoshie O., Bonecchi R., Mantovani A. Differential regulation of chemokine receptors during dendritic cell maturationa model for their trafficking properties. J. Immunol. 1998;161:1083–1086. [PubMed] [Google Scholar]
- Price A.A., Cumberbatch M., Kimber I., Ager A. α6 integrins are required for Langerhans cell migration from the epidermis. J. Exp. Med. 1997;186:1725–1735. doi: 10.1084/jem.186.10.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staquet M.J., Kobayashi Y., Dezutter-Dambuyant C., Schmitt D. Role of specific successive contacts between extracellular matrix proteins and epidermal Langerhans cells in the control of their directed migration. Eur. J. Cell Biol. 1995;66:342–348. [PubMed] [Google Scholar]
- Jakob T., Udey M.C. Regulation of E-cadherin-mediated adhesion in Langerhans cell-like dendritic cells by inflammatory mediators that mobilize Langerhans cells in vivo. J. Immunol. 1998;160:4067–4073. [PubMed] [Google Scholar]
- Kobayashi Y., Matsumoto M., Kotani M., Makino T. Possible involvement of matrix metalloproteinase-9 in Langerhans cell migration and maturation. J. Immunol. 1999;163:5989–5993. [PubMed] [Google Scholar]
- Larsen C.P., Steinman R.M., Witmer-Pack M., Hankins D.F., Morris P.J., Austyn J.M. Migration and maturation of Langerhans cells in skin transplants and explants. J. Exp. Med. 1990;172:1483–1493. doi: 10.1084/jem.172.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havenith C.E., van Miert P.P., Breedijk A.J., Beelen R.H., Hoefsmit E.C. Migration of dendritic cells into the draining lymph nodes of the lung after intratracheal instillation. Am. J. Respir. Cell Mol. Biol. 1993;9:484–488. doi: 10.1165/ajrcmb/9.5.484. [DOI] [PubMed] [Google Scholar]
- Lambrecht B.N., Peleman R.A., Bullock G.R., Pauwels R.A. Sensitization to inhaled antigen by intratracheal instillation of dendritic cells. J. Immunol. 2000;164:2937–2946. doi: 10.1046/j.1365-2222.2000.00818.x. [DOI] [PubMed] [Google Scholar]
- Lambrecht B.N., De Veerman M., Coyle A.J., Gutierrez-Ramos J.C., Thielemans K., Pauwels R.A. Myeloid dendritic cells induce Th2 responses to inhaled antigen, leading to eosinophilic airway inflammation. J. Clin. Invest. 2000;106:551–559. doi: 10.1172/JCI8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W., Pinto C.E., Kradin R.L. The antigen-presenting activities of Ia+ dendritic cells shift dynamically from lung to lymph node after an airway challenge with soluble antigen. J. Exp. Med. 1995;181:1275–1283. doi: 10.1084/jem.181.4.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy K.M., Heimberger A.B., Loh D.Y. Induction by antigen of intrathymic apoptosis of CD4+ CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–1723. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- Salomon B., Lores P., Pioche C., Racz P., Jami J., Klatzmann D. Conditional ablation of dendritic cells in transgenic mice. J. Immunol. 1994;152:537–548. [PubMed] [Google Scholar]
- Lambrecht B.N., Salomon B., Klatzmann D., Pauwels R.A. Dendritic cells are required for the development of chronic eosinophilic airway inflammation in response to inhaled antigen in sensitized mice. J. Immunol. 1998;160:4090–4097. [PubMed] [Google Scholar]
- Vremec D., Shortman K. Dendritic cell subtypes in mouse lymphoid organscross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J. Immunol. 1997;159:565–573. [PubMed] [Google Scholar]
- Vremec D., Zorbas M., Scollay R., Saunders D.J., Ardavin C.F., Wu L., Shortman K. The surface phenotype of dendritic cells purified from mouse thymus and spleeninvestigation of the CD8 expression by a subpopulation of dendritic cells. J. Exp. Med. 1992;176:47–58. doi: 10.1084/jem.176.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon B., Cohen J.L., Masurier C., Klatzmann D. Three populations of mouse lymph node dendritic cells with different origins and dynamics. J. Immunol. 1998;160:708–717. [PubMed] [Google Scholar]
- Elbert K.J., Schafer U.F., Schafers H.J., Kim K.J., Lee V.H., Lehr C.M. Monolayers of human alveolar epithelial cells in primary culture for pulmonary absorption and transport studies. Pharm. Res. 1999;16:601–608. doi: 10.1023/a:1018887501927. [DOI] [PubMed] [Google Scholar]
- Hulbert W.C., Forster B.B., Mehta J.G., Man S.F., Molday R.S., Walker B.A., Walker D.C., Hogg J.C. Study of airway epithelial permeability with dextran. Int. Arch. Allergy Appl. Immunol. 1990;92:148–153. doi: 10.1002/jemt.1060110208. [DOI] [PubMed] [Google Scholar]
- Engering A.J., Cella M., Fluitsma D.M., Hoefsmit E.C., Lanzavecchia A., Pieters J. Mannose receptor mediated antigen uptake and presentation in human dendritic cells. Adv. Exp. Med. Biol. 1997;417:183–187. doi: 10.1007/978-1-4757-9966-8_31. [DOI] [PubMed] [Google Scholar]
- Linehan S.A., Martinez-Pomares L., Stahl P.D., Gordon S. Mannose receptor and its putative ligands in normal murine lymphoid and nonlymphoid organsin situ expression of mannose receptor by selected macrophages, endothelial cells, perivascular microglia, and mesangial cells, but not dendritic cells. J. Exp. Med. 1999;189:1961–1972. doi: 10.1084/jem.189.12.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W., Swiggard W.J., Heufler C., Peng M., Mirza A., Steinman R.M., Nussenzweig M.C. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 1995;375:151–155. doi: 10.1038/375151a0. [DOI] [PubMed] [Google Scholar]
- Pollard A.M., Lipscomb M.F. Characterization of murine lung dendritic cellssimilarities to Langerhans cells and thymic dendritic cells. J. Exp. Med. 1990;172:159–167. doi: 10.1084/jem.172.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valladeau J., Ravel O., Dezutter-Dambuyant C., Moore K., Kleijmeer M., Liu Y., Duvert-Frances V., Vincent C., Schmitt D., Davoust J. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 2000;12:71–81. doi: 10.1016/s1074-7613(00)80160-0. [DOI] [PubMed] [Google Scholar]
- Valladeau J., Duvert-Frances V., Pin J.J., Dezutter-Dambuyant C., Vincent C., Massacrier C., Vincent J., Yoneda K., Banchereau J., Caux C. The monoclonal antibody DCGM4 recognizes Langerin, a protein specific of Langerhans cells, and is rapidly internalized from the cell surface. Eur. J. Immunol. 1999;29:2695–2704. doi: 10.1002/(SICI)1521-4141(199909)29:09<2695::AID-IMMU2695>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Holt P.G., Haining S., Nelson D.J., Sedgwick J.D. Origin and steady-state turnover of class II MHC-bearing dendritic cells in the epithelium of the conducting airways. J. Immunol. 1994;153:256–261. [PubMed] [Google Scholar]
- Gong J.L., McCarthy K.M., Telford J., Tamatani T., Miyasaka M., Schneeberger E.E. Intraepithelial airway dendritic cellsa distinct subset of pulmonary dendritic cells obtained by microdissection. J. Exp. Med. 1992;175:797–807. doi: 10.1084/jem.175.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kradin R.L., Xia W., McCarthy K., Schneeberger E.E. FcR+/− subsets of Ia+ pulmonary dendritic cells in the rat display differences in their abilities to provide accessory co-stimulation for naive (OX-22+) and sensitized (OX-22−) T cells. Am. J. Pathol. 1993;142:811–819. [PMC free article] [PubMed] [Google Scholar]
- Havenith C.E., Breedijk A.J., van Miert P.P., Blijleven N., Calame W., Beelen R.H., Hoefsmit E.C. Separation of alveolar macrophages and dendritic cells via autofluorescencephenotypical and functional characterization. J. Leukoc. Biol. 1993;53:504–510. doi: 10.1002/jlb.53.5.504. [DOI] [PubMed] [Google Scholar]
- Masten B.J., Yates J.L., Pollard Koga A.M., Lipscomb M.F. Characterization of accessory molecules in murine lung dendritic cell functionroles for CD80, CD86, CD54, and CD40L. Am. J. Respir. Cell Mol. Biol. 1997;16:335–342. doi: 10.1165/ajrcmb.16.3.9070619. [DOI] [PubMed] [Google Scholar]
- Inaba K., Swiggard W.J., Inaba M., Meltzer J., Mirza A., Sasagawa T., Nussenzweig M.C., Steinman R.M. Tissue distribution of the DEC-205 protein that is detected by the monoclonal antibody NLDC-145. I. Expression on dendritic cells and other subsets of mouse leukocytes. Cell. Immunol. 1995;163:148–156. doi: 10.1006/cimm.1995.1109. [DOI] [PubMed] [Google Scholar]
- Kronin V., Wu L., Gong S., Nussenzweig M.C., Shortman K. DEC-205 as a marker of dendritic cells with regulatory effects on CD8 T cell responses. Int. Immunol. 2000;12:731–735. doi: 10.1093/intimm/12.5.731. [DOI] [PubMed] [Google Scholar]
- Anjuere F., Martin P., Ferrero I., Fraga M.L., del Hoyo G.M., Wright N., Ardavin C. Definition of dendritic cell subpopulations present in the spleen, Peyer's patches, lymph nodes, and skin of the mouse. Blood. 1999;93:590–598. [PubMed] [Google Scholar]
- Merad M., Fong L., Bogenberger J., Engleman E.G. Differentiation of myeloid dendritic cells into CD8α-positive dendritic cells in vivo. Blood. 2000;96:1865–1872. [PubMed] [Google Scholar]
- Anjuere F., Martinez del Hoyo G., Martin P., Ardavin C. Langerhans cells acquire a CD8+ dendritic cell phenotype on maturation by CD40 ligation. J. Leukoc. Biol. 2000;67:206–209. doi: 10.1002/jlb.67.2.206. [DOI] [PubMed] [Google Scholar]
- Inaba K., Pack M., Inaba M., Sakuta H., Isdell F., Steinman R.M. High levels of a major histocompatibility complex II–self peptide complex on dendritic cells from the T cell areas of lymph nodes. J. Exp. Med. 1997;186:665–672. doi: 10.1084/jem.186.5.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba K., Turley S., Yamaide F., Iyoda T., Mahnke K., Inaba M., Pack M., Subklewe M., Sauter B., Sheff D. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J. Exp. Med. 1998;188:2163–2173. doi: 10.1084/jem.188.11.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.L., de St. Groth B.F. Antigen-pulsed CD8α1 dendritic cells generate an immune response after subcutaneous injection without homing to the draining lymph node. J. Exp. Med. 1999;189:593–598. doi: 10.1084/jem.189.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight S.C., Iqball S., Roberts M.S., Macatonia S., Bedford P.A. Transfer of antigen between dendritic cells in the stimulation of primary T cell proliferation. Eur. J. Immunol. 1998;28:1636–1644. doi: 10.1002/(SICI)1521-4141(199805)28:05<1636::AID-IMMU1636>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]