Immune Control of an SIV Challenge by a T Cell-Based Vaccine in Rhesus Monkeys (original) (raw)
. Author manuscript; available in PMC: 2009 Jul 1.
Published in final edited form as: Nature. 2008 Nov 9;457(7225):87–91. doi: 10.1038/nature07469
Abstract
A recombinant adenovirus serotype 5 (rAd5) vector-based vaccine for HIV-1 has recently failed in a phase 2b efficacy study in humans1, 2. Consistent with these results, preclinical studies have demonstrated that rAd5 vectors expressing SIV Gag failed to reduce peak or setpoint viral loads following SIV challenge of rhesus monkeys that lacked the protective MHC class I allele Mamu-A*013. Here we show that an improved T cell-based vaccine regimen utilizing two serologically distinct adenovirus vectors afforded substantially improved protective efficacy in this stringent challenge model. In particular, a heterologous rAd26 prime, rAd5 boost vaccine regimen expressing SIV Gag elicited cellular immune responses with augmented magnitude, breadth, and polyfunctionality as compared with the homologous rAd5 regimen. Following SIVmac251 challenge, monkeys vaccinated with the heterologous rAd26/rAd5 regimen exhibited a 1.4 log reduction of peak and a 2.4 log reduction of setpoint viral loads as well as decreased AIDS-related mortality as compared with control animals. These data demonstrate that durable partial immune control of a pathogenic SIV challenge for over 500 days can be achieved by a T cell-based vaccine in Mamu-A*01-negative rhesus monkeys in the absence of a homologous Env antigen. These findings have important implications for the development of next generation T cell-based vaccine candidates for HIV-1.
Recombinant Ad5 vector-based vaccines expressing SIV Gag have been shown to afford dramatic control of viral replication following simian-human immunodeficiency virus (SHIV) 89.6P challenge of rhesus monkeys4, 5. However, rAd5-Gag vaccines have failed to reduce peak or setpoint viral loads following SIVmac239 challenge of rhesus monkeys3, highlighting important differences in the stringencies of these challenge models. Heterologous DNA prime, rAd5 boost vaccine regimens have also failed to date to reduce setpoint viral loads following SIV challenge of rhesus monkeys that lacked the protective MHC class I allele Mamu-A*013, 6. The inability of vector-based vaccines to afford durable control of setpoint viral loads following SIV challenge of Mamu-A*01-negative rhesus monkeys has led to substantial debate regarding the viability of the concept of developing T cell-based vaccines for HIV-1.
Pre-existing Ad5-specific NAbs have been reported to reduce the immunogenicity of rAd5 vector-based vaccines in clinical trials7, 8 and may also compromise their safety1. Rare serotype rAd vectors, such as rAd35 and rAd26 vectors9-12, have been developed as potential alternatives. Serologically distinct rAd vectors also allow the potential development of heterologous rAd prime-boost regimens. To investigate the immunogenicity and protective efficacy of such regimens, we immunized 22 Indian-origin rhesus monkeys that lacked the protective MHC class I alleles Mamu-A*0113-15 and Mamu-B*1716 with the following heterologous or homologous rAd prime-boost regimens: (1) rAd26-Gag prime, rAd5-Gag boost (N=6); (2) rAd35-Gag prime, rAd5-Gag boost (N=6); (3) rAd5-Gag prime, rAd5-Gag boost (N=4); and (4) sham controls (N=6). One monkey each in Groups 1, 3, and 4 expressed the protective Mamu-B*08 allele. Monkeys were primed at week 0 and boosted at week 24 with 1011 vp of each vector expressing SIVmac239 Gag. At week 52, all animals received a high-dose i.v. challenge with 100 infectious doses of SIVmac2516.
Prior to challenge, we monitored vaccine-elicited SIV Gag-specific cellular (Fig. 1a-c) and humoral (Fig. 1d) immune responses in these animals. Following the priming immunization, IFN-γ ELISPOT responses to pooled SIV Gag peptides were observed in all vaccinees. Monkeys primed with rAd26-Gag and rAd35-Gag were efficiently boosted by the heterologous rAd5-Gag vector to peak responses of 2,513 and 1,163 spot-forming cells (SFC) per 106 PBMC, respectively, two weeks following the boost immunization (Fig. 1a; green bars). In contrast, monkeys primed with rAd5-Gag were only marginally boosted by a second injection of rAd5-Gag as a result of anti-vector immunity generated by the priming immunization11, 17. Cell-depleted ELISPOT assays demonstrated that these responses were primarily CD8+ T lymphocyte responses, although lower levels of CD4+ T lymphocyte responses were also clearly observed (Fig. 1b). Epitope mapping was then performed by assessing ELISPOT responses against all 125 individual 15 amino acid SIV Gag peptides following the boost immunization. The rAd26/rAd5 regimen elicited a mean of 8.6 detectable Gag epitopes per animal, whereas the rAd35/rAd5 regimen elicited a mean of 4.5 epitopes per animal and the rAd5/rAd5 regimen induced a mean of only 2.2 epitopes per animal (Fig. 1c). These data demonstrate that the heterologous rAd26/rAd5 regimen induced an 8.7-fold greater magnitude and a 3.9-fold increased breadth of Gag-specific cellular immune responses as compared with the homologous rAd5/rAd5 regimen.
Figure 1. Immunogenicity of heterologous rAd prime-boost vaccine regimens.
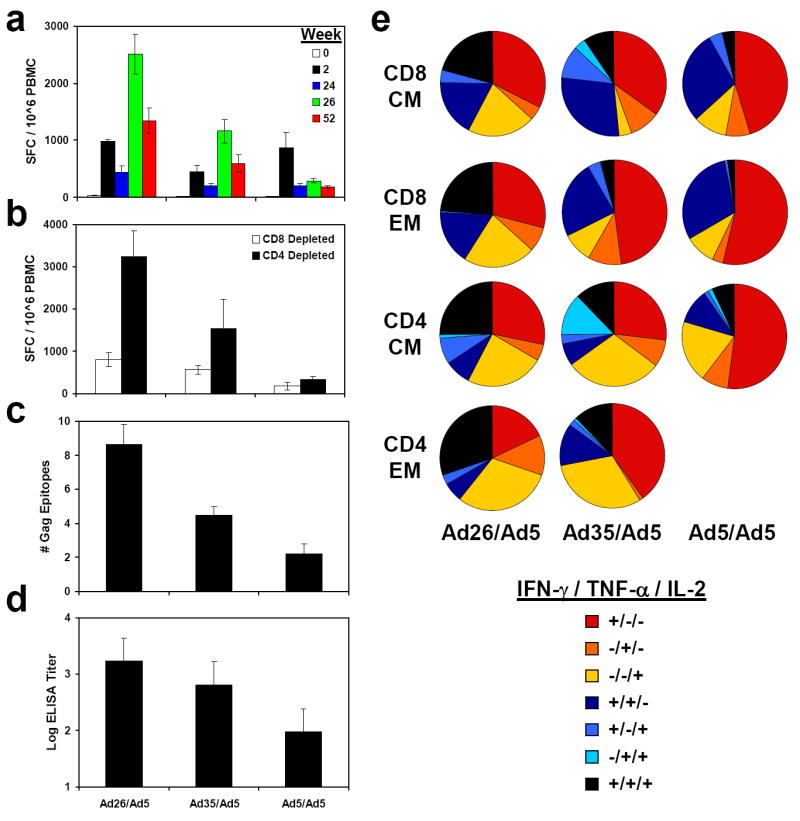
Rhesus monkeys were primed at week 0 and boosted at week 24 with rAd26/rAd5, rAd35/rAd5, or rAd5/rAd5 regimens expressing SIV Gag. a, Gag-specific IFN-γ ELISPOT assays were performed at weeks 0, 2, 24, 26, and 52 following immune priming. b, CD4+ (white bars) and CD8+ (black bars) T lymphocyte responses were evaluated at week 28 by CD8-depleted and CD4-depleted ELISPOT assays, respectively. c, Breadth of responses was determined by Gag epitope mapping at week 28. d, Gag-specific antibody responses were determined by ELISA at week 28. Mean responses with standard errors are shown (a-d). e, Functionality of Gag-specific CD8+ and CD4+ central memory (CM; CD28+CD95+) and effector memory (EM; CD28-CD95+) T lymphocyte responses were assessed by 8-color intracellular cytokine staining (ICS) assays. Proportions of IFN-γ, TNF-α, and IL-2 responses are depicted individually and in all possible combinations for each cellular subpopulation. CD4+ EM responses following rAd5/rAd5 immunization were below the detection limit of the assay.
We next assessed the functionality of the vaccine-elicited, Gag-specific T lymphocyte responses by multiparameter flow cytometry18, 19. Intracellular cytokine staining (ICS) assays were performed to assess IFN-γ, TNF-α, and IL-2 secretion in CD8+ and CD4+ central memory (CM; CD28+CD95+) and effector memory (EM; CD28-CD95+) T lymphocyte subpopulations20, 21. Consistent with our previous findings11, we observed larger proportions of polyfunctional IFN-γ/TNF-α/IL-2-positive (black) and IL-2-positive (yellow) CD8+ and CD4+ T lymphocyte responses in the animals that received the heterologous rAd26/rAd5 regimen as compared with the homologous rAd5/rAd5 regimen (Fig. 1e). In contrast, the rAd5/rAd5 regimen elicited primarily IFN-γ-positive (red) and IFN-γ/TNF-α-positive (blue) responses.
Six months following the boost immunization, all animals were challenged i.v. with SIVmac251. Protective efficacy was evaluated by monitoring plasma SIV RNA levels (Fig. 2a, b), CD4+ T lymphocyte counts (Fig. 2c), and clinical disease progression and mortality (Fig. 2d) following challenge. Monkeys that received the rAd26/rAd5 regimen exhibited mean peak viral loads that were 1.43 logs lower than the mean peak viral loads in the control animals (Fig. 2a, b; P=0.002, two-sided Wilcoxon rank-sum test with multiple comparison adjustments). More importantly, the rAd26/rAd5 vaccinated animals also demonstrated durable partial control of setpoint viral loads, as defined as the mean SIV RNA levels from day 112 to day 420 following challenge. In particular, the rAd26/rAd5 vaccinated monkeys maintained a 2.44 log reduction of setpoint viral loads as compared with the control animals (Fig. 2a, b; P=0.01). If the three Mamu-B*08-positive animals are excluded from the analysis, then the rAd26/rAd5 regimen afforded a 2.66 log reduction of setpoint viral loads as compared with control animals (P=0.008; data not shown). As expected, the homologous rAd5/rAd5 regimen afforded no detectable control of setpoint viral loads, consistent with the previously reported failure of this regimen in this stringent SIV challenge model3.
Figure 2. Protective efficacy of heterologous rAd prime-boost vaccine regimens.
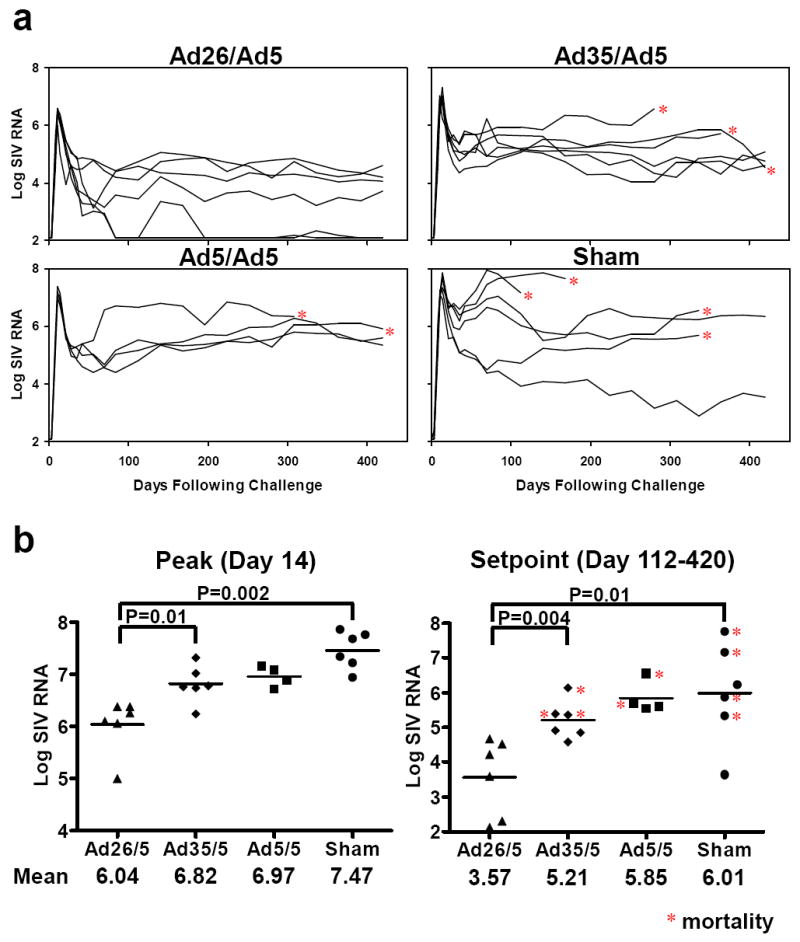
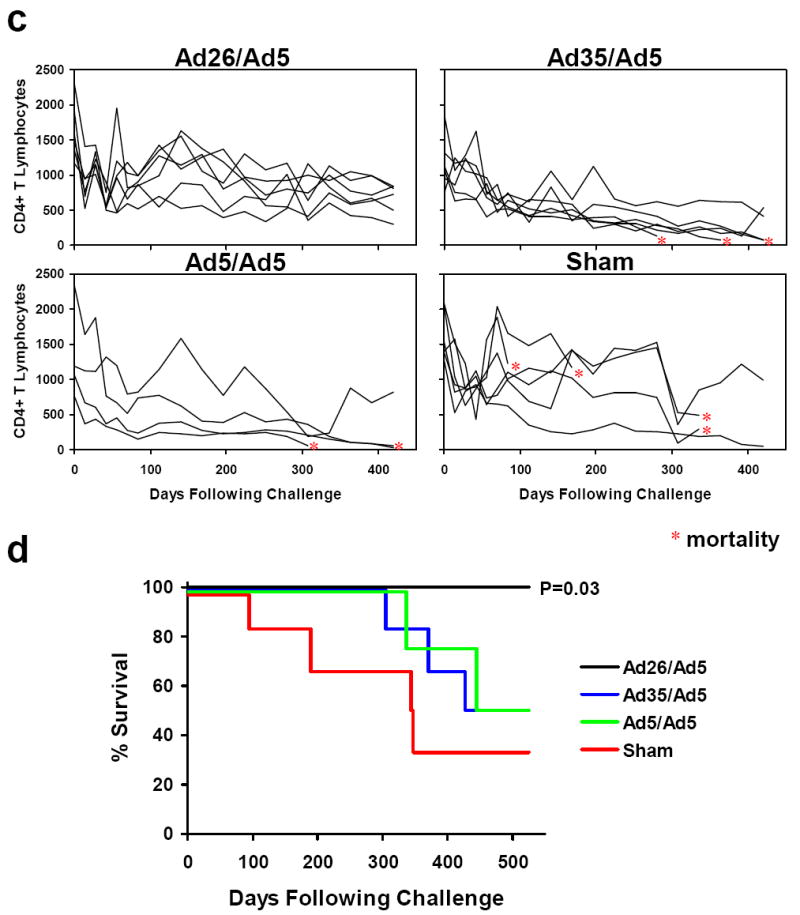
Monkeys were challenged i.v. with SIVmac251, and protective efficacy was monitored by SIV RNA levels (a, b), CD4+ T lymphocyte counts (c), and clinical disease progression and mortality (d) following challenge. Viral loads are depicted longitudinally for each group (a), and peak (day 14) and setpoint (day 112-420) viral loads are summarized for each group (b). Asterisks indicate mortality. Comparisons among groups were performed by two-sided Wilcoxon rank-sum (a, b) and Fisher’s exact (d) tests.
Monkeys that received the rAd26/rAd5 regimen also demonstrated slower declines in CD4+ T lymphocyte counts as compared with the other groups (Fig. 2c). Moreover, the rAd26/rAd5 vaccine afforded a significant reduction in AIDS-related mortality as compared with the controls at day 500 following challenge (Fig. 2d; P=0.03, Fisher’s exact test), whereas the rAd35/rAd5 and rAd5/rAd5 vaccines provided a trend towards a survival advantage. Necropsies revealed that the causes of death were AIDS-defining illnesses, including Pneumocystis pneumonia, Cryptosporidium enteritis, cytomegalovirus pneumonia, SIV encephalitis, and lymphoma.
We next evaluated the evolution of SIV-specific cellular (Fig. 3a, c, e) and humoral (Fig. 3b, d) immune responses in these animals following challenge. The rAd26/rAd5 vaccinees exhibited 10-fold greater anamestic Gag-specific ELISPOT responses as compared with the controls during both acute and chronic infection (Fig. 3a; black bars), suggesting the critical importance of these responses for immune control. In contrast, Pol-, Nef- and Env-specific cellular immune responses were either comparable or lower in magnitude in the rAd26/rAd5 vaccinees as compared with the other groups (Fig. 3a). The rAd26/rAd5 vaccinees thus exhibited less extensive diversification of cellular immune responses against multiple SIV antigens as compared with the other groups, likely reflecting the reduced viral replication in these animals. Phenotypic analysis of the Gag-specific CD8+ T lymphocyte responses in the rAd26/rAd5 vaccinees demonstrated slightly higher expression of IFN-γ and TNF-α but 6.7-fold greater expression of IL-2 as compared with the rAd5/rAd5 vaccinees following challenge (Fig. 3c; green bars). These data suggest that not only quantitative but also qualitative differences among the vaccine regimens may have contributed to protective efficacy.
Figure 3. Cellular and humoral immune responses following challenge.
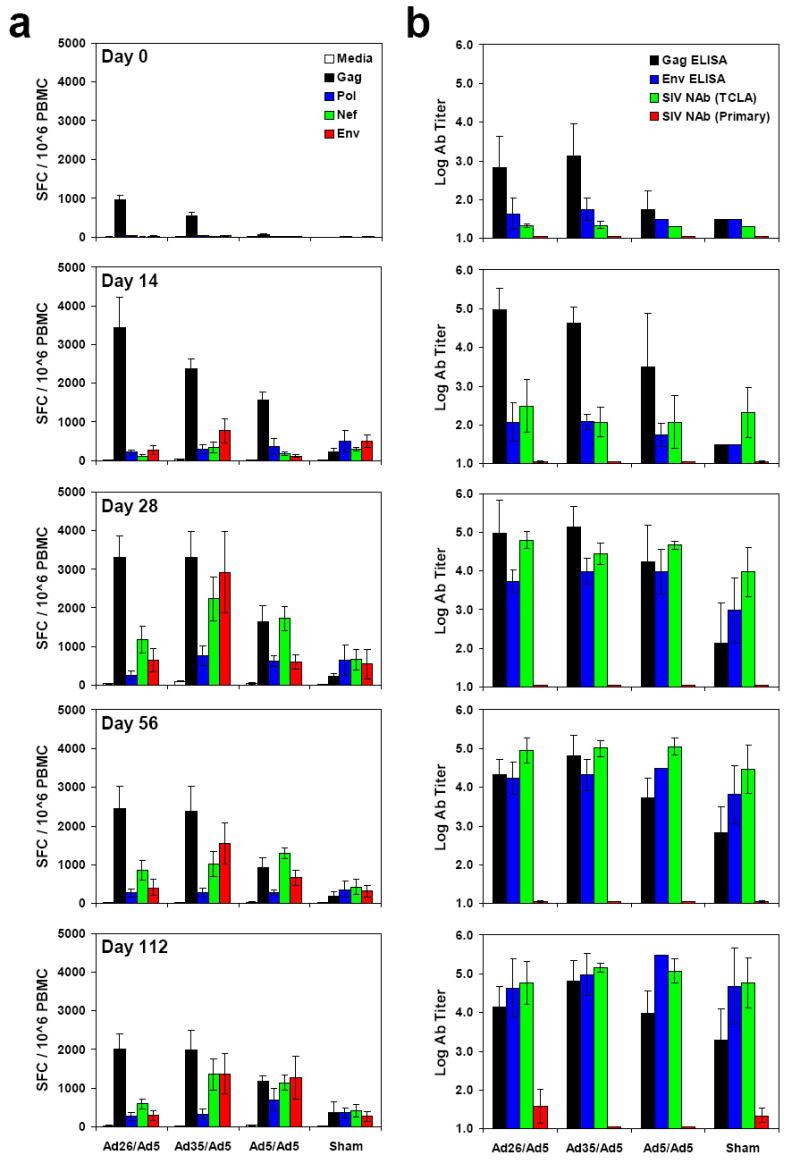
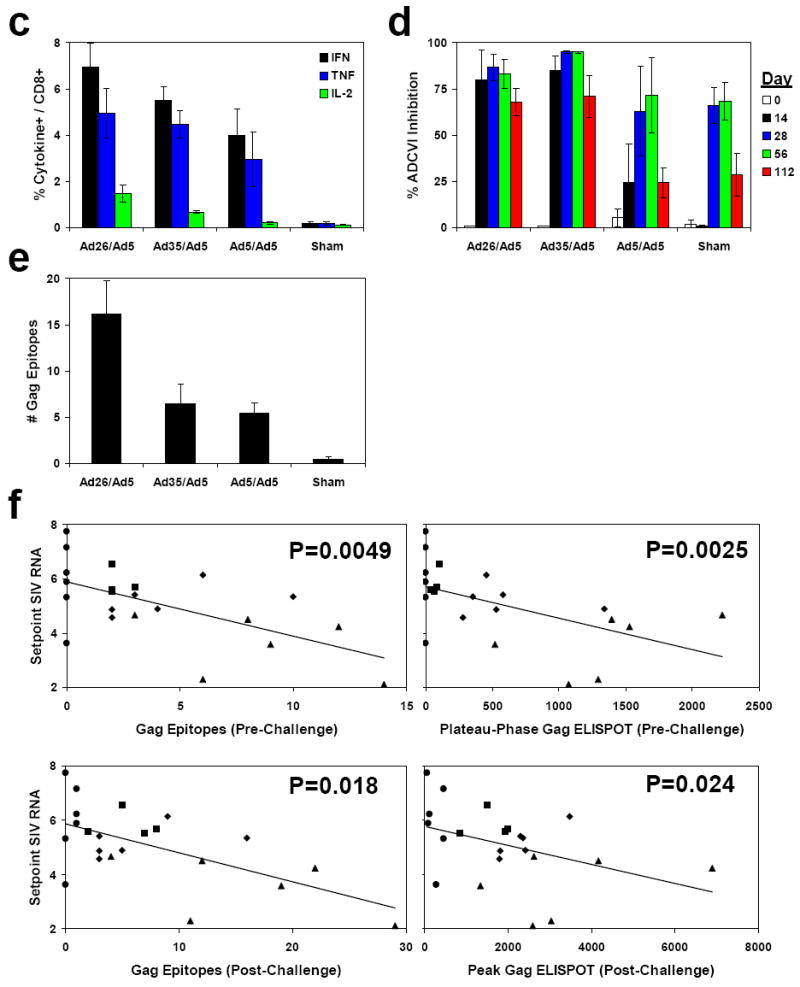
Cellular (a, c, e) and humoral (b, d) immune responses were assessed following challenge. IFN-γ ELISPOT assays (a) and ELISA and NAb assays (b) were performed on days 0, 14, 28, 56, and 112 following challenge. 8-color ICS assays on day 28 (c), ADCVI assays at 1:100 serum dilution at multiple timepoints (d), and Gag epitope mapping studies on day 28 (e) were also performed. Mean responses with standard errors are shown (a-e). f, Correlations between the breadth (left panels) or magnitude (right panels) of pre-challenge (upper panels) or post-challenge (lower panels) Gag-specific cellular immune responses and setpoint viral loads were evaluated by two-sided Spearman rank correlation tests. Monkeys immunized with rAd26/rAd5 (triangles), rAd35/rAd5 (diamonds), rAd5/rAd5 (squares), and sham (circles) vaccine regimens are depicted.
We assessed the breadth of Gag-specific cellular immune responses on day 28 following challenge by epitope mapping using 15 amino acid peptides (Fig. 3e). The monkeys that received the rAd26/rAd5 regimen developed a mean of 16.1 (range 4-29) Gag epitope-specific responses following challenge. In contrast, the control animals developed <1 detectable Gag epitope-specific response following challenge. Importantly, both the breadth and the magnitude of Gag-specific cellular immune responses elicited by vaccination (P=0.0049 and P=0.0025, respectively, Spearman rank-correlation tests) and recalled following challenge (P=0.018 and P=0.024, respectively) correlated with control of setpoint viral loads in these animals (Fig. 3f).
Humoral immune responses were evaluated by Gag- and Env-specific ELISAs as well as by luciferase-based pseudovirus neutralizing antibody (NAb) assays utilizing both TCLA-adapted and primary isolate SIVmac251 viruses22 (Fig. 3b). The vaccinees developed more rapid kinetics of Gag-specific ELISA responses as compared with the controls following challenge, but no clear differences of Env-specific ELISA or NAb responses were observed among groups. The vaccinees also developed more rapid kinetics of antibody-dependent cell-mediated virus inhibition (ADCVI)23 as compared with the controls on day 14 following challenge (Fig. 3d; P=0.01), suggesting the potential functional relevance of nonclassical antibody responses in early SIV infection.
We next monitored CD4+ T lymphocyte dynamics in these animals following challenge. The monkeys that received the rAd26/rAd5 regimen exhibited a relative preservation of central memory CD4+ T lymphocytes6 (Fig. 4a; black bars; P=0.02, Wilcoxon rank-sum test) as well as a striking preservation of CCR5+ central memory CD4+ T lymphocytes24, 25 (Fig. 4b; black bars; P=0.002) as compared with the controls on day 14 following challenge. The rAd26/rAd5 vaccinees also exhibited reduced Ki67+ proliferation of CCR5+ central memory (Fig. 4d; P=0.004) and effector memory (Fig. 4f; P=0.004) CD4+ T lymphocytes as compared with the controls following challenge. In addition, monkeys that received the rAd26/rAd5 regimen maintained markedly higher levels of gastrointestinal CD4+ T lymphocytes in duodenal biopsies on day 21 following challenge as compared with the controls, which showed extensive depletion of this lymphocyte population during acute infection as expected26-28 (Fig. 4g; P=0.02, Wilcoxon rank-sum test).
Figure 4. CD4+ T lymphocyte dynamics following challenge.
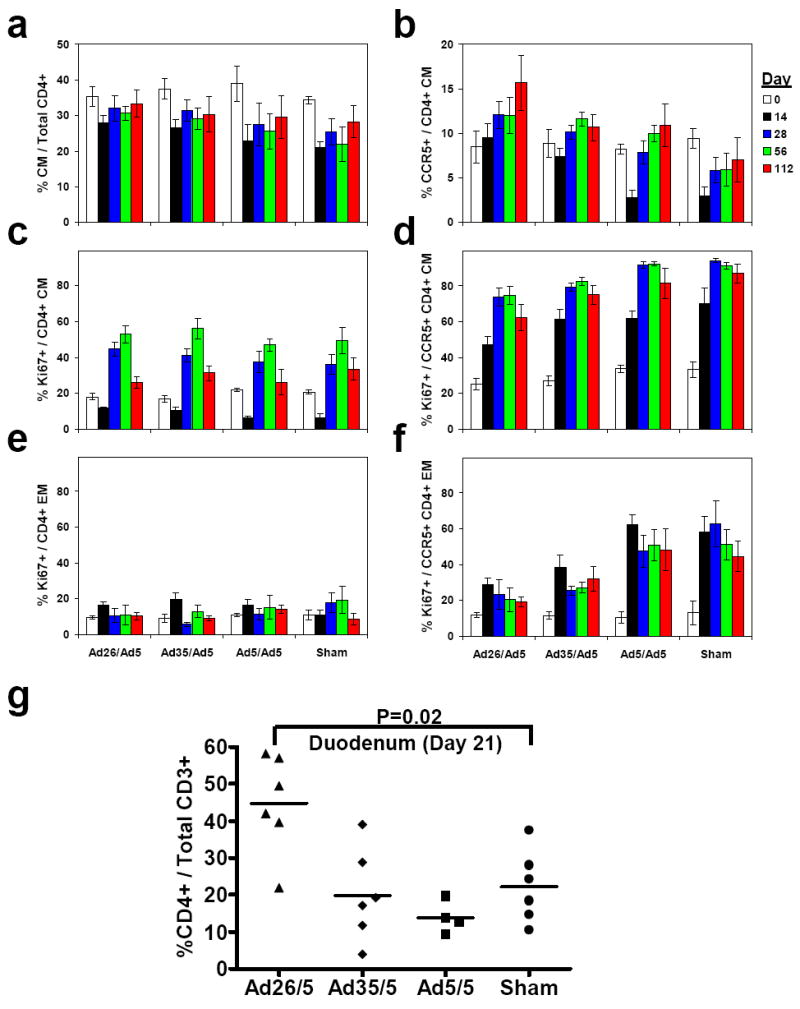
Preservation of central memory (CM; CD28+CD95+) CD4+ T lymphocytes (a) and CCR5+ central memory CD4+ T lymphocytes (b) was assessed on days 0, 14, 28, 56, and 112 following challenge. Ki67 staining of central memory CD4+ (c), CCR5+ central memory CD4+ (d), effector memory (EM; CD28-CD95+) CD4+ (e), and CCR5+ effector memory CD4+ (f) T lymphocytes was also determined. Mean responses with standard errors are shown (a-f). g, Preservation of gastrointestinal CD4+ T lymphocytes was assessed in duodenal biopsies on day 21 following challenge. Comparisons were performed by two-sided Wilcoxon rank-sum tests.
Our data demonstrate that the heterologous rAd26/rAd5 regimen elicited improved magnitude, breadth, and functionality of Gag-specific cellular immune responses as compared with the homologous rAd5/rAd5 regimen and afforded durable partial immune control of a homologous SIVmac251 challenge in rhesus monkeys. To the best of our knowledge, vector-based vaccines have not previously been reported to reduce setpoint viral loads in the stringent system of SIV challenge of Mamu-A*01-negative rhesus monkeys3, 6, although a previous study has shown partial control of setpoint viral loads using a DNA/rAd5 regimen in the less stringent system of Mamu-A*01-positive rhesus monkeys29. In the present study, the breadth and magnitude of vaccine-elicited, Gag-specific cellular immune responses prior to challenge correlated with control of setpoint viral loads following challenge (Fig. 3f), suggesting the critical importance of Gag-specific T lymphocyte responses in controlling viral replication. Protective efficacy was also associated with improved functionality of Gag-specific T lymphocyte responses (Fig. 1e, 3c), reduced diversification of responses against other SIV antigens following challenge (Fig. 3a), and preservation of CCR5+ central memory CD4+ T lymphocytes and gastrointestinal CD4+ T lymphocytes following challenge (Fig. 4b, g), although some of these parameters may reflect the result rather than the cause of the reduced viral replication.
It is important to highlight the fact that the vaccines utilized in the present study expressed only a single SIV Gag antigen and did not include a homologous Env immunogen. The observed protective efficacy was therefore likely mediated by vaccine-elicited Gag-specific cellular immune responses, since it is unlikely that Gag-specific antibodies afforded substantial protection. Consistent with these data, a previous study reported that the breadth of Gag-specific cellular immune responses correlated with control of viral loads in chronically HIV-1-infected humans30. It remains possible, however, that other SIV antigens may also contribute to cellular immune protection if included in a multivalent vaccine. Future studies should address this possibility and evaluate the protective efficacy of optimal vaccine regimens against highly heterologous SIV challenges. The potential role of early ADCVI responses should also be explored, since ADCVI activity emerged more rapidly in the vaccinees as compared with the controls following challenge (Fig. 3d). These responses likely reflect immune preservation in the vaccinees rather than a direct result of vaccination, since ADCVI activity has been reported to be mediated by Env-specific antibodies23.
Despite current controversies regarding the use of rAd vector-based vaccines for HIV-1, our data have important implications regarding the development of next generation T cell-based vaccines by the proof-of-concept demonstration that durable partial immune control of a pathogenic SIV challenge can be achieved in Mamu-A*01-negative rhesus monkeys. It remains unclear, however, whether the observed protection reflected the use of rAd26 vectors, the heterologous rAd prime-boost regimen, or both. Additional studies will therefore be required to evaluate these possibilities and to determine the utility of regimens consisting of two rare serotype rAd vectors. In conclusion, our findings suggest that T cell-based vaccine regimens that elicit augmented magnitude, breadth, and polyfunctionality of cellular immune responses as compared with the homologous rAd5 regimen may afford superior protective efficacy against both HIV-1 and other pathogens.
Methods Summary
Outbred rhesus monkeys were vaccinated with 1011 viral particles (vp) replication-incompetent, E1/E3-deleted rAd5, rAd35, or rAd26 vectors10-12 expressing SIVmac239 Gag at weeks 0 and 24. Sham controls received 1011 vp empty vectors. At week 52, all animals received an i.v. inoculation of 100 infectious doses of SIVmac251 as described6. SIV RNA levels were assessed following challenge on days 0, 3, 7, 10, 14, 21, then weekly until day 42, biweekly until day 84, and monthly thereafter. SIV-specific cellular immune responses were assessed by IFN-γ ELISPOT assays and multiparameter intracellular cytokine staining (ICS) assays11, 20, 21. Gastrointestinal CD4+ T lymphocytes were evaluated following collagenase digestion of duodenal biopsies and percoll gradient purification. SIV-specific humoral immune responses were evaluated by ELISAs, luciferase-based pseudovirus neutralization assays22, and antibody-dependent cell-mediated virus inhibition (ADCVI) assays23.
Methods
Animals, immunizations, and challenge
Outbred rhesus monkeys that did not express the MHC class I alleles Mamu-A*01 and Mamu-B*17 were housed at New England Primate Research Center (NEPRC), Southborough, MA. Immunizations involved 1011 viral particles (vp) replication-incompetent, E1/E3-deleted rAd5, rAd35, or rAd26 vectors10-12 expressing SIVmac239 Gag delivered as 1 ml injections i.m. in both quadriceps muscles at weeks 0 and 24. Sham controls received 1011 vp empty vectors. At week 52, all animals received an i.v. inoculation of 100 infectious doses of SIVmac251 as described6. SIV RNA levels were assessed following challenge on days 0, 3, 7, 10, 14, 21, then weekly until day 42, biweekly until day 84, and monthly thereafter (Siemans Diagnostics). All animal studies were approved by our Institutional Animal Care and Use Committees (IACUC).
Cellular immune assays
SIV-specific cellular immune responses were assessed by IFN-γ ELISPOT assays and multiparameter intracellular cytokine staining (ICS) assays essentially as described11, 20, 21. ELISPOT assays utilized pooled or individual SIV Gag peptides. 8-color ICS assays utilized pooled SIV Gag peptides and the following mAbs (BD Pharmingen): anti-CD3-Alexa700 (SP34), anti-CD4-AmCyan (L200), anti-CD8-APC-Cy7 (SK1), anti-CD28-PerCP-Cy5.5 (L293), anti-CD95-PE (DX2), anti-IFN-γ-PE-Cy7 (B27), anti-IL-2-APC (MQ1-17H12), and anti-TNF-α-FITC (Mab11). Assessment of T lymphocyte dynamics utilized the following mAbs: anti-CD3-Alexa700 (SP34), anti-CD4-AmCyan (L200), anti-CD8-APC-Cy7 (SK1), anti-CD28-PerCP-Cy5.5 (L293), anti-CD95-APC (DX2), anti-CCR5-PE (3A9), anti-HLA-DR-PE-Cy7 (L243), and anti-Ki67-FITC (B56). Gastrointestinal CD4+ T lymphocytes were evaluated following collagenase digestion of duodenal biopsies and percoll gradient purification.
Humoral immune assays
SIV-specific humoral immune responses were evaluated by SIV Gag- and SIV Env gp130-specific ELISAs, luciferase-based pseudovirus neutralization assays (TCLA and primary isolate)22, and antibody-dependent cell-mediated virus inhibition (ADCVI) assays23 essentially as described.
Acknowledgments
We thank R. Vogels, J. Custers, L. Holterman, A. Lemckert, F. Stephens, R. Dolin, N. Letvin, J. Schmitz, M. Lifton, K. Furr, L. Picker, and M. Pensiero for generous advice, assistance, and reagents. The SIV peptide pools were obtained from the NIH AIDS Research and Reference Reagent Program. We acknowledge support from NIH grants AI066305 (D.H.B.), AI066924 (D.H.B.), AI078526 (D.H.B.), AI030034 (D.C.M.), and RR000168 (NEPRC).
Footnotes
Author Information The authors declare no competing financial interests.
References
- 1.Fauci AS. The release of new data from the HVTN 502 (STEP) HIV vaccine study. NIH News. 2007 Nov 7; [Google Scholar]
- 2.Fauci AS. 25 years of HIV. Nature. 2008;453:289–90. doi: 10.1038/453289a. [DOI] [PubMed] [Google Scholar]
- 3.Casimiro DR, et al. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with dna and recombinant adenoviral vaccine vectors expressing Gag. J Virol. 2005;79:15547–55. doi: 10.1128/JVI.79.24.15547-15555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shiver JW, et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 2002;415:331–5. doi: 10.1038/415331a. [DOI] [PubMed] [Google Scholar]
- 5.Shiver JW, Emini EA. Recent advances in the development of HIV-1 vaccines using replication-incompetent adenovirus vectors. Annu Rev Med. 2004;55:355–72. doi: 10.1146/annurev.med.55.091902.104344. [DOI] [PubMed] [Google Scholar]
- 6.Letvin NL, et al. Preserved CD4+ central memory T cells and survival in vaccinated SIV-challenged monkeys. Science. 2006;312:1530–3. doi: 10.1126/science.1124226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Catanzaro AT, et al. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 candidate vaccine delivered by a replication-defective recombinant adenovirus vector. J Infect Dis. 2006;194:1638–49. doi: 10.1086/509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Priddy FH, et al. Safety and immunogenicity of a replication-incompetent adenovirus type 5 HIV-1 clade B gag/pol/nef vaccine in healthy adults. Clinical infectious diseases. 2008;46:1769–81. doi: 10.1086/587993. [DOI] [PubMed] [Google Scholar]
- 9.Barouch DH, et al. Immunogenicity of recombinant adenovirus serotype 35 vaccine in the presence of pre-existing anti-Ad5 immunity. J Immunol. 2004;172:6290–7. doi: 10.4049/jimmunol.172.10.6290. [DOI] [PubMed] [Google Scholar]
- 10.Abbink P, et al. Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J Virol. 2007;81:4654–63. doi: 10.1128/JVI.02696-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu J, et al. Magnitude and phenotype of cellular immune responses elicited by recombinant adenovirus vectors and heterologous prime-boost regimens in rhesus monkeys. Journal of virology. 2008;82:4844–52. doi: 10.1128/JVI.02616-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vogels R, et al. Replication-deficient human adenovirus type 35 vectors for gene transfer and vaccination: efficient human cell infection and bypass of preexisting adenovirus immunity. J Virol. 2003;77:8263–71. doi: 10.1128/JVI.77.15.8263-8271.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mothe BR, et al. Expression of the major histocompatibility complex class I molecule Mamu-A*01 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol. 2003;77:2736–40. doi: 10.1128/JVI.77.4.2736-2740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pal R, et al. ALVAC-SIV-gag-pol-env-based vaccination and macaque major histocompatibility complex class I (A*01) delay simian immunodeficiency virus SIVmac-induced immunodeficiency. J Virol. 2002;76:292–302. doi: 10.1128/JVI.76.1.292-302.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang ZQ, et al. Mamu-A*01 allele-mediated attenuation of disease progression in simian-human immunodeficiency virus infection. J Virol. 2002;76:12845–54. doi: 10.1128/JVI.76.24.12845-12854.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yant LJ, et al. The high-frequency major histocompatibility complex class I allele Mamu-B*17 is associated with control of simian immunodeficiency virus SIVmac239 replication. J Virol. 2006;80:5074–7. doi: 10.1128/JVI.80.10.5074-5077.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santra S, et al. Replication-defective adenovirus serotype 5 vectors elicit durable cellular and humoral immune responses in nonhuman primates. J Virol. 2005;79:6516–22. doi: 10.1128/JVI.79.10.6516-6522.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Betts MR, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–9. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darrah PA, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13:843–50. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- 20.Okoye A, et al. Progressive CD4+ central memory T cell decline results in CD4+ effector memory insufficiency and overt disease in chronic SIV infection. The Journal of experimental medicine. 2007;204:2171–85. doi: 10.1084/jem.20070567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pitcher CJ, et al. Development and homeostasis of T cell memory in rhesus macaque. J Immunol. 2002;168:29–43. doi: 10.4049/jimmunol.168.1.29. [DOI] [PubMed] [Google Scholar]
- 22.Montefiori D. Evaluating neutralizing antibodies against HIV, SIV and SHIV in luciferase reporter gene assays. In: Coligan JE, Kruisbeek AM, Margulies DH, Shevach EM, Strober W, Coico R, editors. Current Protocols in Immunology. John Wiley & Sons; 2004. [DOI] [PubMed] [Google Scholar]
- 23.Forthal DN, Landucci G, Daar ES. Antibody from patients with acute human immunodeficiency virus (HIV) infection inhibits primary strains of HIV type 1 in the presence of natural-killer effector cells. Journal of virology. 2001;75:6953–61. doi: 10.1128/JVI.75.15.6953-6961.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veazey RS, et al. Dynamics of CCR5 expression by CD4(+) T cells in lymphoid tissues during simian immunodeficiency virus infection. Journal of virology. 2000;74:11001–7. doi: 10.1128/jvi.74.23.11001-11007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veazey RS, et al. Increased loss of CCR5+ CD45RA- CD4+ T cells in CD8+ lymphocyte-depleted Simian immunodeficiency virus-infected rhesus monkeys. Journal of virology. 2008;82:5618–30. doi: 10.1128/JVI.02748-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veazey RS, et al. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280:427–31. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- 27.Li Q, et al. Peak SIV replication in resting memory CD4+ T cells depletes gut lamina propria CD4+ T cells. Nature. 2005;434:1148–52. doi: 10.1038/nature03513. [DOI] [PubMed] [Google Scholar]
- 28.Mattapallil JJ, et al. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature. 2005;434:1093–7. doi: 10.1038/nature03501. [DOI] [PubMed] [Google Scholar]
- 29.Wilson NA, et al. Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated low-dose challenge with pathogenic simian immunodeficiency virus SIVmac239. J Virol. 2006;80:5875–85. doi: 10.1128/JVI.00171-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kiepiela P, et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med. 2007;13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]