Progressive accumulation of amyloid-β oligomers in Alzheimer’s disease and APP transgenic mice is accompanied by selective alterations in synaptic scaffold proteins (original) (raw)
. Author manuscript; available in PMC: 2011 Jul 1.
Abstract
The cognitive impairment in patients with Alzheimer’s disease is closely associated with synaptic loss in the neocortex and limbic system. Although the neurotoxic effects of aggregated amyloid-β (Aβ) oligomers in Alzheimer’s disease have been widely studied in experimental models, less is known about the characteristics of these aggregates across the spectrum of Alzheimer’s disease. Here, postmortem frontal cortex samples from control and Alzheimer’s disease patients were fractioned and analyzed for levels of oligomers and synaptic proteins. We found that levels of oligomers correlated with the severity of cognitive impairment (Blessed score and Mini-Mental), and with the loss of synaptic markers. Reduced levels of the synaptic vesicle protein vesicle-associated membrane protein-2 and the postsynaptic protein post-synaptic density-95 (PSD95) correlated with levels of oligomers in the various fractions analyzed. The strongest associations were found with Aβ dimers and pentamers. Co-immunoprecipitation and double-labeling experiments support the possibility that Aβ and PSD95 interact at the synaptic sites. Similarly, in transgenic mice expressing high levels of neuronal amyloid precursor protein (APP), Aβ co-immunoprecipitated with PSD95. This was accompanied by a reduction in the levels of the post-synaptic proteins Shank1 and 3 in Alzheimer’s disease patients and in the brains of APP transgenic mice. In conclusion, this study suggests that the presence of a subpopulation of Aβ oligomers in the brains of patients with Alzheimer’s disease might be related to alterations in selected synaptic proteins and cognitive impairment.
Keywords: oligomers, amyloid, PSD95, Shank, Alzheimer’s disease
Introduction
The cognitive impairment in patients with Alzheimer’s disease (AD) is closely associated with synaptic loss in the neocortex and limbic system [1-3]. Several lines of investigation support the view that increasing levels of amyloid-β 1-42 (Aβ), the proteolytic product of APP metabolism, might be centrally involved in the pathogenesis of AD [4-7]. The mechanisms through which accumulation of Aβ monomers, oligomers and other APP metabolites might lead to synaptic damage and neurodegeneration are under investigation. More specifically, the potential role of neurotoxic Aβ oligomers has emerged as a topic of considerable interest in recent years [8-11].
Under pathological conditions, monomeric forms of Aβ can aggregate to form several different species, which include amyloid fibrils, protofibrils, annular structures, Aβ-derived diffusible ligands (ADDLs) [12] and smaller order oligomeric species (for reviews, see Refs. [13], [14] and [15]). Smaller oligomeric species of synthetic Aβ are different than protofibrils depending on how synthetic Aβ is prepared. Oligomers of Aβ peptides can organize into dimers, trimers, tetramers, and higher order arrays that can form annular structures. Smaller oligomers are divided into those generated from synthetic peptides and those purified from cells, transgenic (tg) mice or AD human brains [13].
Naturally occurring Aβ oligomers can be resistant to SDS, guanidine hydrochloride and Aβ-degrading proteases [16]. An example of a naturally occurring oligomer species is Aβ*56 derived from the brains of APP tg mice, which has been shown to promote age-dependent memory deficits [17]. Aβ*56 and Aβ trimers secreted by cultured cells could turn out to share common synaptotoxic properties [13]. The Aβ dimers, trimers, and higher order oligomers secreted by cultured neurons inhibit LTP, damage spines and interfere with activity-regulated cytoskeleton associated protein (Arc) location [9, 10, 13, 18]. Additional studies have shown that Aβ dimers extracted from human CSF disrupt synaptic plasticity and inhibit hippocampal LTP in vivo [19]. Together, these studies indicate that Aβ oligomers ranging in size from 2-12 subunits might be responsible for the synaptic damage and memory deficits [20]. The mechanisms through which Aβ aggregates might lead to synaptic damage are unclear. A number of recent studies have begun to investigate the possibility that Aβ oligomers might interfere with synaptic function by altering synaptic proteins such as post-synaptic density-95 (PSD95) [21-24] and glutamate receptors [25]. In addition to the role of oligomers, Aβ monomers also accumulate in high levels in the brains of patients with AD and may also contribute to the neurodegenerative process.
Although the neurotoxic effects of the Aβ oligomers have been widely studied in experimental models, less is known about the characteristics of the oligomers across the spectrum of AD and how this correlates with cognition and synaptic proteins. For this purpose, we utilized immunoblot analysis to investigate the relationship between levels of Aβ oligomers and synaptic proteins in fractions from the brains of AD patients and APP tg mice. Our studies show that Aβ oligomers, in particular dimers and pentamers, progressively accumulate in the brains of AD patients as well as in APP tg mice. This was accompanied by reductions in the levels of synaptic scaffold proteins such as PSD95, Shank1 and Shank3.
Results
Levels of Aβ oligomers are associated with cognitive impairment and alterations in synaptic proteins in AD
To analyze the levels of Aβ monomers and oligomers in controls versus MCI and AD cases high resolution immunoblot assays were performed with the cytosolic and membrane fractions obtained by ultracentrifugation with samples extracted either with Buffer A [9] or Buffer B [26, 27] and probed with antibodies against Aβ (clones 82E1 and 6E10, 4G8). When the fractionation procedure was performed with Buffer A (Fig. 1A,B) or Buffer B (Fig. 1C,D) and immunoblots were probed with the anti-Aβ antibodies 82E1 (Fig. 1A,C), 6E10 (Fig. 1B,D) or 4G8 (not shown), we found that the most clear banding pattern, consistent with the estimated weight of the Aβ monomers and multimers, was detected using the membrane fractions of samples prepared with Buffer A and probed with the 82E1 antibody (Fig. 1A). With this approach, bands ranging in approximate molecular weight from 4 to 28 kDa were detected, with the 4 kDa corresponding to monomers, and the higher order bands (8, 12, 16, 20, 24 and 28) probably corresponding to dimers, trimers, tetramers, pentamers, hexamers and heptamers, respectively (Fig. 1A). In brain samples from MCI and AD cases prepared with Buffer A and probed with the 82E1 antibody (Fig. 2A), there was a significant increase in the levels of the bands corresponding to monomers (Fig. 2B), dimers (Fig. 2C) and higher-order oligomers (Fig. 2D, Table 3) when compared to control cases. The greatest difference between the controls and the MCI and AD cases was in the levels of monomeric Aβ (Fig. 2B, Table 3). Further analysis of the human samples by Aβ 1-42 ELISA confirmed undetectable levels of Aβ in the controls, and comparable higher levels in the MCI and AD cases (Table 3) Moreover, the levels of the synaptic proteins VAMP2 and PSD95, and to a lesser extent syntaxin (Fig. 2E-G) and SNAP25 were reduced in MCI and AD cases when compared to neurologically un-impaired controls (Table 3).
Fig. 1.
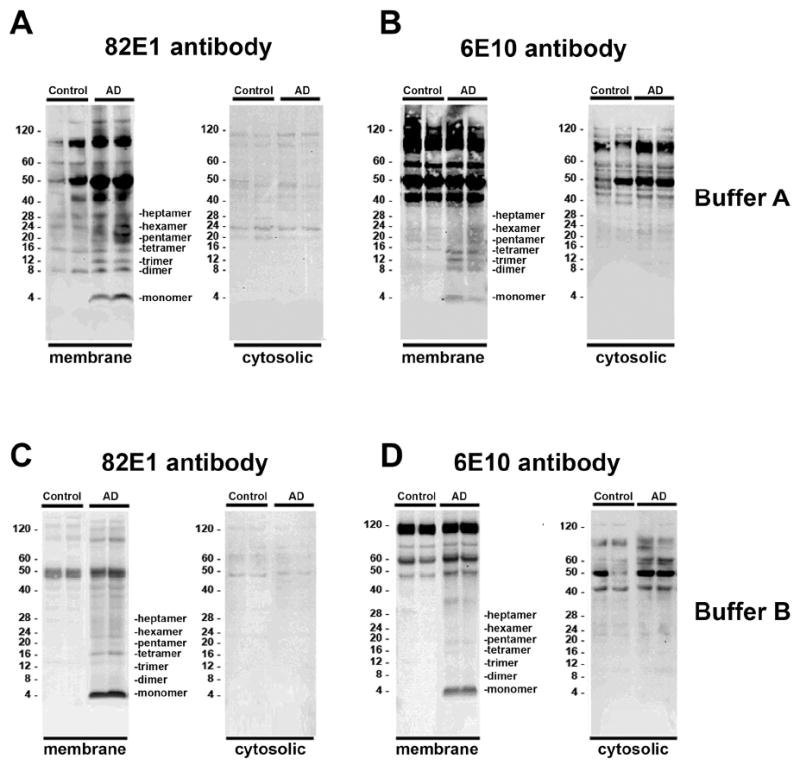
Comparative immunoblot analysis for APP/Aβ in the frontal cortex of control and AD patients. Samples were fractionated into membrane and cytosolic fractions and probed with anti-Aβ antibodies (82E1 and 6E10). A & B. In samples homogenized using Buffer A, compared to non-demented controls, in AD samples multiple bands representing Aβ monomers and multimers were identified at molecular weights ranging from 4 to 28 kDa bands in the membrane fraction. C & D. In samples homogenized using Buffer B, compared to non-demented control, in AD samples the majority of the Aβ was identified as a 4 kDa band in the membrane fraction.
Fig. 2.
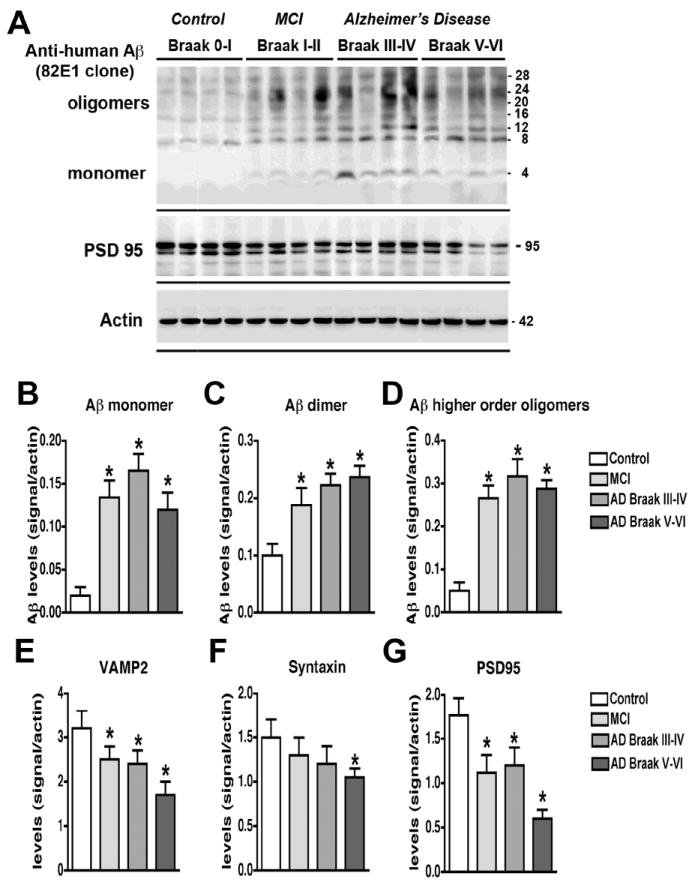
Analysis of the Aβ and synaptic protein bands detected by immunoblot in control and AD brain samples. Samples were homogenized using Buffer A and probed with antibodies against Aβ (82E1) and synaptic proteins (VAMP2, syntaxin, PSD95). A. Representative western blot with the membrane fractions from controls, MCI and AD cases displaying bands corresponding to Aβ monomer (4 kDa) and multimers (8-28 kDa) and PSD95 (95 kDa). B-D. Semi-quantitative analysis of the bands representing Aβ monomer (4 kDa) (B), dimer (8 kDa) (C) and higher order oligomers (12-28 kDa) (D) showing a progressive increase in AD cases. E-G. Semi-quantitative analysis of immunoblots for VAMP2 (E), syntaxin (F), and PSD95 (G) showing a reduction in immunoreactivity in AD cases. N=5 cases per group, *P<0.05 compared to non-demented control by one-way ANOVA with post-hoc Dunnett’s test.
Table 3.
Summary of immunoblot and ELISA analysis of levels of AB and synaptic proteins in control, MCI and AD cases.
| Group | N= | Monomer | Dimer | Trimer | Tetramer | Pentamer | Oligomers Sum | Synapsin I | VAMP2 | Syntaxin 1A | SNAP25 | PSD95 | Aβ pg/ml |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | 5 | 0.051±0.01 | 0.146±0.027 | 0.115±0.013 | 0.126±0.015 | 0.156±0.032 | 0.543±0.04 | 3.690±0.415 | 3.26±0.4 | 1.51±0.291 | 1.625±0.18 | 1.765±0.343 | 0 |
| MCI | 5 | 0.134±0.01* | 0.188±0.01* | 0.208±0.01* | 0.226±0.029* | 0.260±0.019* | 0.882±0.07* | 1.906±0.303* | 2.58±0.1* | 1.34±0.1 | 1.042±0.1* | 1.178±0.1* | 301±42* |
| Moderate AD | 5 | 0.16±0.029* | 0.223±0.012* | 0.246±0.02* | 0.236±0.027* | 0.317±0.036* | 1.020±0.12* | 2.907±0.390 | 2.48±0.187* | 1.25±0.1* | 1.19±0.15* | 1.21±0.15* | 298±34* |
| Advanced AD | 5 | 0.111±0.01* | 0.237±0.011* | 0.205±0.014* | 0.184±0.013* | 0.288±0.018* | 0.940±0.08* | 2.56±0.255* | 1.78±0.21* | 1.04±0.1* | 1.01±0.15* | 0.6±0.23* | 229±41* |
Immunohistochemical analysis with the 82E1 antibody showed, in both patients with AD and in APP tg mice, immunostaining of abundant diffuse and dense core plaques (Fig. 3A-F). In addition, with this antibody there were subtle linear Aβ immunoreactive deposits distributed along the neurons. Double labeling studies utilizing antibodies against PSD95 and 82E1 showed that the linear and punctate deposits along the neurons co-localized with PSD95 in the dendritic processes (Fig. 3G-L). Linear regression analysis was performed to investigate the relationship between the levels of oligomers, synaptic proteins and cognitive impairment. The levels of dimers and pentamers were correlated with the severity of the cognitive impairment (Blessed score and Mini-Mental State Examination [MMSE]) (Fig. S1A, Table 4) and with the Braak stage (Fig. S1B, Table 4). Moreover, levels of dimers and pentamers correlated with the loss of synaptic proteins such as VAMP2 and PSD95 (Fig. S1C,D; Table 4). Consistent with this observation, the levels of the synaptic proteins VAMP2 and PSD95 were significantly correlated with the severity of the cognitive impairment (Fig. S1E, Table 4). Levels of PSD95 were also correlated with the Braak stage (Fig. S1F, Table 4). A total of the six bands, which represent the multimeric forms of Aβ, were significantly correlated with the Blessed and MMSE scores and Braak stage (Table 4).
Fig. 3.
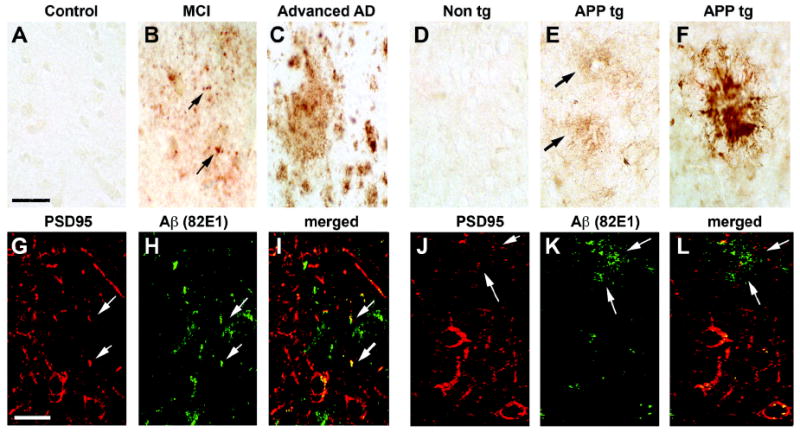
Immunohistochemical analysis of the patterns of Aβ immunoreactivity (82E1) in AD cases and APP tg samples. For panels A-F, vibratome sections were immunolabeled with an anti-Aβ antibody (82E1) and reacted with DAB. For panels G-L, vibratome sections were double-immunolabeled with an anti- Aβ antibody (82E1, green channel) and PSD95 (red channel). A-C. Compared to non-demented controls (A), in the frontal cortex of MCI cases Aβ was detected as discrete granular structures (arrows, B). In advanced AD cases, the antibody detected abundant plaques (C). D-F. Compared to nontg controls (D), in the frontal cortex of APP tg mice the anti-Aβ antibody detected discrete diffuse structures (arrows, E) in the neuropil as well as fibrillar mature plaques (F). G-I. In mild AD cases the discrete Aβ -positive granular structures co-localized with PSD95 (arrows). J-L. In APP tg mice the diffuse Aβ -positive structures co-localized with PSD95 (arrows). Scale bar in panel A equals 50 μm in panels A-F; scale bar in panel G equals 20 μm in panels G-L.
Table 4.
Summary for correlation coefficients between levels of AB; oligomers, synaptic proteins, dementia score and neuropathology.
| Variable | Monomer | Dimer | Trimer | Tetramer | Pentamer | Oligomers Sum | Synapsin I | VAMP2 | Syntaxin | SNAP25 | PSD95 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Blessed score | 0.261 | 0.598* | 0.421 | 0.297 | 0.567* | 0.508* | -0.240 | -0.627* | -0.521* | -0.507* | -0.543* |
| Minimental score | -0.234 | -0.594* | -0.402 | -0.206 | -0.532* | -0.501* | 0.109 | 0.542* | 0.585* | 0.438 | 0.435 |
| Braak score | 0.257 | 0.755* | 0.450 | 0.206 | 0.593* | 0.525* | -0.152 | -0.699* | -0.586* | -0.471 | -0.683* |
| Synapsin I | -0.445 | -0.343 | -0.090 | -0.336 | -0.313 | -0.348 | - | - | - | - | - |
| VAMP2 | -0.157 | -0.727* | -0.565* | -0.162 | -0.427* | -0.396 | - | - | - | - | - |
| Syntaxin | -0.183 | -0.845* | -0.706* | -0.352 | -0.589* | -0.561* | - | - | - | - | - |
| SNAP25 | -0.278 | -0.698* | -0.030 | -0.574* | -0.644* | -0.659* | - | - | - | - | - |
| PSD95 | -0.223 | -0.665* | -0.223 | -0.239 | -0.491* | -0.444* | - | - | - | - | - |
Accumulation of Aβ oligomers and loss of synaptic proteins in APP tg mice
To analyze the levels of Aβ in APP tg mice, immunoblot assays were performed with cytosolic and membrane fractions homogenized with Buffer A (Fig. S2A,B) or Buffer B (Fig. S2C,D), and probed with antibodies against Aβ clones 82E1 (Fig. S2A,C), 6E10 (Fig. S2B,D) and 4G8 (not shown). Similarly to the studies in AD brains (Fig. 1), in the APP tg mice we found a clear banding pattern consistent with the estimated molecular weight of the Aβ monomers and multimers when using the membrane fraction of samples prepared with Buffer A and probed with the 82E1 antibody (Fig. S2A). Compared to nontg mice, in 6-month old APP tg mice we observed abundant levels of monomers, dimers, trimers and, to a lesser extent, other Aβ multimers (Fig. 4A,B). In agreement with the studies in AD patients (Fig. 2), levels of the synaptic proteins VAMP2, syntaxin and PSD95 were significantly reduced in homogenates from APP tg mice compared to nontg controls (Fig. 4C).
Fig. 4.
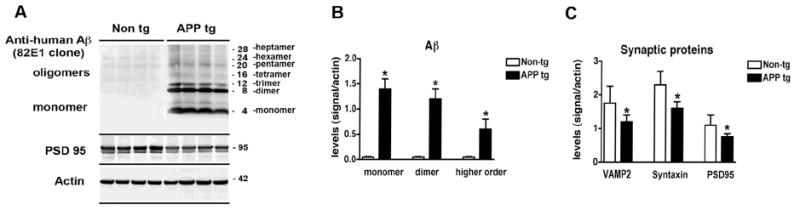
Analysis of the Aβ and synaptic protein bands detected by immunoblot in APP tg mice. Samples were homogenized using Buffer A and probed with antibodies against Aβ (82E1) and synaptic proteins (VAMP2, syntaxin, PSD95). All panels are from the brains of 6-month old mice. A. Representative western blot of the membrane fractions from 6-month old nontg control and APP tg mice displaying bands corresponding to Aβ monomer (4 kDa) and multimers (8-28 kDa) and PSD95 (95 kDa). B. Semi-quantitative analysis of the bands representing Aβ monomer (4 kDa), dimer (8 kDa) and higher order oligomers (12-28 kDa). C. Semi-quantitative analysis of the immunoblots for VAMP2, syntaxin, PSD95. N=8 mice per group, *P<0.01 compared to nontg control by unpaired, two-tailed Student’s t-test.
Interactions between Aβ and PSD95 in the brains of AD patients and APP tg mice
To further investigate the interactions between Aβ and synaptic proteins in AD, co-immunoprecipitation and double-labeling experiments were performed. For this purpose, control and AD brain homogenates extracted with Buffer A were immunoprecipitated with antibodies against Aβ (82E1 clone) or synaptic proteins (PSD95, VAMP2, syntaxin and SNAP25). When samples from human brains were immunoprecipitated with the antibody against Aβ (82E1 clone) and then analyzed by western blot with an antibody against PSD95, the strongest interaction was observed in the AD cases when compared to non-demented controls (Fig. 5A,B). No interacting bands were detected in control experiments with samples immunoprecipitated with a non-immune IgG (Fig. 5A) or when the tissue sample was excluded. No differences were detected between AD and non-demented controls in co-immunoprecipitation experiments with antibodies against Aβ (82E1 clone) and the synaptic proteins syntaxin, SNAP25, or VAMP2 (not shown). Similarly, when brains from the APP tg mice were immunoprecipitated with an antibody against Aβ and analyzed by western blot with an antibody against PSD95, the strongest interaction was observed in the APP tg samples compared to nontg controls (Fig. 5C,D). Similar results were obtained when the reverse co-immunoprecipitation was performed using the antibody against PSD95 followed by western blot analysis with the anti- Aβ antibody (82E1 clone) (Fig. 5E). No significant interactions were detected by co-immunoprecipitation between Aβ (82E1 clone) and the synaptic proteins syntaxin, SNAP25, or VAMP2 (not shown). To further investigate the potential interactions between Aβ and other synaptic proteins in an unbiased manner we immunoprecipitated Aβ with the 82E1 antibody from the brains of APP tg mice and analyzed the copurifying proteins by MudPIT mass spectrometry. Analysis of Aβ immunoprecipates revealed that indeed PSD-95 interacts with Aβ in vivo (Fig. 5F). In contrast, no significant recovery of PSD proteins was obtained in samples precipitated with IgG alone or in control samples.
Fig. 5.
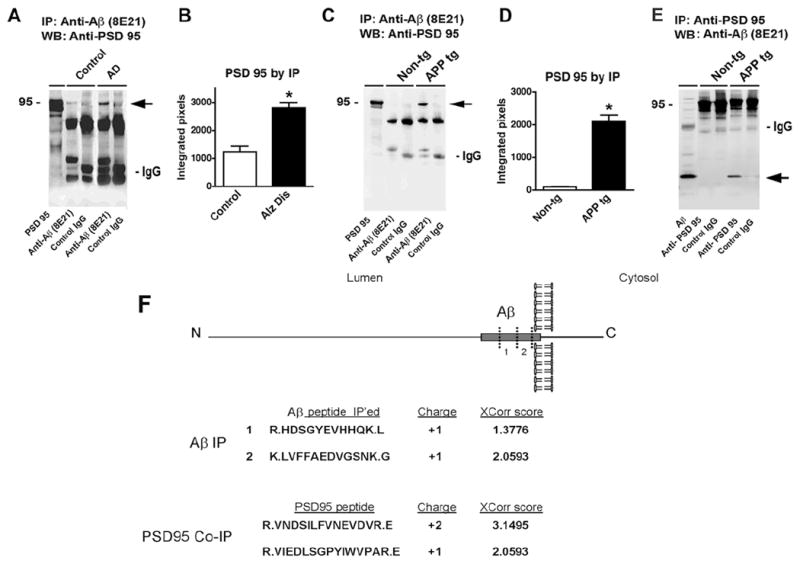
Co-immunoprecipitation studies for Aβ and PSD95 in AD cases and APP tg mice. Samples from the frontal cortex of human non-demented control and AD cases, or from the brains of nontg and APP tg mice were homogenized with Buffer A and membrane fractions were processed for immunoprecipitation. A. Samples from the brains of control and AD cases were immunoprecipitated (IP) with an anti- Aβ antibody (82E1), then analyzed by western blot (WB) with an antibody against PSD95. The reactive band was more intense in the AD cases (arrow); no reactive bands at 95 kDa were observed under control conditions. B. Semi-quantitative analysis of the co-immunoprecipitated band showed higher levels in AD cases. C. Mouse brain cortex samples from 6-month old animals were immunoprecipitated with anti- Aβ antibody (82E1), then analyzed by western blot with an antibody against PSD95. The reactive band was more intense in the APP tg mice (arrow). D. Semi-quantitative analysis of the co-immunoprecipitated band showed higher levels in APP tg mice. E. Mouse brain cortex samples from 6-month old animals were immunoprecipitated with anti-PSD95 antibody, then analyzed by western blot with an antibody against Aβ (82E1). The reactive band was more intense in the APP tg mice (arrow). N=3 cases or mice per group, *P<0.05 compared to control by one-way ANOVA with post-hoc Dunnett’s test. F. Mouse brain cortex samples from 6-month old APP tg mice were immunoprecipitated with an antibody against Aβ (82E1) and the resulting co-precipitates were analyzed by mass spectroscopy. Both Abeta trypitic peptides were identified along with three PSD-95 peptides. Charge and XCorr score of the identified peptides are indicated.
Alterations in proteins involved in the dendritic spine motility apparatus in AD and APP tg mice
Spine motility plays an important role in learning and memory [28] and previous studies have shown that Aβ oligomers might interfere with this function [29]. PSD95 has been shown to play an important role in spine motility by providing a scaffold for other dendritic proteins such as Shank, Homer and actin [30-32]. Given that Aβ oligomers have been shown to interfere with PSD95, then it is possible that abnormalities in spine motility in AD might be associated with alterations in the downstream effectors. To investigate this possibility, immunoblot analysis for post-synaptic proteins was performed in fractionated homogenates from human and mouse brain samples. In the membrane fraction of samples extracted with Buffer A, Shank 1, Shank 3 and pan-Shank were detected as a triple or quadruple band ranging in size from 160-240 kDa, these multiple bands are consistent with the known alternative splicing of Shank. The other PSD proteins such as Homer 1 was detected at approximately 40 kDa, while SAPAP1 was detected as a single band at around 110 kDa (Fig. 6A,B). When compared to controls, in MCI and AD cases there was a significant reduction in the levels of the bands corresponding to Shank 1, 3 and total pan-Shank (Fig. 6C). In contrast, levels of Homer and SAPAP1 were not significantly altered (Fig. 6E). Similarly, compared to nontg controls, in the APP tg mice levels of pan-Shank, Shank 1 and 3 were reduced (Fig. 6D) while Homer and SAPAP1 were not significantly different (Fig. 6F).
Fig. 6.
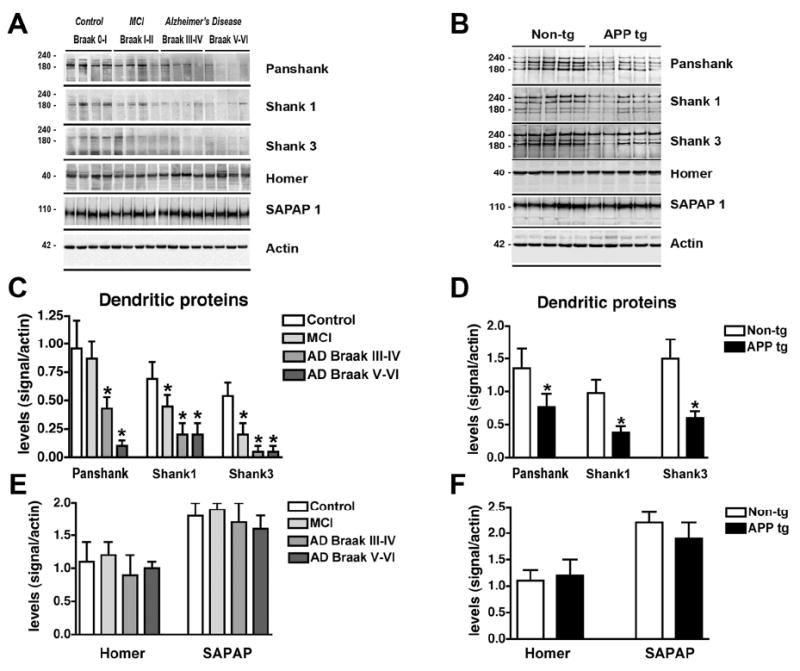
Analysis of the dendritic scaffold proteins by immunoblot in AD brains and APP tg mice. Panel A is from the frontal cortex of control, MCI and AD cases prepared with membrane fractions in Buffer A. Panel B is from nontg and APP tg cortex prepared with membrane fractions in Buffer A. A. Representative western blot analysis of human brain samples probed with antibodies against pan-Shank, Shank1, Shank3, Homer and SAPAP1. B. Representative western blot analysis of mouse brain samples probed with antibodies against pan-Shank, Shank1, Shank3, Homer and SAPAP1. C & D. Semi-quantitative analysis showing a reduction of Shank proteins in MCI and AD compared to non-demented control (C), and a similar reduction in APP tg mice compared to nontg controls (D). E & F. Semi-quantitative analysis showing no changes in Homer or SAPAP1 levels in diseased human (E) or tg mouse brains (F). N=5 cases per group for control, MCI and AD samples, *P<0.05 compared to non-demented controls by one-way ANOVA with post-hoc Dunnett’s test. N=8 mice per group for nontg and APP tg mice, *P<0.01 compared to nontg controls by unpaired, two-tailed Student’s t-test.
To further investigate the effects of Aβ on the dendritic scaffold proteins, primary neuronal cultures were exposed to conditioned media from Chinese hamster ovary (CHO) cells that produce Aβ oligomers. Compared to vehicle-treated controls, after 6 or 24 hrs of exposure to Aβ oligomers, primary neuronal cells displayed a reduction in the numbers of pan-Shank-positive punctae along the dendritic arbor (Fig. 7). This effect was detected after 6 hrs of treatment (Fig. 7C), and after 24 hrs, these effects became more evident (Fig. 7A-E). In addition, a similar reduction in the numbers of PSD95-positive punctae was detected after 24 hrs of treatment with Aβ (Fig. S3). By LDH assay levels of neuronal survival were comparable between cells treated with control conditioned media and Aβ containing media (Fig. S3). Together these results support the possibility that Aβ reduces Shank and PSD95 content in the dendrites and that the effects sare not the result of cell death.
Fig. 7.
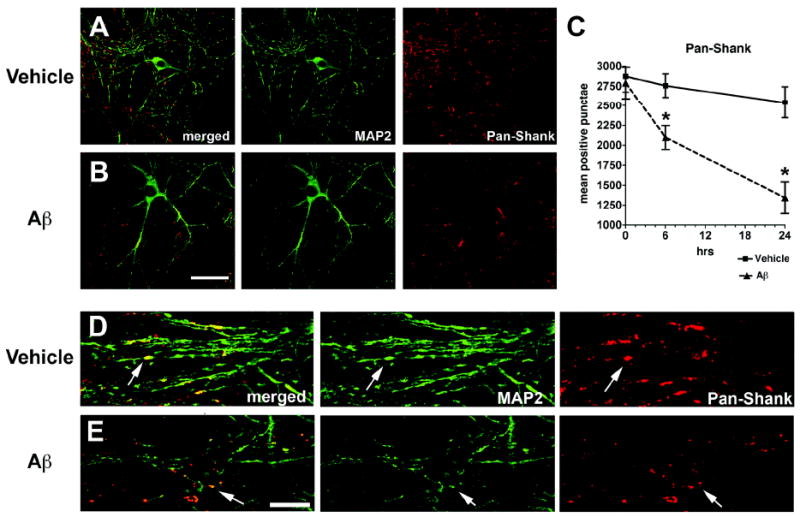
Double immunolabeling analysis for MAP2 and pan-Shank in primary neuronal cultures treated with conditioned media containing Aβ oligomers. Hippocampal neuronal cells from P1 mice were treated for 6 or 24 hrs with conditioned media from APP-expressing CHO cells (80 pM, a sublethal dose). Fixed cells on coverslips were immunolabeled with antibodies against MAP2 (green channel) and pan-Shank (red channel) and analyzed with a laser scanning confocal microscope. All images are from cells treated for 24 hrs; the graph represents data from both 6 and 24 hr timepoints. A & B. Confocal images showing neurons after 24 hrs of treatment with vehicle (A) or Aβ (B). Compared to vehicle-treated cells, Abtreatment resulted in a reduction in pan-Shank-positive punctae along the dendrites. C. Analysis of levels of pan-Shank and MAP2-immunoreactive structures after 6 and 24 hrs of treatment with vehicle or Aβ. D & E. Confocal images at higher power showing the detail of MAP2-labeled dendritic branches and pan-Shank-immunoreactive punctae (arrows) along the dendrites. Scale bar in panel B equals 20 μm for panels A & B; scale bar in panel E equals 10 μm for panels D & E. N=3 samples per condition, *P<0.05 compared to vehicle-treated controls by unpaired, two-tailed Student’s t-test.
Discussion
The present study showed that levels of Aβ oligomers, assessed with the 82E1 antibody in fractionated brain homogenates from patients with AD at different stages, correlated with the severity of the cognitive impairment (Blessed score and MMSE), and with the loss of synaptic markers such as the synaptic vesicle protein VAMP2, and the post-synaptic protein PSD95. The levels of Aβ dimers and pentamers correlated more closely to the severity of the cognitive impairment and the alterations in synaptic proteins.
This is consistent with recent studies in cellular and rodent models showing that small soluble oligomers are toxic because they damage the synapses [20, 33, 34]. Aβ dimers, trimers and pentamers secreted by cultured neurons inhibit LTP and damage spines [18]. In hippocampal slices and in animal models, Aβ oligomers ranging in size from 2-12 subunits impair synapse function [20]. A recent study showed that intraaxonal injection of oligomeric Aβ42 acutely inhibited synaptic transmission at the squid giant synapse by disrupting synaptic vesicles [35]. Interestingly, Aβ dimers recovered from the CSF of patients with AD cause memory deficits and synaptic dysfunction when infused in vivo [19]. Moreover, soluble Aβ dimers purified from AD brains have been shown to inhibit LTP, enhance LTD, and reduce dendritic spine density in rodent hippocampus [36]. Co-administering antibodies specific for the N-terminus of Aβ prevented these synaptic and functional deficits, whereas antibodies against the C-terminus were less effective [37].
Taken together, these results suggest that Aβ dimers and other small oligomers might initiate the cascade of events leading to synaptic damage and cognitive impairment in patients with AD. However, correlational studies can only suggest that such interaction between Aβ and synapses might be at play in AD and additional studies will be needed to confirm this possibility. The mechanisms through which accumulation of Aβ monomers and oligomers might damage the synapses are not completely clear. One possibility is that this effect might be in part mediated by interactions between Aβ and pre- and post-synaptic proteins such as VAMP2 and PSD95, respectively. Another possible interpretation is that once Aβ toxic arrays cross from the pre-synaptic to the post-synaptic site, the damage in the post-synaptic site results in dennervation and secondary changes in the pre-synaptic site. However it is possible that Abeta has toxic effects both in the pre and post-synaptic sites. This study also showed that Aβ co-immunoprecipitated with PSD95 in the brains of patients with AD and in APP tg mice, and that discrete Aβ aggregates co-localized with PSD95 along the dendrites in the brains of AD patients and APP tg mice. In addition, levels of PSD95, Shank1 and Shank3 were reduced. This is consistent with previous studies that have shown alterations in the levels of PSD95 in AD [21, 22, 24]and APP tg models [23, 25]. Moreover, and in agreement with our publication, a recent study showed a reduction in PSD95 levels in the hippocampus of subjects with MCI, which was accompanied by decreased levels of two proteins associated with PSD95, namely the NMDA receptor subunit A (NR2A) and the low-density lipoprotein receptor-1 (LRP1) [24]. In support of a role for Aβ in the mechanisms of post-synaptic damage in AD, previous studies show that soluble Aβ oligomers induce degradation of PSD95 [38], and promote alterations in PSD architecture by depleting the synaptic pool of Homer1b and Shank1 clusters [39]. It is thought that signaling pathways such as PI3K and ERK might mediate the actions of soluble Aβ on Homer1b and Shank levels [39]. In support of this possibility, we found that Aβ co-immunoprecipitated with PSD95 in the brains of patients with AD and in APP tg mice, and that discrete Aβ aggregates co-localized with PSD95 along the dendrites in AD and APP tg mice brains. Further supporting the co-IP results mass spectrometry studies showed that PSD95 fragments can be recovered from the brains of APP tg mice precipitated with and antibody against Aβ but not with an IgG control. However these results should be interpreted with caution given the known tendency of Aβ to bind other proteins. Moreover, interactions between Aβ and PSD95 may be indirectly mediated by other, as yet unidentified, proteins. The distribution of the discrete Aβ aggregates detected with 82E1 are similar to those previously described using the A11 antibody against oligomers [40]. This is consistent with previous studies showing that ADDLs and other naturally occurring Aβ oligomers bind to the post-synaptic density and interfere with dendritic spine function [33, 41]. Studies utilizing real time two-photon microscopy have demonstrated that spine motility plays an important role in learning and memory, and Aβ oligomers are capable of impairing this process [29, 36]. Oligomers might disturb spine motility by disrupting the spine scaffold supported by PSD95, Shank1 and Shank3, however the mechanisms through which Aβ might disturb PSD95 are not completely understood. One possibility might be that soluble monomers and oligomers secreted at the presynaptic side might be uptaken by the postsynaptic side. This is supported by recent studies where fluorescently labeled Aβ oligomers were shown to be taken up via the endocytic pathway in neuronal cell cultures [42]. The second is that oligomers could leak from lysosomal compartments, as has been suggested in previous studies [43, 44].
PSD95 can join N-methyl-D-aspartic acid (NMDA) receptors (NMDA-Rs) to the post-synaptic membrane by the interaction of the first and second PDZ domain with the NR2 subunit of the heteromeric NMDA-R complex [45-48]. In addition to its ability to cluster NMDA-Rs, SAP90/PSD95 is most likely also involved in targeting and NMDA-R signaling, as indicated by the analysis of various tg mouse models [49, 50]. In addition, PSD95 is believed to play a central role in the process of spine motility by serving as a scaffold to bind and organize other integral post-synaptic membrane proteins and PSD components, such as the Shank proteins. The Shank family of proteins are major components of the post-synaptic density [31, 51, 52] and interact with the PDZ ternary complex composed of PSD95, Discs large, and zona occludens-1 (PDZ). This allows Shank to bind indirectly to the multiprotein NMDA-R and α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptor (AMPA-R) complexes via the guanylate kinase-associated protein (GKAP) [53] and to the C-terminus of group I metabotropic glutamate receptors (mGluRs) [54]. The Shank proteins contain a ligand motif for Homer [54], which in turn binds to group I mGluRs, IP3 receptors, and ryanodine receptors [55]. Shanks can also bind to cytoskeletal proteins and regulate spine motility via interactions with cortactin. Shank1 and Shank3 bind to spectrin [56] and F-actin-associated proteins [30-32].
Consistent with the possibility that alterations in PSD95 might lead to downstream changes in dendritic proteins involved in spine function, we found that levels of the post-synaptic proteins Shank1 and 3 were reduced in AD patients and in the brains of APP tg mice when compared to controls. This is consistent with a recent study showing alterations in glutamate receptors and Shank proteins in AD [57]. Interestingly, haploinsufficiency of Shank3 in humans causes a syndrome with dendritic spine dysgenesis [31] that results in a learning disability known as the 22q13 deletion syndrome [58, 59].
Taken together, these studies suggests that in AD, Aβ oligomers might lead to synaptic dysfunction by sequestering PSD95, which in turn might result in alterations to the dendritic spine scaffold and Shank proteins. This could then result in cytoskeletal changes and reduced spine motility. Alternatively, alterations in PSD95/Shank complexes could also result in dysregulation of glutamate receptors. This latter possibility is consistent with recent studies [60] showing that Aβ triggers alterations in the endocytosis of AMPA-R, which compromises synaptic plasticity. Similarly, other studies have shown that cell-derived oligomers decrease dendritic spine density in the hippocampus by an NMDA-dependent signaling pathway [36], suggesting a model in which exposure to Aβ oligomers mimics a state of NMDA-R blockade, either by reducing NMDA-R activation, reducing NMDA-R-dependent calcium influx, or enhancing NMDA-R-dependent activation of calcineurin.
In conclusion, this study showed that the presence of certain species of small Aβ oligomers in the brains of patients with AD correlated with alterations in selected synaptic proteins and cognitive impairment. These results suggest that Aβ could directly interact with post-synaptic proteins, leading to alterations in the spine scaffold system.
Experimental procedures
Please note that additional methodological details are included in the Supplementary Experimental Procedures.
Subjects
A total of 20 human cases were included for the present study. These were divided into several groups: control (neurologically un-impaired), MCI, moderate AD, and advanced AD. A summary of the demographic and clinico-pathological characteristics of these cases is presented in Table 1. The autopsy cases in this study came from patients evaluated at a number of sites associated with the Alzheimer’s Disease Research Center (ADRC) at the University of California, San Diego (UCSD). Written informed consent for neurobehavioral evaluation, autopsy, and for the collection of samples and subsequent analysis was obtained from the patient and caregiver (usually the next of kin) before neuropsychological testing and after the procedures of the study had been fully explained. The study methodologies conformed to the standards set by the Declaration of Helsinki and Federal guidelines for the protection of human subjects. All procedures were reviewed and approved by the UCSD Institutional Review Board.
Table 1.
Summary of clinico-pathological characteristics of the control, MCI and AD cases.
| Group | N= | Age (yrs) | Gender (M/F) | Duration (yrs) | Education (yrs) | CDR score | Blessed score (range) | Minimental score (mean) | Brain weight (grs) | Braak Stage |
|---|---|---|---|---|---|---|---|---|---|---|
| Control | 5 | 84.4±15 | 2/3 | 0 | 13.3±5.8 | 0 | 1-2 | 28.5±3.4 | 1181±73.3 | 0-I |
| MCI | 5 | 89.4±4.8 | 3/2 | 7.0±1.1 | 13.8±3.9 | 0.5 | 1-10 | 25.2±5.7 | 1234±90.1 | I-II |
| Moderate AD | 5 | 89.8±7.9 | 2/3 | 7.5±1.7 | 11.8±6.8 | 1 | 11-24 | 16.6±6.5 | 1097±48.0 | III-IV |
| Advanced AD | 5 | 80.4±6.8 | 2/3 | 8.6±3.9 | 15.6±2.9 | 2 | 25-33 | 11.2±8.4 | 986±146.3 | V-VI |
Neurobehavioral and neuropathological examination
Please refer to the Supplementary Experimental Procedures.
APP tg mouse samples
For studies in animal models, brain samples from 16 six-month old mice (n=8 nontg; n=8 APP tg) were included for immunoblot analysis. The characteristics of thy1-APPmut tg (line 41) [61] have been previously described. The APP tg mice express mutated (London V717I and Swedish K670M/N671L) human APP751 under the control of the murine Thy1 promoter [61]. This tg model was selected because mice produce high levels of AB1-42 and exhibit performance deficits in the water maze, synaptic damage, and early plaque formation, beginning around three months of age [61, 62]. Transgenic lines were maintained by crossing heterozygous tg mice with nontg C57BL/6 × DBA/2 F1 breeders. All mice were heterozygous with respect to the transgene. All experiments were performed in accordance with NIH legislations, all animals were handled in strict accordance with good animal practice and all procedures were completed under the specifications set forth by the UCSD Institutional Animal Care and Use Committee.
Tissue fractionation
For the analysis of Aβ and synaptic proteins, tissues from human and APP tg mice were processed utilizing two different methods. The first method utilized a sucrose-containing buffer that allows for the separation of Aβ oligomers (“Buffer A” containing PBS [pH 7.4], 0.32M sucrose, 50mM HEPES, 25mM MgCl2, 0.5mM DTT, 200μg/ml PMSF, 2μg/ml Pepstatin A, 4μg/ml Leupeptin, 30μg/ml Benzamidine hydrochloride), and the second method utilized a buffer that facilitates separation of the membrane and cytosolic fractions (“Buffer B” containing 1.0mM HEPES, 5.0mM Benzamidine, 2.0mM 2-Mercaptoethanol, 3.0mM EDTA, 0.5mM Magnesium Sulfate, 0.05% Sodium Azide; final pH 8.8).
Tissue Extraction with Buffer A
Briefly as previously described [8, 9], frontal cortex from human and mouse brain samples (0.1g) was homogenized in 0.4ml of Buffer A containing phosphatase and protease inhibitor cocktails (Calbiochem, San Diego, CA). The samples were centrifuged at 1,000 × g for 10 minutes at 4°C. Supernatants were retained and placed into appropriate ultra-centrifuge tubes and the pellets were re-homogenized in 0.3ml of Buffer A and re-centrifuged at 1,000 × g for 10 minutes at 4°C. The second supernatant was collected and combined with the first supernatant and centrifuged at 100,000 × g for one hour at 4°C. This final supernatant was collected to serve as the cytosolic fraction and the remaining pellet was resuspended in 0.2ml of Buffer A and re-homogenized; this was the membrane fraction. The BCA protein assay was used to determine the protein concentration of the samples.
Tissue Extraction with Buffer B
Briefly, as previously described [26, 27], frontal cortex from human and mouse brain samples (0.1g) was homogenized in 0.7ml of Buffer B containing phosphatase and protease inhibitor cocktails (Calbiochem). Samples were centrifuged at 5,000 × g for five minutes at room temperature. Supernatants were retained and placed into appropriate ultra-centrifuge tubes and centrifuged at 100,000 × g for one hour at 4°C. This supernatant was collected, to serve as the cytosolic fraction, and the pellets were resuspended in 0.2ml of Buffer B and re-homogenized; this was the membrane fraction. The BCA protein assay was used to determine the protein concentration of the samples.
Antibodies
For immunoblot and immunohistochemical detection of Aβ, the mouse monoclonal antibodies, 4G8 (Signet Laboratories, Dedham, MA), 82E1 (IBL, Minneapolis, MN) and 6E10 (Signet) were used. For analysis of synaptic proteins, mouse monoclonal antibodies against SNAP25 (Abcam, Cambridge, MA), Syntaxin (Abcam), and PSD95 (UC Davis/NIH Neuro-Monoclonal Antibody (MAb) facility, Davis, CA) were used. The synaptic protein VAMP1 was detected with a rabbit polyclonal IgG (Abcam), and actin levels were determined with the mouse monoclonal C4 antibody (Millipore, Temecula, CA). For detection of pan-Shank, Shank 1, Shank 3 and Pan-SAP90/PSD95 associated proteins (SAPAP), mouse monoclonal antibodies from UC Davis/NIH NeuroMAb facility were used. Homer protein was detected with a rat polyclonal antibody (Millipore). Table 2 presents a summary of the antibodies used for this study.
Table 2.
Antibodies for immunohistochemical and immunoblot analysis.
| Antigen | Antibody | Clone | Concentration | Specificity | Source |
|---|---|---|---|---|---|
| AB | Mouse monoclonal | 82E1 | 1:1000 | aa residues 1-16 | IBL |
| AB | Mouse monoclonal | 6E10 | 1:1000 | aa residues 1-16 | Signet |
| AB | Mouse monoclonal | 4G8 | 1:500 | aa residues 17-24 | Signet |
| AB | Rabbit polyclonal | A11 | 1:2000 | oligomers | UC Irvine |
| Synapsin I | Mouse monoclonal | - | 1:1000 | C-terminus | Millipore |
| VAMP2 | Mouse monoclonal | VAMP | 1:1000 | same | Millipore |
| Syntaxin1A | Mouse monoclonal | SP8 | 1:1000 | aa residues 4-190 | Abcam |
| SNAP25 | Mouse monoclonal | SP12 | 1:1000 | Isoforms A and B | Abcam |
| PSD95 | Mouse monoclonal | K28/43 | 1:1000 | aa residues 77-299 | Antibodies Inc |
| PanShank | Mouse monoclonal | N23B/49 | 1:1000 | aa residues 84-309 | UC Davis |
| Shank1 | Mouse monoclonal | N22/21 | 1:1000 | aa residues 469-691 | UC Davis |
| Shank3 | Mouse monoclonal | N69/46 | 1:1000 | aa residues 840-857 | UC Davis |
| Homer | Rat monoclonal | - | 1:1000 | Homer 1a, 1b, 1c, 2a, 2b, 3 | Millipore |
| SAPAP | Mouse monoclonal | N127/31 | 1:1000 | aa residues 772-992 | UC Davis |
| Actin | Mouse monoclonal | C4 | 1:1000 | aa residues 50-70 | Millipore |
Immunoblot analysis
Western blot analysis was performed essentially as previously described [26, 63]. For additional details, please refer to the Supplementary Experimental Procedures.
Immunoprecipitation assays
Briefly, homogenates from human and mouse brains were prepared in Buffer A as for immunoblot analysis. Samples from the membrane fractions were diluted in immunoprecipitation buffer (20mM Tris-HCl, pH7.5, 150mM NaCl, 1mM EDTA, 1mM NaVO4, 50mM NaF, with protease inhibitors (Roche, Basel, Switzerland) containing 1% Triton X-100, and immunoprecipitation assays were carried out essentially as previously described [64]. The lysates were then centrifuged for 20 minutes at 12,000 rpm, and the protein concentrations were determined with a BCA protein assay kit. Three hundred μg of each of the supernatants was incubated with 1μg of the antibody against synaptic proteins PSD95, VAMP, syntaxin or SNAP25 overnight at 4°C. Then the immunocomplexes were adsorbed to protein A-Sepharose 4B or protein G-Sepharose (Amersham, Piscataway, NJ). After extensive washing with immunoprecipitation buffer, which contained 1% Trion X-100, samples were heated in NuPAGE SDS sample buffer (Invitrogen) for five minutes and subjected to gel electrophoresis on tristricine gels followed by immunoblot analysis with an antibody against either synaptic proteins or Aβ (6E10 or 82E1). Samples were also immunoprecipitated with an antibody against Aβ (6E10 or 82E1), separated on a bis-tris 4-20% gel, then subjected to immunoblot analysis with mouse monoclonal antibodies against synaptic proteins.
Aβ 1-42 ELISA
Levels of Aβ 1-42 in the frontal cortex of patients diagnosed with MCI, moderate AD, advanced AD and age-matched controls were assessed via an enzyme-linked immunosorbent assay (ELISA) which was performed as per manufacturer’s instructions (Life technologies, California).
Lactate Dehydrogenase (LDH) assay
Cell viability was evaluated by the LDH assay. Cells were plated on 96 well plates in complete media. After treatments, assays were than performed following manufacturer’s instructions (Promega). Results were expressed as % cell death.
Primary neuronal cultures
Please refer to the Supplementary Experimental Procedures.
Preparation and treatment with natural Aβ
Natural Aβ was prepared according to Walsh et al. [16] (kindly provided by Dr. Eddie Koo) by incubating control CHO cells or CHO cells expressing APP V717F mutation (also referred as 7PA2 cells) with B27 conditioned media for 16 hours. Total Aβ concentration was determined as previously described [65]. Neurons were treated with 80pM of natural Aβ for 6 and 24 hours. Cells were then fixed with 4%PFA/4%sucrose.
Double labeling and laser scanning confocal microscopy
To evaluate the co-localization between Aβ and synaptic markers, double immunohistochemical analysis was performed as previously described [26]. Vibratome sections were immunolabeled with a monoclonal antibody against PSD95 (1:10,000, UC Davis) detected with the Tyramide Signal Amplification™-Direct (Red) system (1:100, NEN Life Sciences, Boston, MA) and the mouse monoclonal antibody against Aβ (clone 82E1, 1:500) detected with FITC-conjugated secondary antibodies (1:75, Vector Laboratories, Burlingame, CA) [26]. All sections were processed simultaneously under the same conditions and the experiments were performed twice to assess reproducibility. Sections were imaged with a Zeiss 63X (N.A. 1.4) objective on an Axiovert 35 microscope (Zeiss, Germany) with an attached MRC1024 LSCM system (BioRad) [26]. To confirm the specificity of primary antibodies, control experiments were performed where sections were incubated overnight in the absence of primary antibody (deleted) or preimmune serum and primary antibody alone.
Multidimensional Protein Identification Technology (MudPIT), LTQ and Analysis of Tandem Mass Spectra
Please refer to the Supplementary Experimental Procedures.
Statistical analysis
Unless otherwise noted, all data were presented as mean ± SEM. Mean values were compared using Kruskal-Wallis test for Braak scores; non-parametric and one-way analysis of variance (ANOVA) tests were used for all other comparisons. If a significant global result was obtained (overall p value < 0.05), Kruskal-Wallis test was followed by Dunn’s Multiple comparison test and ANOVA was followed by either Student Newman-Keuls or Bonferroni’s multiple comparison tests. Pearson product moment correlations were used to determine the intragroup association of MMSE and BIMC to oligomers and synaptic proteins.
Supplementary Material
Supp Fig s1-3
Acknowledgments
This work was supported by NIH grants AG18440, AG5131, AG022074 and AG11385.
Abbreviations
AD
Alzheimer’s disease
VAMP2
vesicle-associated membrane protein-2
PSD95
post-synaptic density-95
tg
transgenic
APP
amyloid precursor protein
Aβ
amyloid-β 1-42
ADDL
Aβ-derived diffusible ligands
Arc
activity-regulated cytoskeleton associated protein
MCI
mild cognitive impairment
ADRC
Alzheimer’s Disease Research Center
BIMC
Blessed Information-Memory-Concentration
MMSE
Mini-Mental State Examination
CDR
Clinical Dementia Rating
H&E
haematoxylin and eosin
CERAD
Consortium to Establish a Registry for Alzheimers Disease
nontg
non-transgenic
SAPAP
SAP90/PSD95 associated proteins
CHO cells
Chinese hamster ovary cells
ANOVA
non-parametric and one-way analysis of variance
NMDA-R
N-methyl-D-aspartic acid receptor
PDZ
PSD95, Discs large, and zona occludens-1 complex
AMPA-R
α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor
GKAP
guanylate kinase-associated protein
mGluR
metabotropic glutamate receptor
Footnotes
Supporting information Supplementary Experimental Procedures (and relevant references) and supplementary figures with legends are included in one Supplementary Material file.
References
- 1.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 2.DeKosky S, Scheff S. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. AnnNeurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 3.DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5:417–421. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- 4.Sisodia SS, Price DL. Role of the beta-amyloid protein in Alzheimer’s disease. Faseb J. 1995;9:366–370. doi: 10.1096/fasebj.9.5.7896005. [DOI] [PubMed] [Google Scholar]
- 5.Selkoe D. Amyloid β protein precursor and the pathogenesis of Alzheimer’s disease. Cell. 1989;58:611–612. doi: 10.1016/0092-8674(89)90093-7. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe D. Amyloid β-protein deposition as a seminal pathogenic event in AD: an hypothesis. NeurobiolAging. 1990;11:299. [Google Scholar]
- 7.Selkoe D. Physiological production of the β-amyloid protein and the mechanisms of Alzheimer’s disease. Trends Neurosci. 1993;16:403–409. doi: 10.1016/0166-2236(93)90008-a. [DOI] [PubMed] [Google Scholar]
- 8.Klein WL. Abeta toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41:345–352. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- 9.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 10.Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- 11.Glabe CC. Amyloid accumulation and pathogensis of Alzheimer’s disease: significance of monomeric, oligomeric and fibrillar Abeta. Subcell Biochem. 2005;38:167–177. doi: 10.1007/0-387-23226-5_8. [DOI] [PubMed] [Google Scholar]
- 12.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 15.Teplow DB. Structural and kinetic features of amyloid beta-protein fibrillogenesis. Amyloid. 1998;5:121–142. doi: 10.3109/13506129808995290. [DOI] [PubMed] [Google Scholar]
- 16.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 17.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 18.Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: prevention by systemic passive immunization. J Neurosci. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Synaptic changes in Alzheimer’s disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simon AM, Schiapparelli L, Salazar-Colocho P, Cuadrado-Tejedor M, Escribano L, Lopez de Maturana R, Del Rio J, Perez-Mediavilla A, Frechilla D. Overexpression of wild-type human APP in mice causes cognitive deficits and pathological features unrelated to Abeta levels. Neurobiol Dis. 2009;33:369–378. doi: 10.1016/j.nbd.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer’s disease. J Neurosci Res. 2010;88:469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Almeida CG, Tampellini D, Takahashi RH, Greengard P, Lin MT, Snyder EM, Gouras GK. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 26.Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 27.Cole G, Dobkins K, Hansen L, Terry R, Saitoh T. Decreased levels of protein kinase C in Alzheimer brain. Brain Res. 1988;452:165–170. doi: 10.1016/0006-8993(88)90021-2. [DOI] [PubMed] [Google Scholar]
- 28.Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- 29.Shrestha BR, Vitolo OV, Joshi P, Lordkipanidze T, Shelanski M, Dunaevsky A. Amyloid beta peptide adversely affects spine number and motility in hippocampal neurons. Mol Cell Neurosci. 2006;33:274–282. doi: 10.1016/j.mcn.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 30.Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- 31.Boeckers TM, Bockmann J, Kreutz MR, Gundelfinger ED. ProSAP/Shank proteins - a family of higher order organizing molecules of the postsynaptic density with an emerging role in human neurological disease. J Neurochem. 2002;81:903–910. doi: 10.1046/j.1471-4159.2002.00931.x. [DOI] [PubMed] [Google Scholar]
- 32.Qualmann B, Boeckers TM, Jeromin M, Gundelfinger ED, Kessels MM. Linkage of the actin cytoskeleton to the postsynaptic density via direct interactions of Abp1 with the ProSAP/Shank family. J Neurosci. 2004;24:2481–2495. doi: 10.1523/JNEUROSCI.5479-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Viola KL, Velasco PT, Klein WL. Why Alzheimer’s is a disease of memory: the attack on synapses by A beta oligomers (ADDLs) J Nutr Health Aging. 2008;12:51S–57S. doi: 10.1007/BF02982587. [DOI] [PubMed] [Google Scholar]
- 34.Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gongz Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moreno H, Yu E, Pigino G, Hernandez AI, Kim N, Moreira JE, Sugimori M, Llinas RR. Synaptic transmission block by presynaptic injection of oligomeric amyloid beta. Proc Natl Acad Sci U S A. 2009;106:5901–5906. doi: 10.1073/pnas.0900944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roselli F, Tirard M, Lu J, Hutzler P, Lamberti P, Livrea P, Morabito M, Almeida OF. Soluble beta-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses. J Neurosci. 2005;25:11061–11070. doi: 10.1523/JNEUROSCI.3034-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roselli F, Hutzler P, Wegerich Y, Livrea P, Almeida OF. Disassembly of shank and homer synaptic clusters is driven by soluble beta-amyloid(1-40) through divergent NMDAR-dependent signalling pathways. PLoS One. 2009;4:e6011. doi: 10.1371/journal.pone.0006011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 41.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chafekar SM, Baas F, Scheper W. Oligomer-specific Abeta toxicity in cell models is mediated by selective uptake. Biochim Biophys Acta. 2008;1782:523–531. doi: 10.1016/j.bbadis.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Yang AJ, Chandswangbhuvana D, Margol L, Glabe CG. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J Neurosci Res. 1998;52:691–698. doi: 10.1002/(SICI)1097-4547(19980615)52:6<691::AID-JNR8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 44.Glabe C. Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer’s disease. J Mol Neurosci. 2001;17:137–145. doi: 10.1385/JMN:17:2:137. [DOI] [PubMed] [Google Scholar]
- 45.Cho KO, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- 46.Kornau HC, Schenker LT, Kennedy MB, Seeburg PH. Domain interaction between NMDA receptor subunits and the postsynaptic density protein PSD-95. Science. 1995;269:1737–1740. doi: 10.1126/science.7569905. [DOI] [PubMed] [Google Scholar]
- 47.Niethammer M, Kim E, Sheng M. Interaction between the C terminus of NMDA receptor subunits and multiple members of the PSD-95 family of membrane-associated guanylate kinases. J Neurosci. 1996;16:2157–2163. doi: 10.1523/JNEUROSCI.16-07-02157.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kistner U, Wenzel BM, Veh RW, Cases-Langhoff C, Garner AM, Appeltauer U, Voss B, Gundelfinger ED, Garner CC. SAP90, a rat presynaptic protein related to the product of the Drosophila tumor suppressor gene dlg-A. J Biol Chem. 1993;268:4580–4583. [PubMed] [Google Scholar]
- 49.Sprengel R, Suchanek B, Amico C, Brusa R, Burnashev N, Rozov A, Hvalby O, Jensen V, Paulsen O, Andersen P, Kim JJ, Thompson RF, Sun W, Webster LC, Grant SG, Eilers J, Konnerth A, Li J, McNamara JO, Seeburg PH. Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell. 1998;92:279–289. doi: 10.1016/s0092-8674(00)80921-6. [DOI] [PubMed] [Google Scholar]
- 50.Migaud M, Charlesworth P, Dempster M, Webster LC, Watabe AM, Makhinson M, He Y, Ramsay MF, Morris RG, Morrison JH, O’Dell TJ, Grant SG. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- 51.Sheng M, Kim E. The Shank family of scaffold proteins. J Cell Sci. 2000;113(Pt 11):1851–1856. doi: 10.1242/jcs.113.11.1851. [DOI] [PubMed] [Google Scholar]
- 52.Ehlers MD. Molecular morphogens for dendritic spines. Trends Neurosci. 2002;25:64–67. doi: 10.1016/s0166-2236(02)02061-1. [DOI] [PubMed] [Google Scholar]
- 53.Kim E, Naisbitt S, Hsueh YP, Rao A, Rothschild A, Craig AM, Sheng M. GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD- 95/SAP90 family of channel clustering molecules. J Cell Biol. 1997;136:669–678. doi: 10.1083/jcb.136.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- 55.Xiao B, Tu JC, Petralia RS, Yuan JP, Doan A, Breder CD, Ruggiero A, Lanahan AA, Wenthold RJ, Worley PF. Homer regulates the association of group 1 metabotropic glutamate receptors with multivalent complexes of homer-related, synaptic proteins. Neuron. 1998;21:707–716. doi: 10.1016/s0896-6273(00)80588-7. [DOI] [PubMed] [Google Scholar]
- 56.Boeckers TM, Kreutz MR, Winter C, Zuschratter W, Smalla KH, Sanmarti-Vila L, Wex H, Langnaese K, Bockmann J, Garner CC, Gundelfinger ED. Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. Ann Anat. 2001;183:101. doi: 10.1016/S0940-9602(01)80024-8. [DOI] [PubMed] [Google Scholar]
- 57.Gong Y, Lippa CF, Zhu J, Lin Q, Rosso AL. Disruption of glutamate receptors at Shank-postsynaptic platform in Alzheimer’s disease. Brain Res. 2009 doi: 10.1016/j.brainres.2009.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonaglia MC, Giorda R, Borgatti R, Felisari G, Gagliardi C, Selicorni A, Zuffardi O. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet. 2001;69:261–268. doi: 10.1086/321293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003;40:575–584. doi: 10.1136/jmg.40.8.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rockenstein E, Mallory M, Mante M, Sisk A, Masliah E. Early formation of mature amyloid-b proteins deposits in a mutant APP transgenic model depends on levels of Ab1-42. J neurosci Res. 2001;66:573–582. doi: 10.1002/jnr.1247. [DOI] [PubMed] [Google Scholar]
- 62.Rockenstein E, Mallory M, Mante M, Alford M, Windisch M, Moessler H, Masliah E. Effects of Cerebrolysin on amyloid-beta deposition in a transgenic model of Alzheimer’s disease. J Neural Transm Suppl. 2002:327–336. [PubMed] [Google Scholar]
- 63.Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, Dallapiccola B, Valente EM, Masliah E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J Neurochem. 2009;108:1561–1574. doi: 10.1111/j.1471-4159.2009.05932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hashimoto M, Rockenstein E, Mante M, Mallory M, Masliah E. β-Synuclein inhibits alpha-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron. 2001;32:213–223. doi: 10.1016/s0896-6273(01)00462-7. [DOI] [PubMed] [Google Scholar]
- 65.Levites Y, Das P, Price RW, Rochette MJ, Kostura LA, McGowan EM, Murphy MP, Golde TE. Anti-Abeta42- and anti-Abeta40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J Clin Invest. 2006;116:193–201. doi: 10.1172/JCI25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp Fig s1-3