Usher syndrome: animal models, retinal function of Usher proteins, and prospects for gene therapy (original) (raw)
. Author manuscript; available in PMC: 2009 May 11.
Abstract
Usher syndrome is a deafness-blindness disorder. The blindness occurs from a progressive retinal degeneration that begins after deafness and after the retina has developed. Three clinical subtypes of Usher syndrome have been identified, with mutations in any one of six different genes giving rise to type 1, in any one of three different genes to type 2, and in one identified gene causing Usher type 3. Mutant mice for most of the genes have been studied; while they have clear inner ear defects, retinal phenotypes are relatively mild and have been difficult to characterize. The retinal functions of the Usher proteins are still largely unknown. Protein binding studies have suggested many interactions among the proteins, and a model of interaction among all the proteins in the photoreceptor synapse has been proposed. However this model is not supported by localization data from some laboratories, or the indication of any synaptic phenotype in mutant mice. An earlier suggestion, based on patient pathologies, of Usher protein function in the photoreceptor cilium continues to gain support from immunolocalization and mutant mouse studies, which are consistent with Usher protein interaction in the photoreceptor ciliary/periciliary region. So far, the most characterized Usher protein is myosin VIIa. It is present in the apical RPE and photoreceptor ciliary/periciliary region, where it is required for organelle transport and clearance of opsin from the connecting cilium, respectively. Usher syndrome is amenable to gene replacement therapy, but also has some specific challenges. Progress in this treatment approach has been achieved by correction of mutant phenotypes in _Myo7a_-null mouse retinas, following lentiviral delivery of MYO7A.
1. Clinical subtypes and genetics of Usher syndrome
Retinitis pigmentosa in combination with deafness was reported ~150 years ago (von Graefe, 1858; Leibreich, 1861), and became known as Usher syndrome, as a result of a report by Charles Usher in 1914 (Usher, 1914). Usher syndrome is inherited in an autosomal recessive manner (Usher, 1914), and is responsible for over half of the cases involving deafness and blindness (Vernon, 1969). It affects about 1 in 23,000 in the U.S. (Boughman et al., 1983), 1 in 29,000 in Scandinavia and 1 in 12,500 in Germany (Otterstedde et al., 2001). Since the frequency of retinitis pigmentosa is 1 per 4000 persons (Berson, 1993), Usher syndrome accounts for about 17% of all cases of RP in the US.
Usher syndrome is clinically and genetically heterogeneous. Three clinical subtypes, which are distinguished from each other primarily by the extent and onset of the deafness, have been defined (Smith et al., 1994). Usher type 1 patients are profoundly deaf from birth, and have vestibular dysfunction, which results in retarded motor development. The deafness in Usher 2 is less severe, and vestibular function has been described as normal. Usher 3 patients also have milder deafness, but, unlike in Usher 2, the hearing loss is progressive, and about half have vestibular dysfunction (Sadeghi et al., 2005). RP, which is clinically similar to nonsyndromic retinitis pigmentosa, develops in all types of Usher syndrome (Fishman et al., 1983; Smith et al., 1994).
As listed in Table 1, Usher 1 can be caused by mutations in any one of six different genes, and mutations in any one of three different genes can result in Usher 2. Only one gene has been identified for Usher 3. Usher 1 and 2 are the most common forms of Usher syndrome, with Usher 3 contributing to a large proportion of cases only in isolated areas, such as Finland (Pakarinen et al., 1996) and Birmingham, UK (Hope et al., 1997). In some cases, mutations in these genes do not give rise to both blindness and deafness. Cases of nonsyndromic deafness have been linked to mutations in the Usher 1B, 1C, 1D, 1F and 2D genes (Liu et al., 1997b; Liu et al., 1997c; Weil et al., 1997; Ahmed et al., 2002; Bork et al., 2001; Mburu et al., 2003). Conversely, mutations in the Usher 2A and 3 genes can cause autosomal recessive RP without reported hearing loss (Rivolta et al., 2000; Seyedahmadi et al., 2004a; Seyedahmadi et al., 2004b).
Table 1.
Usher syndrome genes and proteins
| Subtype | Gene | Protein (function1) | Animal models |
|---|---|---|---|
| Mouse (zebrafish) | |||
| Usher 1B | MYO7A | myosin VIIa (actin motor) | shaker1 (mariner) |
| Usher 1C | USH1C | harmonin (PDZ-domain protein) | deaf circler |
| Usher 1D | CDH23 | cadherin23 (adhesion protein) | waltzer (sputnik) |
| Usher 1E | unknown | ||
| Usher 1F | PCDH15 | protocadherin15 (adhesion protein) | Ames waltzer (orbiter, Pcdh15a) |
| Usher 1G | USH1G | sans (scaffold) | Jackson shaker |
| Usher 2A | USH2A | usherin (transmembrane linkage) | knockout |
| Usher 2C | GPR98 | VLGR1 (G-protein coupled receptor) | Vlgr1/del7TM |
| Usher 2D | DFNB31 | whirlin (PDZ-domain protein) | whirler |
| Usher 3 | CLRN1 | clarin (synaptic shaping) | none reported |
2. Animal models of Usher syndrome
Many of the mouse models for Usher syndrome were identified by early mouse geneticists due to their characteristic circling and head-tossing behavior that results from vestibular dysfunction. They were given various names that describe this aberrant behavior. Thus, shaker1 (sh1), deaf circler (dfcr), waltzer (v), Ames waltzer (av), and Jackson shaker (js) mice have mutations in genes that are homologous to the Usher 1 genes, 1B, 1C, 1D, 1F, and 1G, respectively (Gibson et al., 1995; Johnson et al., 2003; Di Palma et al., 2001; Alagramam et al., 2001a; Kikkawa et al., 2003). Ush2a knockout mice do not exhibit this behavior (Liu et al., 2007), which is consistent with normal vestibular function generally found in Usher 2 patients. Interestingly, whirler (wi) mice, the model for Usher 2D, do run in circles (as their name indicates) and toss their heads about (Lane, 1963), suggesting that some Usher 2 patients may indeed possess vestibular dysfunction.
Despite a relatively faithful manifestation of the hearing and balance disorders found in Usher syndrome, these mice do not possess severe mutant phenotypes in their retinas. None of the Usher 1 mouse models undergo retinal degeneration. Some evidence of slight degeneration was noted in the periphery of dfcr retinas (Johnson et al., 2003); however, none was observed in an examination of animals in excess of one year old, raised under a controlled light/dark cycle (C. Lillo and D.S. Williams, unpublished observations). Mice expressing mutant Gpr98, lacking the transmembrane and cytoplasmic domains (Vlgr1/del7TM), also have normal retinal histology (McGee et al., 2006). A small loss of photoreceptor cells was measured in old double shaker1/waltzer mutant mice (Lillo et al., 2003), but the only Usher mouse model that has so far been reported to possess any convincing retinal degeneration is the Ush2a knockout mouse. Individuals have normal photoreceptor morphology and numbers at 10 months of age, but by 20 months over half the photoreceptor cells have been lost (Liu et al., 2007).
Of the mice that show no retinal degeneration, reduced electroretinogram amplitudes have been reported for some of the alleles of shaker1, waltzer and Ames waltzer mice (Libby & Steel, 2001; Libby et al., 2003; Haywood-Watson et al., 2006), as well as _Gpr98_-mutant mice (McGee et al., 2006) and knock-in mice expressing the Acadian USH1C mutant gene (Lentz et al., 2007a; Lentz et al., 2007b). At least in the shaker1, waltzer and Ames waltzer mice the a- and b-wave amplitudes were reduced by a similar proportion, indicating the defect lay within the photoreceptor cell response. Additional mutant retinal phenotypes have been described for the shaker1 mice. These mice possess increased opsin concentration in the photoreceptor connecting cilium (Liu et al., 1999), aberrant RPE melanosome localization and motility (Liu et al., 1998; Gibbs et al., 2004), and defective phagosome localization and digestion (Gibbs et al., 2003), consistent with multiple roles for myosin VIIa in the retina (see below).
There are now a number of zebrafish models with mutant or knocked down genes that represent Usher orthologues. Mariner expresses mutant Myo7a, and, like shaker1 mice, exhibits circling behavior and possesses sensory hair cells with morphological and functional defects (Ernest et al., 2000). In mariner retinas, the apical localization of RPE melanosomes has been reported to be defective (Biehlmaier et al., 2007), comparable to that in shaker1 retinas (Liu et al., 1998). A distinction is that, in many fish, including zebrafish, the apical migration of RPE melanosome has a significant impact on light adaptation (Burnside, 2001), so that in mariner this defect may be functionally more relevant (Biehlmaier et al., 2007). Zebrafish have two orthologues of the Usher 1F gene, with orbiter having mutant Pcdh15a. Mutation and knock down studies have shown that Pcdh15a is required for normal auditory and vestibular function, and Pcdh15b is required for normal photoreceptor outer segment organization and retinal function (Seiler et al., 2005). Sputnik zebrafish express mutant Cdh23, and have reduced or absent mechanotransduction (Nicolson et al., 1998; Seiler & Nicolson, 1999; Sollner et al., 2004), although there are not reports on their vision. Morphilino knock down studies of the Usher 1C, 2A and 2C orthologues result in swimming and balance defects in larvae, similar to those observed in mariner, and reduced visual function with increased photoreceptor cell death (Phillips et al., 2007).
The utility of these zebrafish models for retinal studies related to Usher syndrome is unclear at present. Some of the orthologous genes possess regions that are not well conserved with the Usher genes, so that their proteins may not have the same function (e.g. Gibert et al., 2005). However, there appear to be at least some overlapping functions (e.g. the role of myosin VIIa in RPE melanosome localization), and the presence of photoreceptor cell death in the zebrafish models provides an important similarity to the human condition, which is lacking in most of the mouse models.
3. Functions of Usher proteins in the retina
The proteins encoded by the known Usher genes are listed in Table 1. MYO7A was predicted to encode an unconventional myosin; i.e. a molecular motor that uses energy from ATP hydrolysis to move along actin filaments. Direct experiments have now demonstrated that myosin VIIa is a bona fide actin-based motor (Udovichenko et al., 2002; Inoue & Ikebe, 2003). The USH1C gene generates a number of different isoforms, belonging to three different classes of harmonin. The isoforms each contain two or three PDZ domains, so that harmonin is predicted to be a scaffolding protein (Verpy et al., 2000). The Usher 1D and 1F genes are both predicted to encode cadherins (Bolz et al., 2001; Bork et al., 2001; Ahmed et al., 2001; Alagramam et al., 2001b). The Usher 1G protein, sans, is another putative scaffolding protein (Weil et al., 2003). Usherin, encoded by USH2A, was reported to be an extracellular matrix protein that binds type IV collagen (Bhattacharya et al., 2004), however, more recently, a longer variant of usherin, an exceptionally large protein of ~600 kD, with a membrane anchor and short cytoplasmic, PDZ-binding motif, has been detected in photoreceptor cells (Liu et al., 2007). VLGR1 appears to be a G-protein coupled receptor with a large N-terminal region (Weston et al., 2004). Whirlin is another PDZ-domain protein and potential scaffold (Mburu et al., 2003). Lastly, the only reported Usher 3 gene encodes clarin1, which has been speculated to function in synaptic shaping and maintenance, based on loose homology with a protein known to function in this manner in the cerebellum (Adato et al., 2002).
It has been proposed that many of the proteins might function together in a common cellular mechanism. Such a unifying hypothesis is attractive, and, certainly, the similarities of clinical phenotype within the different types of Usher syndrome suggest that the mutated genes of each type might affect a common cellular mechanism. Experimental evidence to support this notion has come from studies indicating that some of the Usher 1 proteins can interact with each other.
In the first studies to test protein binding among Usher proteins, two groups found binding of the cytoplasmic region of the ear-specific isoform of cadherin23 to the second PDZ domain of harmonin (Boeda et al., 2002; Siemens et al., 2002). One of the groups also reported binding between the first PDZ domain (PDZ1) of harmonin and myosin VIIa (Boeda et al., 2002), while the other group reported that the PDZ1 domain of harmonin binds the isoform of cadherin23 that is found in tissues other than the ear, such as kidney, brain and retina (Siemens et al., 2002). Subsequent binding studies linked more of the Usher proteins together, with harmonin playing a central role in the network (Weil et al., 2003). In the cochlear hair cells, these binding studies have been supported by mutant phenotypes, exhibiting defects in stereociliary development and organization. Myosin VIIa and protocadherin15 have thus been shown to bind each other and interact functionally in the cochlear hair cells (Senften et al., 2006).
The extent of interactions among Usher proteins in the retina is less clear. Protein interaction in vitro is not necessarily relevant in the physiological environment of an intact tissue. It has been proposed that all the Usher 1 and Usher 2 proteins (including NBC3, which was considered previously, but is no longer a candidate for an Usher 2 protein) form a large complex in the photoreceptor synapse (Reiners et al., 2003; Reiners et al., 2005b; Reiners et al., 2005a; Kremer et al., 2006; Reiners et al., 2006; Reiners & Wolfrum, 2006; van Wijk et al., 2006). A weakness of this proposal is the lack of solid evidence that most of these proteins actually reside in the synaptic region. Immunofluorescence images have been presented to demonstrate localization in the photoreceptor synaptic layer of the retina (Reiners et al., 2003; Reiners et al., 2005b; Reiners et al., 2005a; Kremer et al., 2006; Reiners et al., 2006; Reiners & Wolfrum, 2006; van Wijk et al., 2006), however, this localization is not consistent with that reported by other groups (e.g. Gibbs & Williams, 2004; Lillo et al., 2005; Liu et al., 2007). Nor is it supported by retinal phenotypes of mutant mice.
None of the Usher mouse models has a mutant phenotype that could be attributed to photoreceptor synaptic function. The ultrastructure and physiology of the synapses appear normal. Shaker1, waltzer, Ames waltzer, whirler and usherin knockout mice have all been shown to have normal a-wave to b-wave ratios in their electroretinograms (Libby et al., 2003; Libby & Steel, 2001; Haywood-Watson et al., 2006; Sun et al., 2006; Liu et al., 2007), indicating that the photoreceptor cell response (a-wave) is passed on faithfully to the rest of the retina (which generates the b-wave).
The one location where a number of Usher proteins appear to be found together is in the ciliary and periciliary region of the photoreceptor cells (Fig. 1). While some of the Usher proteins might thus interact in this region, it is noteworthy that this is the one region in the photoreceptor cells that harmonin appears to be absent (e.g. Fig. 4E in Reiners et al. 2003; Lillo et al., 2005). Harmonin has been presented as the central scaffold of Usher protein networks in the inner ear and in the photoreceptor synapse, so that a network in the ciliary and periciliary region of the photoreceptor cells would require a different organization. Sans, usherin and whirlin all appear to have scaffolding properties and may thus play such a role.
Figure 1.
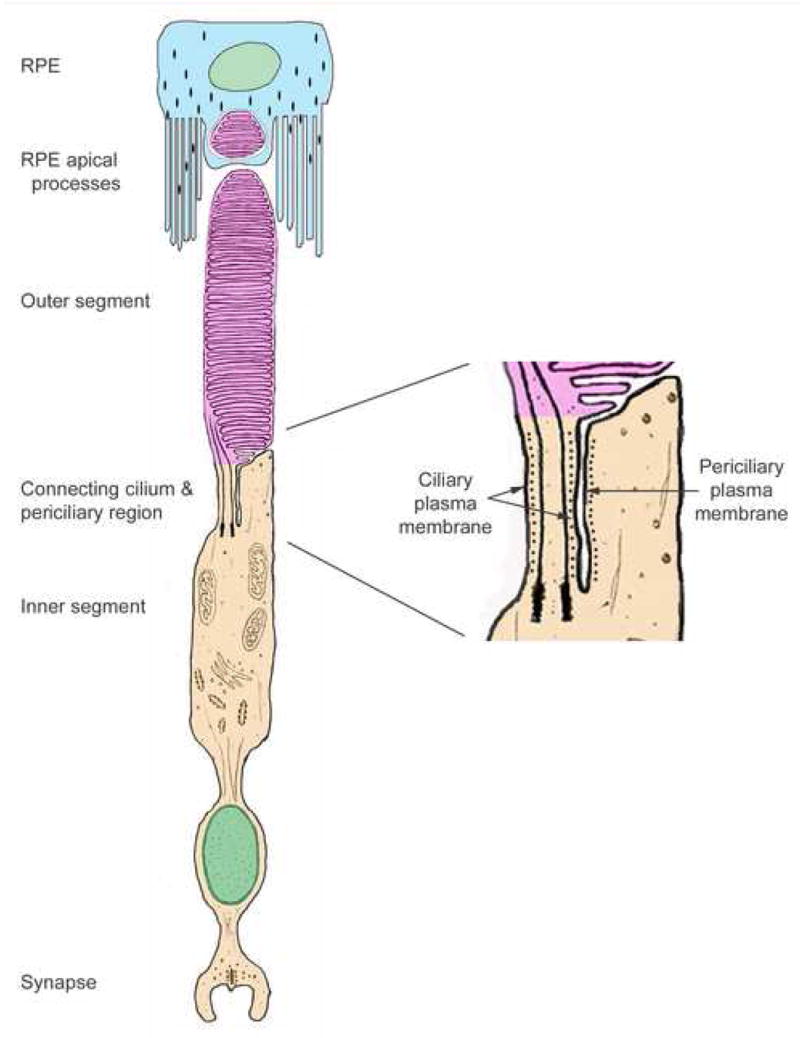
Diagram of a photoreceptor and RPE cell. The region of the connecting cilium and pericilium is enlarged on the right, with the black dots indicating where myosin VIIa has been detected by immunogold labeling (Liu et al., 1997a). Evidence of the presence of a number of other Usher proteins along the periciliary membrane has now been reported (Liu et al., 2007; Yang et al., 2006; Sun et al., 2006; Wolfrum et al., 2007; Goodyear & Richardson, 1999; McGee et al., 2006). Modified from (Williams, 2002).
The first Usher protein to be localized in this ciliary region was myosin VIIa (Liu et al., 1997a). This localization was corroborated by the identification of a mutant phenotype – abnormal accumulation of opsin in the connecting cilium – in studies of shaker1 mice (Liu et al., 1999). While the emphasis on these earlier localization studies was on the localization of myosin VIIa within the connecting cilium, in retrospect, it is clear from published and unpublished immunoelectron micrographs that the protein can be also detected in the periciliary part of the inner segment (see, for example, Figs. 12–14 in Liu et al., 1997a) (Fig. 1). Full-length usherin has been localized to the periciliary membrane (Liu et al., 2007), and this localization has been reported to be dependent on the PDZ-domain protein, whirlin (Yang et al., 2006; Liu et al., 2007), suggesting that the Usher 1D protein is present here also. Abstracts reporting periciliary localization of protocadherin15 (Sun et al., 2006) and sans (Wolfrum et al., 2007) have also been presented. Earlier, the “ankle link antigen”, now identified as VLGR1 (McGee et al., 2006), was detected in this region (Goodyear & Richardson, 1999)
Hence, the connecting cilium region of the photoreceptor cells is a likely focus for Usher protein interaction and function in the retina. Interestingly, the importance of the photoreceptor cilium in retinal pathogenesis was suggested some time ago as a result of Usher patient studies. Abnormal cilia were described in the photoreceptor cells of postmortem Usher 2 retinas (Hunter et al., 1986; Barrong et al., 1992). Various abnormalities that could be related to cilium defects in other tissues of Usher patients were also reported. They include impaired olfaction (Zrada et al., 1996), decreased sperm motility (Hunter et al., 1986), abnormal nasal cilia (Arden & Fox, 1979; Bonneau et al., 1993), bronchitis (Bonneau et al., 1993), and asthma (Baris et al., 1994). Consistent with a general ciliary function, myosin VIIa was detected in cilia of a variety of tissues (Wolfrum et al., 1998).
Although more is known about the retinal function of myosin VIIa than for any other Usher protein, its role in the photoreceptor connecting cilium and periciliary region is not clear. Lack of myosin VIIa causes an abnormal accumulation of opsin in the connecting cilium, and a retarded rate of the disk membrane renewal, thus indicating impairment of delivery of opsin to the site of disk membrane morphogenesis (Liu et al., 1999). Nevertheless, this defect is not as severe as lack of heterotrimeric kinesin-2 function, for deletion of this motor activity appears to completely block the delivery of opsin to the outer segment, and cause rapid photoreceptor cell death (Marszalek et al., 2000; Jimeno et al., 2006). Hence, kinesin-2 appears more likely to provide motor transport of opsin along the connecting cilium. Myosin VIIa could play an auxiliary role with kinesin-2, consistent with a general theme in which a myosin provides local transport in conjunction with longer-range transport that is powered by a microtubule motor. Alternatively, its effect on opsin transport could be less direct, perhaps acting through structural elements of the cilium and pericilium.
In the ciliary region, myosin VIIa could interact with other Usher proteins, given their colocalization, however, by far the majority of retinal myosin VIIa is located in the apical RPE (Hasson et al., 1995), where it appears to be the only Usher protein. Here, its roles include organelle transport, most likely in concert with microtubule motors. A combination of in vivo studies and analyses of primary cultures of RPE cells from mutant mice indicate that myosin VIIa transports both phagosomes (Gibbs et al., 2003) and melanosomes (Futter et al., 2004; Gibbs et al., 2004; Klomp et al., 2007; Lopes et al., 2007). Phagosomes take longer to travel to the basal RPE and are digested more slowly in RPE cells lacking myosin VIIa (Gibbs et al., 2003). Live cell imaging of melanosomes shows that melanosomes travel further and faster (at the speed of a microtubule motor) in the absence of myosin VIIa, which moves melanosomes at ~200 nm/sec and appears to effect their tethering in the apical processes of the RPE (Gibbs et al., 2004; Klomp et al., 2007; Lopes et al., 2007).
4. Prospects for retinal gene therapy for Usher syndrome
Gene therapy is a potential approach to prevent blindness in Usher syndrome. Indeed, Usher syndrome is potentially a highly suitable form of retinal degeneration for treatment by gene therapy. First, all types appear to be inherited recessively and caused by loss of gene function, so that therapy need only focus on adding gene function. Second, because of the associated impaired hearing at birth, Usher syndrome patients can be more readily identified prior to the onset of retinal degeneration. Nevertheless, there are some drawbacks. Some of the Usher genes are very large –USH2A and GPR98 encode retinal proteins of 600–700 kD (Liu et al., 2007; McMillan et al., 2002) – making their delivery problematic. Moreover, as noted above, there is presently a paucity of mutant phenotypes that can be assayed to test for efficacy of treatment in animal models. When testing gene therapy, it is important not only to monitor the expression of the gene, but also to determine if the resulting protein is functional.
Some types of Usher syndrome are therefore currently not amenable to gene therapy. However, there has been some progress with treatment for Usher 1B. MYO7A cDNA has been delivered to retinas and cultured primary RPE cells of _Myo7a-_null mice, using a lentiviral vector. Using a promoter containing elements of the native MYO7A promoter, appropriate levels of myosin VIIa were obtained in the RPE cells, correction of mutant phenotypes – melanosome motility and phagosome digestion in cultured RPE cells, and melanosome localization and opsin clearance from the connecting cilium in vivo – was achieved (Hashimoto et al., 2007).
Concluding comment
Usher syndrome has turned out to be more genetically complex than clinicians would have first envisaged. Nevertheless, the identification of the genes involved now appears to be nearing completion. Understanding the retinal roles of the Usher proteins represents a current major challenge. A clearer picture of the localization, interactions and functions of the Usher proteins in the retina will facilitate progress towards developing gene therapy prevention of Usher blindness.
Acknowledgments
I thank Dan Gibbs for assistance with Fig. 1, and for helpful suggestions on the manuscript. My position and laboratory are funded by NIH grant EY07042.
Abbreviations
RPE
retinal pigment epithelium
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adato A, Vreugde S, Joensuu T, Avidan N, Hamalainen R, Belenkiy O, Olender T, Bonne-Tamir B, Ben-Asher E, Espinos C, Millan JM, Lehesjoki AE, Flannery JG, Avraham KB, Pietrokovski S, Sankila EM, Beckmann JS, Lancet D. USH3A transcripts encode clarin-1, a four-transmembrane-domain protein with a possible role in sensory synapses. Eur J Hum Genet. 2002;10(6):339–350. doi: 10.1038/sj.ejhg.5200831. [DOI] [PubMed] [Google Scholar]
- Ahmed ZM, Riazuddin S, Bernstein SL, Ahmed Z, Khan S, Griffith AJ, Morell RJ, Friedman TB, Wilcox ER. Mutations of the protocadherin gene PCDH15 cause Usher syndrome type 1F. Am J Hum Genet. 2001;69(1):25–34. doi: 10.1086/321277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Smith TN, Riazuddin S, Makishima T, Ghosh M, Bokhari S, Menon PS, Deshmukh D, Griffith AJ, Friedman TB, Wilcox ER. Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum Genet. 2002;110(6):527–531. doi: 10.1007/s00439-002-0732-4. [DOI] [PubMed] [Google Scholar]
- Alagramam KN, Murcia CL, Kwon HY, Pawlowski KS, Wright CG, Woychik RP. The mouse Ames waltzer hearing-loss mutant is caused by mutation of Pcdh15, a novel protocadherin gene. Nat Genet. 2001a;27(1):99–102. doi: 10.1038/83837. [DOI] [PubMed] [Google Scholar]
- Alagramam KN, Yuan H, Kuehn MH, Murcia CL, Wayne S, Srisailpathy CR, Lowry RB, Knaus R, Van Laer L, Bernier FP, Schwartz S, Lee C, Morton CC, Mullins RF, Ramesh A, Van Camp G, Hageman GS, Woychik RP, Smith RJ, Hagemen GS. Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum Mol Genet. 2001b;10(16):1709–1718. doi: 10.1093/hmg/10.16.1709. [DOI] [PubMed] [Google Scholar]
- Arden GB, Fox B. Increased incidence of abnormal nasal cilia in patients with retinitis pigmentosa. Nature. 1979;279(5713):534–536. doi: 10.1038/279534a0. [DOI] [PubMed] [Google Scholar]
- Baris B, Ataman M, Sener C, Kalyoncu F. Bronchial asthma in a patient with Usher syndrome: case report. J Asthma. 1994;31(6):487–490. doi: 10.3109/02770909409089492. [DOI] [PubMed] [Google Scholar]
- Barrong SD, Chaitin MH, Fliesler SJ, Possin DE, Jacobson SG, Milam AH. Ultrastructure of connecting cilia in different forms of retinitis pigmentosa. Arch Ophthalmol. 1992;110(5):706–710. doi: 10.1001/archopht.1992.01080170128040. [DOI] [PubMed] [Google Scholar]
- Berson EL. Retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1993;34:1659–1676. [PubMed] [Google Scholar]
- Bhattacharya G, Kalluri R, Orten DJ, Kimberling WJ, Cosgrove D. A domain-specific usherin/collagen IV interaction may be required for stable integration into the basement membrane superstructure. J Cell Sci. 2004;117(Pt 2):233–242. doi: 10.1242/jcs.00850. [DOI] [PubMed] [Google Scholar]
- Biehlmaier O, Hodel C, Neuhaus SCF. Localization and function of myosin VIIa in the retina of wild-type and mutant zebrafish. ARVO abstracts, #4495 2007 [Google Scholar]
- Boeda B, El-Amraoui A, Bahloul A, Goodyear R, Daviet L, Blanchard S, Perfettini I, Fath KR, Shorte S, Reiners J, Houdusse A, Legrain P, Wolfrum U, Richardson G, Petit C. Myosin VIIa, harmonin and cadherin 23, three Usher I gene products that cooperate to shape the sensory hair cell bundle. Embo J. 2002;21(24):6689–6699. doi: 10.1093/emboj/cdf689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG, Seeliger M, del CSCM, Vila MC, Molina OP, Gal A, Kubisch C. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet. 2001;27(1):108–112. doi: 10.1038/83667. [DOI] [PubMed] [Google Scholar]
- Bonneau D, Raymond F, Kremer C, Klossek JM, Kaplan J, Patte F. Usher syndrome type I associated with bronchiectasis and immotile nasal cilia in two brothers. J Med Genet. 1993;30(3):253–254. doi: 10.1136/jmg.30.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, Wayne S, Bellman S, Desmukh D, Ahmed Z, Khan SN, Kaloustian VM, Li XC, Lalwani A, Bitner-Glindzicz M, Nance WE, Liu XZ, Wistow G, Smith RJ, Griffith AJ, Wilcox ER, Friedman TB, Morell RJ. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am J Hum Genet. 2001;68(1):26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boughman J, Vernon M, Shaver K. Usher syndrome: definition and estimate of prevalence from two high-risk populations. J Chronic Dis. 1983;36:595–603. doi: 10.1016/0021-9681(83)90147-9. [DOI] [PubMed] [Google Scholar]
- Burnside B. Light and circadian regulation of retinomotor movement. Prog Brain Res. 2001;131:477–485. doi: 10.1016/s0079-6123(01)31038-5. [DOI] [PubMed] [Google Scholar]
- Chaib H, Kaplan J, Gerber S, Vincent C, Ayadi H, Slim R, Munnich A, Weissenbach J, Petit C. A newly identified locus for Usher syndrome type I, USH1E, maps to chromosome 21q21. Human Molecular Genetics. 1997;6(1):27–31. doi: 10.1093/hmg/6.1.27. [DOI] [PubMed] [Google Scholar]
- Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, Steel KP, Noben-Trauth K. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001;27(1):103–107. doi: 10.1038/83660. [DOI] [PubMed] [Google Scholar]
- Ebermann I, Scholl HP, Charbel Issa P, Becirovic E, Lamprecht J, Jurklies B, Millan JM, Aller E, Mitter D, Bolz H. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet. 2007;121(2):203–211. doi: 10.1007/s00439-006-0304-0. [DOI] [PubMed] [Google Scholar]
- Ernest S, Rauch GJ, Haffter P, Geisler R, Petit C, Nicolson T. Mariner is defective in myosin VIIA: a zebrafish model for human hereditary deafness. Hum Mol Genet. 2000;9(14):2189–2196. doi: 10.1093/hmg/9.14.2189. [DOI] [PubMed] [Google Scholar]
- Eudy JD, Weston MD, Yao S, Hoover DM, Rehm HL, Ma-Edmonds M, Yan D, Ahmad I, Cheng JJ, Ayuso C, Cremers C, Davenport S, Moller C, Talmadge CB, Beisel KW, Tamayo M, Morton CC, Swaroop A, Kimberling WJ, Sumegi J. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998a;280(5370):1753–1757. doi: 10.1126/science.280.5370.1753. [DOI] [PubMed] [Google Scholar]
- Eudy JD, Yao S, Weston MD, Ma-Edmonds M, Talmadge CB, Cheng JJ, Kimberling WJ, Sumegi J. Isolation of a gene encoding a novel member of the nuclear receptor superfamily from the critical region of Usher syndrome type IIa at 1q41. Genomics. 1998b;50(3):382–384. doi: 10.1006/geno.1998.5345. [DOI] [PubMed] [Google Scholar]
- Fishman GA, Kumar A, Joseph ME, Torok N, Anderson RJ. Usher’s syndrome. Ophthalmic and neuro-otologic findings suggesting genetic heterogeneity. Arch Ophthalmol. 1983;101(9):1367–1374. doi: 10.1001/archopht.1983.01040020369005. [DOI] [PubMed] [Google Scholar]
- Futter CE, Ramalho JS, Jaissle GB, Seeliger MW, Seabra MC. The role of Rab27a in the regulation of melanosome distribution within retinal pigment epithelial cells. Mol Biol Cell. 2004;15(5):2264–2275. doi: 10.1091/mbc.E03-10-0772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber S, Bonneau D, Gilbert B, Munnich A, Dufier JL, Rozet JM, Kaplan J. USH1A: Chronicle of a Slow Death. Am J Hum Genet. 2006;78(2):357–359. doi: 10.1086/500275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs D, Azarian SM, Lillo C, Kitamoto J, Klomp AE, Steel KP, Libby RT, Williams DS. Role of myosin VIIa and Rab27a in the motility and localization of RPE melanosomes. J Cell Sci. 2004;117(Pt 26):6473–6483. doi: 10.1242/jcs.01580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs D, Kitamoto J, Williams DS. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the Usher syndrome 1B protein. Proc Natl Acad Sci U S A. 2003;100(11):6481–6486. doi: 10.1073/pnas.1130432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs D, Williams DS. Usher 1 protein complexes in the retina. Invest Ophthalmol Vis Sci. 2004;45 e-letter (May 26) [Google Scholar]
- Gibert Y, McMillan DR, Kayes-Wandover K, Meyer A, Begemann G, White PC. Analysis of the very large G-protein coupled receptor gene (Vlgr1/Mass1/USH2C) in zebrafish. Gene. 2005;353(2):200–206. doi: 10.1016/j.gene.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Gibson F, Walsh J, Mburu P, Varela A, Brown KA, Antonio M, Beisel KW, Steel KP, Brown SDM. A type VII myosin encoded by mouse deafness gene shaker-1. Nature. 1995;374:62–64. doi: 10.1038/374062a0. [DOI] [PubMed] [Google Scholar]
- Goodyear R, Richardson G. The ankle-link antigen: an epitope sensitive to calcium chelation associated with the hair-cell surface and the calycal processes of photoreceptors. J Neurosci. 1999;19(10):3761–3772. doi: 10.1523/JNEUROSCI.19-10-03761.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Gibbs D, Lillo C, Azarian SM, Legacki E, Zhang XM, Yang XJ, Williams DS. Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B. Gene Ther. 2007;14(7):584–594. doi: 10.1038/sj.gt.3302897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson T, Heintzelman MB, Santos-Sacchi J, Corey DP, Mooseker MS. Expression in cochlea and retina of myosin VIIa, the gene product defective in Usher syndrome type 1B. Proc Natl Acad Sci USA. 1995;92:9815–9819. doi: 10.1073/pnas.92.21.9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haywood-Watson RJ, 2nd, Ahmed ZM, Kjellstrom S, Bush RA, Takada Y, Hampton LL, Battey JF, Sieving PA, Friedman TB. Ames Waltzer deaf mice have reduced electroretinogram amplitudes and complex alternative splicing of Pcdh15 transcripts. Invest Ophthalmol Vis Sci. 2006;47(7):3074–3084. doi: 10.1167/iovs.06-0108. [DOI] [PubMed] [Google Scholar]
- Hmani M, Ghorbel A, Boulila-Elgaied A, Ben Zina Z, Kammoun W, Drira M, Chaabouni M, Petit C, Ayadi H. A novel locus for Usher syndrome type II, USH2B, maps to chromosome 3 at p23–24.2. European Journal of Human Genetics. 1999;7(3):363–367. doi: 10.1038/sj.ejhg.5200307. [DOI] [PubMed] [Google Scholar]
- Hope CI, Bundey S, Proops D, Fielder AR. Usher syndrome in the city of Birmingham--prevalence and clinical classification. British Journal of Ophthalmology. 1997;81(1):46–53. doi: 10.1136/bjo.81.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DG, Fishman GA, Mehta RS, Kretzer FL. Abnormal sperm and photoreceptor axonemes in Usher’s Syndrome. Arch Opthalmol. 1986;104:385–389. doi: 10.1001/archopht.1986.01050150085033. [DOI] [PubMed] [Google Scholar]
- Inoue A, Ikebe M. Characterization of the motor activity of mammalian myosin VIIA. J Biol Chem. 2003;278:5478–5487. doi: 10.1074/jbc.M210489200. [DOI] [PubMed] [Google Scholar]
- Jimeno D, Feiner L, Lillo C, Teofilo K, Goldstein LS, Pierce E, Williams DS. Analysis of kinesin-2 function in photoreceptor cells using synchronous Cre-loxP knockout of Kif3a with RHO-Cre. Invest Ophthalmol Vis Sci. 2006;47(11):5039–5046. doi: 10.1167/iovs.06-0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Gagnon LH, Webb LS, Peters LL, Hawes NL, Chang B, Zheng QY. Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum Mol Genet. 2003;12(23):3075–3086. doi: 10.1093/hmg/ddg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J, Gerber S, Bonneau D, Rozet JM, Delrieu O, Briard ML, Dollfus H, Ghazi I, Dufier JL, Frezal J, et al. A gene for Usher syndrome type I (USH1A) maps to chromosome 14q. Genomics. 1992;14(4):979–987. doi: 10.1016/s0888-7543(05)80120-x. [DOI] [PubMed] [Google Scholar]
- Kikkawa Y, Shitara H, Wakana S, Kohara Y, Takada T, Okamoto M, Taya C, Kamiya K, Yoshikawa Y, Tokano H, Kitamura K, Shimizu K, Wakabayashi Y, Shiroishi T, Kominami R, Yonekawa H. Mutations in a new scaffold protein Sans cause deafness in Jackson shaker mice. Hum Mol Genet. 2003;12(5):453–461. doi: 10.1093/hmg/ddg042. [DOI] [PubMed] [Google Scholar]
- Kimberling WJ, Moller CG, Davenport S, Priluck IA, Beighton PH, Greenberg J, Reardon W, Weston MD, Kenyon JB, Grunkemeyer JA, et al. Linkage of Usher syndrome type I gene (USH1B) to the long arm of chromosome 11. Genomics. 1992;14(4):988–994. doi: 10.1016/s0888-7543(05)80121-1. [DOI] [PubMed] [Google Scholar]
- Klomp AE, Teofilo K, Legacki E, Williams DS. Analysis of the linkage of MYRIP and MYO7A to melanosomes by RAB27A in retinal pigment epithelial cells. Cell Motil Cytoskeleton. 2007;64(6):474–487. doi: 10.1002/cm.20198. [DOI] [PubMed] [Google Scholar]
- Kremer H, van Wijk E, Marker T, Wolfrum U, Roepman R. Usher syndrome: molecular links of pathogenesis, proteins and pathways. Hum Mol Genet. 2006;15(Spec No 2):R262–270. doi: 10.1093/hmg/ddl205. [DOI] [PubMed] [Google Scholar]
- Lane PW. Whirler Mice: a Recessive Behavior Mutation in Linkage Group Viii. J Hered. 1963;54:263–266. doi: 10.1093/oxfordjournals.jhered.a107262. [DOI] [PubMed] [Google Scholar]
- Leibreich R. Abkunft und Eben unter Blutsverwandten als Grund von Retinitis Pigmentosa. Deutsch Klin. 1861;13:53–55. [Google Scholar]
- Lentz J, Pan F, Ng SS, Deininger P, Keats B. Ush1c216A knock-in mouse survives Katrina. Mutat Res. 2007a;616(1–2):139–144. doi: 10.1016/j.mrfmmm.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Lentz JJ, Gordon WC, Farris H, Sampath S, Deininger PD, Bazan NG, Keats BJ. A knock-in mouse model of Usher type 1C. ARVO abstracts, #1341 2007b [Google Scholar]
- Libby RT, Kitamoto J, Holme RH, Williams DS, Steel KP. Cdh23 mutations in the mouse are associated with retinal dysfunction but not retinal degeneration. Exp Eye Res. 2003;77(6):731–739. doi: 10.1016/j.exer.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Libby RT, Steel KP. Electroretinographic anomalies in mice with mutations in Myo7a, the gene involved in human Usher syndrome type 1B. Invest Ophthalmol Vis Sci. 2001;42(3):770–778. [PubMed] [Google Scholar]
- Lillo C, Kitamoto J, Liu X, Quint E, Steel KP, Williams DS. Mouse models for Usher syndrome 1B. Adv Exp Med Biol. 2003;533:143–150. doi: 10.1007/978-1-4615-0067-4_18. [DOI] [PubMed] [Google Scholar]
- Lillo C, Siemens J, Kazmierczak P, Mueller U, Williams DS. Roles and interactions of three USH1 proteins in the retina and inner ear. ARVO abstracts, #5176 2005 [Google Scholar]
- Liu X, Bulgakov OV, Darrow KN, Pawlyk B, Adamian M, Liberman MC, Li T. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc Natl Acad Sci U S A. 2007;104(11):4413–4418. doi: 10.1073/pnas.0610950104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ondek B, Williams DS. Mutant myosin VIIa causes defective melanosome distribution in the RPE of shaker-1 mice. Nat Genet. 1998;19(2):117–118. doi: 10.1038/470. [DOI] [PubMed] [Google Scholar]
- Liu X, Udovichenko IP, Brown SDM, Steel KP, Williams DS. Myosin VIIa participates in opsin transport through the photoreceptor cilium. J Neurosci. 1999;19(15):6267–6274. doi: 10.1523/JNEUROSCI.19-15-06267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Vansant G, Udovichenko IP, Wolfrum U, Williams DS. Myosin VIIa, the product of the Usher 1B syndrome gene, is concentrated in the connecting cilia of photoreceptor cells. Cell Motil Cytoskel. 1997a;37(3):240–252. doi: 10.1002/(SICI)1097-0169(1997)37:3<240::AID-CM6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat Genet. 1997b;16(2):188–190. doi: 10.1038/ng0697-188. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Walsh J, Tamagawa Y, Kitamura K, Nishizawa M, Steel KP, Brown SD. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat Genet. 1997c;17(3):268–289. doi: 10.1038/ng1197-268. [DOI] [PubMed] [Google Scholar]
- Lopes VS, Ramalho JS, Owen DM, Karl MO, Strauss O, Futter CE, Seabra MC. The Ternary Rab27a-Myrip-Myosin VIIa Complex Regulates Melanosome Motility in the Retinal Pigment Epithelium. Traffic. 2007;8(5):486–499. doi: 10.1111/j.1600-0854.2007.00548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalek JR, Liu X, Roberts EA, Chui D, Marth JD, Williams DS, Goldstein LS. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell. 2000;102(2):175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- Mburu P, Mustapha M, Varela A, Weil D, El-Amraoui A, Holme RH, Rump A, Hardisty RE, Blanchard S, Coimbra RS, Perfettini I, Parkinson N, Mallon AM, Glenister P, Rogers MJ, Paige AJ, Moir L, Clay J, Rosenthal A, Liu XZ, Blanco G, Steel KP, Petit C, Brown SD. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat Genet. 2003;34(4):421–428. doi: 10.1038/ng1208. [DOI] [PubMed] [Google Scholar]
- McGee J, Goodyear RJ, McMillan DR, Stauffer EA, Holt JR, Locke KG, Birch DG, Legan PK, White PC, Walsh EJ, Richardson GP. The very large G-protein-coupled receptor VLGR1: a component of the ankle link complex required for the normal development of auditory hair bundles. J Neurosci. 2006;26(24):6543–6553. doi: 10.1523/JNEUROSCI.0693-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan DR, Kayes-Wandover KM, Richardson JA, White PC. Very large G protein-coupled receptor-1, the largest known cell surface protein, is highly expressed in the developing central nervous system. J Biol Chem. 2002;277(1):785–792. doi: 10.1074/jbc.M108929200. [DOI] [PubMed] [Google Scholar]
- McMillan DR, White PC. Loss of the transmembrane and cytoplasmic domains of the very large G-protein-coupled receptor-1 (VLGR1 or Mass1) causes audiogenic seizures in mice. Mol Cell Neurosci. 2004;26(2):322–329. doi: 10.1016/j.mcn.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Mustapha M, Chouery E, Torchard-Pagnez D, Nouaille S, Khrais A, Sayegh FN, Megarbane A, Loiselet J, Lathrop M, Petit C, Weil D. A novel locus for Usher syndrome type I, USH1G, maps to chromosome 17q24-25. Hum Genet. 2002;110(4):348–350. doi: 10.1007/s00439-002-0690-x. [DOI] [PubMed] [Google Scholar]
- Nicolson T, Rusch A, Friedrich RW, Granato M, Ruppersberg JP, Nusslein-Volhard C. Genetic analysis of vertebrate sensory hair cell mechanosensation: the zebrafish circler mutants. Neuron. 1998;20(2):271–283. doi: 10.1016/s0896-6273(00)80455-9. [DOI] [PubMed] [Google Scholar]
- Otterstedde CR, Spandau U, Blankenagel A, Kimberling WJ, Reisser C. A new clinical classification for Usher’s syndrome based on a new subtype of Usher’s syndrome type I. Laryngoscope. 2001;111(1):84–86. doi: 10.1097/00005537-200101000-00014. [DOI] [PubMed] [Google Scholar]
- Pakarinen L, Tuppurainen K, Laippala P, Mantyjarvi M, Puhakka H. The ophthalmological course of Usher syndrome type III. International Ophthalmology. 1996;19(5):307–311. doi: 10.1007/BF00130927. [DOI] [PubMed] [Google Scholar]
- Phillips J, Chiem JL, Westerfield M. Zebrafish Usher genes are necessary for retinal cell function and survival. ARVO abstracts, #4494 2007 [Google Scholar]
- Pieke-Dahl S, Moller CG, Kelley PM, Astuto LM, Cremers CW, Gorin MB, Kimberling WJ. Genetic heterogeneity of Usher syndrome type II: localisation to chromosome 5q. J Med Genet. 2000;37(4):256–262. doi: 10.1136/jmg.37.4.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiners J, Marker T, Jurgens K, Reidel B, Wolfrum U. Photoreceptor expression of the Usher syndrome type 1 protein protocadherin 15 (USH1F) and its interaction with the scaffold protein harmonin (USH1C) Mol Vis. 2005a;11:347–355. [PubMed] [Google Scholar]
- Reiners J, Nagel-Wolfrum K, Jurgens K, Marker T, Wolfrum U. Molecular basis of human Usher syndrome: deciphering the meshes of the Usher protein network provides insights into the pathomechanisms of the Usher disease. Exp Eye Res. 2006;83(1):97–119. doi: 10.1016/j.exer.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Reiners J, Reidel B, El-Amraoui A, Boeda B, Huber I, Petit C, Wolfrum U. Differential distribution of harmonin isoforms and their possible role in Usher-1 protein complexes in mammalian photoreceptor cells. Invest Ophthalmol Vis Sci. 2003;44(11):5006–5015. doi: 10.1167/iovs.03-0483. [DOI] [PubMed] [Google Scholar]
- Reiners J, van Wijk E, Marker T, Zimmermann U, Jurgens K, te Brinke H, Overlack N, Roepman R, Knipper M, Kremer H, Wolfrum U. Scaffold protein harmonin (USH1C) provides molecular links between Usher syndrome type 1 and type 2. Hum Mol Genet. 2005b;14(24):3933–3943. doi: 10.1093/hmg/ddi417. [DOI] [PubMed] [Google Scholar]
- Reiners J, Wolfrum U. Molecular analysis of the supramolecular usher protein complex in the retina. Harmonin as the key protein of the Usher syndrome. Adv Exp Med Biol. 2006;572:349–353. doi: 10.1007/0-387-32442-9_49. [DOI] [PubMed] [Google Scholar]
- Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: Association with recessive retinitis pigmentosa without hearing loss. American Journal of Human Genetics. 2000;66(6):1975–1978. doi: 10.1086/302926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghi M, Cohn ES, Kimberling WJ, Tranebjaerg L, Moller C. Audiological and vestibular features in affected subjects with USH3: a genotype/phenotype correlation. Int J Audiol. 2005;44(5):307–316. doi: 10.1080/14992020500060610. [DOI] [PubMed] [Google Scholar]
- Sankila EM, Pakarinen L, Kaariainen H, Aittomaki K, Karjalainen S, Sistonen P, de la Chapelle A. Assignment of an Usher syndrome type III (USH3) gene to chromosome 3q. Hum Mol Genet. 1995;4(1):93–98. doi: 10.1093/hmg/4.1.93. [DOI] [PubMed] [Google Scholar]
- Seiler C, Finger-Baier KC, Rinner O, Makhankov YV, Schwarz H, Neuhauss SC, Nicolson T. Duplicated genes with split functions: independent roles of protocadherin15 orthologues in zebrafish hearing and vision. Development. 2005;132(3):615–623. doi: 10.1242/dev.01591. [DOI] [PubMed] [Google Scholar]
- Seiler C, Nicolson T. Defective calmodulin-dependent rapid apical endocytosis in zebrafish sensory hair cell mutants. J Neurobiol. 1999;41(3):424–434. [PubMed] [Google Scholar]
- Senften M, Schwander M, Kazmierczak P, Lillo C, Shin JB, Hasson T, Geleoc GS, Gillespie PG, Williams D, Holt JR, Muller U. Physical and functional interaction between protocadherin 15 and myosin VIIa in mechanosensory hair cells. J Neurosci. 2006;26(7):2060–2071. doi: 10.1523/JNEUROSCI.4251-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedahmadi BJ, Berson EL, Dryja TP. USH3A mutations in patients with a prior diagnosis of Usher syndrome type I, Usher syndrome type II, and nonsyndromic recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2004a;45 E-Abstract 4726. [Google Scholar]
- Seyedahmadi BJ, Rivolta C, Keene JA, Berson EL, Dryja TP. Comprehensive screening of the USH2A gene in Usher syndrome type II and non-syndromic recessive retinitis pigmentosa. Exp Eye Res. 2004b;79(2):167–173. doi: 10.1016/j.exer.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Siemens J, Kazmierczak P, Reynolds A, Sticker M, Littlewood-Evans A, Muller U. The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions. Proc Natl Acad Sci U S A. 2002;99(23):14946–14951. doi: 10.1073/pnas.232579599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Berlin CI, Hejtmancik JF, Keats BJ, Kimberling WJ, Lewis RA, Moller CG, Pelias MZ, Tranebjaerg L. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am J Med Genet. 1994;50(1):32–38. doi: 10.1002/ajmg.1320500107. [DOI] [PubMed] [Google Scholar]
- Smith RJ, Lee EC, Kimberling WJ, Daiger SP, Pelias MZ, Keats BJ, Jay M, Bird A, Reardon W, Guest M, et al. Localization of two genes for Usher syndrome type I to chromosome 11. Genomics. 1992;14(4):995–1002. doi: 10.1016/s0888-7543(05)80122-3. [DOI] [PubMed] [Google Scholar]
- Sollner C, Rauch GJ, Siemens J, Geisler R, Schuster SC, Muller U, Nicolson T. Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature. 2004;428(6986):955–959. doi: 10.1038/nature02484. [DOI] [PubMed] [Google Scholar]
- Sun X, Pawlyk B, Adamian M, Michaud N, Bulgakov O, Li T. Functional and structural deficits of cone photoreceptors in mice lacking PCDH15, a protein encoded by the Ush1F gene. ARVO abstracts, #5770 2006 [Google Scholar]
- Udovichenko IP, Gibbs D, Williams DS. Actin-based motor properties of native myosin VIIa. J Cell Sci. 2002;115(Pt 2):445–450. doi: 10.1242/jcs.115.2.445. [DOI] [PubMed] [Google Scholar]
- Usher C. On the inheritance of retinitis pigmentosa with notes of cases. R Lond Ophthalmol Hosp Rep. 1914;19:130–236. [Google Scholar]
- van Wijk E, van der Zwaag B, Peters T, Zimmermann U, Te Brinke H, Kersten FF, Marker T, Aller E, Hoefsloot LH, Cremers CW, Cremers FP, Wolfrum U, Knipper M, Roepman R, Kremer H. The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum Mol Genet. 2006;15(5):751–765. doi: 10.1093/hmg/ddi490. [DOI] [PubMed] [Google Scholar]
- Vernon M. Usher’s syndrome--deafness and progressive blindness. Clinical cases, prevention, theory and literature survey. J Chronic Dis. 1969;22(3):133–151. doi: 10.1016/0021-9681(69)90055-1. [DOI] [PubMed] [Google Scholar]
- Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, Slim R, Petit C. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet. 2000;26(1):51–55. doi: 10.1038/79171. [DOI] [PubMed] [Google Scholar]
- von Graefe A. Vereinzelte Beobachtungen und Bemerkungen Exceptionelle Verhalten des Gesichtsfeldes bei Pigmentenartung des Netzhaut. Arch Klin Ophthalmol. 1858;4:250–253. [Google Scholar]
- Wayne S, Derkaloustian VM, Schloss M, Polomeno R, Scott DA, Hejtmancik JF, Sheffield VC, Smith RJH. Localization of the usher syndrome type id gene (ush1d) to chromosome 10. Human Molecular Genetics. 1996;5(10):1689–1692. doi: 10.1093/hmg/5.10.1689. [DOI] [PubMed] [Google Scholar]
- Weil D, Blanchard S, Kaplan J, Guilford P, Gibson F, Walsh J, Mburu P, Varela A, Levilliers J, Weston MD, Kelley PM, Kimberling WJ, Wagenaar M, Levi-Acobas F, Larget-Piet D, Munnich A, Steel KP, Brown SDM, Petit C. Defective myosin VIIA gene responsible for Usher syndrome type 1B. Nature. 1995;374:60–61. doi: 10.1038/374060a0. [DOI] [PubMed] [Google Scholar]
- Weil D, El-Amraoui A, Masmoudi S, Mustapha M, Kikkawa Y, Laine S, Delmaghani S, Adato A, Nadifi S, Zina BZ, Hamel C, Gal A, Ayadi H, Yonekawa H, Petit C. Usher syndrome type I G (USH1G) is caused by mutations in the gene encoding SANS, a protein that associates with the USH1C protein, harmonin. Human Mol Genet. 2003;12(5):463–471. doi: 10.1093/hmg/ddg051. [DOI] [PubMed] [Google Scholar]
- Weil D, Kussel P, Blanchard S, Levy G, Levi-Acobas F, Drira M, Ayadi H, Petit C. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet. 1997;16(2):191–193. doi: 10.1038/ng0697-191. [DOI] [PubMed] [Google Scholar]
- Weston MD, Luijendijk MW, Humphrey KD, Moller C, Kimberling WJ. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet. 2004;74(2):357–366. doi: 10.1086/381685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DS. Transport to the photoreceptor outer segment by myosin VIIa and kinesin II. Vision Res. 2002;42(4):455–462. doi: 10.1016/s0042-6989(01)00228-0. [DOI] [PubMed] [Google Scholar]
- Wolfrum U, Liu X, Schmitt A, Udovichenko IP, Williams DS. Myosin VIIa as a common component of cilia and microvilli. Cell Motil Cytoskeleton. 1998;40(3):261–271. doi: 10.1002/(SICI)1097-0169(1998)40:3<261::AID-CM5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Wolfrum U, Overlack N, Wijk E, Reidel B, Goldman T, Roepman R, Kremer H, Maerker T. The molecular arrangement of an Usher syndrome protein network at the photoreceptor cilium and its role in the intersegmental transport in photoreceptors. ARVO abstracts, #3066 2007 [Google Scholar]
- Yang J, Liu X, Zhao Y, Adamian M, Pawlyk B, Li T. Subcellular localization of whirlin and its interaction with USH2A in the photoreceptors. ARVO abstracts, #2848 2006 [Google Scholar]
- Zrada SE, Braat K, Doty RL, Laties AM. Olfactory loss in Usher syndrome: another sensory deficit? Am J Med Genet. 1996;64(4):602–603. doi: 10.1002/ajmg.1320640402. [DOI] [PubMed] [Google Scholar]