Viruses Associated with Human Cancer (original) (raw)
. Author manuscript; available in PMC: 2009 Mar 1.
Published in final edited form as: Biochim Biophys Acta. 2007 Dec 23;1782(3):127–150. doi: 10.1016/j.bbadis.2007.12.005
Abstract
It is estimated that viral infections contribute to 15–20% of all human cancers. As obligatory intracellular parasites, viruses encode proteins that reprogram host cellular signaling pathways that control proliferation, differentiation, cell death, genomic integrity, and recognition by the immune system. These cellular processes are governed by complex and redundant regulatory networks and are surveyed by sentinel mechanisms that ensure that aberrant cells are removed from the proliferative pool. Given that the genome size of a virus is highly restricted to ensure packaging within an infectious structure, viruses must target cellular regulatory nodes with limited redundancy and need to inactivate surveillance mechanisms that would normally recognize and extinguish such abnormal cells. In many cases, key proteins in these same regulatory networks are subject to mutation in non-virally associated diseases and cancers. Oncogenic viruses have thus served as important experimental models to identify and molecularly investigate such cellular networks. These include the discovery of oncogenes and tumor suppressors, identification of regulatory networks that are critical for maintenance of genomic integrity, and processes that govern immune surveillance.
Keywords: Human T-Cell Leukemia Virus (HTLV-1), Hepatitis C Virus (HCV), Human Papillomavirus (HPV), Hepatitis B Virus (HBV), Epstein-Barr Virus (EBV), Kaposi’s Sarcoma-Associated Herpesvirus (KSHV)/Human Herpes Virus 8 (HHV8)
1. Introduction
With 10.9 million new cases and 6.7 million deaths per year, cancer is a devastating disease, presenting an immense disease burden to affected individuals and their families as well as health care systems [1]. Development of treatment and prevention strategies to manage this disease critically depends on our understanding of cancer cells and the mechanism(s) through which they arise. In general terms, carcinogenesis represents a complex, multi-step process. During the past 30 years it has become exceedingly apparent that several viruses play significant roles in the multistage development of human neoplasms; in fact, approximately 15% to 20% of cancers are associated with viral infections [2, 3]. Oncogenic viruses can contribute to different steps of the carcinogenic process, and the association of a virus with a given cancer can be anywhere from 15% to 100% [3]. In addition to elucidating the etiology of several human cancers, the study of oncogenic viruses has been invaluable to the discovery and analysis of key cellular pathways that are commonly rendered dysfunctional during carcinogenesis in general.
2. Historic Context
The belief in the infectious nature of cancer originated in classical times as evidenced by accounts of “cancer houses” in which many dwellers developed a certain cancer. Observations that married couples sometimes could be affected by similar cancer types and that cancer appeared to be transmitted from mother to child lent further support to an infectious etiology of tumors. However, during the 19th century, extensive investigations failed to demonstrate a carcinogenic role for bacteria, fungi, or parasites leading to the belief that cancer is not caused by an infectious agent. Despite the prevailing dogma, a small number of researchers hypothesized that the failure to detect an infectious cause of cancer did not necessarily mean that the general idea of the infectious nature of cancer was invalid. Rather, they hypothesized that the causal organism had merely not yet been found and that smaller entities not detectable by standard microscopy may indeed be the culprits. Despite increasing evidence to suggest that infectious entities of sub-microscopic size may be associated with cancer, acceptance of this hypothesis took many years. M’Fadyan and Hobday described the cell-free transmission of oral dog warts with cell free extracts in 1898 [4], and Ciuffo published similar transmission studies with human warts in 1907 [5]. The significance of these findings was not fully appreciated since warts are benign hyperplasias and not malignant tumors. In 1908, Ellermann and Bang demonstrated that leukemia in birds could be transmitted from animal to animal via extracts of leukemic cells or serum from diseased birds [6]. However, at the time it was not realized that this was the first successful transmission of a naturally occurring tumor, as leukemia was not yet accepted as a cancer. In 1911, Peyton Rous produced solid tumors in chickens using cell-free extracts from a transplantable sarcoma [7]. This study was also met with considerable skepticism due to the fact that infectious cancers of birds were not considered valid models for human cancers. In fact, the importance of this study was not fully appreciated until the finding that murine leukemias could be induced by viruses [8, 9]. Over the next two decades numerous additional animal oncogenic viruses were isolated, Rous was awarded the Noble Prize for his pioneering work in 1966, and the importance of the early work on animal tumor viruses was finally recognized. In fact, the enthusiasm for these findings contributed in no small part to President Nixon signing the National Cancer Act into law in 1971 and declaring the “War on Cancer”.
After the successes of the animal tumor virus field, scientists began the search for human tumor viruses. However, initial attempts to isolate transmissible carcinogenic viruses from human tumors proved disappointing, once again raising doubts about the existence of human cancer viruses. The discovery of Epstein-Barr virus (EBV) by electron microscopy (EM) in cells cultured from Burkitt’s lymphoma (BL) in 1964 [10] and the discovery of hepatitis B virus (HBV) in human sera positive for hepatitis B surface antigen in 1970 [11], together with the development of animal and cell culture model systems, resulted in a renewed interest in the roles of viruses in human cancer. The search for additional human tumor viruses continued, and, despite several setbacks, the ultimate acknowledgment of the causal relationship between viruses and human cancer occurred during the early 1980s, due in large part to three major discoveries during that time. In 1983 and 1984, human papillomavirus (HPV) 16 and 18 were isolated from human cervical cancer specimens [12, 13]. Additionally, although the link between HBV and liver cancer had been suspected for decades, the results of a large-scale epidemiological study provided a compelling link between persistent HBV infection and liver carcinogenesis [14]. The third major discovery was the isolation of the human T-cell leukemia virus (HTLV-I) from T-cell lymphoma/leukemia patients [15, 16]. Since their initial discovery, associations of these viruses with cancers at other anatomical sites have been discovered. Moreover, new links between viruses, most notably hepatitis C virus (HCV) [17] and human herpes-virus 8 (HHV8)/Kaposi’s sarcoma herpesvirus (KSHV) [18], and human cancers have been discovered. Today, viruses are accepted as bona-fide causes of human cancers, and it has been estimated that between 15 and 20% of all human cancers may have a viral etiology [2, 3].
3. General Aspects of Viral Carcinogenesis
The infectious nature of oncogenic viruses sets them apart from other carcinogenic agents. As such, a thorough study of both the pathogenesis of viral infection and the host response is crucial to a full understanding of the resulting cancers. Such an understanding, in turn, has increased our knowledge of cellular pathways involved in growth and differentiation and neoplasia as a whole.
Even though human oncogenic viruses belong to different virus families and utilize diverse strategies to contribute to cancer development, they share many common features. One key feature is their ability to infect, but not kill, their host cell. In contrast to many other viruses that cause disease, oncogenic viruses have the tendency to establish long-term persistent infections. Consequently, they have evolved strategies for evading the host immune response, which would otherwise clear the virus during these persistent infections. Despite the viral etiology of several cancers, it appears that the viruses often may contribute to, but are not sufficient for, carcinogenesis; in fact, the majority of tumor virus infected individuals do not develop cancer, and in those patients that do develop cancer many years may pass between initial infection and tumor appearance. Additional co-factors, such as host immunity and chronic inflammation, as well as additional host cellular mutations, must therefore also play an important role in the transformation process. Additionally, there is an obvious geographical distribution of many virus-associated cancers, which is possibly due to either geographical restriction of the virus or access to essential cofactors. Thus, the long-term interactions between virus and host are key features of the oncogenic viruses, as they set the stage for a variety of molecular events that may contribute to eventual virus-mediated tumorigenesis [19].
4. Criteria for Defining an Etiologic Role for Viruses in Cancer
In many cases, viral carcinogenesis is associated with an abortive, non-productive infection. Hence, the original Koch postulate for the infectious etiology of disease [20] cannot be applied, as oftentimes no disease causing infectious entity can be isolated from a tumor. Therefore, it is often difficult to establish a viral cause for a human cancer. As a result, different guidelines have been proposed to aid in establishing a causal relationship between viruses and human cancers (Tables 1 and 2) [21–23]. Although some of the guidelines are difficult to meet and others are not applicable to all viruses, the guidelines are nonetheless quite useful when evaluating a putative association between a virus and a human malignancy.
Table 1.
Evans and Mueller guidelines [21]
| Epidemiologic guidelines Geographic distribution of viral infection corresponds with that of the tumor, adjusting for the presence of known cofactors Viral markers are higher in case subjects than in matched control subjects Viral markers precede tumor development, with a higher incidence of tumors in persons with markers than those without Tumor incidence is decreased by viral infection prevention |
|---|
| Virologic guidelines Virus can transform cells in vitro Viral genome is present in tumor cells, but not in normal cells Virus induces the tumor in an experimental animal |
Table 2.
Hill criteria for causality [22,23]
- Strength of association (how often is the virus associated with the tumor?)
- Consistency (has the association been observed repeatedly?)
- Specificity of association (is the virus uniquely associated with the tumor?)
- Temporal relationship (does virus infection precede tumorigenesis?)
- Biologic gradient (is there a dose response with viral load?)
- Biologic plausibility (is it biologically plausible that the virus could cause the tumor?)
- Coherence (does the association make sense with what is known about the tumor?)
- Experimental evidence (is there supporting laboratory data?)
5. Human Oncogenic Viruses
Human tumor viruses belong to a number of virus families, including the RNA virus families Retroviridae and Flaviviridae and the DNA virus families Hepadnaviridae, Herpesviridae, and Papillomaviridae. To date, viruses that are compellingly associated with human malignancies include; (i) HTLV-1 (adult T-cell leukemia (ATL)) [15], (reviewed in [24]); (ii) HPV (cervical cancer, skin cancer in patients with epidermodysplasia verruciformis (EV), head and neck cancers, and other anogenital cancers) [12, 13], (reviewed in [25–28]); iii) HHV-8 (Kaposi’s sarcoma (KS), primary effusion lymphoma, and Castleman’s disease) [18], (reviewed in [29, 30]) (iv) EBV (Burkitt’s Lymphoma (BL), nasopharyngeal carcinoma (NPC), post-transplant lymphomas, and Hodgkin’s disease) [10], (reviewed in [31–33]); and (v) HBV and HCV (hepatocellular carcinoma (HCC)) [11, 17], (reviewed in [34–38]). Viruses with potential roles in human malignancies include; (i) simian vacuolating virus 40 (SV40) (brain cancer, bone cancer, and mesothelioma) [39]; (ii) BK virus (BKV) (prostate cancer) [40], (reviewed in [41]); (iii) JC virus (JCV) (brain cancer) [42], (reviewed in [41]); (iv) human endogenous retroviruses (HERVs) (germ cell tumors, breast cancer, ovarian cancer, and melanoma) [43, 44]; (v) human mammary tumor virus (HMTV) (breast cancer) (reviewed in [45]; and (vi) Torque teno virus (TTV) (gastrointestinal cancer, lung cancer, breast cancer, and myeloma) [46]. General information about viruses with known and potential associations with human cancer is provided in Tables 3 and 4, respectively. Studies of the RNA and DNA tumor viruses has led to the discovery of oncogenes and tumor suppressors and has greatly added to our understanding of the etiology of carcinogenesis, both virally and non-virally induced.
Table 3.
Properties of human tumor viruses
| Virus | Viral Taxonomy | Genome | Cell Tropism | Human Cancers |
|---|---|---|---|---|
| EBV | Herpesviridae | dsDNA 172kb ~90 ORFs | Oropharyngeal epithelial cells, B-cells | BL, NPC, lymphomas |
| HBV | Hepadnoviridae | dsDNA 3.2kb 4 ORFs | Hepatocytes, white blood cells | HCC |
| HCV | Flaviviridae | dsRNA 9.4 kb 9 ORFs | Hepatocytes | HCC |
| HPV | Papillomavirdae | dsDNA 8 kb 8–10 ORFs | Squamous epithelial cells | Cervical, oral, and anogenital cancer |
| HTLV-1 | Retroviridae | dsRNA 9.0 kb 6 ORFs | T-cells | ATL |
| KSHV | Herpesviridae | dsDNA 165kb ~90ORFs | B-cells | Kaposi sarcoma, primary effusion lymphoma |
Table 4.
Properties of viruses implicated in human cancers
| Virus | Viral Taxonomy | Genome | Human Cancers |
|---|---|---|---|
| BKV | Polyomaviridae | dsDNA ~5.2 kb | Prostate? |
| JCV | Polyomaviridae | dsDNA ~5.2 kb | Brain? |
| SV40 | Polyomaviridae | dsDNA ~5.2 kb | Brain, bone, mesothelioma? |
| HERVs | Retroviridae | dsRNA/DNA? | Seminomas, breast, ovarian, melanoma? |
| HMTV | Retroviridae | dsRNA/DNA? | Breast? |
| TTV | Circoviridae | ssDNA 3.8 kb | Gastrointestinal, lung, breast, and myleoma? |
5.1. RNA Tumor Viruses
Although retroviruses have been associated with many animal tumors, to date, only one human retrovirus, HTLV-1, has been associated with human cancers. The biology of HTLV-1 will be discussed in more detail in the next section. Studies with animal retroviruses have been instrumental establishing the concept of oncogenic viruses and led to the discovery of oncogenes and tumor suppressors as well as other key regulators of cellular signal transduction pathways. Hence, animal retroviruses warrant some discussion in this review.
The advent of modern tumor virology came about with the development of an in vitro transformation assay for Rous Sarcoma Virus (RSV) [47]. This assay allowed for the genetic analysis of the retroviral life cycle and retrovirus-induced transformation in cell culture (reviewed in [48–52]). Retroviruses are classified as either simple or complex viruses based on the organization of their genomes. Shortly after infection, the viral RNA genome is reverse-transcribed by the virally encoded reverse transcriptase into a double-stranded DNA copy, which then integrates into the host chromosome and is expressed under the control of viral transcriptional regulatory sequences. Once integrated, proviruses are rarely lost from the host chromosome. As a consequence of integration, and of particular relevance when considering oncogenesis, is the ability of simple retroviruses to acquire and transduce cellular genetic material or to activate or inactivate cellular genes via provirus insertion. It was the analysis of this group, the transducing retroviruses, that led to the finding that the RSV transforming gene, v-src, hybridized to cellular sequences; ultimately this finding led to the discovery of proto-oncogenes, a group of cellular genes that mediate viral carcinogenesis and have critical roles in the control of cell growth and differentiation (reviewed in [48–52]). Since this initial discovery, numerous animal retroviruses with oncogenic properties have been discovered, and, as is the case with src, the transduced retroviral oncogenes are derived from cellular sequences and are not necessary for viral replication [50, 52].
The erroneous recombination events that allow for acquisition of host cell derived coding sequences often leave viral genomes mutated and the virus defective for replication. As such, these viruses are dependent on replication competent helper viruses to provide the necessary replication functions in trans. Normal cellular transcriptional and translational controls are lost once an acquired cellular sequence is incorporated into the viral genome, and the over-expression of a proto-oncogene under the control of strong viral promoters can cause malignant transformation. Moreover, since the acquired proto-oncogene is not necessary for viral replication but is replicated through the same error prone mechanisms as the viral genome, retrovirally acquired proto-oncogenes are subject to frequent mutation and some “activating” proto-oncogene mutations endow the infected cell with a growth advantage and hence are selected for over time. Such oncogene transducing retroviruses efficiently transform cells in culture and cause tumors in experimental animals with very short latency periods. However, this mechanism is relatively rarely seen in animals in the wild and has not been documented in humans. Nonetheless, this ‘oncogene piracy’ has proven to be quite useful in laboratory studies of the actions of oncogenes in cancer. Most remarkably, mutations in cellular oncogenes that arise in human tumors as a consequence of mutagenic insults are often similar or identical to those discovered in transducing carcinogenic retroviruses [53].
A second group, the cis-acting retroviruses, does not contain host cell derived sequences but transforms cells by integrating in the vicinity of a cellular proto-oncogene or tumor suppressor. Unlike the transducing retroviruses, the cis-acting retroviruses retain all of their viral genes and thus can replicate without the aid of a helper virus. Cis-acting retroviruses cause malignancy in only a percentage of infected animals after a longer latency period than that required for transducing retroviruses, and they generally do not efficiently transform cells in culture. Insertional mutagenesis is a common mechanism observed for rodent, feline, and avian retroviruses, such as avian leukosis virus and mouse mammary tumor virus (MMTV). Whereas there is no compelling evidence that such a mechanism significantly contributes to human carcinogenesis, cloning of affected genes led to the discovery of numerous oncogenes, such as int-1 [54, 55], int-2 [55], Pim-1 [56], bmi-1 [57], Tpl-1 [58], and Tpl-2 [59], that importantly contribute to the development of human neoplasms. Moreover, the finding that the Friend murine leukemia virus had integrated into both alleles of the p53 gene in an erythroleukemic cell line provided critical evidence that p53 was a tumor suppressor rather than an oncogene as was originally suspected [60].
5.1.1. Human T-Cell Leukemia Virus (HTLV-1)
Of more significance to human carcinogenesis, the third mechanism of retroviral oncogenesis does not involve transmission of a mutated version of a cellular proto-oncogene or dysregulated expression of proto-oncogenes or tumor suppressor genes near or at the integration site. The latency period between the initial infection and development of a neoplasm with this group of viruses is often in the range of several years to several decades. The best-studied example of these viruses is HTLV-1, the first human retrovirus to be discovered that is clearly associated with a human malignancy [15, 16]. HTLV-1 is delta-type complex retrovirus and is the etiologic agent of ATL and tropical spastic paraparesis/HTLV-1-associated myelopathy (TSP/HAM). HTLV-1 is endemic to Japan, South America, Africa, and the Caribbean [61, 62]. While it is estimated that approximately 20 million people worldwide are infected with HTLV-1 [63], only a small percentage (2–6%) will develop ATL [64]. The long clinical latency, together with the relatively low cumulative lifetime risk of a carrier developing ATL, indicates that HTLV-1 infection is not sufficient to elicit T-cell transformation. While the exact cellular events remain unclear, a variety of steps, including virus, host cell, and immune factors, are implicated in the leukemogenesis of ATL [65] (Figure 1).
Figure 1.

Schematic depiction of the major biological activities that contribute to the transforming activities of HTLV-1. See text for details.
A number of studies indicate that the multifunctional viral accessory protein Tax is the major transforming protein of HTLV-1 [66–73]. Tax modulates expression of viral genes though the viral long terminal repeats (LTRs), and also dysregulates multiple cellular transcriptional signaling pathways including nuclear factor kappa B (NF-κB) [74–85], serum responsive factor (SRF) [86–90], cyclic AMP response element-binding protein (CREB) [91–94], and activator protein 1 (AP-1) [95, 96]. Rather than binding to promoter or enhancer sequences directly, Tax interacts with cellular transcriptional co-activators such as p300/CBP [94, 97–104], and P/CAF [105]. In addition to its role in transcriptional regulation, Tax is also able to functionally inactivate p53 [106, 107], p16INK4A [108, 109], and the mitotic checkpoint protein, Mitotic Arrest Deficient (MAD) 1 [110–112]. Additionally, the C-terminal PDZ domain-binding motif of Tax, which interacts with the tumor suppressor hDLG [113], is important for transformation of rat fibroblasts [114] and inducing interleukin-2-independent growth of mouse T-cells [115].
Unlike other well-established DNA tumor viruses, which generally require continuous expression of viral oncoproteins to sustain transformation (reviewed in [116]), tax transcripts are detected in only 40% of ATLs [117], suggesting that Tax may be needed to initiate transformation, but may not be necessary for maintenance of the transformed phenotype. Tax is the main target of the host’s cytotoxic T lymphocyte (CTL) response; therefore, the repression of Tax expression allows infected host cells to evade immunesurveillance and allows for the preferential selection of these cells during the progression of ATL [117]. There are several mechanisms by which ATL cells lose Tax expression, including the loss of the viral promoter for tax transcription, the 5′-LTR [117], mutation of the tax gene [118], and epigenetic changes in the 5′-LTR [119, 120].
Tax has also been implicated in inducing genomic instability, most prominently aneuploidy, which, as is the case in many cancers, is a hallmark of ATL [112]. Recent studies have shown that HTLV-1 Tax expression causes multipolar mitoses, from which aneuploidy can arise, in two ways [121–123]. First, Tax targets the cellular TAX1BP2 protein, which normally blocks centriole replication, thus causing numerical centrosome aberrations [121]. Second, it has been proposed that Tax engages RANBP1 during mitosis and fragments spindle poles, thereby provoking multipolar, asymmetrical chromosome segregation. Together, these mechanisms help explain the long-standing observations of aneuploidy and multipolar spindles in ATL cells (“flower cells”) [124]. In addition, several ATL cell lines have been demonstrated to lack an intact mitotic spindle assembly checkpoint [112], which may be related to binding to MAD1 [110, 111]. Evidence that Tax may act as a mitotic mutator gene, greatly increasing the incidence of mitotic abnormalities, is consistent with reports that Tax binds to and activates the anaphase-promoting complex/cyclosome (APC/C), thereby promoting premature securin degradation and mitotic exit, thus contributing to aneuploidy [125–127]. However, this concept is not fully accepted as a recent study found that Tax did not increase securin degradation [128].
As mentioned previously, alterations of the 5′-LTR, such as deletions or hypermethylation, are common in ATL cells. As a result, the transcription of viral genes encoded on the plus strand is often repressed. On the other hand, the 3′-LTR is conserved and hypomethylated in all ATLs [120]. The HBZ mRNA is transcribed from the 3′-LTR [129, 130] and is expressed in all ATL cells [131]. Suppression of HBZ gene transcription inhibits the proliferation of ATL cells; additionally HBZ gene expression promotes the proliferation of a human T-cell line [131]. It appears that HBZ may have a bimodal function at the mRNA and protein levels, as the RNA form of HBZ supports T cell proliferation through regulation of the E2F1 pathway, whereas HBZ protein suppresses Tax-mediated viral transcription through the 5′-LTR [129].
5.1.2. Hepatitis C Virus (HCV)
HCV is the etiologic agent of posttransfusion and sporadic non-A, non-B hepatitis [17] and infects approximately 2% of the population worldwide, although the prevalence of HCV infection varies by geographical location [132]. Persistent infection with HCV is associated with hepatitis, hepatic steatosis, cirrhosis, and hepatocellular carcinoma (HCC) [133–137]. HCV is a single stranded RNA virus of the Hepacivirus genus in the Flaviviridae family and is the only positive-stranded RNA virus among the human oncogenic viruses. Its approximately 9.6 kb genome contains an open reading frame (ORF) that codes for a 3000 amino acid residue polyprotein precursor [17] that is cleaved by cellular and viral proteases into three structural proteins (core, E1, E2) and seven nonstructural proteins (p7, NS2, NS3, NS4a, NS4B, NS5A, and NS5B) [138].
In the vast majority of infected individuals, HCV establishes a persistent and life-long infection via highly effective viral immune evasion strategies [139–143]. The formation of double stranded RNA (dsRNA) intermediates during HCV genome replication induces cellular dsRNA-sensing machinery, which in turn leads to the activation of proteins involved in antiviral response, including interferons (IFNs), interferon regulatory factors (IRFs), signal transducers and activators of transcription (STATs), interferon stimulated genes (ISGs) and NF-κB [144]. The HCV core, E2, NS3, and NS5A proteins counteract this cellular response through a variety of mechanisms, including NS5A and E2 mediated suppression of dsRNA-activated kinase PKR [139, 142]. HCV is also very effective in subverting T-cell mediated adaptive immunity [140, 141]. Although the underlying mechanisms are still unclear, it is believed that the persistence of HCV infection is due in part to the selection of quasispecies that have escaped the host immune response [139].
Infection with HCV causes active inflammation and fibrosis, which can progress to cirrhosis and ultimately lead to tumor development. Numerous co-factors for the development of HCV-associated HCC exist, including co-infection with HBV and excessive alcohol consumption [145]. While it is currently thought that chronic inflammation and cirrhosis play key roles in HCV-induced carcinogenesis, the exact underlying mechanisms are not fully understood [146]. Moreover, the multiple functions of HCV proteins and their impact on cell signaling have led to the idea that both viral and host factors also play a role in HCC. Core, NS3, NS4B, and NS5A have each been shown to be transforming in murine fibroblasts [147] and transgenic mice expressing HCV core protein develop HCC [148, 149]. In addition, HCV proteins have also been reported to activate cellular oncoproteins and inactivate tumor suppressors, such as p53 [150], CREB2/LZIP [151], and the retinoblastoma protein (pRB) [152]. Finally, HCV causes genome instability, suggesting that certain HCV proteins may have a mutator function [153]. A summary of the role of HCV in hepatocellular carcinogenesis is depicted on Figure 2.
Figure 2.

Schematic depiction of the major biological activities that contribute to the transforming activities of HCV. See text for details.
5.2. DNA Tumor Viruses
The human DNA tumor viruses are a diverse group with varied structures, genome organizations, and replication strategies. Certain DNA tumor viruses, such as HPV, EBV, HBV, and KSHV, cause malignancies in their natural hosts, whereas other DNA tumor viruses, such as human adenoviruses, can transform cultured cells and only cause tumors in heterologous animal models. Unlike oncogenes encoded by animal retroviruses, DNA tumor virus oncogenes are of viral, not cellular, origin and are necessary for replication of the virus (reviewed in [154]). In addition to elucidating the etiology of several human diseases, analysis of the DNA tumor virus oncoproteins has revealed mechanisms controlling mammalian cell growth, ultimately leading to the discovery of cellular tumor suppressor genes.
Studies on the small DNA tumor viruses, which include the adenoviruses, polyomaviruses, and papillomaviruses, have been instrumental in elucidating the underlying molecular mechanisms of virus-induced cell transformation. Although these viruses are evolutionarily distinct, the striking similarities in their transforming functions emphasize the mutual need of these viruses to utilize the host cell’s replication machinery for efficient viral replication. Much of the understanding of the actions of the small DNA tumor virus oncoproteins has been derived from the study of the physical associations of the viral oncoproteins with a variety of cellular tumor suppressors, most notably the associations of adenovirus E1A (Ad E1A), SV40 large T antigen (TAg) and HPV E7 with the pRB family and adenovirus E1B (Ad E1B), SV40 TAg, and HPV E6 with p53 (reviewed in [27, 155, 156]).
The first major cellular tumor suppressor that was found to be targeted by small DNA tumor virus oncoproteins is pRB, which was identified as the 105 kDa protein associated with Ad E1A in adenovirus-transformed cells [157, 158], and has subsequently been identified as a cellular target for SV40 Tag [159] and HPV16 E7 [160]. The binding of Ad E1A, SV40 TAg, or HPV E7 to pRB results in the disruption or alteration of cellular complexes that normally contain pRB, resulting in inactivation of growth regulatory functions [161–164]. Cells expressing E1A, SV40 TAg, or HPV E7 display increased levels of free E2F with associated loss of cell-cycle dependent regulation of E2F responsive genes [165, 166]. E2F responsive genes play key roles in cell cycle progression, and thus the association of small DNA tumor virus oncoproteins with pRB ultimately results in S-phase induction, an essential aspect of these viruses’ life cycles. Despite the fact that the core pRB binding sites of SV40 TAg, HPV E7, and Ad E1A are similar, the functional consequences of viral oncoprotein association are different. Whereas Ad E1A inactivates pRB by binding both hypo- and hyper- phosphorylated forms [163], SV40 TAg specifically interacts with G1-specific, growth suppressive hypophosphorylated form [167] and HPV16 E7 preferentially binds hypophosphorylated pRB [168, 169] and targets pRB for proteasome mediated degradation [170–172]. The second major cellular tumor suppressor that is targeted by small DNA tumor virus oncoproteins is p53, which was originally detected as a cellular protein complexed with SV40 TAg in SV40-transformed cells [173, 174], and has since been shown to also complex with high-risk HPV E6 and Ad E1B [175, 176]. The p53 protein was initially classified as an oncogene because it was cloned from a cancer cell line and contained a point mutation, and it was only later discovered that the wild type form has tumor suppressor activity [177, 178].
The interaction between SV40 TAg, HPV E6, and Ad E1B with p53 results in the inhibition of p53’s tumor suppressor activities [179–181]. p53 is a sequence specific, DNA binding transcriptional activator [182, 183]. It is not required for normal cellular proliferation, but rather integrates signal transduction pathways that sense cellular stress, and thus has been referred to as a “guardian of the genome” [184]. Unlike the case with SV40 TAg, HPV E7, and Ad E1A, there is no structural similarity between SV40 TAg, HPV E6, and Ad E1B; this lack of structural similarity is reflected in the differences between their interactions with p53. The metabolic half-life of p53 is extended in cells that express SV40 TAg or Ad E1B; consequently p53 levels are higher in these cells than in normal cells [185, 186]. On the other hand, p53 levels in high-risk HPV E6 expressing cells are lower than in their normal counterparts [187, 188], due to induction of p53 degradation through ubiquitin-mediated proteolysis [189].
5.2.1. Human Papillomavirus (HPV)
Papillomaviruses (PVs) are a group of small, non-enveloped, double-stranded DNA viruses that constitute the Papillomaviridae family. These viruses infect squamous epithelia of a variety of species [190]; to date, approximately 200 human papillomavirus (HPV) types have been described [191]. HPVs cause a range of epithelial hyperplastic lesions and can be classified into two groups: mucosal and cutaneous. These groups can be further divided into low- and high-risk, depending on the associated lesion’s propensity for malignant progression.
The cutaneous HPVs 5 and 8 may be considered high-risk, as they are associated with a rare, genetically determined skin disease, epidermodysplasia verruciformis (EV). These lesions can progress to skin cancers particularly in sun-exposed areas of the body. Skin cancer in EV patients was the first HPV cancer type that was associated with HPV infections [192–196]; More recent studies have revealed that HPV5 and 8 as well as related cutaneous HPVs may contribute to the development of non-melanoma skin cancers (NMSC), particularly in immunecompromised patients [197, 198]. Infections with cutaneous HPVs appear extremely frequently in the general population and some of these viruses, even the presumed high-risk HPV5, may be part of the normal “flora” of the skin as they can be detected in follicles of plucked hair [199, 200]. The mechanistic contributions of cutaneous HPVs and their interplay with other co-factors that may result in NMSC development remains the subject of intense study (reviewed in [201]). It has been shown that E6 proteins of cutaneous HPVs can target the proapoptotic Bcl-2 family member, Bak, for degradation. Bak plays an important role in signaling apoptosis in response the UV irradiation and hence it has been postulated that cutaneous HPV expressing cells may be less prone to undergo apoptosis after UV induced DNA damage [202]. This may lead to survival and expansion of HPV containing cells with extensive genomic aberrations, which may contribute to transformation. Some cutaneous HPVs exhibit bona fide transforming activities in cultured cells [203–206]. Moreover, skin hyperplasia and skin tumors develop in transgenic mice that express early region genes of cutaneous HPVs [207, 208]. HPV genomes are often found in only a subset of the cancer cells suggesting that either cutaneous HPVs may contribute to initiation of carcinogenesis but are not be required for maintenance of the transformed phenotype, or that they contribute to transformation through non cell autonomous mechanisms (reviewed in [201]).
The concept of low-risk and high-risk HPVs has been most clearly established with the mucosal HPVs. The low-risk mucosal HPVs, such as HPV6 and 11, cause genital warts, whereas the high-risk mucosal HPVs, such as HPV16 and 18, cause squamous intraepithelial lesions that can progress to invasive squamous cell carcinoma. HPV is also associated with oral and other anogenital malignancies, however, it is most commonly associated with cervical cancer; in fact, over 99% of all cervical cancers are associated with high-risk HPV infections. Both epidemiological and molecular evidence strongly supports the link between infection with high-risk HPVs and the development of cervical cancer. Nonetheless, the incidence of malignant progression of high-risk HPV associated lesions is relatively low; malignant progression usually occurs with other risk factors, such as decreased immune function, and/or after a long latency period after other genomic alterations in the host cell DNA have occurred. Smoking and prolonged use of birth control pills have also been implicated as risk factors for progression (reviewed in [209]). Maybe most intriguing, Fanconi anemia (FA) patients often develop squamous cell carcinomas at anatomical sites that are susceptible to HPV infections. It has been reported that oral carcinomas in FA patients are more frequently HPV positive than in the general population [210]. Hence, certain aberrations in the FA pathway may predispose to malignant progression of HPV associated lesions. Interestingly, the ability of HPV E7 to induce genomic instability is increased in FA derived cells, thus providing a potential mechanistic rationalization for these findings [211]. The molecular mechanisms by which high-risk HPV causes cervical cancer have been studied extensively, and numerous viral and host interactions that may contribute to transformation and malignancy have been described (reviewed in [27]).
During carcinogenic progression the HPV genome frequently integrates into a host cell chromosome and, as a result, the viral oncoproteins, E6 and E7, are the only viral proteins that are consistently expressed in HPV positive cervical carcinomas. Viral genome integration is a terminal event for the viral life cycle and often results in deletion and/or mutation of other viral genes. Most significantly, expression of the transcriptional repressor E2 is often lost during integration. Moreover, the viral E6/E7 genes are expressed from spliced mRNAs that contain cellular sequences which results in increased mRNA stability. Hence, HPV genome integration is often associated with higher, dysregulated E6/E7 expression [212]. Persistent expression of E6 and E7 is necessary for maintenance of the transformed phenotype of cervical carcinoma cells [213, 214]. The expression of high-risk HPV E6 and E7 immortalizes primary human keratinocytes [215, 216], and when grown as organotypic “raft” cultures these cells display histopathological hallmarks of high-grade premalignant lesions [217]. However, these cells remain non-tumorigenic at low passages after immortalization; tumorigenic progression is not observed until long-term passaging in vitro or after the transduction of an additional oncogene [218–220]. Moreover, transgenic mice with expression of HPV E6 and E7 require low-dose estrogen for the development of cervical cancer [221]. This situation mimics the progression of high-risk HPV-positive cervical lesions, a process that occurs with a low frequency and requires the acquisition of additional host cellular mutations (reviewed in [222]).
HPVs have transforming properties in a number of rodent cell lines, and the transforming potential correlates with their clinical classification as high-risk and low-risk. Mutational analyses have revealed that E7 encodes the major transforming function [223–226], while E6 does not score as a major transforming activity in most assays. High-risk HPV E7 also scores in the classical oncogene cooperation assay in baby rat kidney cells [223, 227], whereas E6 can cooperate with ras in baby mouse kidney cells [228]. High-risk HPVs can extend the life span of primary human genital epithelial cells, and E6 and E7 are necessary and sufficient for this activity [226, 229–233]
As mentioned previously, the transforming activities of the high-risk E6 and E7 oncoproteins is related to their ability to associate with and dysregulate cellular regulatory protein complexes, most notably p53 and pRB (reviewed in [155]). As p53 and pRB normally control cellular proliferation, differentiation, and apoptosis, the abrogation of their normal biological activities places such a cell at a risk of malignant progression.
In addition, high-risk HPV E6 and E7 expressing cells have a decreased ability to maintain genomic integrity [234]. The high-risk HPV E7 oncoprotein acts as a mitotic mutator and induces multiple forms of mitotic abnormalities, including anaphase bridges, unaligned or lagging chromosomes, and most notably multipolar mitoses [235]. Multipolar mitoses are histopathological hallmarks of high-risk HPV associated cervical lesions and cancer [236] and are caused by the ability of high-risk HPV to uncouple centrosome duplication from the cell division cycle [237, 238]. Hence, HPV E6/E7 oncoproteins mechanistically contribute to initiation and progression of cervical cancer (Figure 3).
Figure 3.

Schematic depiction of the major biological activities that contribute to the transforming activities of high-risk mucosal HPVs. See text for details.
5.2.2. Hepatitis B Virus (HBV)
It is estimated that over 400 million people worldwide are chronic carriers of HBV [239]. The vast majority of infections are asymptomatic, and the non-cytopathic HBV [240] does not cause a significant immune response after the initial infection, presumably at least in part because HBV entry and expansion do not induce the expression of any cellular genes [142, 241]. Nonetheless, HBV is a major etiological factor in the development of HCC, as 15–40% of infected individuals will develop chronic active hepatitis (CAH) which can in turn lead to cirrhosis, liver failure, or HCC [242, 243]. This development is accelerated by the exposure to environmental carcinogens including aflatoxin B, cigarette smoke, and alcohol [146]. CAH is characterized by liver cell necrosis, inflammation, and fibrosis, and it is believed that the resulting cirrhosis may eventually lead to HCC due to the fact that the rapid regeneration of hepatocytes following constant necrosis may lead to the accumulation of mutations and the subsequent selection of cells with a carcinogenic phenotype [244]. Indeed, patients with cirrhosis are more likely to develop HCC than patients without cirrhosis [245, 246]. While it appears that both CAH and cirrhosis contribute to the development of liver carcinogenesis, there is also evidence, such as the correlation between serum HBV DNA level and risk of HCC [247], suggesting a direct oncogenic contribution of HBV to the carcinogenic process. Therefore, it appears that the cause of HBV-associated HCC is a combination of HBV encoded oncogenic activities along with the synergistic effects of chronic inflammation.
HBV has a circular, partially double-stranded, DNA genome with four overlapping open reading frames (ORFs) that encode for the envelope (preS/S), core (preC/C), polymerase, and X proteins [248]. Like retroviruses, the replication of HBV is dependent on reverse transcription; unlike retroviruses, integration of the viral genome into the host chromosome is not necessary for viral replication but does allow for persistence of the viral genome (reviewed in [249]). The integration event precedes tumor development, as the HBV genome is often found integrated in the host chromosome of patients with both CAH and HCC [250, 251]. HBV integration is a dynamic process, as chronic inflammation together with increased proliferation of hepatocytes may result in rearrangements of integrated viral and adjacent cellular sequences [245]. Moreover, the integration event can result in chromosomal deletions and transpositions of viral sequences from one chromosome to another [252, 253]; consequently, HBV integration may result in genomic instability [254, 255] as well as activation of proto-oncogenes [256–260]. However, integration of the HBV genome is not a necessary prerequisite for malignant progression as approximately 20% of patients with HBV-associated HCC do not display evidence of integration [250].
Examination of viral DNA sequences present in HCC has provided insight into additional oncogenic mechanisms of HBV. Sequences encoding the HBV X protein (HBx) and/or truncated envelope PreS2/S viral proteins are expressed in the majority of HCC tumor cells. Additionally, a novel viral hepatitis B spliced protein (HBSP) has been identified in HBV-infected patients [261]. However, the mere expression of such proteins does not confirm their role in HCC development and further studies are necessary to determine their potential contributions to HCC development.
HBx is a 154 amino acid multifunctional regulatory protein that is highly conserved among all of the mammalian hepadnaviruses [262], indicating that it likely plays an important role in the viral life cycle. The expression of HBx is maintained throughout all stages of carcinogenesis, including in cells with integrated HBV genomes [263–268]. Studies of HBx in cultured cells and transgenic animals support a role for HBx in transformation and have demonstrated that HBx functions as a regulatory protein for viral replication and, in the case of woodchuck hepatitis virus (WHV), is required for efficient infectivity [269–272]. In addition, in vitro and in vivo studies indicate that HBx plays an important role in the control of cell proliferation and viability [273–275]. The findings from studies of the biological functions of HBX can be summarized as follows: 1) The HBx protein acts on cellular promoters via protein-protein interactions and exhibits pleiotropic effects that modulate various cell responses to genotoxic stress, protein degradation, and signaling pathways (reviewed in [276]), ultimately affecting cell proliferation and viability [277, 278]. Specifically, HBx stimulates signal transduction pathways such as MAPK/ERK and can also upregulate the expression of genes such as c-Myc, c-Jun, NF-κB, AP-1, Ap-2, RPB5 subunit of RNA polymerase II, TATA binding protein, and CREB [279]. 2) HBx regulates proteasomal function [280–282]. 3) HBx affects mitochondrial function [283, 284]. 4) HBx protein modulates calcium homeostasis [285, 286]. 5) Expression of HBx causes genomic instability [287].
In addition to HBx, the HBV genome encodes a second group of regulatory proteins: the PreS2 activators [large surface proteins (LHBs) and truncated middle surface proteins (MHBst)] [288–291]. More than one-third of HBV-integrates in HBV related HCC encode functional MHBst transactivators [292, 293], supporting the biological significance of PreS2 activators. The PreS2 activators are derived from the HBV surface gene ORF, which consists of a single open reading frame divided into three coding regions, preS1, preS2, and S. Large (LHBs; preS1+preS2+S), middle (MHBs; preS2+S), and small (SHBs; S) envelope glycoproteins can be synthesized through alternate translational initiation [288, 290, 291]. The LHBs and MHBst display transactivation activities; their transcriptional activator function is based on the cytoplasmic orientation of the PreS2 domain. PreS2 activators upregulate COX-2 and cyclin A and induce cell cycle progression [294]. Consistent with this notion, transgenic mice expressing MHBst in the liver displayed increased hepatocyte proliferation rate and an increased occurrence of liver tumors [295].
In addition to sequences encoding for HBx and truncated envelope PreS2/S viral proteins, a novel viral hepatitis B spliced protein (HBSP) has been identified in HBV-infected patients [261]. The spliced HBV RNA encoding for HBSP can be reverse transcribed and encapsidated in defective HBV particles or expressed as HBSP [261]. HBSP induces apoptosis without cell-cycle block in an in vitro tissue culture model, and HBSP antibodies are present in the serum of 45% of chronic hepatitis patients [296]. Moreover, there seems to be a correlation between the presence of HBSP antibodies, viral replication and liver fibrosis [296].
HBV-associated liver carcinogenesis is viewed a multi-factorial process (Figure 4). The integration of the HBV genome into the host chromosome at early stages of clonal tumor expansion has been demonstrated to both affect a variety of cellular genes as well as exert insertional mutagenesis, while chronic liver inflammation confers the accumulation of mutations in the host genome. Additionally, HBV encoded HBx, PreS2 activators, and HBSP may exert oncogenic functions. However, exactly how these viral factors contribute to higher risks of HCC development remains unclear. Further studies are necessary to reveal the molecular mechanisms underlying HBV-associated HCC development. Moreover, since host genomic background plays a role in the final disease outcome, an evaluation of the genetic factors in both the host and viral genome that cause a predisposition to hepatocarcinogenesis will help elucidate the carcinogenic mechanisms involved in HCC development.
Figure 4.
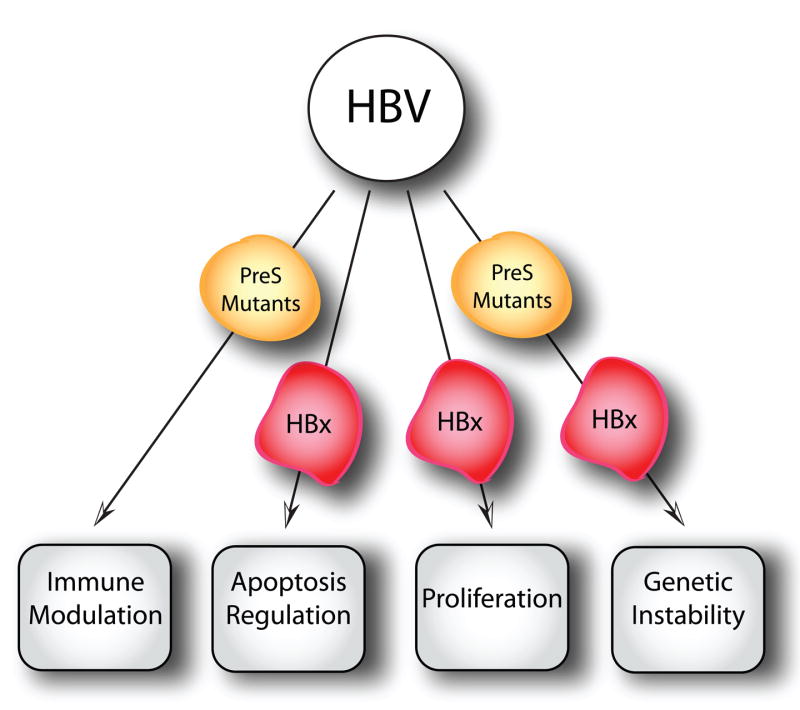
Schematic depiction of the major biological activities that contribute to the transforming activities of HBV. See text for details.
5.2.3. Epstein-Barr Virus (EBV)
EBV is a ubiquitous double-stranded DNA virus of the γherpesviruses subfamily of the lymphocryptovirus (LCV) genus. Worldwide, more than 95% of the population is infected with EBV [297, 298]; the majority of EBV infections occurs during childhood without causing overt symptoms. Post adolescent infection with EBV frequently results in mononucleosis, a self-limiting lymphoproliferative disease. EBV infects and replicates in the oral epithelium, and resting B lymphocytes trafficking through the oral pharynx become latently infected. Infected B lymphocytes resemble antigen activated B cells, and EBV gene expression in these cells is limited to a B cell growth program, termed Latency III, that includes LMP1, LMP2a/b, EBNAs -1, -2, -3a-3b, -3c, and –LP, miRNAs, BARTs, and EBERs. These cells are eliminated by a robust immune response to EBNA3 proteins, resulting in Latency I, a reservoir of latently infected resting memory B cells expressing only EBNA1 and LMP2. The differentiation of memory cells to plasma cells results in reactivation of the replication phase of the viral life cycle that includes expression of latency III gene products. In addition, there is likely another amplification step by re-infection of the epithelial cells followed by shedding virus in the saliva to the next host.
All phases of the EBV life cycle are associated with human disease. In immunecompromised individuals, infected cells increase in number and eventually B-cell growth control pathways are activated, inducing transformation and leading to malignancies such as NPC, BL, post-transplant lymphomas, and gastric carcinomas [3]. EBV-associated malignancies follow distinct geographical distributions and occur at particularly high frequency in certain racial groups, indicating that host genetic factors may influence disease risk [299, 300]. EBV encodes several viral proteins that have transforming potential, including EBV latent membrane protein 1 and 2 (LMP1 and LMP2) and EBV nuclear antigen 2 and 3 (EBNA2 and EBNA3). LMP1 can transform a variety of cell types, including rodent fibroblasts [301], and is essential for the ability of EBV to immortalize B cells [302]. The multiple transmembrane-spanning domains and the carboxyl terminus of LMP1 can interact with several tumor necrosis factor receptor associated factors (TRAFs) [303, 304]; this interaction results in high levels of activity of NF-κB, Jun, and p38 in LMP1-expressing epithelial and B cells [305–307]. Through NF-κB, LMP1 provides survival signals by inducing Bcl-2 family members, c-FLIP, c-IAPs, and adhesion molecules [308]. LMP1 also upregulates the expression of numerous anti-apoptotic and adhesion genes and activates the expression of IRF-7 [309], matrix metalloproteinase-9 (MMP-9) and fibroblast growth factor-2 (FGF-2) [310]. A second viral membrane protein, LMP2, is dispensable for transformation of naïve B cells but is required for transformation of post-germinal center B cells. LMP2 interacts with Lyn and Syk to mimic B-cell-receptor (BCR) signaling, including activation of the PI3K/AKT survival pathway [311].
Other viral genes involved in transformation include EBNA2 and EBNA3. EBNA2, one of the first latent proteins to be detected after EBV infection together with EBNA-LP, is a promiscuous transcriptional activator of both cellular and viral genes [312, 313] and is essential for B-cell transformation [314]. EBNA3A, 3B, and 3C are hydrophilic nuclear transcriptional regulators. EBNA3A and EBNA3C are essential for B-cell transformation in vitro, whereas EBNA3B is dispensable [315]. All three EBNA3 proteins can suppress EBNA2-mediated transactivation [316].
An excellent cell culture model system exists for the study of EBV. In vitro infection of human peripheral blood B cells results in the long-term growth of EBV transformed lymphoblastoid cell lines (LCLs). These cell lines express Latency III gene products which coopt cellular pathways to effect cell growth. LMP1 mimics CD40 to activate NF-κB, LMP2 mimics the B cell antigen receptor, and EBNA-2, -LP, -3A, -3B, and -3C mimic activated Notch. These pathways are constitutively active and drive proliferation through a normal cell cycle cascade. Moreover, there are no additional mutations to the RB or p53 pathways. Finally, if EBV signals are removed, the cells stop proliferating; when LMP1 or EBNA2 expression is restored, the cells begin to proliferate again. A summary of the role of EBV in carcinogenesis is depicted on Figure 5.
Figure 5.

Schematic depiction of the major biological activities that contribute to the transforming activities of EBV. See text for details.
5.2.4. Kaposi’s Sarcoma-associated herpesvirus (KSHV)/Human Herpes Virus 8 (HHV8)
HHV8, also known as KSHV, is a recently discovered [18] double-stranded human rhadinovirus of the γ-herpesvirus subfamily and, like other γ-herpesviruses, establishes lifelong latency in B cells. KSHV is associated with all forms of KS, primary effusion lymphomas (PELs), and multicentric Castleman’s disease (MCD) [18, 317–319]. The neoplastic potential of KSHV, especially in immunecompromised individuals, is well established: epidemiological studies link KSHV to human malignancies [3], KSHV transforms endothelial cells [320], and KSHV encoded transforming genes have been identified. The current model of KSHV-induced malignancy involves a combination of proliferation, survival, and transformation mediated by latently expressed viral proteins together with a paracrine mechanism that is exerted directly or indirectly by the lytically expressed v-cytokines and viral G-protein coupled receptor (vGPCR) (Figure 6). HIV-1 infected individuals are at the highest risk for developing KS. Interestingly, KS lesions and tumors appear to regress in patients who receive Highly Active Antiretroviral Therapy (HAART), suggesting that KSHV gene expression may be insufficient to initiate or maintain transformation [321, 322].
Figure 6.
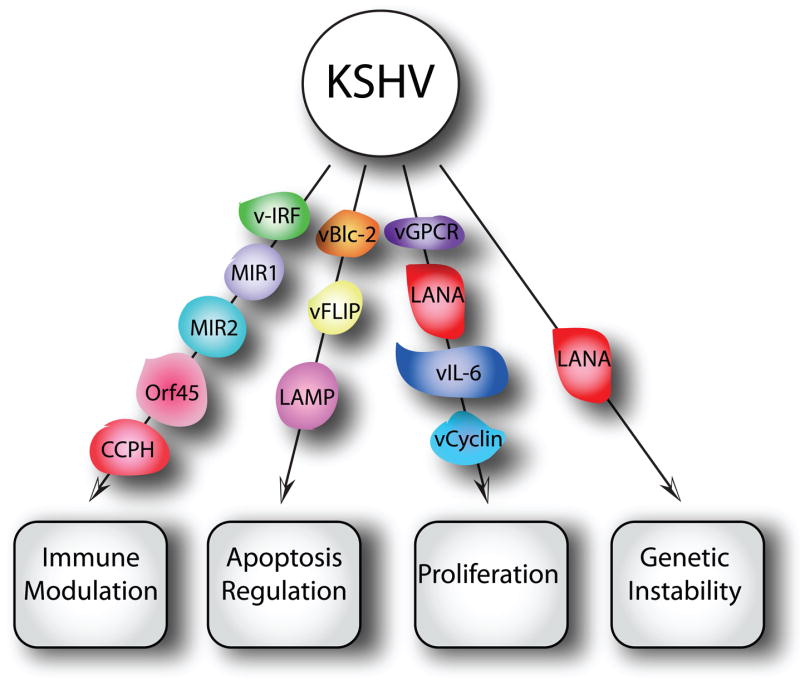
Schematic depiction of the major biological activities that contribute to the transforming activities of HHV-8/KSHV. See text for details.
KS is an angioproliferative disease involving numerous angiogenic factors, endothelial cell (EC) growth factors, and pro-inflammatory cytokines, including viral factors such as vIL-6, vCCL-1, 2, and 3, and viral G protein-coupled receptor (vGPCR). VEGF is induced by vIL6 and subsequently promotes angiogenesis, and mouse cell lines that stably express vIL-6 and secrete high levels of VEGF and are tumorigenic in nude mice [323]. A notable property of the v-chemokines are their pro-angiogenic activities [324, 325]. Finally, vGPCR may contribute to KSHV-associated neoplasia by inducing and sustaining cell proliferation [326–330].
Latency expressed KSHV proteins that promote cell proliferation and survival and thus may contribute to cellular transformation include the ORF73–71 locus-encoded latency associated antigen (LANA, ORF73), viral cyclin (v-cyclin, ORF72), viral FLICE inhibitory protein (vFLIP, ORF71), viral interferon regulatory factor 1 (vIRF-1), and the Kaposin/K12 gene. LANA stimulates cellular proliferation and survival [331, 332], v-cyclin induces cell cycle progression [333], vFLIP and vIRF3 mediate pro-survival signaling [334, 335], and kaposins induce cytokine expression and cell growth [336].
In addition to the viral cytokines, vGPCR, and the five latency genes mentioned, there are two KSHV-encoded constitutive signaling membrane proteins, variable ITAM-containing protein (VIP) and latency associated membrane protein (LAMP), that play a role in KSHV-associated malignancies. VIP is encoded by the K1 ORF and can transform rodent fibroblasts, which, when injected into nude mice, induce multiple and disseminated tumors [337]. VIP can also functionally substitute for the saimiri transformation protein (STP) of herpesvirus saimiri (HVS) to induce lymphomas in common marmoset monkeys and transgenic animals expressing VIP develop lymphomas and sarcomas [338]. In addition to its direct transforming functions, VIP can induce angiogenic factors and inflammatory cytokines [116]. LAMP, which is encoded by the K15 ORF, exhibits potential mitogenic and survival signaling via Src-family kinases and NF-κB activation and may promote cell survival via interaction with the Bcl-2 related anti-apoptotic protein HAX-1 [339].
Finally, KSHV also contains several immune-evasion genes, including MIR1, MIR2, vIRFs, Orf45, and complement control protein homolog (CCPH). The KSHV K3 and K5 proteins, MIR1 and MIR2, downregulate MHC I expression, thus inhibiting viral antigen presentation. The KSHV vIRFs and Orf45 inhibit the host interferon response, while CCPH inhibits complement-mediated lysis of infected cells (reviewed in [327, 340]). Together, these immune evasion proteins ensure life-long viral persistence in the host, and consequently contribute to KSHV-associated pathogenesis.
6. Viruses Implicated in Human Cancers
In addition to the above viruses and cancers, there also exist a number of other cancers that may have an infectious etiology. A causal role for these viruses in human malignancies remains to be fully addressed; current knowledge regarding these viruses and their potential role in human cancer will be summarized in the following sections.
6.1. Polyomaviruses
The Polyomaviridae family is a group of non-enveloped, small double-stranded DNA viruses that have been isolated from humans, monkeys, rodents, and birds. The first polyomavirus to be discovered, mouse polyoma virus [9, 341], is one of the most aggressive carcinogenic viruses and causes tumors in almost every tissue (poly-oma) of susceptible mouse strains [342, 343]. Although it does not cause malignancies in humans, mouse polyoma virus has been thoroughly studied and has been instrumental in the establishment of systems for the study of numerous eukaryotic cellular processes [344]. The second polyomavirus discovered, SV40, was isolated as a contaminant of early batches of the polio vaccine, which was produced in African green monkey kidney cells [39]. SV40 naturally infects the rhesus monkey where it establishes a low-level infection and persists in the kidneys without any noticeable affects. SV40 is closely related to two human polyomaviruses, BKV and JCV, which were discovered in 1971 from the urine of a renal transplant patient [40] and the brain of a patient with progressive multifocal leukoencephalopathy [42], respectively. Infection with BKV and JCV is widespread in humans and is usually subclinical. However, in immunesuppressed patients, BKV is associated with renal nephropathy [345, 346] and JCV can cause progressive multifocal leukoencephalopathy (PML) [347, 348]. More recently two additional novel putative human polyomaviruses, termed KI and WU, have been isolated [349, 350].
SV40, BKV, and JCV can all transform cells in vitro and induce tumors in rodents; additionally, the injection of polyomavirus transformed cells in animal models or the ectopic expression of polyomavirus regulatory proteins in transgenic animals also induces tumors (reviewed in [40, 41, 351]). Together, these findings demonstrate the oncogenic potential of the polyomaviruses. However, the association of these polyomaviruses with human malignancy remains controversial.
A number of studies have implicated SV40 in a range of human cancers, including mesothelioma, osteosarcoma, non-Hodgkin lymphoma (NHL), and a variety of childhood brain tumors. On the other hand, other studies have failed to demonstrate an association of SV40 with human cancer, and the question of whether the release of SV40 into the human population through the polio vaccine contributed to the development of human cancers remains a contentious issue. Although in the 1950s approximately 100 million people were exposed to SV40 through the polio vaccine in the US alone, there is currently no evidence to suggest that SV40 infection is widespread among the population or that there is an increase in tumor burden among individuals who received the contaminated vaccine. Recently, studies have suggested that flawed detection methods may account for many of the positive correlations of SV40 with human tumors. In summary, the studies performed to date have failed to provide conclusive proof implicating SV40 as a human pathogen [352, 353].
Like SV40, a role for BKV and JCV in human tumors has been suggested, however, no conclusive proof exists that either virus directly causes or acts as a cofactor in human cancer. The search for a correlation between BKV and JCV and human cancer is complicated due to the fact that both viruses are ubiquitous in the population. With respect to JCV, there have been reports of JCV variants in a variety of human brain tumors; these studies have not, though, demonstrated any causal effects (reviewed in [354]). There is also correlative data regarding the potential role of BKV in human cancer. BKV persistently infects epithelial cells in the urinary tract, and some studies have linked BK virus infections to prostate cancer [355].
6.2. Adenoviruses
Members of the Adenoviridae family cause lytic and persistent infection in a range of mammalian and avian hosts. In humans, more than 50 different adenovirus serotypes have been described. They can be divided into six species, designated A–F [356]. Generally, clinically inapparent infection with these viruses occurs during early childhood, and tonsillar lymphocytes or peripheral blood lymphocytes remain persistently infected [357, 358]. Although certain Ad serotypes are highly transforming in cell culture and in animal models, early studies indicated that these viruses may not associated with human cancers [359–361]. This issue has been recently readdressed, however, and one study reported detection of adenovirus DNA in pediatric brain tumors [362]. However, a possible causal relationship of Ad infection with the pathogenesis of malignant brain tumors still remains unclear.
The transforming and oncogenic potential of adenoviruses has been traditionally ascribed to the E1A and E1B oncoproteins, which are similar to SV40 TAg and high-risk HPV E7/E6 target pRb and p53, respectively. However, more recent studies have suggested that, at least with some serotypes, E4 ORFs may contribute to cellular transformation [363], potentially through a “hit-and-run” mechanism [364]. Hence, the possible contribution of adenovirus infections to some aspects of human tumor development may need to be carefully re-evaluated.
6.3. Human Endogenous Retroviruses (HERVs)
HERVs are sequences within the genome that resemble infectious retroviruses (reviewed in [43, 44, 365]) that result from ancestral exogenous retroviral infections that became incorporated into the germ line DNA. Today, HERVs constitute ~8% of genome [366] and possess a similar genomic organization to exogenous complex retroviruses such as human immunodeficiency virus (HIV) and HTLV. Evolutionary pressure has ensured the inactivation of HERVs, leading to the idea that HERVs are merely “junk DNA”. However, recent evidence suggests that some HERVs may have both physiological and pathological roles, including roles in human malignancy.
While the exact role(s) of HERVs has yet to be fully elucidated, some studies have suggested that HERVs might play a role in carcinogenesis. Specifically, some HERVs have been implicated in human malignancy, due mainly to the increased expression of the HERV mRNA [367], functional protein [368], and retrovirus-like particles [369] in certain cancers. HERVs may also be linked to the generation of new promoters [370] or the activation of proto-oncogenes [371].
Current studies focus on the association of HERV-K with a number of cancers, including germ cell tumors, in particular seminomas [368, 372], breast cancer, myeloproliferative disease, ovarian cancer [373], melanoma [374, 375], and prostate cancer [376]. Some seminomas have been reported to express HERV-K proteins and sometimes release defective viral particles [369]. HERV-K proteins Rec and Np9 can bind the promyelocytic leukemia zinc finger (PLZF) protein [377], and hence it has been suggested that HERV-encoded proteins may contribute to the carcinogenic process.
None of these studies, however, provides compelling evidence that HERV-K expression contributes to tumorigenesis and it is equally possible that HERV gene expression may merely represent a corollary of cell transformation.
Recently, it has been discovered that recurrent chromosomal translocations importantly contribute to the development of human solid tumors. Interestingly, some of these translocations result in HERV-K regulatory sequences being placed upstream of the coding sequences of ETS transcription factor family members. Since HERV-K regulatory sequences are androgen responsive, such translocations cause aberrant, androgen-induced expression of these ETS transcription factors [376]. This is an exciting finding as the result of such translocations is conceptually similar to insertional mutagenesis, where retroviruses contribute to carcinogenesis by integrating in the vicinity of cellular proto-oncogenes, thereby causing their aberrant expression.
6.4. Human Mammary Tumor Virus (HMTV)/Pogo Virus
The idea of the existence of a human breast cancer virus followed Bittner’s demonstration that a virus, MMTV, can cause breast cancer in mice [378]. While studies in the last 7 decades have failed to provide compelling evidence proving this model, a possible viral etiology for some breast cancers remains an active and controversial area of research. A number of reports have suggested that an MMTV-related virus, human mammary tumor virus (HMTV), sometimes also referred to as the Pogo virus, may be associated with human breast cancers (reviewed in [45]).
A 660-bp sequence similar to the MMTV env gene was reportedly detected in 38% of American women’s breast cancers [379]; subsequent polymerase chain reaction (PCR) amplifications detected other gene segments homologous to MMTV [380]. Moreover, env and LTR HMTV genes have been found inserted into several chromosomes [381], reminiscent of MMTV, which induces oncogenesis via insertional mutagenesis. The entire HMTV genome, which contains hormone response elements, has also been detected in fresh breast cancers [381]; additionally, primary breast cancer cells produce HMTV particles in vitro [382]; additionally, HMTV particles with morphogenic and molecular characteristics similar to MMTV were reported in primary breast cancer cells [382]. However, other groups have been unable to detect such sequences in breast cancers [383].
Since many HERVs are similar to MMTV, it is not clear whether HMTV represents an infectious entity, and/or if it can be acquired from infected mice as has been suggested by an epidemiological study, which reported that regions where Mus domesticus infestations are prevalent have the highest incidence of human breast cancer [384]. In possible support of an infectious etiology, human MMTV receptor-related proteins have been cloned [385]. Moreover, it has recently been reported that MMTV replicates rapidly and successfully in human breast cancer cells [386].
Even if HMTV-like sequences are indeed expressed in some human breast cancers, there is presently no compelling evidence that unambiguously supports a mechanistic contribution of such viruses to breast cancer development.
6.5. Xenotropic murine leukemia virus-related virus (XMRV)
The finding that the familial prostate cancer susceptibility locus Hpc1 is linked to mutations in the structural gene for RNase L [387], an effector in the interferon induced innate viral response [388], led to the suggestion that inherited defects in RNase L might allow for infection with an oncogenic virus, thus leading to the development of PC. Xenotropic murine leukemia virus-related virus (XMRV) is a recently described gamma retrovirus that was discovered using PCR cloned codas from tumors from PC patients with a mutation in RNase L [389]. XMRV protein was detected in the stoma and hematopoetic cells of the prostate tumors, but was not detected in the actual cancer cells [389]. Therefore, it is unclear whether XMRV may be causally associated with prostate cancer. However, it is possible that XMRV may contribute to tumorigenesis through an indirect mechanism.
6.6. Torque teno virus (TTV)
Torque teno virus (TTV) is a single-stranded circular DNA virus of the Circoviridae family and was first found in a patient with non-A-E-hepatitis [46]. Since its initial discovery, a large number of TTV types and subtypes that display substantial genomic variation have been described [390–395]. TTV is acquired in early childhood [396, 397] and remains prevalent in adults [391, 398, 399]. Numerous attempts to link TT viruses to pathogenicity, particularly liver diseases, have thus far been unsuccessful [400–402]. However, one study did suggest that a high TTV load may have prognostic significance in HCV-associated liver disease, but the issue of whether high TTV load mediates HCV-associated disease progression remains to be addressed. Another study demonstrated a relatively high prevalence of TTV-related DNA sequences in human cancers, specifically gastrointestinal cancer, lung cancer, breast cancer, and myeloma [403]. No causal relationship between TTV infection and carcinogenesis can be drawn from this study, as normal control tissues were not included.
7. Concluding Remarks/Perspectives
The study of virus-associated cancers has provided many critical insights into key mechanisms of carcinogenesis. The initial work that showed that some animal cancers were caused by retroviruses firmly established the concept that cancers can be caused by infectious agents. Analyses of retroviral integration sites and retrovirally transmitted transforming genes yielded a treasure trove of information on oncogenes and tumor suppressors that were later found to be similarly mutated in human malignancies. While there is no firm evidence that human tumors are caused by retroviral transmission of activated oncogenes, these studies, nevertheless, provided much of the foundation of modern oncology.
The main difference between these early studies with transforming animal retroviruses and human tumor viruses is that oncogenes of human tumor viruses are viral genes, rather than mutated versions of cellular genes that were accidentally assimilated during the viral replication cycle. These human tumorvirus oncogenes play central roles in viral life cycles and their oncogenic potential is a manifestation of these activities.
Some viruses, most notably the high-risk HPVs, play essential roles in the initiation as well as progression of cancers and continued expression of their viral transforming activities is necessary for the maintenance of the transformed phenotype. This concept is supported by studies that showed that the major targets of the HPV E6 and E7 oncoproteins, the p53 and pRB tumor suppressors, are retained in a dormant, yet functional state, in HPV positive cervical cancer cell lines, whereas the pRB and p53 tumor suppressor sustained mutations in HPV negative cervical carcinoma lines [188]. Hence, these proteins are predicted to be excellent targets for therapy. This concept is impressively supported by numerous studies that showed that cervical cancer cell lines that are manipulated to lose E6/E7 oncogene expression promptly undergo growth arrest, senescence, and/or apoptosis and that this is related to reactivation of the p53 and pRB tumor suppressor pathways [213].
Since HPV E6 and E7 lack intrinsic enzymatic activities, the development of E6/E7 specific antivirals is difficult. HPV E6 and E7, like many viral oncoproteins, function by associating with host cellular proteins, including enzymes such as ubiquitin ligases and histone modifying enzymes. It will be important to determine whether targeting such enzymes will have a specific therapeutic benefit [404]. Last, but not least, viral proteins that are consistently expressed in virus-associated tumors are attractive targets for development of therapeutic vaccination strategies.
It will also be interesting to explore whether the alterations in cell signaling networks due to viral oncogene expression renders infected cells uniquely dependent on (“addicted to”) specific cellular signal transduction pathways. The concept of synthetic lethality in yeast predicts that such pathways may be excellent candidates for therapeutic intervention [405]. The availability of genome wide RNAi and cDNA libraries should make such approaches feasible. Given that the pathways targeted by viral oncoproteins are often mutated in non-virus associated cancers, one might predict that such studies may reveal viable therapeutic targets for numerous malignancies.
In some cases, however, the virus may only contribute to one specific step in the carcinogenic process and its continued expression may thus not be necessary for the maintenance of the transformed state. This is sometimes referred to as “hit-and-run” carcinogenesis. In these cases, it is very difficult to conclusively demonstrate that the virus indeed did contribute to the development of a given tumor. The relatively recent realization that some viruses encode proteins that subvert genomic stability lends some mechanistic credence to this model (reviewed in [27]). Accordingly, a virus-infected cell would be significantly more promiscuous for accumulation of genetic aberrations that eventually may lead to tumor formation. One might also imagine that viral proteins that alter the histone code may permanently modify the epigenetic imprint of a host cell and the resulting alterations in gene expression may contribute to cellular transformation.
Another possibility is that viral gene expression is only maintained in some tumor cells. This has been observed with some HPV containing non-melanoma skin cancers [201]. While this is consistent with a “hit-and-run” mechanism, it may also indicate that viral oncogene expression might provide a growth promoting activity indirectly through secreted factors that are necessary for optimal tumor growth.
Conversely, the mere presence of viral sequences in a given cancer type does not necessarily imply that the virus mechanistically contributes to the genesis of the tumor. It may simply represent the fact that tumor cells represent a particularly fertile ground for viral replication. Tumors often contain a larger population of proliferative cells than the surrounding normal tissue and hence might be particularly permissive for viral replication. In addition, they often are subject to decreased surveillance by the immune system. Given the availability of new tools to discover virus sequences in tumors, this issue will represent a major challenge for the future.
Host defense mechanisms play a significant role in modulating viral carcinogenesis. Their protective role is most clearly illustrated by the fact that Epstein-Barr virus in most cases establishes life long asymptomatic latency and tumors arise when immunesurveillance of the host organism is weakened or fails. Similarly, non-melanoma skin cancers associated with cutaneous HPV infections are a frequent complication in organ transplant patients.
However, in other cases the host response to viral infection and/or their cellular consequences significantly contributes to the carcinogenic process. While infection of liver cells with hepatitis viruses does not appear to cause a major immune response to the initial infection it causes extensive inflammation. This causes cell damage and production of genotoxic reactive oxygen species, which in concert with rapid proliferation due to regenerative processes and potentially genomic destabilization due to viral gene expression, may lead to accumulation of mutations that eventually lead to carcinogenic progression.
The infectious etiology of certain cancers affords the unique possibility for prophylactic vaccination. Since vaccination with a weakened live virus is not an option for oncogene carrying viruses, it is necessary to develop “subunit vaccines” consisting of immunogenic recombinant viral proteins. Such a vaccine has been developed for HBV and has proven highly efficacious. HBV vaccines are relatively inexpensive; consequently, they are widely used and have had a remarkable public health impact. Prophylactic vaccines to prevent infections with the most abundant low-risk and/or high-risk HPVs (HPV6, HPV11 and/or HPV16, HPV18) are now commercially available. These vaccines consist of recombinant L1 proteins that form virus-like particles (VLPs), which induce a vigorous type-specific immune response and show great promise (reviewed in [406]). Vaccination has to be before the onset of sexual activity and it will be important to determine how long the protection will last. However, since HPV-induced cervical carcinogenesis is a slow process, it will be several decades before there will be a noticeable decrease in cervical cancer cases due to vaccination [407]. It will also be important to determine whether other high-risk HPVs that are not targeted by the current vaccine preparations will fill the void and become more abundant in the population. This would require development and inclusion of VLPs corresponding to additional HPV types to the vaccine preparations. Perhaps most significant, these vaccines are extraordinarily expensive to produce. Since most cervical cancer cases and deaths occur in medically underserved patients, it is thus unlikely that the population that would profit the most from a prophylactic approach will gain access to these vaccines in the near future.
Acknowledgments
We apologize to those authors whose important contributions could not be appropriately discussed and cited due to space limitations. We thank E. Cahir-McFarland and M. Imperiale for critical comments on parts of this manuscript. The research on viral oncology in the authors’ laboratory is supported by PHS grants CA066980, CA081135; M M-D is supported by PF-07-072-01-MBC from the American Cancer Society. This article is dedicated to the memory of Konrad Lerch.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.zur Hausen H. Viruses in human cancers. Current Science. 2001;81:523–27. [Google Scholar]
- 3.Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–44. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 4.M’Fadyan J, Hobday F. Note on the experimental “transmission of warts in the dog”. J Comp Pathol Ther. 1898;11:341–43. [Google Scholar]
- 5.Ciuffo G. Innesto positivo con filtrato di verruca volgare. Giorn Ital Mal Venereol. 1907;48:12–17. [Google Scholar]
- 6.Ellermann V, Bang O. Experimentelle Leukamie bei Huhnern. Centralbt f Bakt Abt I (orig) 1908;46:595–609. [Google Scholar]
- 7.Rous P. Transmission of a malignant new growth by means of a cell-free filtrate. J Am Med Assoc. 1911;56:198. [PubMed] [Google Scholar]
- 8.Gross L. Susceptibility of newborn mice of an otherwise apparently ‘resistent’ strain to inoculation with leukemia. Proc Soc Exp Biol Med. 1950;73:246–248. [Google Scholar]
- 9.Stewart SE, Eddy BE, Borgese N. Neoplasms in mice inoculated with a tumor agent carried in tissue culture. J Natl Cancer Inst. 1958;20:1223–1243. doi: 10.1093/jnci/20.6.1223. [DOI] [PubMed] [Google Scholar]
- 10.Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet. 1964;i doi: 10.1016/s0140-6736(64)91524-7. [DOI] [PubMed] [Google Scholar]
- 11.Dane DS, Cameron CH, Briggs M. Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet. 1970;1:695–8. doi: 10.1016/s0140-6736(70)90926-8. [DOI] [PubMed] [Google Scholar]
- 12.Durst M, Gissmann L, Ikenberg H, zur Hausen H. A papillomavirus DNA from a cervical carcinoma and its prevalence in cancer biopsy samples from different geographic regions. Proc Natl Acad Sci U S A. 1983;80:3812–5. doi: 10.1073/pnas.80.12.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boshart M, Gissmann L, Ikenberg H, Kleinheinz A, Scheurlen W, zur Hausen H. A new type of papillomavirus DNA, its presence in genital cancer biopsies and in cell lines derived from cervical cancer. Embo J. 1984;3:1151–7. doi: 10.1002/j.1460-2075.1984.tb01944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet. 1981;2:1129–33. doi: 10.1016/s0140-6736(81)90585-7. [DOI] [PubMed] [Google Scholar]
- 15.Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci U S A. 1980;77:7415–9. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshida M, Miyoshi I, Hinuma Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci U S A. 1982;79:2031–5. doi: 10.1073/pnas.79.6.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–62. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 18.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–9. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 19.Cohen SM. In: Microbes and Malignancy: Infection as a Cause of Human Cancers. Parsonnet J, editor. Oxford University Press; Oxford, UK: 1999. pp. 89–106. [Google Scholar]
- 20.Koch R. Vernhandlungen des X Internationalen Medicinischen Congresses. Vol. 1. Berlin: 1890. pp. 35–47. [Google Scholar]
- 21.Evans AS, Mueller NE. Viruses and cancer. Causal associations. Ann Epidemiol. 1990;1:71–92. doi: 10.1016/1047-2797(90)90020-s. [DOI] [PubMed] [Google Scholar]
- 22.Hill AB. The Environment and Disease: Association or Causation? Proc R Soc Med. 1965;58:295–300. [PMC free article] [PubMed] [Google Scholar]
- 23.Hill AB, Hill ID. Bradford Hill’s Principles of Medical Statistics. 12. Edward Arnold; London, UK: 1991. [Google Scholar]
- 24.Giam CZ, Jeang KT. HTLV-1 Tax and adult T-cell leukemia. Front Biosci. 2007;12:1496–507. doi: 10.2741/2163. [DOI] [PubMed] [Google Scholar]
- 25.zur Hausen H. Papillomavirus infections--a major cause of human cancers. Biochim Biophys Acta. 1996;1288:F55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]
- 26.zur Hausen H. Immortalization of human cells and their malignant conversion by high risk human papillomavirus genotypes. Semin Cancer Biol. 1999;9:405–11. doi: 10.1006/scbi.1999.0144. [DOI] [PubMed] [Google Scholar]
- 27.Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78:11451–60. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- 29.Nicholas J. Human herpesvirus 8-encoded proteins with potential roles in virus-associated neoplasia. Front Biosci. 2007;12:265–81. doi: 10.2741/2063. [DOI] [PubMed] [Google Scholar]
- 30.Cathomas G. Kaposi’s sarcoma-associated herpesvirus (KSHV)/human herpesvirus 8 (HHV-8) as a tumour virus. Herpes. 2003;10:72–7. [PubMed] [Google Scholar]
- 31.Pagano JS. Epstein-Barr virus: the first human tumor virus and its role in cancer. Proc Assoc Am Physicians. 1999;111:573–80. doi: 10.1046/j.1525-1381.1999.t01-1-99220.x. [DOI] [PubMed] [Google Scholar]
- 32.Tao Q, Young LS, Woodman CB, Murray PG. Epstein-Barr virus (EBV) and its associated human cancers--genetics, epigenetics, pathobiology and novel therapeutics. Front Biosci. 2006;11:2672–713. doi: 10.2741/2000. [DOI] [PubMed] [Google Scholar]
- 33.Klein E, Kis LL, Klein G. Epstein-Barr virus infection in humans: from harmless to life endangering virus-lymphocyte interactions. Oncogene. 2007;26:1297–305. doi: 10.1038/sj.onc.1210240. [DOI] [PubMed] [Google Scholar]
- 34.Jin DY. Molecular pathogenesis of hepatitis C virus-associated hepatocellular carcinoma. Front Biosci. 2007;12:222–33. doi: 10.2741/2060. [DOI] [PubMed] [Google Scholar]
- 35.Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006;25:3834–47. doi: 10.1038/sj.onc.1209562. [DOI] [PubMed] [Google Scholar]
- 36.Park NH, Song IH, Chung YH. Chronic hepatitis B in hepatocarcinogenesis. Postgrad Med J. 2006;82:507–15. doi: 10.1136/pgmj.2006.047431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kremsdorf D, Soussan P, Paterlini-Brechot P, Brechot C. Hepatitis B virus-related hepatocellular carcinoma: paradigms for viral-related human carcinogenesis. Oncogene. 2006;25:3823–33. doi: 10.1038/sj.onc.1209559. [DOI] [PubMed] [Google Scholar]
- 38.Lee AT, Lee CG. Oncogenesis and transforming viruses: the hepatitis B virus and hepatocellularcarcinoma--the etiopathogenic link. Front Biosci. 2007;12:234–45. doi: 10.2741/2061. [DOI] [PubMed] [Google Scholar]
- 39.Sweet BH, Hilleman MR. The vacuolating virus, S.V. 40. Proc Soc Exp Biol Med. 1960;105:420–7. doi: 10.3181/00379727-105-26128. [DOI] [PubMed] [Google Scholar]
- 40.Gardner SD, Field AM, Coleman DV, Hulme B. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet. 1971;1:1253–7. doi: 10.1016/s0140-6736(71)91776-4. [DOI] [PubMed] [Google Scholar]
- 41.Imperiale MJ. Oncogenic transformation by the human polyomaviruses. Oncogene. 2001;20:7917–23. doi: 10.1038/sj.onc.1204916. [DOI] [PubMed] [Google Scholar]
- 42.Padgett BL, Walker DL, ZuRhein GM, Eckroade RJ, Dessel BH. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet. 1971;1:1257–60. doi: 10.1016/s0140-6736(71)91777-6. [DOI] [PubMed] [Google Scholar]
- 43.Bannert N, Kurth R. Retroelements and the human genome: new perspectives on an old relation. Proc Natl Acad Sci U S A. 2004;101(Suppl 2):14572–9. doi: 10.1073/pnas.0404838101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gifford R, Tristem M. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes. 2003;26:291–315. doi: 10.1023/a:1024455415443. [DOI] [PubMed] [Google Scholar]
- 45.Holland JF, Pogo BGT. Mouse mammary tumor virus-like infection and human breast cancer. Clinical Cancer Research. 2004;10:5647–5649. doi: 10.1158/1078-0432.CCR-04-1234. [DOI] [PubMed] [Google Scholar]
- 46.Nishizawa T, Okamoto H, Konishi K, Yoshizawa H, Miyakawa Y, Mayumi M. A novel DNA virus (TTV) associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochem Biophys Res Commun. 1997;241:92–7. doi: 10.1006/bbrc.1997.7765. [DOI] [PubMed] [Google Scholar]
- 47.Temin HM, Rubin H. Characteristics of an assay for Rous sarcoma virus and Rous sarcoma cells in tissue culture. Virology. 1958;6:669–88. doi: 10.1016/0042-6822(58)90114-4. [DOI] [PubMed] [Google Scholar]
- 48.Kurth R. Oncogenes in retroviruses and cells. Naturwissenschaften. 1983;70:439–50. doi: 10.1007/BF01079610. [DOI] [PubMed] [Google Scholar]
- 49.Bradshaw RA, Prentis S. Oncogenes and growth factors. Elsevier; Amsterdam: 1987. [Google Scholar]
- 50.Varmus HE. The molecular genetics of cellular oncogenes. Annu Rev Genet. 1984;18:553–612. doi: 10.1146/annurev.ge.18.120184.003005. [DOI] [PubMed] [Google Scholar]
- 51.Weinberg RA. The action of oncogenes in the cytoplasm and nucleus. Science. 1985;230:770–6. doi: 10.1126/science.2997917. [DOI] [PubMed] [Google Scholar]
- 52.Weinberg RA, Wigler M. Oncogenes and the molecular origins of cancer. Cold Spring Harbor Press; Cold Sprinh, NY: 1989. [Google Scholar]
- 53.Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature. 1982;297:474–8. doi: 10.1038/297474a0. [DOI] [PubMed] [Google Scholar]
- 54.Nusse R, van Ooyen A, Cox D, Fung YK, Varmus H. Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature. 1984;307:131–6. doi: 10.1038/307131a0. [DOI] [PubMed] [Google Scholar]
- 55.Peters G, Kozak C, Dickson C. Mouse mammary tumor virus integration regions int-1 and int-2 map on different mouse chromosomes. Mol Cell Biol. 1984;4:375–8. doi: 10.1128/mcb.4.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cuypers HT, Selten G, Quint W, Zijlstra M, Maandag ER, Boelens W, van Wezenbeek P, Melief C, Berns A. Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region. Cell. 1984;37:141–50. doi: 10.1016/0092-8674(84)90309-x. [DOI] [PubMed] [Google Scholar]
- 57.van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, Berns A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell. 1991;65:737–52. doi: 10.1016/0092-8674(91)90382-9. [DOI] [PubMed] [Google Scholar]
- 58.Bear SE, Bellacosa A, Lazo PA, Jenkins NA, Copeland NG, Hanson C, Levan G, Tsichlis PN. Provirus insertion in Tpl-1, an Ets-1-related oncogene, is associated with tumor progression in Moloney murine leukemia virus-induced rat thymic lymphomas. Proc Natl Acad Sci U S A. 1989;86:7495–9. doi: 10.1073/pnas.86.19.7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Makris A, Patriotis C, Bear SE, Tsichlis PN. Structure of a Moloney murine leukemia virus-virus-like 30 recombinant: implications for transduction of the c-Ha-ras proto-oncogene. J Virol. 1993;67:1286–91. doi: 10.1128/jvi.67.3.1286-1291.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hicks GG, Mowat M. Integration of Friend murine leukemia virus into both alleles of the p53 oncogene in an erythroleukemic cell line. J Virol. 1988;62:4752–5. doi: 10.1128/jvi.62.12.4752-4755.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gessian A, Yanagihara R, Franchini G, Garruto RM, Jenkins CL, Ajdukiewicz AB, Gallo RC, Gajdusek DC. Highly divergent molecular variants of human T-lymphotropic virus type I from isolated populations in Papua New Guinea and the Solomon Islands. Proc Natl Acad Sci U S A. 1991;88:7694–8. doi: 10.1073/pnas.88.17.7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wong-Staal F, Gallo RC. Human T-lymphotropic retroviruses. Nature. 1985;317:395–403. doi: 10.1038/317395a0. [DOI] [PubMed] [Google Scholar]
- 63.Proietti FA, Carneiro-Proietti AB, Catalan-Soares BC, Murphy EL. Global epidemiology of HTLV-I infection and associated diseases. Oncogene. 2005;24:6058–68. doi: 10.1038/sj.onc.1208968. [DOI] [PubMed] [Google Scholar]
- 64.Matsuoka M, Jeang KT. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer. 2007;7:270–80. doi: 10.1038/nrc2111. [DOI] [PubMed] [Google Scholar]
- 65.Matsuoka M. Human T-cell leukemia virus type I and adult T-cell leukemia. Oncogene. 2003;22:5131–40. doi: 10.1038/sj.onc.1206551. [DOI] [PubMed] [Google Scholar]
- 66.Nerenberg M, Hinrichs SH, Reynolds RK, Khoury G, Jay G. The tat gene of human T-lymphotropic virus type 1 induces mesenchymal tumors in transgenic mice. Science. 1987;237:1324–9. doi: 10.1126/science.2888190. [DOI] [PubMed] [Google Scholar]
- 67.Grassmann R, Dengler C, Muller-Fleckenstein I, Fleckenstein B, McGuire K, Dokhelar MC, Sodroski JG, Haseltine WA. Transformation to continuous growth of primary human T lymphocytes by human T-cell leukemia virus type I X-region genes transduced by a Herpesvirus saimiri vector. Proc Natl Acad Sci U S A. 1989;86:3351–5. doi: 10.1073/pnas.86.9.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grassmann R, Berchtold S, Radant I, Alt M, Fleckenstein B, Sodroski JG, Haseltine WA, Ramstedt U. Role of human T-cell leukemia virus type 1 X region proteins in immortalization of primary human lymphocytes in culture. J Virol. 1992;66:4570–5. doi: 10.1128/jvi.66.7.4570-4575.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pozzatti R, Vogel J, Jay G. The human T-lymphotropic virus type I tax gene can cooperate with the ras oncogene to induce neoplastic transformation of cells. Mol Cell Biol. 1990;10:413–7. doi: 10.1128/mcb.10.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tanaka A, Takahashi C, Yamaoka S, Nosaka T, Maki M, Hatanaka M. Oncogenic transformation by the tax gene of human T-cell leukemia virus type I in vitro. Proc Natl Acad Sci U S A. 1990;87:1071–5. doi: 10.1073/pnas.87.3.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Smith MR, Greene WC. Functional analyses of the type I human T-cell leukemia virus tax gene product. Trans Assoc Am Physicians. 1991;104:78–91. [PubMed] [Google Scholar]
- 72.Yamaoka S, Tobe T, Hatanaka M. Tax protein of human T-cell leukemia virus type I is required for maintenance of the transformed phenotype. Oncogene. 1992;7:433–7. [PubMed] [Google Scholar]
- 73.Rosin O, Koch C, Schmitt I, Semmes OJ, Jeang KT, Grassmann R. A human T-cell leukemia virus Tax variant incapable of activating NF-kappaB retains its immortalizing potential for primary T-lymphocytes. J Biol Chem. 1998;273:6698–703. doi: 10.1074/jbc.273.12.6698. [DOI] [PubMed] [Google Scholar]
- 74.Chu ZL, DiDonato JA, Hawiger J, Ballard DW. The tax oncoprotein of human T-cell leukemia virus type 1 associates with and persistently activates IkappaB kinases containing IKKalpha and IKKbeta. J Biol Chem. 1998;273:15891–4. doi: 10.1074/jbc.273.26.15891. [DOI] [PubMed] [Google Scholar]
- 75.Sun SC, Ballard DW. Persistent activation of NF-kappaB by the tax transforming protein of HTLV-1: hijacking cellular IkappaB kinases. Oncogene. 1999;18:6948–58. doi: 10.1038/sj.onc.1203220. [DOI] [PubMed] [Google Scholar]
- 76.Leachman SA, Tigelaar RE, Shlyankevich M, Slade MD, Irwin M, Chang E, Wu TC, Xiao W, Pazhani S, Zelterman D, Brandsma JL. Granulocyte-macrophage colony-stimulating factor priming plus papillomavirus E6 DNA vaccination: effects on papilloma formation and regression in the cottontail rabbit papillomavirus--rabbit model. J Virol. 2000;74:8700–8. doi: 10.1128/jvi.74.18.8700-8708.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jin DY, Giordano V, Kibler KV, Nakano H, Jeang KT. Role of adapter function in oncoprotein-mediated activation of NF-kappaB. Human T-cell leukemia virus type I Tax interacts directly with IkappaB kinase gamma. J Biol Chem. 1999;274:17402–5. doi: 10.1074/jbc.274.25.17402. [DOI] [PubMed] [Google Scholar]
- 78.Iha H, Kibler KV, Yedavalli VR, Peloponese JM, Haller K, Miyazato A, Kasai T, Jeang KT. Segregation of NF-kappaB activation through NEMO/IKKgamma by Tax and TNFalpha: implications for stimulus-specific interruption of oncogenic signaling. Oncogene. 2003;22:8912–23. doi: 10.1038/sj.onc.1207058. [DOI] [PubMed] [Google Scholar]
- 79.Fu DX, Kuo YL, Liu BY, Jeang KT, Giam CZ. Human T-lymphotropic virus type I tax activates I-kappa B kinase by inhibiting I-kappa B kinase-associated serine/threonine protein phosphatase 2A. J Biol Chem. 2003;278:1487–93. doi: 10.1074/jbc.M210631200. [DOI] [PubMed] [Google Scholar]
- 80.Xiao G, Fong A, Sun SC. Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation. J Biol Chem. 2004;279:30099–105. doi: 10.1074/jbc.M401428200. [DOI] [PubMed] [Google Scholar]
- 81.Uhlik M, Good L, Xiao G, Harhaj EW, Zandi E, Karin M, Sun SC. NF-kappaB-inducing kinase and IkappaB kinase participate in human T-cell leukemia virus I Tax-mediated NF-kappaB activation. J Biol Chem. 1998;273:21132–6. doi: 10.1074/jbc.273.33.21132. [DOI] [PubMed] [Google Scholar]
- 82.Xiao G, Sun SC. Activation of IKKalpha and IKKbeta through their fusion with HTLV-I tax protein. Oncogene. 2000;19:5198–203. doi: 10.1038/sj.onc.1203894. [DOI] [PubMed] [Google Scholar]
- 83.Xiao G, Harhaj EW, Sun SC. Domain-specific interaction with the I kappa B kinase (IKK)regulatory subunit IKK gamma is an essential step in tax-mediated activation of IKK. J Biol Chem. 2000;275:34060–7. doi: 10.1074/jbc.M002970200. [DOI] [PubMed] [Google Scholar]
- 84.Sun SC, Yamaoka S. Activation of NF-kappaB by HTLV-I and implications for cell transformation. Oncogene. 2005;24:5952–64. doi: 10.1038/sj.onc.1208969. [DOI] [PubMed] [Google Scholar]
- 85.Jeang KT. Functional activities of the human T-cell leukemia virus type I Tax oncoprotein: cellular signaling through NF-kappa B. Cytokine Growth Factor Rev. 2001;12:207–17. doi: 10.1016/s1359-6101(00)00028-9. [DOI] [PubMed] [Google Scholar]
- 86.Alexandre C, Charnay P, Verrier B. Transactivation of Krox-20 and Krox-24 promoters by the HTLV-1 Tax protein through common regulatory elements. Oncogene. 1991;6:1851–7. [PubMed] [Google Scholar]
- 87.Alexandre C, Verrier B. Four regulatory elements in the human c-fos promoter mediate transactivation by HTLV-1 Tax protein. Oncogene. 1991;6:543–51. [PubMed] [Google Scholar]
- 88.Fujii M, Niki T, Mori T, Matsuda T, Matsui M, Nomura N, Seiki M. HTLV-1 Tax induces expression of various immediate early serum responsive genes. Oncogene. 1991;6:1023–9. [PubMed] [Google Scholar]
- 89.Fujii M, Sassone-Corsi P, Verma IM. c-fos promoter trans-activation by the tax1 protein of human T-cell leukemia virus type I. Proc Natl Acad Sci U S A. 1988;85:8526–30. doi: 10.1073/pnas.85.22.8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nagata K, Ohtani K, Nakamura M, Sugamura K. Activation of endogenous c-fos proto-oncogene expression by human T-cell leukemia virus type I-encoded p40tax protein in the human T-cell line, Jurkat. J Virol. 1989;63:3220–6. doi: 10.1128/jvi.63.8.3220-3226.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhao LJ, Giam CZ. Human T-cell lymphotropic virus type I (HTLV-I) transcriptional activator, Tax, enhances CREB binding to HTLV-I 21-base-pair repeats by protein-protein interaction. Proc Natl Acad Sci U S A. 1992;89:7070–4. doi: 10.1073/pnas.89.15.7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cox JM, Sloan LS, Schepartz A. Conformation of Tax-response elements in the human T-cell leukemia virus type I promoter. Chem Biol. 1995;2:819–26. doi: 10.1016/1074-5521(95)90088-8. [DOI] [PubMed] [Google Scholar]
- 93.Franklin AA, Kubik MF, Uittenbogaard MN, Brauweiler A, Utaisincharoen P, Matthews MA, Dynan WS, Hoeffler JP, Nyborg JK. Transactivation by the human T-cell leukemia virus Tax protein is mediated through enhanced binding of activating transcription factor-2 (ATF-2) ATF-2 response and cAMP element-binding protein (CREB) J Biol Chem. 1993;268:21225–31. [PubMed] [Google Scholar]
- 94.Kwok RP, Laurance ME, Lundblad JR, Goldman PS, Shih H, Connor LM, Marriott SJ, Goodman RH. Control of cAMP-regulated enhancers by the viral transactivator Tax through CREB and the co-activator CBP. Nature. 1996;380:642–6. doi: 10.1038/380642a0. [DOI] [PubMed] [Google Scholar]
- 95.Iwai K, Mori N, Oie M, Yamamoto N, Fujii M. Human T-cell leukemia virus type 1 tax protein activates transcription through AP-1 site by inducing DNA binding activity in T cells. Virology. 2001;279:38–46. doi: 10.1006/viro.2000.0669. [DOI] [PubMed] [Google Scholar]
- 96.Peloponese JM, Jr, Jeang KT. Role for Akt/protein kinase B and activator protein-1 in cellular proliferation induced by the human T-cell leukemia virus type 1 tax oncoprotein. J Biol Chem. 2006;281:8927–38. doi: 10.1074/jbc.M510598200. [DOI] [PubMed] [Google Scholar]
- 97.Giebler HA, Loring JE, van Orden K, Colgin MA, Garrus JE, Escudero KW, Brauweiler A, Nyborg JK. Anchoring of CREB binding protein to the human T-cell leukemia virus type 1 promoter: a molecular mechanism of Tax transactivation. Mol Cell Biol. 1997;17:5156–64. doi: 10.1128/mcb.17.9.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lenzmeier BA, Giebler HA, Nyborg JK. Human T-cell leukemia virus type 1 Tax requires direct access to DNA for recruitment of CREB binding protein to the viral promoter. Mol Cell Biol. 1998;18:721–31. doi: 10.1128/mcb.18.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bex F, Yin MJ, Burny A, Gaynor RB. Differential transcriptional activation by human T-cell leukemia virus type 1 Tax mutants is mediated by distinct interactions with CREB binding protein and p300. Mol Cell Biol. 1998;18:2392–405. doi: 10.1128/mcb.18.4.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harrod R, Kuo YL, Tang Y, Yao Y, Vassilev A, Nakatani Y, Giam CZ. p300 and p300/cAMP-responsive element-binding protein associated factor interact with human T-cell lymphotropic virus type-1 Tax in a multi-histone acetyltransferase/activator-enhancer complex. J Biol Chem. 2000;275:11852–7. doi: 10.1074/jbc.275.16.11852. [DOI] [PubMed] [Google Scholar]
- 101.Scoggin KE, Ulloa A, Nyborg JK. The oncoprotein Tax binds the SRC-1-interacting domain of CBP/p300 to mediate transcriptional activation. Mol Cell Biol. 2001;21:5520–30. doi: 10.1128/MCB.21.16.5520-5530.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Harrod R, Tang Y, Nicot C, Lu HS, Vassilev A, Nakatani Y, Giam CZ. An exposed KID-like domain in human T-cell lymphotropic virus type 1 Tax is responsible for the recruitment of coactivators CBP/p300. Mol Cell Biol. 1998;18:5052–61. doi: 10.1128/mcb.18.9.5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yan JP, Garrus JE, Giebler HA, Stargell LA, Nyborg JK. Molecular interactions between the coactivator CBP and the human T-cell leukemia virus Tax protein. J Mol Biol. 1998;281:395–400. doi: 10.1006/jmbi.1998.1951. [DOI] [PubMed] [Google Scholar]
- 104.Lemasson I, Lewis MR, Polakowski N, Hivin P, Cavanagh MH, Thebault S, Barbeau B, Nyborg JK, Mesnard JM. Human T-cell leukemia virus type 1 (HTLV-1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV-1 transcription. J Virol. 2007;81:1543–53. doi: 10.1128/JVI.00480-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Okada M, Jeang KT. Differential requirements for activation of integrated and transiently transfected human T-cell leukemia virus type 1 long terminal repeat. J Virol. 2002;76:12564–73. doi: 10.1128/JVI.76.24.12564-12573.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Reid RL, Lindholm PF, Mireskandari A, Dittmer J, Brady JN. Stabilization of wild-type p53 in human T-lymphocytes transformed by HTLV-I. Oncogene. 1993;8:3029–36. [PubMed] [Google Scholar]
- 107.Pise-Masison CA, Choi KS, Radonovich M, Dittmer J, Kim SJ, Brady JN. Inhibition of p53 transactivation function by the human T-cell lymphotropic virus type 1 Tax protein. J Virol. 1998;72:1165–70. doi: 10.1128/jvi.72.2.1165-1170.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Suzuki T, Narita T, Uchida-Toita M, Yoshida M. Down-regulation of the INK4 family of cyclin-dependent kinase inhibitors by tax protein of HTLV-1 through two distinct mechanisms. Virology. 1999;259:384–91. doi: 10.1006/viro.1999.9760. [DOI] [PubMed] [Google Scholar]
- 109.Low KG, Dorner LF, Fernando DB, Grossman J, Jeang KT, Comb MJ. Human T-cell leukemia virus type 1 Tax releases cell cycle arrest induced by p16INK4a. J Virol. 1997;71:1956–62. doi: 10.1128/jvi.71.3.1956-1962.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jin DY, Spencer F, Jeang KT. Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell. 1998;93:81–91. doi: 10.1016/s0092-8674(00)81148-4. [DOI] [PubMed] [Google Scholar]
- 111.Iwanaga Y, Kasai T, Kibler K, Jeang KT. Characterization of regions in hsMAD1 needed for binding hsMAD2. A polymorphic change in an hsMAD1 leucine zipper affects MAD1-MAD2 interaction and spindle checkpoint function. J Biol Chem. 2002;277:31005–13. doi: 10.1074/jbc.M110666200. [DOI] [PubMed] [Google Scholar]
- 112.Kasai T, Iwanaga Y, Iha H, Jeang KT. Prevalent loss of mitotic spindle checkpoint in adult T-cell leukemia confers resistance to microtubule inhibitors. J Biol Chem. 2002;277:5187–93. doi: 10.1074/jbc.M110295200. [DOI] [PubMed] [Google Scholar]
- 113.Suzuki T, Ohsugi Y, Uchida-Toita M, Akiyama T, Yoshida M. Tax oncoprotein of HTLV-1 binds to the human homologue of Drosophila discs large tumor suppressor protein, hDLG, and perturbs its function in cell growth control. Oncogene. 1999;18:5967–72. doi: 10.1038/sj.onc.1203008. [DOI] [PubMed] [Google Scholar]
- 114.Hirata A, Higuchi M, Niinuma A, Ohashi M, Fukushi M, Oie M, Akiyama T, Tanaka Y, Gejyo F, Fujii M. PDZ domain-binding motif of human T-cell leukemia virus type 1 Tax oncoprotein augments the transforming activity in a rat fibroblast cell line. Virology. 2004;318:327–36. doi: 10.1016/j.virol.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 115.Tsubata C, Higuchi M, Takahashi M, Oie M, Tanaka Y, Gejyo F, Fujii M. PDZ domain-binding motif of human T-cell leukemia virus type 1 Tax oncoprotein is essential for the interleukin 2 independent growth induction of a T-cell line. Retrovirology. 2005;2:46. doi: 10.1186/1742-4690-2-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang L, Dittmer DP, Tomlinson CC, Fakhari FD, Damania B. Immortalization of primary endothelial cells by the K1 protein of Kaposi’s sarcoma-associated herpesvirus. Cancer Res. 2006;66:3658–66. doi: 10.1158/0008-5472.CAN-05-3680. [DOI] [PubMed] [Google Scholar]
- 117.Takeda S, Maeda M, Morikawa S, Taniguchi Y, Yasunaga J, Nosaka K, Tanaka Y, Matsuoka M. Genetic and epigenetic inactivation of tax gene in adult T-cell leukemia cells. Int J Cancer. 2004;109:559–67. doi: 10.1002/ijc.20007. [DOI] [PubMed] [Google Scholar]
- 118.Furukawa Y, Kubota R, Tara M, Izumo S, Osame M. Existence of escape mutant in HTLV-I tax during the development of adult T-cell leukemia. Blood. 2001;97:987–93. doi: 10.1182/blood.v97.4.987. [DOI] [PubMed] [Google Scholar]
- 119.Koiwa T, Hamano-Usami A, Ishida T, Okayama A, Yamaguchi K, Kamihira S, Watanabe T. 5′-long terminal repeat-selective CpG methylation of latent human T-cell leukemia virus type 1 provirus in vitro and in vivo. J Virol. 2002;76:9389–97. doi: 10.1128/JVI.76.18.9389-9397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Taniguchi Y, Nosaka K, Yasunaga J, Maeda M, Mueller N, Okayama A, Matsuoka M. Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology. 2005;2:64. doi: 10.1186/1742-4690-2-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ching YP, Chan SF, Jeang KT, Jin DY. The retroviral oncoprotein Tax targets the coiled-coil centrosomal protein TAX1BP2 to induce centrosome overduplication. Nat Cell Biol. 2006;8:717–24. doi: 10.1038/ncb1432. [DOI] [PubMed] [Google Scholar]
- 122.Peloponese JM, Jr, Haller K, Miyazato A, Jeang KT. Abnormal centrosome amplification in cells through the targeting of Ran-binding protein-1 by the human T cell leukemia virus type-1 Tax oncoprotein. Proc Natl Acad Sci U S A. 2005;102:18974–9. doi: 10.1073/pnas.0506659103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nitta T, Kanai M, Sugihara E, Tanaka M, Sun B, Nagasawa T, Sonoda S, Saya H, Miwa M. Centrosome amplification in adult T-cell leukemia and human T-cell leukemia virus type 1 Tax-induced human T cells. Cancer Sci. 2006;97:836–41. doi: 10.1111/j.1349-7006.2006.00254.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kamihira S, Atogami S, Sohda H, Momita S, Toryia K, Ikeda S, Yamada Y, Tomonaga M. DNA aneuploidy of adult T-cell leukemia cells. Leuk Res. 1994;18:79–84. doi: 10.1016/0145-2126(94)90122-8. [DOI] [PubMed] [Google Scholar]
- 125.Liu B, Liang MH, Kuo YL, Liao W, Boros I, Kleinberger T, Blancato J, Giam CZ. Human T-lymphotropic virus type 1 oncoprotein tax promotes unscheduled degradation of Pds1p/securin and Clb2p/cyclin B1 and causes chromosomal instability. Mol Cell Biol. 2003;23:5269–81. doi: 10.1128/MCB.23.15.5269-5281.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu B, Hong S, Tang Z, Yu H, Giam CZ. HTLV-I Tax directly binds the Cdc20-associated anaphase-promoting complex and activates it ahead of schedule. Proc Natl Acad Sci U S A. 2005;102:63–8. doi: 10.1073/pnas.0406424101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kuo YL, Giam CZ. Activation of the anaphase promoting complex by HTLV-1 tax leads to senescence. Embo J. 2006;25:1741–52. doi: 10.1038/sj.emboj.7601054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sheleg SV, Peloponese JM, Chi YH, Li Y, Eckhaus M, Jeang KT. Evidence for cooperative transforming activity of the human pituitary tumor transforming gene and human T-cell leukemia virus type 1 Tax. J Virol. 2007;81:7894–901. doi: 10.1128/JVI.00555-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol. 2002;76:12813–22. doi: 10.1128/JVI.76.24.12813-12822.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Larocca D, Chao LA, Seto MH, Brunck TK. Human T-cell leukemia virus minus strand transcription in infected T-cells. Biochem Biophys Res Commun. 1989;163:1006–13. doi: 10.1016/0006-291x(89)92322-x. [DOI] [PubMed] [Google Scholar]
- 131.Satou Y, Yasunaga J, Yoshida M, Matsuoka M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci U S A. 2006;103:720–5. doi: 10.1073/pnas.0507631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5:558–67. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- 133.Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med. 2001;345:41–52. doi: 10.1056/NEJM200107053450107. [DOI] [PubMed] [Google Scholar]
- 134.Saito I, Miyamura T, Ohbayashi A, Harada H, Katayama T, Kikuchi S, Watanabe Y, Koi S, Onji M, Ohta Y, et al. Hepatitis C virus infection is associated with the development of hepatocellular carcinoma. Proc Natl Acad Sci U S A. 1990;87:6547–9. doi: 10.1073/pnas.87.17.6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Alter MJ. Epidemiology of hepatitis C in the West. Semin Liver Dis. 1995;15:5–14. doi: 10.1055/s-2007-1007259. [DOI] [PubMed] [Google Scholar]
- 136.Poynard T, Yuen MF, Ratziu V, Lai CL. Viral hepatitis C. Lancet. 2003;362:2095–100. doi: 10.1016/s0140-6736(03)15109-4. [DOI] [PubMed] [Google Scholar]
- 137.Pawlotsky JM. Pathophysiology of hepatitis C virus infection and related liver disease. Trends Microbiol. 2004;12:96–102. doi: 10.1016/j.tim.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 138.Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436:933–8. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 139.Gale M, Jr, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436:939–45. doi: 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- 140.Bowen DG, Walker CM. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature. 2005;436:946–52. doi: 10.1038/nature04079. [DOI] [PubMed] [Google Scholar]
- 141.Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–29. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- 142.Wieland SF, Chisari FV. Stealth and cunning: hepatitis B and hepatitis C viruses. J Virol. 2005;79:9369–80. doi: 10.1128/JVI.79.15.9369-9380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gremion C, Cerny A. Hepatitis C virus and the immune system: a concise review. Rev Med Virol. 2005;15:235–68. doi: 10.1002/rmv.466. [DOI] [PubMed] [Google Scholar]
- 144.Sen GC, Sarkar SN. Transcriptional signaling by double-stranded RNA: role of TLR3. Cytokine Growth Factor Rev. 2005;16:1–14. doi: 10.1016/j.cytogfr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 145.Heathcote EJ. Prevention of hepatitis C virus-related hepatocellular carcinoma. Gastroenterology. 2004;127:S294–302. doi: 10.1053/j.gastro.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 146.Fattovich G, Stroffolini T, Zagni I, Donato F. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology. 2004;127:S35–50. doi: 10.1053/j.gastro.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 147.Liang TJ, Heller T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology. 2004;127:S62–71. doi: 10.1053/j.gastro.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 148.Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T, Koike K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–7. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- 149.Lerat H, Honda M, Beard MR, Loesch K, Sun J, Yang Y, Okuda M, Gosert R, Xiao SY, Weinman SA, Lemon SM. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122:352–65. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- 150.Macdonald A, Harris M. Hepatitis C virus NS5A: tales of a promiscuous protein. J Gen Virol. 2004;85:2485–502. doi: 10.1099/vir.0.80204-0. [DOI] [PubMed] [Google Scholar]
- 151.Jin DY, Wang HL, Zhou Y, Chun AC, Kibler KV, Hou YD, Kung H, Jeang KT. Hepatitis C virus core protein-induced loss of LZIP function correlates with cellular transformation. Embo J. 2000;19:729–40. doi: 10.1093/emboj/19.4.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Munakata T, Nakamura M, Liang Y, Li K, Lemon SM. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C virus NS5B RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A. 2005;102:18159–64. doi: 10.1073/pnas.0505605102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Smirnova IS, Aksenov ND, Kashuba EV, Payakurel P, Grabovetsky VV, Zaberezhny AD, Vonsky MS, Buchinska L, Biberfeld P, Hinkula J, Isaguliants MG. Hepatitis C virus core protein transforms murine fibroblasts by promoting genomic instability. Cell Oncol. 2006;28:177–90. doi: 10.1155/2006/864648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Damania B. DNA tumor viruses and human cancer. Trends Microbiol. 2007;15:38–44. doi: 10.1016/j.tim.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 155.Munger K, Howley PM. Human papillomavirus immortalization and transformation functions. Virus Res. 2002;89:213–28. doi: 10.1016/s0168-1702(02)00190-9. [DOI] [PubMed] [Google Scholar]
- 156.Felsani A, Mileo AM, Paggi MG. Retinoblastoma family proteins as key targets of the small DNA virus oncoproteins. Oncogene. 2006;25:5277–85. doi: 10.1038/sj.onc.1209621. [DOI] [PubMed] [Google Scholar]
- 157.Harlow E, Whyte P, Franza BR, Jr, Schley C. Association of adenovirus early-region 1A proteins with cellular polypeptides. Mol Cell Biol. 1986;6:1579–89. doi: 10.1128/mcb.6.5.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA, Harlow E. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature. 1988;334:124–9. doi: 10.1038/334124a0. [DOI] [PubMed] [Google Scholar]
- 159.DeCaprio JA, Ludlow JW, Figge J, Shew JY, Huang CM, Lee WH, Marsilio E, Paucha E, Livingston DM. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–83. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- 160.Dyson N, Howley PM, Munger K, Harlow E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science. 1989;243:934–7. doi: 10.1126/science.2537532. [DOI] [PubMed] [Google Scholar]
- 161.Nevins JR. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science. 1992;258:424–9. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- 162.Ewen ME, Ludlow JW, Marsilio E, DeCaprio JA, Millikan RC, Cheng SH, Paucha E, Livingston DM. An N-terminal transformation-governing sequence of SV40 large T antigen contributes to the binding of both p110Rb and a second cellular protein, p120. Cell. 1989;58:257–67. doi: 10.1016/0092-8674(89)90840-4. [DOI] [PubMed] [Google Scholar]
- 163.Whyte P, Williamson NM, Harlow E. Cellular targets for transformation by the adenovirus E1A proteins. Cell. 1989;56:67–75. doi: 10.1016/0092-8674(89)90984-7. [DOI] [PubMed] [Google Scholar]
- 164.Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. Embo J. 1989;8:4099–105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Reichel R, Kovesdi I, Nevins JR. Activation of a preexisting cellular factor as a basis for adenovirus E1A-mediated transcription control. Proc Natl Acad Sci U S A. 1988;85:387–90. doi: 10.1073/pnas.85.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chellappan S, Kraus VB, Kroger B, Munger K, Howley PM, Phelps WC, Nevins JR. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc Natl Acad Sci U S A. 1992;89:4549–53. doi: 10.1073/pnas.89.10.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ludlow JW, DeCaprio JA, Huang CM, Lee WH, Paucha E, Livingston DM. SV40 large T antigen binds preferentially to an underphosphorylated member of the retinoblastoma susceptibility gene product family. Cell. 1989;56:57–65. doi: 10.1016/0092-8674(89)90983-5. [DOI] [PubMed] [Google Scholar]
- 168.Imai Y, Matsushima Y, Sugimura T, Terada M. Purification and characterization of human papillomavirus type 16 E7 protein with preferential binding capacity to the underphosphorylated form of retinoblastoma gene product. J Virol. 1991;65:4966–72. doi: 10.1128/jvi.65.9.4966-4972.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Dyson N, Guida P, Munger K, Harlow E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J Virol. 1992;66:6893–902. doi: 10.1128/jvi.66.12.6893-6902.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620–4. [PubMed] [Google Scholar]
- 171.Jones DL, Thompson DA, Munger K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology. 1997;239:97–107. doi: 10.1006/viro.1997.8851. [DOI] [PubMed] [Google Scholar]
- 172.Huh K, Zhou X, Hayakawa H, Cho JY, Libermann TA, Jin J, Harper JW, Munger K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol. 2007;81:9737–47. doi: 10.1128/JVI.00881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature. 1979;278:261–3. doi: 10.1038/278261a0. [DOI] [PubMed] [Google Scholar]
- 174.Linzer DI, Levine AJ. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell. 1979;17:43–52. doi: 10.1016/0092-8674(79)90293-9. [DOI] [PubMed] [Google Scholar]
- 175.Sarnow P, Ho YS, Williams J, Levine AJ. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell. 1982;28:387–94. doi: 10.1016/0092-8674(82)90356-7. [DOI] [PubMed] [Google Scholar]
- 176.Werness BA, Levine AJ, Howley PM. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science. 1990;248:76–9. doi: 10.1126/science.2157286. [DOI] [PubMed] [Google Scholar]
- 177.Finlay CA, Hinds PW, Levine AJ. The p53 proto-oncogene can act as a suppressor of transformation. Cell. 1989;57:1083–93. doi: 10.1016/0092-8674(89)90045-7. [DOI] [PubMed] [Google Scholar]
- 178.Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, White R, Vogelstein B. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–21. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 179.Yew PR, Berk AJ. Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature. 1992;357:82–5. doi: 10.1038/357082a0. [DOI] [PubMed] [Google Scholar]
- 180.Mietz JA, Unger T, Huibregtse JM, Howley PM. The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T-antigen and by HPV-16 E6 oncoprotein. Embo J. 1992;11:5013–20. doi: 10.1002/j.1460-2075.1992.tb05608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lechner MS, Mack DH, Finicle AB, Crook T, Vousden KH, Laimins LA. Human papillomavirus E6 proteins bind p53 in vivo and abrogate p53-mediated repression of transcription. Embo J. 1992;11:3045–52. doi: 10.1002/j.1460-2075.1992.tb05375.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Funk WD, Pak DT, Karas RH, Wright WE, Shay JW. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol Cell Biol. 1992;12:2866–71. doi: 10.1128/mcb.12.6.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kern SE, Pietenpol JA, Thiagalingam S, Seymour A, Kinzler KW, Vogelstein B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science. 1992;256:827–30. doi: 10.1126/science.1589764. [DOI] [PubMed] [Google Scholar]
- 184.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–6. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 185.Oren M, Maltzman W, Levine AJ. Post-translational regulation of the 54K cellular tumor antigen in normal and transformed cells. Mol Cell Biol. 1981;1:101–10. doi: 10.1128/mcb.1.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Reich NC, Oren M, Levine AJ. Two distinct mechanisms regulate the levels of a cellular tumor antigen, p53. Mol Cell Biol. 1983;3:2143–50. doi: 10.1128/mcb.3.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hubbert NL, Sedman SA, Schiller JT. Human papillomavirus type 16 E6 increases the degradation rate of p53 in human keratinocytes. J Virol. 1992;66:6237–41. doi: 10.1128/jvi.66.10.6237-6241.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Scheffner M, Munger K, Byrne JC, Howley PM. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc Natl Acad Sci U S A. 1991;88:5523–7. doi: 10.1073/pnas.88.13.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 190.de Villiers EM. Taxonmic Classification of Papillomaviruses. Papillomavirus Report. 2001;12:57–63. [Google Scholar]
- 191.de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- 192.Jablonska S, Fabjanska L, Formas I. On the viral etiology of epidermodysplasia verruciformis. Dermatologica. 1966;132:369–85. doi: 10.1159/000254437. [DOI] [PubMed] [Google Scholar]
- 193.Pass F, Reissig M, Shah KV, Eisinger M, Orth G. Identification of an immunologically distinct papillomavirus from lesions of epidermodysplasia verruciformis. J Natl Cancer Inst. 1977;59:1107–12. doi: 10.1093/jnci/59.4.1107. [DOI] [PubMed] [Google Scholar]
- 194.Pfister H, Nurnberger F, Gissmann L, zur Hausen H. Characterization of a human papillomavirus from epidermodysplasia verruciformis lesions of a patient from Upper-volta. Int J Cancer. 1981;27:645–50. doi: 10.1002/ijc.2910270511. [DOI] [PubMed] [Google Scholar]
- 195.Ramoz N, Rueda LA, Bouadjar B, Montoya LS, Orth G, Favre M. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat Genet. 2002;32:579–81. doi: 10.1038/ng1044. [DOI] [PubMed] [Google Scholar]
- 196.Orth G, Jablonska S, Favre M, Croissant O, Jarzabek-Chorzelska M, Rzesa G. Characterization of two types of human papillomaviruses in lesions of epidermodysplasia verruciformis. Proc Natl Acad Sci U S A. 1978;75:1537–41. doi: 10.1073/pnas.75.3.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Shamanin V, Glover M, Rausch C, Proby C, Leigh IM, zur Hausen H, de Villiers EM. Specific types of human papillomavirus found in benign proliferations and carcinomas of the skin in immunosuppressed patients. Cancer Res. 1994;54:4610–3. [PubMed] [Google Scholar]
- 198.Shamanin V, zur Hausen H, Lavergne D, Proby CM, Leigh IM, Neumann C, Hamm H, Goos M, Haustein UF, Jung EG, Plewig G, Wolff H, de Villiers EM. Human papillomavirus infections in nonmelanoma skin cancers from renal transplant recipients and nonimmunosuppressed patients. J Natl Cancer Inst. 1996;88:802–11. doi: 10.1093/jnci/88.12.802. [DOI] [PubMed] [Google Scholar]
- 199.Boxman IL, Berkhout RJ, Mulder LH, Wolkers MC, Bouwes Bavinck JN, Vermeer BJ, ter Schegget J. Detection of human papillomavirus DNA in plucked hairs from renal transplant recipients and healthy volunteers. J Invest Dermatol. 1997;108:712–5. doi: 10.1111/1523-1747.ep12292090. [DOI] [PubMed] [Google Scholar]
- 200.Boxman IL, Mulder LH, Russell A, Bouwes Bavinck JN, Green A, Ter Schegget J. Human papillomavirus type 5 is commonly present in immunosuppressed and immunocompetent individuals. Br J Dermatol. 1999;141:246–9. doi: 10.1046/j.1365-2133.1999.02972.x. [DOI] [PubMed] [Google Scholar]
- 201.Akgul B, Cooke JC, Storey A. HPV-associated skin disease. J Pathol. 2006;208:165–75. doi: 10.1002/path.1893. [DOI] [PubMed] [Google Scholar]
- 202.Jackson S, Harwood C, Thomas M, Banks L, Storey A. Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev. 2000;14:3065–73. doi: 10.1101/gad.182100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Iftner T, Bierfelder S, Csapo Z, Pfister H. Involvement of human papillomavirus type 8 genes E6 and E7 in transformation and replication. J Virol. 1988;62:3655–61. doi: 10.1128/jvi.62.10.3655-3661.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Caldeira S, Zehbe I, Accardi R, Malanchi I, Dong W, Giarre M, de Villiers EM, Filotico R, Boukamp P, Tommasino M. The E6 and E7 proteins of the cutaneous human papillomavirus type 38 display transforming properties. J Virol. 2003;77:2195–206. doi: 10.1128/JVI.77.3.2195-2206.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Akgul B, Garcia-Escudero R, Ghali L, Pfister HJ, Fuchs PG, Navsaria H, Storey A. The E7 protein of cutaneous human papillomavirus type 8 causes invasion of human keratinocytes into the dermis in organotypic cultures of skin. Cancer Res. 2005;65:2216–23. doi: 10.1158/0008-5472.CAN-04-1952. [DOI] [PubMed] [Google Scholar]
- 206.Akgul B, Ghali L, Davies D, Pfister H, Leigh IM, Storey A. HPV8 early genes modulate differentiation and cell cycle of primary human adult keratinocytes. Exp Dermatol. 2007;16:590–9. doi: 10.1111/j.1600-0625.2007.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Schaper ID, Marcuzzi GP, Weissenborn SJ, Kasper HU, Dries V, Smyth N, Fuchs P, Pfister H. Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res. 2005;65:1394–400. doi: 10.1158/0008-5472.CAN-04-3263. [DOI] [PubMed] [Google Scholar]
- 208.Dong W, Kloz U, Accardi R, Caldeira S, Tong WM, Wang ZQ, Jansen L, Durst M, Sylla BS, Gissmann L, Tommasino M. Skin hyperproliferation and susceptibility to chemical carcinogenesis in transgenic mice expressing E6 and E7 of human papillomavirus type 38. J Virol. 2005;79:14899–908. doi: 10.1128/JVI.79.23.14899-14908.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Lowy DR, Howley PM. In: Fields’ Virology. Knipe DM, Howley PM, editors. Lippincott Williams and Wilkins; New York: 2001. pp. 2231–2264. [Google Scholar]
- 210.Kutler DI, Wreesmann VB, Goberdhan A, Ben-Porat L, Satagopan J, Ngai I, Huvos AG, Giampietro P, Levran O, Pujara K, Diotti R, Carlson D, Huryn LA, Auerbach AD, Singh B. Human papillomavirus DNA and p53 polymorphisms in squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst. 2003;95:1718–21. doi: 10.1093/jnci/djg091. [DOI] [PubMed] [Google Scholar]
- 211.Spardy N, Duensing A, Charles D, Haines N, Nakahara T, Lambert PF, Duensing S. The human papillomavirus type 16 E7 oncoprotein activates the Fanconi Anemia (FA) pathway and causes accelerated chromosomal instability in FA cells. J Virol. 2007 doi: 10.1128/JVI.01121-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Jeon S, Allen-Hoffmann BL, Lambert PF. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol. 1995;69:2989–97. doi: 10.1128/jvi.69.5.2989-2997.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci U S A. 2000;97:12513–8. doi: 10.1073/pnas.97.23.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wells SI, Francis DA, Karpova AY, Dowhanick JJ, Benson JD, Howley PM. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21(CIP)-dependent pathways. Embo J. 2000;19:5762–71. doi: 10.1093/emboj/19.21.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hawley-Nelson P, Vousden KH, Hubbert NL, Lowy DR, Schiller JT. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. Embo J. 1989;8:3905–10. doi: 10.1002/j.1460-2075.1989.tb08570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989;63:4417–21. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.McCance DJ, Kopan R, Fuchs E, Laimins LA. Human papillomavirus type 16 alters human epithelial cell differentiation in vitro. Proc Natl Acad Sci U S A. 1988;85:7169–73. doi: 10.1073/pnas.85.19.7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.DiPaolo JA, Woodworth CD, Popescu NC, Notario V, Doniger J. Induction of human cervical squamous cell carcinoma by sequential transfection with human papillomavirus 16 DNA and viral Harvey ras. Oncogene. 1989;4:395–9. [PubMed] [Google Scholar]
- 219.Durst M, Gallahan D, Jay G, Rhim JS. Glucocorticoid-enhanced neoplastic transformation of human keratinocytes by human papillomavirus type 16 and an activated ras oncogene. Virology. 1989;173:767–71. doi: 10.1016/0042-6822(89)90595-3. [DOI] [PubMed] [Google Scholar]
- 220.Pei XF, Meck JM, Greenhalgh D, Schlegel R. Cotransfection of HPV-18 and v-fos DNA induces tumorigenicity of primary human keratinocytes. Virology. 1993;196:855–60. doi: 10.1006/viro.1993.1546. [DOI] [PubMed] [Google Scholar]
- 221.Arbeit JM, Howley PM, Hanahan D. Chronic estrogen-induced cervical and vaginal squamous carcinogenesis in human papillomavirus type 16 transgenic mice. Proc Natl Acad Sci U S A. 1996;93:2930–5. doi: 10.1073/pnas.93.7.2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–50. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 223.Phelps WC, Yee CL, Munger K, Howley PM. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell. 1988;53:539–47. doi: 10.1016/0092-8674(88)90570-3. [DOI] [PubMed] [Google Scholar]
- 224.Tanaka A, Noda T, Yajima H, Hatanaka M, Ito Y. Identification of a transforming gene of human papillomavirus type 16. J Virol. 1989;63:1465–9. doi: 10.1128/jvi.63.3.1465-1469.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Vousden KH, Doniger J, DiPaolo JA, Lowy DR. The E7 open reading frame of human papillomavirus type 16 encodes a transforming gene. Oncogene Res. 1988;3:167–75. [PubMed] [Google Scholar]
- 226.Watanabe S, Yoshiike K. Transformation of rat 3Y1 cells by human papillomavirus type-18 DNA. Int J Cancer. 1988;41:896–900. doi: 10.1002/ijc.2910410622. [DOI] [PubMed] [Google Scholar]
- 227.Matlashewski G, Schneider J, Banks L, Jones N, Murray A, Crawford L. Human papillomavirus type 16 DNA cooperates with activated ras in transforming primary cells. Embo J. 1987;6:1741–6. doi: 10.1002/j.1460-2075.1987.tb02426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Storey A, Banks L. Human papillomavirus type 16 E6 gene cooperates with EJ-ras to immortalize primary mouse cells. Oncogene. 1993;8:919–24. [PubMed] [Google Scholar]
- 229.Durst M, Dzarlieva-Petrusevska RT, Boukamp P, Fusenig NE, Gissmann L. Molecular and cytogenetic analysis of immortalized human primary keratinocytes obtained after transfection with human papillomavirus type 16 DNA. Oncogene. 1987;1:251–6. [PubMed] [Google Scholar]
- 230.Pirisi L, Yasumoto S, Feller M, Doniger J, DiPaolo JA. Transformation of human fibroblasts and keratinocytes with human papillomavirus type 16 DNA. J Virol. 1987;61:1061–6. doi: 10.1128/jvi.61.4.1061-1066.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Schlegel R, Phelps WC, Zhang YL, Barbosa M. Quantitative keratinocyte assay detects two biological activities of human papillomavirus DNA and identifies viral types associated with cervical carcinoma. Embo J. 1988;7:3181–7. doi: 10.1002/j.1460-2075.1988.tb03185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Watanabe S, Kanda T, Yoshiike K. Human papillomavirus type 16 transformation of primary human embryonic fibroblasts requires expression of open reading frames E6 and E7. J Virol. 1989;63:965–9. doi: 10.1128/jvi.63.2.965-969.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Woodworth CD, Bowden PE, Doniger J, Pirisi L, Barnes W, Lancaster WD, DiPaolo JA. Characterization of normal human exocervical epithelial cells immortalized in vitro by papillomavirus types 16 and 18 DNA. Cancer Res. 1988;48:4620–8. [PubMed] [Google Scholar]
- 234.White AE, Livanos EM, Tlsty TD. Differential disruption of genomic integrity and cell cycle regulation in normal human fibroblasts by the HPV oncoproteins. Genes Dev. 1994;8:666–77. doi: 10.1101/gad.8.6.666. [DOI] [PubMed] [Google Scholar]
- 235.Duensing S, Munger K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002;62:7075–82. [PubMed] [Google Scholar]
- 236.Crum CP, Ikenberg H, Richart RM, Gissman L. Human papillomavirus type 16 and early cervical neoplasia. N Engl J Med. 1984;310:880–3. doi: 10.1056/NEJM198404053101403. [DOI] [PubMed] [Google Scholar]
- 237.Duensing S, Lee LY, Duensing A, Basile J, Piboonniyom S, Gonzalez S, Crum CP, Munger K. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc Natl Acad Sci U S A. 2000;97:10002–7. doi: 10.1073/pnas.170093297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Duensing S, Duensing A, Crum CP, Munger K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001;61:2356–60. [PubMed] [Google Scholar]
- 239.Ganem D, Prince AM. Hepatitis B virus infection--natural history and clinical consequences. N Engl J Med. 2004;350:1118–29. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 240.Hoofnagle JH, Dusheiko GM, Seeff LB, Jones EA, Waggoner JG, Bales ZB. Seroconversion from hepatitis B e antigen to antibody in chronic type B hepatitis. Ann Intern Med. 1981;94:744–8. doi: 10.7326/0003-4819-94-6-744. [DOI] [PubMed] [Google Scholar]
- 241.Wieland S, Thimme R, Purcell RH, Chisari FV. Genomic analysis of the host response to hepatitis B virus infection. Proc Natl Acad Sci U S A. 2004;101:6669–74. doi: 10.1073/pnas.0401771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Feitelson MA. Hepatitis B virus in hepatocarcinogenesis. J Cell Physiol. 1999;181:188–202. doi: 10.1002/(SICI)1097-4652(199911)181:2<188::AID-JCP2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 243.Lok AS. Prevention of hepatitis B virus-related hepatocellular carcinoma. Gastroenterology. 2004;127:S303–9. doi: 10.1053/j.gastro.2004.09.045. [DOI] [PubMed] [Google Scholar]
- 244.Singh M, Kumar V. Transgenic mouse models of hepatitis B virus-associated hepatocellular carcinoma. Rev Med Virol. 2003;13:243–53. doi: 10.1002/rmv.392. [DOI] [PubMed] [Google Scholar]
- 245.Brechot C. Pathogenesis of hepatitis B virus-related hepatocellular carcinoma: old and new paradigms. Gastroenterology. 2004;127:S56–61. doi: 10.1053/j.gastro.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 246.Tanaka H, Iwasaki Y, Nouso K, Kobayashi Y, Nakamura S, Matsumoto E, Toshikuni N, Kaneyoshi T, Ohsawa T, Takaguchi K, Fujio K, Senoh T, Ohnishi T, Sakaguchi K, Shiratori Y. Possible contribution of prior hepatitis B virus infection to the development of hepatocellular carcinoma. J Gastroenterol Hepatol. 2005;20:850–6. doi: 10.1111/j.1440-1746.2005.03823.x. [DOI] [PubMed] [Google Scholar]
- 247.Kajiya Y, Hamasaki K, Nakata K, Miyazoe S, Takeda Y, Higashi S, Ohkubo K, Ichikawa T, Nakao K, Kato Y, Eguchi K. A long-term follow-up analysis of serial core promoter and precore sequences in Japanese patients chronically infected by hepatitis B virus. Dig Dis Sci. 2001;46:509–15. doi: 10.1023/a:1005582812466. [DOI] [PubMed] [Google Scholar]
- 248.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Feitelson MA, Lee J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007;252:157–70. doi: 10.1016/j.canlet.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 250.Brechot C, Gozuacik D, Murakami Y, Paterlini-Brechot P. Molecular bases for the development of hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC) Semin Cancer Biol. 2000;10:211–31. doi: 10.1006/scbi.2000.0321. [DOI] [PubMed] [Google Scholar]
- 251.Minami M, Daimon Y, Mori K, Takashima H, Nakajima T, Itoh Y, Okanoue T. Hepatitis B virus-related insertional mutagenesis in chronic hepatitis B patients as an early drastic genetic change leading to hepatocarcinogenesis. Oncogene. 2005;24:4340–8. doi: 10.1038/sj.onc.1208628. [DOI] [PubMed] [Google Scholar]
- 252.Dandri M, Burda MR, Burkle A, Zuckerman DM, Will H, Rogler CE, Greten H, Petersen J. Increase in de novo HBV DNA integrations in response to oxidative DNA damage or inhibition of poly(ADP-ribosyl)ation. Hepatology. 2002;35:217–23. doi: 10.1053/jhep.2002.30203. [DOI] [PubMed] [Google Scholar]
- 253.Wang HP, Zhang L, Dandri M, Rogler CE. Antisense downregulation of N-myc1 in woodchuck hepatoma cells reverses the malignant phenotype. J Virol. 1998;72:2192–8. doi: 10.1128/jvi.72.3.2192-2198.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Aoki H, Kajino K, Arakawa Y, Hino O. Molecular cloning of a rat chromosome putative recombinogenic sequence homologous to the hepatitis B virus encapsidation signal. Proc Natl Acad Sci U S A. 1996;93:7300–4. doi: 10.1073/pnas.93.14.7300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Hino O, Kajino K, Umeda T, Arakawa Y. Understanding the hypercarcinogenic state in chronic hepatitis: a clue to the prevention of human hepatocellular carcinoma. J Gastroenterol. 2002;37:883–7. doi: 10.1007/s005350200149. [DOI] [PubMed] [Google Scholar]
- 256.Horikawa I, Barrett JC. cis-Activation of the human telomerase gene (hTERT) by the hepatitis B virus genome. J Natl Cancer Inst. 2001;93:1171–3. doi: 10.1093/jnci/93.15.1171. [DOI] [PubMed] [Google Scholar]
- 257.Ferber MJ, Montoya DP, Yu C, Aderca I, McGee A, Thorland EC, Nagorney DM, Gostout BS, Burgart LJ, Boix L, Bruix J, McMahon BJ, Cheung TH, Chung TK, Wong YF, Smith DI, Roberts LR. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene. 2003;22:3813–20. doi: 10.1038/sj.onc.1206528. [DOI] [PubMed] [Google Scholar]
- 258.Paterlini-Brechot P, Saigo K, Murakami Y, Chami M, Gozuacik D, Mugnier C, Lagorce D, Brechot C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911–6. doi: 10.1038/sj.onc.1206492. [DOI] [PubMed] [Google Scholar]
- 259.Murakami Y, Saigo K, Takashima H, Minami M, Okanoue T, Brechot C, Paterlini-Brechot P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut. 2005;54:1162–8. doi: 10.1136/gut.2004.054452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Tamori A, Yamanishi Y, Kawashima S, Kanehisa M, Enomoto M, Tanaka H, Kubo S, Shiomi S, Nishiguchi S. Alteration of gene expression in human hepatocellular carcinoma with integrated hepatitis B virus DNA. Clin Cancer Res. 2005;11:5821–6. doi: 10.1158/1078-0432.CCR-04-2055. [DOI] [PubMed] [Google Scholar]
- 261.Soussan P, Garreau F, Zylberberg H, Ferray C, Brechot C, Kremsdorf D. In vivo expression of a new hepatitis B virus protein encoded by a spliced RNA. J Clin Invest. 2000;105:55–60. doi: 10.1172/JCI8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Diao J, Garces R, Richardson CD. X protein of hepatitis B virus modulates cytokine and growth factor related signal transduction pathways during the course of viral infections and hepatocarcinogenesis. Cytokine Growth Factor Rev. 2001;12:189–205. doi: 10.1016/s1359-6101(00)00034-4. [DOI] [PubMed] [Google Scholar]
- 263.Wollersheim M, Debelka U, Hofschneider PH. A transactivating function encoded in the hepatitis B virus X gene is conserved in the integrated state. Oncogene. 1988;3:545–52. [PubMed] [Google Scholar]
- 264.Paterlini P, Poussin K, Kew M, Franco D, Brechot C. Selective accumulation of the X transcript of hepatitis B virus in patients negative for hepatitis B surface antigen with hepatocellular carcinoma. Hepatology. 1995;21:313–21. [PubMed] [Google Scholar]
- 265.Wei Y, Etiemble J, Fourel G, Vitvitski-Trepo L, Buendia MA. Hepadna virus integration generates virus-cell cotranscripts carrying 3′ truncated X genes in human and woodchuck liver tumors. J Med Virol. 1995;45:82–90. doi: 10.1002/jmv.1890450116. [DOI] [PubMed] [Google Scholar]
- 266.Su Q, Schroder CH, Hofmann WJ, Otto G, Pichlmayr R, Bannasch P. Expression of hepatitis B virus X protein in HBV-infected human livers and hepatocellular carcinomas. Hepatology. 1998;27:1109–20. doi: 10.1002/hep.510270428. [DOI] [PubMed] [Google Scholar]
- 267.Sirma H, Giannini C, Poussin K, Paterlini P, Kremsdorf D, Brechot C. Hepatitis B virus X mutants, present in hepatocellular carcinoma tissue abrogate both the antiproliferative and transactivation effects of HBx. Oncogene. 1999;18:4848–59. doi: 10.1038/sj.onc.1202867. [DOI] [PubMed] [Google Scholar]
- 268.Peng Z, Zhang Y, Gu W, Wang Z, Li D, Zhang F, Qiu G, Xie K. Integration of the hepatitis B virus X fragment in hepatocellular carcinoma and its effects on the expression of multiple molecules: a key to the cell cycle and apoptosis. Int J Oncol. 2005;26:467–73. [PubMed] [Google Scholar]
- 269.Chen HS, Kaneko S, Girones R, Anderson RW, Hornbuckle WE, Tennant BC, Cote PJ, Gerin JL, Purcell RH, Miller RH. The woodchuck hepatitis virus X gene is important for establishment of virus infection in woodchucks. J Virol. 1993;67:1218–26. doi: 10.1128/jvi.67.3.1218-1226.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Zoulim F, Saputelli J, Seeger C. Woodchuck hepatitis virus X protein is required for viral infection in vivo. J Virol. 1994;68:2026–30. doi: 10.1128/jvi.68.3.2026-2030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Melegari M, Wolf SK, Schneider RJ. Hepatitis B virus DNA replication is coordinated by core protein serine phosphorylation and HBx expression. J Virol. 2005;79:9810–20. doi: 10.1128/JVI.79.15.9810-9820.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Tang H, Delgermaa L, Huang F, Oishi N, Liu L, He F, Zhao L, Murakami S. The transcriptional transactivation function of HBx protein is important for its augmentation role in hepatitis B virus replication. J Virol. 2005;79:5548–56. doi: 10.1128/JVI.79.9.5548-5556.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Brechot C, Thiers V, Kremsdorf D, Nalpas B, Pol S, Paterlini-Brechot P. Persistent hepatitis B virus infection in subjects without hepatitis B surface antigen: clinically significant or purely “occult”? Hepatology. 2001;34:194–203. doi: 10.1053/jhep.2001.25172. [DOI] [PubMed] [Google Scholar]
- 274.Klein NP, Bouchard MJ, Wang LH, Kobarg C, Schneider RJ. Src kinases involved in hepatitis B virus replication. Embo J. 1999;18:5019–27. doi: 10.1093/emboj/18.18.5019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Yen TS. Hepadnaviral X Protein:Review of Recent Progress. J Biomed Sci. 1996;3:20–30. doi: 10.1007/BF02253575. [DOI] [PubMed] [Google Scholar]
- 276.Bouchard MJ, Schneider RJ. The enigmatic X gene of hepatitis B virus. J Virol. 2004;78:12725–34. doi: 10.1128/JVI.78.23.12725-12734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Arbuthnot P, Capovilla A, Kew M. Putative role of hepatitis B virus X protein in hepatocarcinogenesis: effects on apoptosis, DNA repair, mitogen-activated protein kinase and JAK/STAT pathways. J Gastroenterol Hepatol. 2000;15:357–68. doi: 10.1046/j.1440-1746.2000.02069.x. [DOI] [PubMed] [Google Scholar]
- 278.Ahn JY, Jung EY, Kwun HJ, Lee CW, Sung YC, Jang KL. Dual effects of hepatitis B virus X protein on the regulation of cell-cycle control depending on the status of cellular p53. J Gen Virol. 2002;83:2765–72. doi: 10.1099/0022-1317-83-11-2765. [DOI] [PubMed] [Google Scholar]
- 279.Pang R, Tse E, Poon RT. Molecular pathways in hepatocellular carcinoma. Cancer Lett. 2006;240:157–69. doi: 10.1016/j.canlet.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 280.Huang J, Kwong J, Sun EC, Liang TJ. Proteasome complex as a potential cellular target of hepatitis B virus X protein. J Virol. 1996;70:5582–91. doi: 10.1128/jvi.70.8.5582-5591.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Hu Z, Zhang Z, Doo E, Coux O, Goldberg AL, Liang TJ. Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex. J Virol. 1999;73:7231–40. doi: 10.1128/jvi.73.9.7231-7240.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Zhang Z, Torii N, Furusaka A, Malayaman N, Hu Z, Liang TJ. Structural and functional characterization of interaction between hepatitis B virus X protein and the proteasome complex. J Biol Chem. 2000;275:15157–65. doi: 10.1074/jbc.M910378199. [DOI] [PubMed] [Google Scholar]
- 283.Rahmani Z, Huh KW, Lasher R, Siddiqui A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J Virol. 2000;74:2840–6. doi: 10.1128/jvi.74.6.2840-2846.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Huh KW, Siddiqui A. Characterization of the mitochondrial association of hepatitis B virus X protein, HBx. Mitochondrion. 2002;1:349–59. doi: 10.1016/s1567-7249(01)00040-x. [DOI] [PubMed] [Google Scholar]
- 285.Chami M, Ferrari D, Nicotera P, Paterlini-Brechot P, Rizzuto R. Caspase-dependent alterations of Ca2+ signaling in the induction of apoptosis by hepatitis B virus X protein. J Biol Chem. 2003;278:31745–55. doi: 10.1074/jbc.M304202200. [DOI] [PubMed] [Google Scholar]
- 286.Bouchard MJ, Wang LH, Schneider RJ. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science. 2001;294:2376–8. doi: 10.1126/science.294.5550.2376. [DOI] [PubMed] [Google Scholar]
- 287.Forgues M, Difilippantonio MJ, Linke SP, Ried T, Nagashima K, Feden J, Valerie K, Fukasawa K, Wang XW. Involvement of Crm1 in hepatitis B virus X protein-induced aberrant centriole replication and abnormal mitotic spindles. Mol Cell Biol. 2003;23:5282–92. doi: 10.1128/MCB.23.15.5282-5292.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Hildt E, Hofschneider PH. The PreS2 activators of the hepatitis B virus: activators of tumour promoter pathways. Recent Results Cancer Res. 1998;154:315–29. doi: 10.1007/978-3-642-46870-4_23. [DOI] [PubMed] [Google Scholar]
- 289.Murakami S. Hepatitis B virus X protein: structure, function and biology. Intervirology. 1999;42:81–99. doi: 10.1159/000024969. [DOI] [PubMed] [Google Scholar]
- 290.Kekule AS, Lauer U, Meyer M, Caselmann WH, Hofschneider PH, Koshy R. The preS2/S region of integrated hepatitis B virus DNA encodes a transcriptional transactivator. Nature. 1990;343:457–61. doi: 10.1038/343457a0. [DOI] [PubMed] [Google Scholar]
- 291.Hildt E, Saher G, Bruss V, Hofschneider PH. The hepatitis B virus large surface protein (LHBs) is a transcriptional activator. Virology. 1996;225:235–9. doi: 10.1006/viro.1996.0594. [DOI] [PubMed] [Google Scholar]
- 292.Schluter V, Meyer M, Hofschneider PH, Koshy R, Caselmann WH. Integrated hepatitis B virus X and ′ truncated preS/S sequences derived from human hepatomas encode functionally active transactivators. Oncogene. 1994;9:3335–44. [PubMed] [Google Scholar]
- 293.Zhong S, Chan JY, Yeo W, Tam JS, Johnson PJ. Hepatitis B envelope protein mutants in human hepatocellular carcinoma tissues. J Viral Hepat. 1999;6:195–202. doi: 10.1046/j.1365-2893.1999.00154.x. [DOI] [PubMed] [Google Scholar]
- 294.Wang HC, Huang W, Lai MD, Su IJ. Hepatitis B virus pre-S mutants, endoplasmic reticulum stress and hepatocarcinogenesis. Cancer Sci. 2006;97:683–8. doi: 10.1111/j.1349-7006.2006.00235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Hildt E, Munz B, Saher G, Reifenberg K, Hofschneider PH. The PreS2 activator MHBs(t) of hepatitis B virus activates c-raf-1/Erk2 signaling in transgenic mice. Embo J. 2002;21:525–35. doi: 10.1093/emboj/21.4.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Soussan P, Tuveri R, Nalpas B, Garreau F, Zavala F, Masson A, Pol S, Brechot C, Kremsdorf D. The expression of hepatitis B spliced protein (HBSP) encoded by a spliced hepatitis B virus RNA is associated with viral replication and liver fibrosis. J Hepatol. 2003;38:343–8. doi: 10.1016/s0168-8278(02)00422-1. [DOI] [PubMed] [Google Scholar]
- 297.I.A.f.R.o. Cancer, Epstein-Barr virus and Kaposi sarcoma herpesvirus/human herpesvirus 8. IARC Monograph. 1997;70 [PMC free article] [PubMed] [Google Scholar]
- 298.Evans AS, Niederman JC. Viral Infections of humans. Plenum Press; New York: 1989. [Google Scholar]
- 299.Chan SH. Aetiology of nasopharyngeal carcinoma. Ann Acad Med Singapore. 1990;19:201–7. [PubMed] [Google Scholar]
- 300.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58:1347–63. [PMC free article] [PubMed] [Google Scholar]
- 301.Wang D, Liebowitz D, Kieff E. An EBV membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell. 1985;43:831–40. doi: 10.1016/0092-8674(85)90256-9. [DOI] [PubMed] [Google Scholar]
- 302.Rickinson A, Kieff E. In: Fields Virology. Knipe DM, Howley PM, editors. Lippincott Williams and Wilkins; Philadelphia, PA: 2001. pp. 2575–2627. [Google Scholar]
- 303.Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C, Kieff E. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell. 1995;80:389–99. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]
- 304.Kieff E, Rickinson A. In: Fields Virology. Knipe DM, Howley PM, editors. Lippincott Williams and Wilkins; Philadelphia, PA: 2001. pp. 2511–2573. [Google Scholar]
- 305.Miller WE, Cheshire JL, Baldwin AS, Jr, Raab-Traub N. The NPC derived C15 LMP1 protein confers enhanced activation of NF-kappa B and induction of the EGFR in epithelial cells. Oncogene. 1998;16:1869–77. doi: 10.1038/sj.onc.1201696. [DOI] [PubMed] [Google Scholar]
- 306.Eliopoulos AG, Young LS. Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) Oncogene. 1998;16:1731–42. doi: 10.1038/sj.onc.1201694. [DOI] [PubMed] [Google Scholar]
- 307.Eliopoulos AG, Gallagher NJ, Blake SM, Dawson CW, Young LS. Activation of the p38 mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J Biol Chem. 1999;274:16085–96. doi: 10.1074/jbc.274.23.16085. [DOI] [PubMed] [Google Scholar]
- 308.Rowe M, Peng-Pilon M, Huen DS, Hardy R, Croom-Carter D, Lundgren E, Rickinson AB. Upregulation of bcl-2 by the Epstein-Barr virus latent membrane protein LMP1: a B-cell-specific response that is delayed relative to NF-kappa B activation and to induction of cell surface markers. J Virol. 1994;68:5602–12. doi: 10.1128/jvi.68.9.5602-5612.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Zhang L, Pagano JS. Interferon regulatory factor 7 is induced by Epstein-Barr virus latent membrane protein 1. J Virol. 2000;74:1061–8. doi: 10.1128/jvi.74.3.1061-1068.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Wakisaka N, Murono S, Yoshizaki T, Furukawa M, Pagano JS. Epstein-barr virus latent membrane protein 1 induces and causes release of fibroblast growth factor-2. Cancer Res. 2002;62:6337–44. [PubMed] [Google Scholar]
- 311.Caldwell RG, Wilson JB, Anderson SJ, Longnecker R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals. Immunity. 1998;9:405–11. doi: 10.1016/s1074-7613(00)80623-8. [DOI] [PubMed] [Google Scholar]
- 312.Grossman SR, Johannsen E, Tong X, Yalamanchili R, Kieff E. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc Natl Acad Sci U S A. 1994;91:7568–72. doi: 10.1073/pnas.91.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Tong X, Wang F, Thut CJ, Kieff E. The Epstein-Barr virus nuclear protein 2 acidic domain can interact with TFIIB, TAF40, and RPA70 but not with TATA-binding protein. J Virol. 1995;69:585–8. doi: 10.1128/jvi.69.1.585-588.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Cohen JI, Kieff E. An Epstein-Barr virus nuclear protein 2 domain essential for transformation is a direct transcriptional activator. J Virol. 1991;65:5880–5. doi: 10.1128/jvi.65.11.5880-5885.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Tomkinson B, Kieff E. Second-site homologous recombination in Epstein-Barr virus: insertion of type 1 EBNA 3 genes in place of type 2 has no effect on in vitro infection. J Virol. 1992;66:780–9. doi: 10.1128/jvi.66.2.780-789.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Robertson ES, Grossman S, Johannsen E, Miller C, Lin J, Tomkinson B, Kieff E. Epstein-Barr virus nuclear protein 3C modulates transcription through interaction with the sequence-specific DNA-binding protein J kappa. J Virol. 1995;69:3108–16. doi: 10.1128/jvi.69.5.3108-3116.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8) Proc Natl Acad Sci U S A. 1996;93:14862–7. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Ablashi DV, Chatlynne LG, Whitman JE, Jr, Cesarman E. Spectrum of Kaposi’s sarcoma-associated herpesvirus, or human herpesvirus 8, diseases. Clin Microbiol Rev. 2002;15:439–64. doi: 10.1128/CMR.15.3.439-464.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Schulz TF. The pleiotropic effects of Kaposi’s sarcoma herpesvirus. J Pathol. 2006;208:187–98. doi: 10.1002/path.1904. [DOI] [PubMed] [Google Scholar]
- 320.Flore O, Rafii S, Ely S, O’Leary JJ, Hyjek EM, Cesarman E. Transformation of primary human endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Nature. 1998;394:588–92. doi: 10.1038/29093. [DOI] [PubMed] [Google Scholar]
- 321.Lebbe C, Blum L, Pellet C, Blanchard G, Verola O, Morel P, Danne O, Calvo F. Clinical and biological impact of antiretroviral therapy with protease inhibitors on HIV-related Kaposi’s sarcoma. Aids. 1998;12:F45–9. doi: 10.1097/00002030-199807000-00002. [DOI] [PubMed] [Google Scholar]
- 322.Gill J, Bourboulia D, Wilkinson J, Hayes P, Cope A, Marcelin AG, Calvez V, Gotch F, Boshoff C, Gazzard B. Prospective study of the effects of antiretroviral therapy on Kaposi sarcoma--associated herpesvirus infection in patients with and without Kaposi sarcoma. J Acquir Immune Defic Syndr. 2002;31:384–90. doi: 10.1097/00126334-200212010-00003. [DOI] [PubMed] [Google Scholar]
- 323.Aoki Y, Jaffe ES, Chang Y, Jones K, Teruya-Feldstein J, Moore PS, Tosato G. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood. 1999;93:4034–43. [PubMed] [Google Scholar]
- 324.Stine JT, Wood C, Hill M, Epp A, Raport CJ, Schweickart VL, Endo Y, Sasaki T, Simmons G, Boshoff C, Clapham P, Chang Y, Moore P, Gray PW, Chantry D. KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood. 2000;95:1151–7. [PubMed] [Google Scholar]
- 325.Boshoff C, Endo Y, Collins PD, Takeuchi Y, Reeves JD, Schweickart VL, Siani MA, Sasaki T, Williams TJ, Gray PW, Moore PS, Chang Y, Weiss RA. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science. 1997;278:290–4. doi: 10.1126/science.278.5336.290. [DOI] [PubMed] [Google Scholar]
- 326.Yang TY, Chen SC, Leach MW, Manfra D, Homey B, Wiekowski M, Sullivan L, Jenh CH, Narula SK, Chensue SW, Lira SA. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi’s sarcoma. J Exp Med. 2000;191:445–54. doi: 10.1084/jem.191.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Moore PS, Chang Y. Kaposi’s sarcoma-associated herpesvirus immunoevasion and tumorigenesis: two sides of the same coin? Annu Rev Microbiol. 2003;57:609–39. doi: 10.1146/annurev.micro.57.030502.090824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Damania B. Oncogenic gamma-herpesviruses: comparison of viral proteins involved in tumorigenesis. Nat Rev Microbiol. 2004;2:656–68. doi: 10.1038/nrmicro958. [DOI] [PubMed] [Google Scholar]
- 329.Cesarman E, Mesri EA, Gershengorn MC. Viral G protein-coupled receptor and Kaposi’s sarcoma: a model of paracrine neoplasia? J Exp Med. 2000;191:417–22. doi: 10.1084/jem.191.3.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Nicholas J. Human herpesvirus-8-encoded signalling ligands and receptors. J Biomed Sci. 2003;10:475–89. doi: 10.1007/BF02256109. [DOI] [PubMed] [Google Scholar]
- 331.Bajaj BG, Verma SC, Lan K, Cotter MA, Woodman ZL, Robertson ES. KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology. 2006;351:18–28. doi: 10.1016/j.virol.2006.03.037. [DOI] [PubMed] [Google Scholar]
- 332.Friborg J, Jr, Kong W, Hottiger MO, Nabel GJ. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature. 1999;402:889–94. doi: 10.1038/47266. [DOI] [PubMed] [Google Scholar]
- 333.Swanton C, Mann DJ, Fleckenstein B, Neipel F, Peters G, Jones N. Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature. 1997;390:184–7. doi: 10.1038/36606. [DOI] [PubMed] [Google Scholar]
- 334.Glykofrydes D, Niphuis H, Kuhn EM, Rosenwirth B, Heeney JL, Bruder J, Niedobitek G, Muller-Fleckenstein I, Fleckenstein B, Ensser A. Herpesvirus saimiri vFLIP provides an antiapoptotic function but is not essential for viral replication, transformation, or pathogenicity. J Virol. 2000;74:11919–27. doi: 10.1128/jvi.74.24.11919-11927.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Esteban M, Garcia MA, Domingo-Gil E, Arroyo J, Nombela C, Rivas C. The latency protein LANA2 from Kaposi’s sarcoma-associated herpesvirus inhibits apoptosis induced by dsRNA-activated protein kinase but not RNase L activation. J Gen Virol. 2003;84:1463–70. doi: 10.1099/vir.0.19014-0. [DOI] [PubMed] [Google Scholar]
- 336.McCormick C, Ganem D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science. 2005;307:739–41. doi: 10.1126/science.1105779. [DOI] [PubMed] [Google Scholar]
- 337.Lee H, Veazey R, Williams K, Li M, Guo J, Neipel F, Fleckenstein B, Lackner A, Desrosiers RC, Jung JU. Deregulation of cell growth by the K1 gene of Kaposi’s sarcoma-associated herpesvirus. Nat Med. 1998;4:435–40. doi: 10.1038/nm0498-435. [DOI] [PubMed] [Google Scholar]
- 338.Prakash O, Tang ZY, Peng X, Coleman R, Gill J, Farr G, Samaniego F. Tumorigenesis and aberrant signaling in transgenic mice expressing the human herpesvirus-8 K1 gene. J Natl Cancer Inst. 2002;94:926–35. doi: 10.1093/jnci/94.12.926. [DOI] [PubMed] [Google Scholar]
- 339.Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, Ye H, Du MQ, Boshoff C. K15 protein of Kaposi’s sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function. J Virol. 2002;76:802–16. doi: 10.1128/JVI.76.2.802-816.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Damania B. Modulation of cell signaling pathways by Kaposi’s sarcoma-associated herpesvirus (KSHVHHV-8) Cell Biochem Biophys. 2004;40:305–22. doi: 10.1385/CBB:40:3:305. [DOI] [PubMed] [Google Scholar]
- 341.Gross L. A filterable agent, recovered from Ak leukemic extracts, causing salivary gland carcinomas in C3H mice. Proc Soc Exp Biol Med. 1953;83:414–21. doi: 10.3181/00379727-83-20376. [DOI] [PubMed] [Google Scholar]
- 342.Eddy BE. In: Virology Monographs. Gard CHS, Meyers KF, editors. Springer-Verlag; New York: 1969. pp. 1–114. [Google Scholar]
- 343.Gross L. Oncogenic Viruses. Pergamon Press; Oxford, New York, Toronto, Sydney, Paris, Frankfurt: 1983. [Google Scholar]
- 344.Benjamin TL. Polyoma virus: old findings and new challenges. Virology. 2001;289:167–73. doi: 10.1006/viro.2001.1124. [DOI] [PubMed] [Google Scholar]
- 345.Hirsch HH, Knowles W, Dickenmann M, Passweg J, Klimkait T, Mihatsch MJ, Steiger J. Prospective study of polyomavirus type BK replication and nephropathy in renal-transplant recipients. N Engl J Med. 2002;347:488–96. doi: 10.1056/NEJMoa020439. [DOI] [PubMed] [Google Scholar]
- 346.Randhawa PS, Demetris AJ. Nephropathy due to polyomavirus type BK. N Engl J Med. 2000;342:1361–3. doi: 10.1056/NEJM200005043421809. [DOI] [PubMed] [Google Scholar]
- 347.Berger JR, Koralnik IJ. Progressive multifocal leukoencephalopathy and natalizumab--unforeseen consequences. N Engl J Med. 2005;353:414–6. doi: 10.1056/NEJMe058122. [DOI] [PubMed] [Google Scholar]
- 348.Safak M, Khalili K. An overview: Human polyomavirus JC virus and its associated disorders. J Neurovirol. 2003;9(Suppl 1):3–9. doi: 10.1080/13550280390195360. [DOI] [PubMed] [Google Scholar]
- 349.Allander T, Andreasson K, Gupta S, Bjerkner A, Bogdanovic G, Persson MA, Dalianis T, Ramqvist T, Andersson B. Identification of a third human polyomavirus. J Virol. 2007;81:4130–6. doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Gaynor AM, Nissen MD, Whiley DM, Mackay IM, Lambert SB, Wu G, Brennan DC, Storch GA, Sloots TP, Wang D. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007;3:e64. doi: 10.1371/journal.ppat.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.White MK, Khalili K. Polyomaviruses and human cancer: molecular mechanisms underlying patterns of tumorigenesis. Virology. 2004;324:1–16. doi: 10.1016/j.virol.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 352.Shah KV. SV40 and human cancer: a review of recent data. Int J Cancer. 2007;120:215–23. doi: 10.1002/ijc.22425. [DOI] [PubMed] [Google Scholar]
- 353.Poulin DL, DeCaprio JA. Is there a role for SV40 in human cancer? J Clin Oncol. 2006;24:4356–65. doi: 10.1200/JCO.2005.03.7101. [DOI] [PubMed] [Google Scholar]
- 354.Gallia GL, Gordon J, Khalili K. Tumor pathogenesis of human neurotropic JC virus in the CNS. J Neurovirol. 1998;4:175–81. doi: 10.3109/13550289809114517. [DOI] [PubMed] [Google Scholar]
- 355.Das D, Shah RB, Imperiale MJ. Detection and expression of human BK virus sequences in neoplastic prostate tissues. Oncogene. 2004;23:7031–46. doi: 10.1038/sj.onc.1207920. [DOI] [PubMed] [Google Scholar]
- 356.Benko MB, Harrach B, Russell WC. In: Van Regenmortel MHV, Fauquet CM, Bishop DHL, Carsten EB, Estes MK, Lemon SM, Maniloff J, Mayo MA, McGeoch DJ, Pringle R, Wickner RB, editors. Virus Taxonomy; Seventh Report of the International Committee on Taxonomy of Viruses; New York: Academic Press; 2000. [Google Scholar]
- 357.Garnett CT, Erdman D, Xu W, Gooding LR. Prevalence and quantitation of species C adenovirus DNA in human mucosal lymphocytes. J Virol. 2002;76:10608–16. doi: 10.1128/JVI.76.21.10608-10616.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Horvath J, Palkonyay L, Weber J. Group C adenovirus DNA sequences in human lymphoid cells. J Virol. 1986;59:189–92. doi: 10.1128/jvi.59.1.189-192.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Mackey JK, Rigden PM, Green M. Do highly oncogenic group A human adenoviruses cause human cancer? Analysis of human tumors for adenovirus 12 transforming DNA sequences. Proc Natl Acad Sci U S A. 1976;73:4657–61. doi: 10.1073/pnas.73.12.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Wold WS, Mackey JK, Rigden P, Green M. Analysis of human cancer DNA’s for DNA sequence of human adenovirus serotypes 3, 7, 11, 14, 16, and 21 in group B1. Cancer Res. 1979;39:3479–84. [PubMed] [Google Scholar]
- 361.Green M, Wold WS, Mackey JK, Rigden P. Analysis of human tonsil and cancer DNAs and RNAs for DNA sequences of group C (serotypes 1, 2, 5, and 6) human adenoviruses. Proc Natl Acad Sci U S A. 1979;76:6606–10. doi: 10.1073/pnas.76.12.6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Kosulin K, Haberler C, Hainfellner JA, Amann G, Lang S, Lion T. Investigation of adenovirus occurrence in pediatric tumor entities. J Virol. 2007;81:7629–35. doi: 10.1128/JVI.00355-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Javier RT. Adenovirus type 9 E4 open reading frame 1 encodes a transforming protein required for the production of mammary tumors in rats. J Virol. 1994;68:3917–24. doi: 10.1128/jvi.68.6.3917-3924.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Nevels M, Tauber B, Spruss T, Wolf H, Dobner T. “Hit-and-run” transformation by adenovirus oncogenes. J Virol. 2001;75:3089–94. doi: 10.1128/JVI.75.7.3089-3094.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Lower R. The pathogenic potential of endogenous retroviruses: facts and fantasies. Trends Microbiol. 1999;7:350–6. doi: 10.1016/s0966-842x(99)01565-6. [DOI] [PubMed] [Google Scholar]
- 366.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 367.Andersson AC, Svensson AC, Rolny C, Andersson G, Larsson E. Expression of human endogenous retrovirus ERV3 (HERV-R) mRNA in normal and neoplastic tissues. Int J Oncol. 1998;12:309–13. doi: 10.3892/ijo.12.2.309. [DOI] [PubMed] [Google Scholar]
- 368.Sauter M, Schommer S, Kremmer E, Remberger K, Dolken G, Lemm I, Buck M, Best B, Neumann-Haefelin D, Mueller-Lantzsch N. Human endogenous retrovirus K10: expression of Gag protein and detection of antibodies in patients with seminomas. J Virol. 1995;69:414–21. doi: 10.1128/jvi.69.1.414-421.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Lower R, Lower J, Tondera-Koch C, Kurth R. A general method for the identification of transcribed retrovirus sequences (R-U5 PCR) reveals the expression of the human endogenous retrovirus loci HERV-H and HERV-K in teratocarcinoma cells. Virology. 1993;192:501–11. doi: 10.1006/viro.1993.1066. [DOI] [PubMed] [Google Scholar]
- 370.Schulte AM, Lai S, Kurtz A, Czubayko F, Riegel AT, Wellstein A. Human trophoblast and choriocarcinoma expression of the growth factor pleiotrophin attributable to germ-line insertion of an endogenous retrovirus. Proc Natl Acad Sci U S A. 1996;93:14759–64. doi: 10.1073/pnas.93.25.14759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Bera TK, Tsukamoto T, Panda DK, Huang T, Guzman RC, Hwang SI, Nandi S. Defective retrovirus insertion activates c-Ha-ras protooncogene in an MNU-induced rat mammary carcinoma. Biochem Biophys Res Commun. 1998;248:835–40. doi: 10.1006/bbrc.1998.9059. [DOI] [PubMed] [Google Scholar]
- 372.Sauter M, Roemer K, Best B, Afting M, Schommer S, Seitz G, Hartmann M, Mueller-Lantzsch N. Specificity of antibodies directed against Env protein of human endogenous retroviruses in patients with germ cell tumors. Cancer Res. 1996;56:4362–5. [PubMed] [Google Scholar]
- 373.Wang-Johanning F, Liu J, Rycaj K, Huang M, Tsai K, Rosen DG, Chen DT, Lu DW, Barnhart KF, Johanning GL. Expression of multiple human endogenous retrovirus surface envelope proteins in ovarian cancer. Int J Cancer. 2007;120:81–90. doi: 10.1002/ijc.22256. [DOI] [PubMed] [Google Scholar]
- 374.Muster T, Waltenberger A, Grassauer A, Hirschl S, Caucig P, Romirer I, Fodinger D, Seppele H, Schanab O, Magin-Lachmann C, Lower R, Jansen B, Pehamberger H, Wolff K. An endogenous retrovirus derived from human melanoma cells. Cancer Res. 2003;63:8735–41. [PubMed] [Google Scholar]
- 375.Buscher K, Trefzer U, Hofmann M, Sterry W, Kurth R, Denner J. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 2005;65:4172–80. doi: 10.1158/0008-5472.CAN-04-2983. [DOI] [PubMed] [Google Scholar]
- 376.Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, Yu J, Wang L, Montie JE, Rubin MA, Pienta KJ, Roulston D, Shah RB, Varambally S, Mehra R, Chinnaiyan AM. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–9. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 377.Denne M, Sauter M, Armbruester V, Licht JD, Roemer K, Mueller-Lantzsch N. Physical and functional interactions of human endogenous retrovirus proteins Np9 and rec with the promyelocytic leukemia zinc finger protein. J Virol. 2007;81:5607–16. doi: 10.1128/JVI.02771-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Bittner JJ. Some possible effects of nursing on the mammary gland tumor incidence in mice. Science. 1936;84:162. doi: 10.1126/science.84.2172.162. [DOI] [PubMed] [Google Scholar]
- 379.Wang Y, Holland JF, Bleiweiss IJ, Melana S, Liu X, Pelisson I, Cantarella A, Stellrecht K, Mani S, Pogo BG. Detection of mammary tumor virus env gene-like sequences in human breast cancer. Cancer Res. 1995;55:5173–9. [PubMed] [Google Scholar]
- 380.Wang Y, Pelisson I, Melana SM, Holland JF, Pogo BG. Detection of MMTV-like LTR and LTR-env gene sequences in human breast cancer. Int J Oncol. 2001;18:1041–4. doi: 10.3892/ijo.18.5.1041. [DOI] [PubMed] [Google Scholar]
- 381.Liu B, Wang Y, Melana SM, Pelisson I, Najfeld V, Holland JF, Pogo BG. Identification of a proviral structure in human breast cancer. Cancer Res. 2001;61:1754–9. [PubMed] [Google Scholar]
- 382.Melana SM, Nepomnaschy I, Sakalian M, Abbott A, Hasa J, Holland JF, Pogo BG. Characterization of viral particles isolated from primary cultures of human breast cancer cells. Cancer Res. 2007;67:8960–5. doi: 10.1158/0008-5472.CAN-06-3892. [DOI] [PubMed] [Google Scholar]
- 383.Bindra A, Muradrasoli S, Kisekka R, Nordgren H, Warnberg F, Blomberg J. Search for DNA of exogenous mouse mammary tumor virus-related virus in human breast cancer samples. J Gen Virol. 2007;88:1806–9. doi: 10.1099/vir.0.82767-0. [DOI] [PubMed] [Google Scholar]
- 384.Stewart TH, Sage RD, Stewart AF, Cameron DW. Breast cancer incidence highest in the range of one species of house mouse, Mus domesticus. Br J Cancer. 2000;82:446–51. doi: 10.1054/bjoc.1999.0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Stewart AF. Identification of human homologues of the mouse mammary tumor virus receptor. Arch Virol. 2002;147:577–81. doi: 10.1007/s007050200007. [DOI] [PubMed] [Google Scholar]
- 386.Indik S, Gunzburg WH, Kulich P, Salmons B, Rouault F. Rapid spread of mouse mammary tumor virus in cultured human breast cells. Retrovirology. 2007;4:73. doi: 10.1186/1742-4690-4-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Carpten J, Nupponen N, Isaacs S, Sood R, Robbins C, Xu J, Faruque M, Moses T, Ewing C, Gillanders E, Hu P, Bujnovszky P, Makalowska I, Baffoe-Bonnie A, Faith D, Smith J, Stephan D, Wiley K, Brownstein M, Gildea D, Kelly B, Jenkins R, Hostetter G, Matikainen M, Schleutker J, Klinger K, Connors T, Xiang Y, Wang Z, De Marzo A, Papadopoulos N, Kallioniemi OP, Burk R, Meyers D, Gronberg H, Meltzer P, Silverman R, Bailey-Wilson J, Walsh P, Isaacs W, Trent J. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet. 2002;30:181–4. doi: 10.1038/ng823. [DOI] [PubMed] [Google Scholar]
- 388.Zhou A, Hassel BA, Silverman RH. Expression cloning of 2-5A-dependent RNAase: a uniquely regulated mediator of interferon action. Cell. 1993;72:753–65. doi: 10.1016/0092-8674(93)90403-d. [DOI] [PubMed] [Google Scholar]
- 389.Urisman A, Molinaro RJ, Fischer N, Plummer SJ, Casey G, Klein EA, Malathi K, Magi-Galluzzi C, Tubbs RR, Ganem D, Silverman RH, DeRisi JL. Identification of a novel Gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathog. 2006;2:e25. doi: 10.1371/journal.ppat.0020025. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 390.Ukita M, Okamoto H, Nishizawa T, Tawara A, Takahashi M, Iizuka H, Miyakawa Y, Mayumi M. The entire nucleotide sequences of two distinct TT virus (TTV) isolates (TJN01 and TJN02) remotely related to the original TTV isolates. Arch Virol. 2000;145:1543–59. doi: 10.1007/s007050070075. [DOI] [PubMed] [Google Scholar]
- 391.Okamoto H, Takahashi M, Nishizawa T, Ukita M, Fukuda M, Tsuda F, Miyakawa Y, Mayumi M. Marked genomic heterogeneity and frequent mixed infection of TT virus demonstrated by PCR with primers from coding and noncoding regions. Virology. 1999;259:428–36. doi: 10.1006/viro.1999.9770. [DOI] [PubMed] [Google Scholar]
- 392.Hallett RL, Clewley JP, Bobet F, McKiernan PJ, Teo CG. Characterization of a highly divergent TT virus genome. J Gen Virol. 2000;81:2273–9. doi: 10.1099/0022-1317-81-9-2273. [DOI] [PubMed] [Google Scholar]
- 393.Takahashi K, Iwasa Y, Hijikata M, Mishiro S. Identification of a new human DNA virus (TTV-like mini virus, TLMV) intermediately related to TT virus and chicken anemia virus. Arch Virol. 2000;145:979–93. doi: 10.1007/s007050050689. [DOI] [PubMed] [Google Scholar]
- 394.Biagini P, Attoui H, Gallian P, Touinssi M, Cantaloube JF, de Micco P, de Lamballerie X. Complete sequences of two highly divergent european isolates of TT virus. Biochem Biophys Res Commun. 2000;271:837–41. doi: 10.1006/bbrc.2000.2721. [DOI] [PubMed] [Google Scholar]
- 395.Tanaka Y, Primi D, Wang RY, Umemura T, Yeo AE, Mizokami M, Alter HJ, Shih JW. Genomic and molecular evolutionary analysis of a newly identified infectious agent (SEN virus) and its relationship to the TT virus family. J Infect Dis. 2001;183:359–67. doi: 10.1086/318091. [DOI] [PubMed] [Google Scholar]
- 396.Davidson F, MacDonald D, Mokili JL, Prescott LE, Graham S, Simmonds P. Early acquisition of TT virus (TTV) in an area endemic for TTV infection. J Infect Dis. 1999;179:1070–6. doi: 10.1086/314730. [DOI] [PubMed] [Google Scholar]
- 397.Gerner P, Oettinger R, Gerner W, Falbrede J, Wirth S. Mother-to-infant transmission of TT virus: prevalence, extent and mechanism of vertical transmission. Pediatr Infect Dis J. 2000;19:1074–7. doi: 10.1097/00006454-200011000-00009. [DOI] [PubMed] [Google Scholar]
- 398.Ball JK, Curran R, Berridge S, Grabowska AM, Jameson CL, Thomson BJ, Irving WL, Sharp PM. TT virus sequence heterogeneity in vivo: evidence for co-infection with multiple genetic types. J Gen Virol. 1999;80(Pt 7):1759–68. doi: 10.1099/0022-1317-80-7-1759. [DOI] [PubMed] [Google Scholar]
- 399.Niel C, Saback FL, Lampe E. Coinfection with multiple TT virus strains belonging to different genotypes is a common event in healthy Brazilian adults. J Clin Microbiol. 2000;38:1926–30. doi: 10.1128/jcm.38.5.1926-1930.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Yotsuyanagi H, Shintani Y, Moriya K, Fujie H, Tsutsumi T, Kato T, Nishioka K, Takayama T, Makuuchi M, Iino S, Kimura S, Koike K. Virologic analysis of non-B, non-C hepatocellular carcinoma in Japan: frequent involvement of hepatitis B virus. J Infect Dis. 2000;181:1920–8. doi: 10.1086/315512. [DOI] [PubMed] [Google Scholar]
- 401.Kim SR, Hayashi Y, Kudo M, Imoto S, Song KB, Ando K, Shintani S, Koterazawa T, Kim KI, Taniguchi M. TTV positivity and transfusion history in non-B, non-C hepatocellular carcinoma compared with HBV- and HCV-positive cases. Intervirology. 2000;43:13–5. doi: 10.1159/000025017. [DOI] [PubMed] [Google Scholar]
- 402.Pineau P, Meddeb M, Raselli R, Qin LX, Terris B, Tang ZY, Tiollais P, Mazzaferro V, Dejean A. Effect of TT virus infection on hepatocellular carcinoma development: results of a Euro-Asian survey. J Infect Dis. 2000;181:1138–42. doi: 10.1086/315321. [DOI] [PubMed] [Google Scholar]
- 403.de Villiers EM, Schmidt R, Delius H, zur Hausen H. Heterogeneity of TT virus related sequences isolated from human tumour biopsy specimens. J Mol Med. 2002;80:44–50. doi: 10.1007/s001090100281. [DOI] [PubMed] [Google Scholar]
- 404.Finzer P, Kuntzen C, Soto U, zur Hausen H, Rosl F. Inhibitors of histone deacetylase arrest cell cycle and induce apoptosis in cervical carcinoma cells circumventing human papillomavirus oncogene expression. Oncogene. 2001;20:4768–76. doi: 10.1038/sj.onc.1204652. [DOI] [PubMed] [Google Scholar]
- 405.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH. Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997;278:1064–8. doi: 10.1126/science.278.5340.1064. [DOI] [PubMed] [Google Scholar]
- 406.Villa LL. Prophylactic HPV vaccines: reducing the burden of HPV-related diseases. Vaccine. 2006;24(Suppl 1):S23–8. doi: 10.1016/j.vaccine.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 407.Frazer IH. Prevention of cervical cancer through papillomavirus vaccination. Nat Rev Immunol. 2004;4:46–54. doi: 10.1038/nri1260. [DOI] [PubMed] [Google Scholar]