Induction of cyclin E–cdk2 kinase activity, E2F-dependent transcription and cell growth by Myc are genetically separable events (original) (raw)
Abstract
The c-myc gene has been implicated in three distinct genetic programs regulating cell proliferation: control of cyclin E–cdk2 kinase activity, E2F-dependent transcription and cell growth. We have now used p27–/– fibroblasts to dissect these downstream signalling pathways. In these cells, activation of Myc stimulates transcription of E2F target genes, S-phase entry and cell growth without affecting cyclin E–cdk2 kinase activity. Both cyclin D2 and E2F2, potential direct target genes of Myc, are induced in p27–/– MycER cells. Ectopic expression of E2F2, but not of cyclin D2, induces S-phase entry, but, in contrast to Myc, does not stimulate cell growth. Our results show that stimulation of cyclin E–cdk2 kinase, of E2F-dependent transcription and of cell growth by Myc can be genetically separated from each other.
Keywords: cell growth/c-Myc/cyclin E kinase/E2F2/p27
Introduction
The proto-oncogene c-myc encodes a transcription factor of the helix–loop–helix/leucine zipper family of proteins. Expression of c-myc is frequently enhanced in human tumours, either due to mutations in the c-myc gene itself or due to mutations in signalling pathways that control c-myc expression, such as mutations of the APC gene (He et al., 1998). One possible explanation for this frequent deregulation is the unusual ability of Myc protein to interfere with genetic programs that control cellular proliferation (Obaya et al., 1999). For example, activation of conditional alleles of Myc (Eilers et al., 1989; Littlewood et al., 1995) can be sufficient to induce growth factor-independent proliferation in established rodent cell lines (Eilers et al., 1991).
c-myc has been implicated in at least three distinct genetic pathways controlling progression through the G1 phase. First, activation of Myc rapidly induces activation of cyclin E–cdk2 kinase and loss of p27kip1 from cdk2 complexes (Steiner et al., 1995; Santoni-Rugiu et al., 2000); this is required for Myc to promote proliferation (Rudolph et al., 1996). Conversely, levels of p27kip1 are elevated and cyclin E–cdk2 kinase activity is suppressed in cells expressing dominant-negative alleles of Myc and in c-_myc_–/– fibroblasts (Berns et al., 1997; Mateyak et al., 1999). In murine fibroblasts expressing Myc, p27kip1 is initially sequestered in cyclin D–cdk4 complexes (Bouchard et al., 1999; Perez-Roger et al., 1999); the direct transcriptional induction of the cyclin D2 and, potentially, of the cdk4 gene by Myc contributes to the formation of these complexes (Bouchard et al., 1999; Coller et al., 2000; Hermeking et al., 2000). In primary cells or in response to TGF-β signalling, suppression of the p15ink4b gene by Myc further contributes to sequestration of p27 (Warner et al., 1999). Subsequently, p27 is degraded (Müller et al., 1997) and the transcriptional stimulation of the Cul-1 gene by Myc has been implicated in this process (O’Hagan et al., 2000b). As a result, expression of Myc suppresses the growth inhibitory function of physiological levels of p27kip1 (Vlach et al., 1996). Similarly, suppression of the expression of p27kip1 by antisense oligonucleotides can induce proliferation in serum-starved fibroblasts in a manner reminiscent of induction of MycER (Coats et al., 1996). Together, these findings have led to the hypothesis that Myc controls G1 progression by suppressing the function of p27kip1 (Amati et al., 1998).
Secondly, activation of Myc induces the transcriptional activity of the E2F/DP family of transcription factors (Jansen-Dürr et al., 1993; Leone et al., 1997). Tran scriptional activation by E2F/DP family proteins is inhibited by association with members of the pocket protein family. Since the interaction between E2F/DP proteins and pocket proteins is controlled by cdk- dependent phosphorylation, activation of E2F-dependent transcription may be an indirect consequence of regulation of cdk activity by Myc. Alternatively, Myc has been shown to activate the promoter of the E2F2 gene, and constitutive expression of Myc upregulates expression of E2F2 protein (Sears et al., 1997, 1999). Therefore, induction of E2F transcriptional activity may be an independent and direct effector pathway of Myc.
Thirdly, Myc has also been implicated in cell growth, i.e. an increase in cell mass, both in Drosophila (Johnston et al., 1999) and in mammalian cells (Rosenwald et al., 1993; Mateyak et al., 1997; Iritani and Eisenman, 1999). An involvement of Myc in cell growth had also been suspected from the identity of putative target genes of Myc, many of which appear to control cellular metabolism and protein translation rather than cell cycle progression per se (Johnston et al., 1998; Dang, 1999; Coller et al., 2000; Greasley et al., 2000; O’Hagan et al., 2000a).
Together, the findings have prompted the question as to whether only one pathway is the primary effector pathway downstream of Myc, with other events occurring as secondary consequences of Myc-induced proliferation. Now, we report experiments aimed at genetically dissecting signalling pathways downstream of Myc. Our results allow the conclusion that activation of cyclin E–cdk2 kinase, induction of E2F-dependent transcription and stimulation of cell growth define genetically separable pathways by which Myc promotes G1 progression.
Results
Immortalization and induction of apoptosis by Myc are independent of p27
Previous work had linked the ability of Myc to stimulate G1 progression to its ability to antagonize the cell cycle arrest by physiological levels of p27kip1 (Amati et al., 1998). In order to determine whether loss of p27kip1 affects Myc function in primary cells, mouse embryo fibroblasts (MEFs) were isolated from both p27+/+ and p27–/– embryos. The genotype of the embryos was verified by Southern blotting and by western blotting of cellular extracts with antibodies directed against murine p27 (not shown). Cells were plated and infected in parallel with retroviruses expressing either a resistance gene alone (pbabe-puro; Morgenstern and Land, 1990) or together with a human c-myc cDNA. Western blots confirmed that both p27+/+ and p27–/– cells expressed human Myc protein at equal levels when infected with the corresponding virus (Figure 1A). When infected cells were plated at low density in selective medium, colonies emerged after 10–14 days. The number of colonies was greatly enhanced in plates infected with a virus expressing c-myc relative to vector control cells. In several independent experiments, expression of Myc stimulated colony formation in both p27+/+ and p27–/– cells to a similar extent (Figure 1A). Pools of infected p27–/– and p27+/+ cells proliferated with no sign of senescence upon infection with retroviruses expressing Myc, but arrested and showed a senescent phenotype (not shown) upon infection with control viruses (Figure 1B). In the absence of serum, both p27+/+ and p27–/– cells expressing c-myc underwent apoptosis within 48 h (Figure 1C). FACScan experiments confirmed that DNA fragmentation took place in ∼70% of both p27+/+ and p27–/– cells expressing c-myc (not shown). We concluded that primary p27–/– and p27+/+ cells were indistinguishable with respect to immortalization and induction of apoptosis by Myc.

Fig. 1. Immortalization and induction of apoptosis by Myc in primary p27+/+ and p27–/– MEFs. (A) Number of colonies growing upon infection of either p27–/– or p27+/+ cells with control (‘pbabe’) or Myc-expressing virus (‘pbabe-Myc’), after selection with puromycin and incubation for 2 weeks. The graph shows a quantitation of a representative experiment. The western blot documents equal expression of Myc proteins in p27–/– and p27+/+ cells. (B) Growth curve of pools of p27+/+ and p27–/– cells recovered after infection with the indicated viruses; 5 × 104 drug-resistant cells were plated at the start of the experiment. (C) Induction of apoptosis by Myc. p27+/+ and p27–/– MEFs, recovered after infection with the indicated viruses and selection, were plated in the absence or presence of serum; photographs were taken 48 h after plating.
Activation of cyclin E–cdk2 kinase by Myc depends on p27
The extensive fragmentation of DNA that occurred in primary cells infected with Myc upon serum starvation precluded a detailed analysis as to whether Myc stimulated cell cycle progression under these conditions. Therefore, cell lines were established from both p27–/– and p27+/+ cells using a standard 3T3 protocol. Both lines that were established maintained expression of p19_ARF_; however, in contrast to primary cells both p27–/– and p27+/+ 3T3 cells constitutively expressed high levels of p53 that were independent of DNA damage induced by adriamycin. These findings indicate that both lines had undergone a mutation in the p53 gene during immortalization (Figure 2A).


Fig. 2. Induction of cell cycle progression and cell growth by Myc. (A) Western blots documenting DNA-damage-independent expression of p53 in p27–/– and p27+/+ 3T3 cell lines and two MycER clones, and expression of p19ARF in MycER clones. Where indicated, cells were treated with 0.5 µg/ml adriamycin for the indicated periods of time (24 h in the upper panel). (B) Percentage of cells incorporating BrdU in p27–/– and p27+/+ MycER cell lines. Cells were serum starved for 48 h before addition of either 200 nM 4-OHT or 10% FCS. The percentage of cells incorporating BrdU was determined 20 h later. (C) Cell cycle distribution of the indicated cell lines. Cells were starved for 48 h before re-induction. Samples were taken at the indicated time points after addition of either 200 nM 4-OHT or 10% FCS. (D) FSC profiles indicating growth of p27–/– and p27+/+ MycER cells after activation of Myc or addition of FCS for 24 h.
p27+/+ and p27–/– 3T3 lines were infected with retroviruses expressing MycER proteins (Eilers et al., 1989; Littlewood et al., 1995). Both pools and clones were analysed for expression of MycER protein by western blotting. Clones and pools with detectable expression were expanded and subsequently analysed (see below). More detailed data were obtained first from one clonal p27+/+ and one clonal p27–/– MycER cell line.
BrdU (5-bromo-2-deoxyuridine) labelling was used to determine whether the ability of Myc to promote progression into S phase depends on p27. As described previously (Coats et al., 1999), both p27+/+ and p27–/– cells accumulate in G0 upon growth factor starvation (Figure 2B) and can be re-induced to enter the cell cycle upon addition of growth factors. In some experiments, we noted a higher percentage of cells incorporating BrdU in p27–/– cells relative to p27+/+ cells upon serum stimulation. Activation of Myc by addition of 4-hydroxy-tamoxifen (4-OHT) induced S-phase entry to a similar extent in both cell types (Figure 2B); time course experiments revealed no significant difference in the length of G1 and S phase (not shown). To determine whether stimulated cells progressed to mitosis, FACScan profiles were recorded at different time points after stimulation with either serum (10% fetal calf serum, FCS) or 4-OHT (Figure 2C). Both p27+/+ and p27–/– cells efficiently progressed into S and G2 phase when stimulated by addition of serum; counting of mitotic figures from 4′,6-diamidine-2-phenylindole (DAPI)-stained preparations revealed a relatively synchronous passage through mitosis 20 h after stimulation (not shown). In contrast, cells stimulated by addition of 4-OHT efficiently progressed into S phase but did not progress through mitosis (Figure 2C), and no significant increase in the number of mitotic figures was seen in either p27+/+ or p27–/– MycER cells (not shown).
Analysis of forward scatter (FSC) profiles to determine cell size from the same samples showed that addition of either FCS or 4-OHT also promoted growth of both p27+/+ and p27–/– cells (Figure 2D). Activation of Myc also promoted cell growth in the presence of either hydroxyurea or aphidicolin, whereas addition of either drug effectively blocked Myc-induced DNA synthesis (see Supplementary data, available at The EMBO Journal Online). The findings confirm previous observations that activation of Myc can promote cell growth in the absence of overt cell cycle progression (Schuhmacher et al., 1999). Finally, we noted that activation of Myc causes an increase in cell size in all phases of the cell cycle (see below, Figure 6F), similar to observations made in B cells of mice harbouring an Ig-Myc transgene (Iritani and Eisenman, 1999).


Fig. 6. Ectopic expression of cyclin D2 and of E2F2 in p27–/– MycER cells. (A) Western blots documenting expression of cyclin D2 (left) and of E2F2 (right) in pools of p27–/– MycER cells after infection with either control retroviruses (top), or retroviruses encoding cyclin D2 (lower left) or E2F2 (lower right). Cells were either growing exponentially or serum starved for 24 or 48 h. After 48 h, 200 nM 4-OHT was added and samples were harvested after the indicated times. (B) Western blot documenting the expression of cyclin A in p27–/– MycER control infected cells (top), and in cells expressing cyclin D2 (middle) or E2F2 (bottom) constitutively. (C) BrdU incorporation. The indicated cells were serum starved for 48 h before addition of 4-OHT. BrdU incorporation was measured 20 h later. (D) Cell cycle distribution as determined by FACScan of the indicated p27–/– MycER cell lines after addition of 4-OHT. (E) Top: FSC profiles of p27–/– MycER/cyclin D2 and of p27–/– MycER/E2F2 cells, relative to control cells, after 48 h of serum deprivation. Bottom: FSC profiles of serum-deprived p27–/– MycER/cyclin D2 (left) and of p27–/– MycER/E2F2 (right) cells after addition of 4-OHT, relative to non-induced control cells. Cells were deprived of serum for 48 h before addition of 4-OHT; samples were analysed after 20 h. (F) FSC profiles of cells gated for individual cell cycle phases. p27–/– MycER/E2F2 cells (as in E) were serum starved (control) and MycER was activated by addition of 4-OHT for 20 h.
Cyclin E–cdk2 kinase complexes are inactive in primary serum-starved p27–/– fibroblasts and are activated in response to addition of serum (Coats et al., 1999). In agreement with these observations, little kinase activity was detected in cyclin E immunoprecipitates from serum-starved p27+/+ or p27–/– MycER cells (Figure 3A). Re-addition of serum induced kinase activity in both cell lines to a similar extent. In contrast, addition of 4-OHT activated cyclin E–cdk2 kinase only in p27+/+, but not in p27–/– cells. A quantitation of the results of two representative experiments is shown in Figure 3B.

Fig. 3. Failure of Myc to activate cyclin E-dependent kinase in p27–/– cells. (A) Autoradiogram of cyclin E-dependent kinase assays. p27+/+ and p27–/– MycER cells were serum starved for 48 h and re-stimulated by addition of either 4-OHT or FCS as above. Samples were taken at the indicated time points and cyclin E-kinase activity was determined using histone H1 as substrate. (B) Quantitation of cyclin E-kinase activity (mean values from two independent experiments). (C) Western blots documenting the amount of cyclin E and p130 proteins, and cyclin E–p130 complexes in serum-starved p27+/+ and p27–/– MycER cells after induction of Myc. Samples were taken at the indicated time points. (D) Western blots documenting expression of cdk2 and cyclin A and quantitation of cyclin E-dependent kinase (relative to non-induced cells) in pools of p27–/– and p27+/+ MycER cells and in several independent clones of p27–/– MycER cells.
In serum-starved primary p27–/– cells, inhibition of cyclin E-dependent kinase activity is due to complex formation with the pocket protein p130 (Coats et al., 1999). Indeed, significant amounts of cyclin E–p130 complexes were detected in serum-starved p27–/– but not in p27+/+ MycER cells (Figure 3C); immunoprecipitations with α-p130 antibodies showed that such complexes were inactive both before and after activation of Myc (not shown). Cyclin E–p130 complexes remained stable after induction of Myc in p27–/– MycER cells (Figure 3C). Consistent with a view in which Myc specifically antagonizes association of p27_kip1_ with cyclin E, activation of Myc actually promoted the formation of a small amount of cyclin E–p130 complexes in p27+/+ MycER (Figure 3C) and in RAT1 MycER cells (not shown). We noted that in the first clones picked for analysis, levels of p130 appeared higher in the p27–/– than in the p27+/+ cells. However, analysis of the pools and clones, shown in Figure 3D, revealed that there was not a systematic difference in p130 protein levels between p27–/– and p27+/+ cells (data not shown).
Bandshift experiments showed that a fraction of complexes containing cyclin E, cdk2 and p130 were bound to E2F proteins in serum-starved p27–/– MycER cells, whereas no cyclin E–cdk2–p130–E2F complexes were detectable in serum-starved p27+/+ MycER cells (not shown). Activation of Myc did not alter the composition of E2F complexes in p27–/– MycER cells (as determined in bandshift analysis), demonstrating that activation of Myc did not antagonize association of p130 with either cyclin E–cdk2 complexes or E2F (not shown). Con sistent with this view, expression of elevated levels of p130 by microinjection inhibited cell cycle induction by Myc in p27–/– cells; analysis of mutants of p130 (Castano et al., 1998) revealed that binding to both E2F and cdk2 contributed to inhibition (not shown).
The data show that Myc can promote activation of cell cycle progression in the absence of activation of cyclin E–cdk2 kinase in p27–/– cells. To exclude the possibility that this phenotype is limited to the specific clones analysed, we tested both pools of p27+/+ and p27–/– MycER cells, and several additional clones of p27–/– MycER cells that expressed varying levels of MycER proteins. In all cases, activation of Myc upregulated expression of cyclin A, a marker protein of cell cycle progression (see below). However, cyclin E kinase was upregulated only in pools of p27+/+ MycER cells, but neither in pools nor in independent clones of p27–/– MycER cells (Figure 3D), demonstrating that the failure of Myc to induce cyclin E–cdk2 kinase activity is indeed due to the lack of p27.
Recently, a second cyclin E(2) has been identified (Lauper et al., 1998; Gudas et al., 1999); thus, activation of cyclin E2–cdk2 kinase might substitute for cyclin E–cdk2 kinase in p27–/– cells. However, in several experiments only low cyclin E2-dependent kinase activity was found that was induced upon addition of serum, but not significantly altered in response to activation of MycER (not shown). We concluded that Myc promotes cell cycle progression and cell growth in the absence of cyclin E–cdk2 activation in p27–/– cells.
Finally, we noted that ectopic expression of constitutive Myc induced extensive apoptosis in pools of serum-starved p27–/– and p27+/+ 3T3 cells, and that expression levels of Myc in stable cell lines derived from such pools were very low. These findings precluded a clear analysis of the mitogenic effects of constitutive Myc in these cell lines.
Induction of target genes of E2F by Myc is independent of p27
In order to understand how activation of Myc promotes cell cycle progression in the absence of activation of cyclin E–cdk2 kinase, we measured the expression of target genes of E2F after activation of Myc by reverse transcription PCR (RT–PCR) assays (Vigo et al., 1999). In both p27+/+ and p27–/– MycER cells, we observed induction of multiple target genes of E2F after activation of Myc and no significant difference was observed between both cell lines (Figure 4A). Induction of cyclin A expression was confirmed by western blotting (see below). The results demonstrate that activation of Myc can induce transcription of E2F target genes in the absence of induction of cyclin E–cdk2 kinase. A previous analysis of the cyclin A promoter had shown that induction of cyclin A expression by Myc is mediated via regulation of E2F activity (Rudolph et al., 1996).
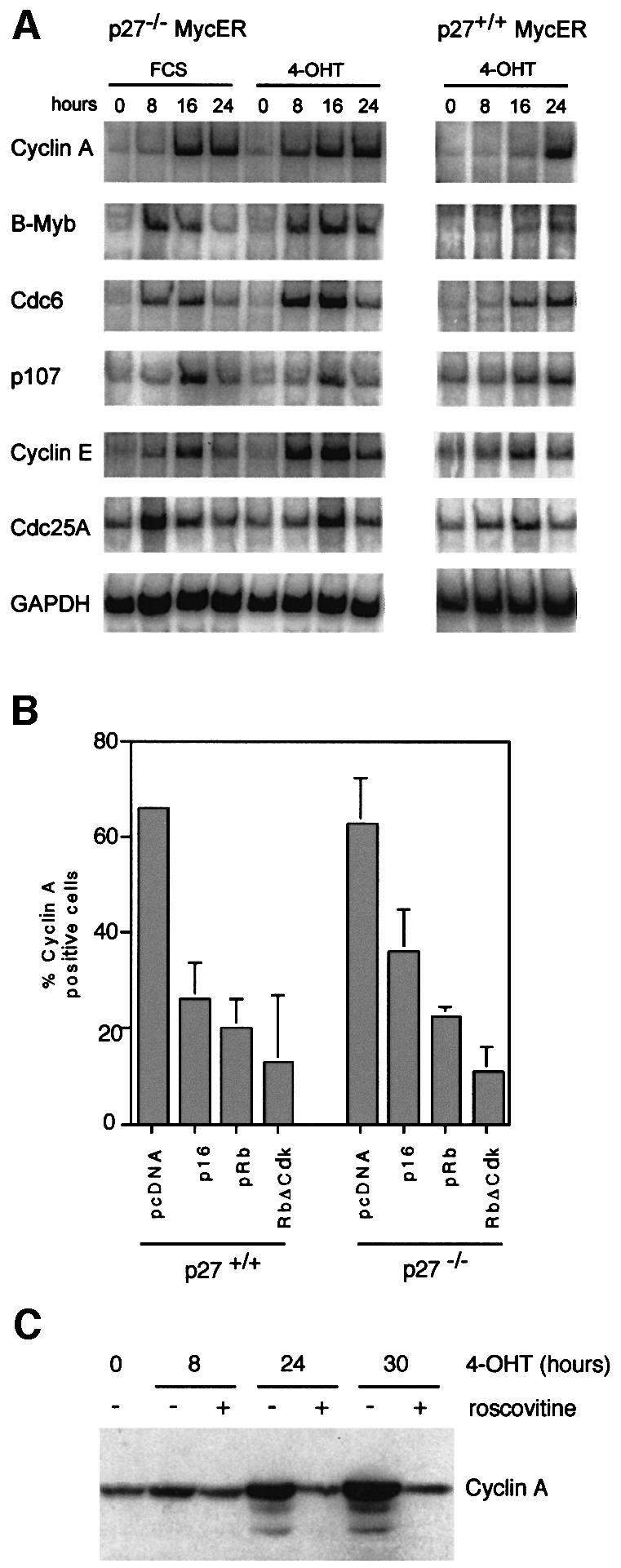
Fig. 4. Activation of E2F-dependent genes by Myc is independent of p27. (A) RT–PCR documenting the expression of target genes of E2F. Total RNA was prepared from p27–/– and p27+/+ MycER cells after stimulation with FCS or 4-OHT, and subjected to RT–PCR analysis as detailed in Materials and methods. (B) Percentage of cyclin A-positive cells after microinjection of expression plasmids encoding the indicated proteins. Cells were serum starved for 48 h before microinjection. Four hours later, cells were re-stimulated by the addition of 4-OHT. Cyclin A expression was detected by immunofluorescence 20 h later. (C) Western blot documenting expression of cyclin A in p27–/– MycER cells at the indicated time points after the addition of 200 nM 4-OHT to serum-starved cells. Where indicated, 25 µM roscovitine was added together with 4-OHT.
In RAT1 cells, both cdk2 and cdk4 kinase activities are required for Myc to activate cyclin A expression (Rudolph et al., 1996). Since p27 is sequestered by cdk4 complexes in response to activation of Myc, and since p27 is a substrate of cdk2, loss of p27 might influence the requirement for either cdk2 or cdk4 kinase activity. To test this, we microinjected expression plasmids encoding p16_ink4a_, pRb or non-phosphorylatable alleles of pRb into p27+/+ and p27–/– MycER cells (Figure 4B). In both cell lines, expression of any protein inhibited induction of cyclin A expression by Myc, demonstrating that cdk4 kinase activity is required for Myc to activate cyclin A expression. Similar experiments using dominant-negative alleles of cdk2 yielded equivocal results in p27–/– cells (not shown). Therefore, we used roscovitine, a specific chemical inhibitor of cdk2 kinase (Alessi et al., 1998). Addition of roscovitine abolished induction of cyclin A expression by Myc in both p27+/+ and p27–/– MycER cells (data shown for p27–/– cells in Figure 4C) at low micromolar concentrations. We concluded that the loss of p27 does not completely abolish the requirement for either cdk2 or cdk4 kinase activity in Myc-induced G1 progression.
Previous work has shown that ectopic expression of E2F1 can induce S-phase entry in the absence of detectable cdk2 activity, suggesting that this might be the mechanism by which Myc acts to promote S-phase entry in p27–/– cells (Leone et al., 1999). Surprisingly, however, the total cdk2 activity strongly increased after activation of Myc in both p27+/+ and p27–/– cells (Figure 5A; a quantitation of the results of two representative experiments is shown in Figure 5B). Precipitation with α-cyclin A antibodies showed identical results (not shown). Thus, cyclin A–cdk2 complexes that formed after induction of Myc were active in both p27+/+ and p27–/– cells. To explain the difference compared with cyclin E–cdk2 complexes, we tested whether cyclin A–p130 complexes could be detected; indeed, immunoprecipitation experiments detected the presence of cyclin A in α-p130 immunoprecipitates and vice versa (data not shown).

Fig. 5. Activation of cyclin A–cdk2 kinase activity by Myc in p27–/– MycER cells. (A) Autoradiogram of cdk2 kinase assays. p27+/+ and p27–/– MycER cells were serum starved for 48 h and re-stimulated by the addition of either 4-OHT or FCS as before. Samples were taken at the indicated time points and cdk2-kinase activity was determined using histone H1 as substrate. Under these conditions, cyclin A regulates most of the cdk2 activity. (B) Quantitation of cdk2-kinase activity (mean values from two independent experiments). (C) Western blots documenting the amount of p130, cyclin A and cdk2 in p27–/– MycER cells after stimulation with either 4-OHT or FCS as indicated. Cellular lysates were either left untreated (–) or depleted three times with α-cdk2 antibodies (+) before loading on the gel. (D) Western blots documenting the amount of p130 and of cyclin A in p27–/– MycER cells after stimulation with either 4-OHT or FCS as indicated. Cellular lysates were either left untreated (–) or depleted three times with α-p130 antibodies (+) before loading on the gel.
In contrast to observations made with p27_kip1_ (Poon et al., 1995), depletion of cell lysates by an α-cdk2 antibody not only removed cdk2, but also p130 in serum-starved cells, demonstrating that the amount of p130 is limiting in these cells (Figure 5C). Indeed, p130 is depleted by α-cdk2 antibodies both before and after activation of Myc or addition of serum in p27–/– MycER cells. At early time points after activation, newly synthesized cyclin A is also quantitatively bound to cdk2 (see 8 and 12 h after activation of Myc); taken together, both observations suggest that there is not enough p130 in these cells to inhibit quantitatively newly formed cyclin A–cdk2 complexes. Consistent with this view, depletion with an α-p130 antibody failed to deplete cyclin A from p27–/– MycER cells (Figure 5D). The data suggest that the strong increase in cyclin A protein is sufficient to account for the preferential activation of cyclin A–cdk2 versus cyclin E–cdk2 complexes. They do not exclude, however, that differences in affinity between cyclin A–cdk2 and cyclin E–cdk2 complexes towards p130 contribute to the different behaviour of these complexes upon induction of Myc.
Neither activation of E2F nor cyclin E–cdk2 kinase accounts for the growth-promoting effect of Myc
In response to activation of Myc, p27 is sequestered by cyclin D1 and cyclin D2–cdk4 complexes; the accumulation of these complexes is driven in part by the direct transcriptional activation of the cyclin D2 gene by Myc (Bouchard et al., 1999; Perez-Roger et al., 1999; Coller et al., 2000). Since D-type cyclin complexes can phosphorylate pocket proteins, induction of cyclin D2 could account for the transcriptional activation of E2F-dependent genes. An alternative mechanism is suggested by the finding that the E2F2 gene is a potential direct target gene of Myc (Sears et al., 1997, 1999). Indeed, western blotting from p27–/– MycER cells demonstrated that expression of both cyclin D2 and E2F2 is upregulated in response to activation of MycER (Figure 6A). In contrast, expression of cdk4 was high and constitutive under all conditions tested (data not shown).
To test whether induction of either gene can account for the mitogenic effect of Myc in p27–/– cells, we superinfected p27–/– MycER cells either with control retroviruses or with retroviruses expressing cyclin D2 or E2F2, together with a phleomycin-resistance gene (Figure 6A). Pools of infected cells expressing either cyclin D2 or E2F2 were serum starved for 48 h, before MycER was activated by addition of 4-OHT. From these pools, expression of cyclin D2, cyclin A and E2F2, cell cycle progression and cell size were determined (Figure 6B–E). Expression of cyclin D2 did not by itself maintain expression of cyclin A and did not stimulate cell cycle progression in serum-starved cells (Figure 6B and C). Expression of cyclin D2 slightly facilitated emergence from quiescence and cell cycle progression upon activation of Myc (Figure 6D). In contrast, cells expressing E2F2 maintained higher levels of cyclin A even upon serum starvation than exponentially growing control cells, supporting the view that induction of E2F2 expression in p27–/– cells can account for the effects of Myc on E2F-dependent gene expression (Figure 6B). Also, an increased percentage of cells expressing E2F2 remained in cycle upon serum starvation, demonstrating that induction of E2F2, but not cyclin D2, can partially account for the S-phase-promoting effects of Myc in p27–/– cells (Figure 6C and D).
However, cells that express E2F2 constitutively were significantly smaller than control cells after serum deprivation (Figure 6E), suggesting that constitutive expression of E2F2 promotes cell cycle progression without promoting cell growth. In contrast, activation of Myc promoted cell growth both in cells expressing cyclin D2 and in cells expressing E2F2 (Figure 6E). Importantly, activation of Myc caused a shift in the FSC profile of cells in each phase of the cell cycle (Figure 6F). In contrast, the size of apoptotic cells with a subG1 DNA content was equal before and after activation of Myc. These data further support the notion that Myc-stimulated cell growth is independent of cell cycle progression per se (also see above).
To exclude the possibility that the inability of E2F2 to stimulate obvious cell growth was due to the use of a non-regulatable protein, we established pools of p27+/+ and p27–/–E2F2-ER cells (data shown for p27–/– cells in Figure 7A) (Vigo et al., 1999). Cells were arrested by serum deprivation before E2F2 was activated by addition of 4-OHT. Activation of E2F2 in serum-starved cells efficiently induced expression of cyclin A and stimulated S-phase entry (Figure 7A and B), but did not lead to an increase in cell size at any time point after addition of 4-OHT (Figure 7C). Similar results were obtained in p27+/+ E2F2-ER cells (not shown).

Fig. 7. Activation of E2F2 promotes cell cycle progression, but not cell growth. (A) Left: western blot documenting expression of cyclin A before and after addition of 4-OHT, in pools of p27–/– cells infected with either a retrovirus expressing an E2F2-ER chimera (Vigo et al., 1999) or control virus. Cells were serum starved for 72 h and induced by addition of 200 nM 4-OHT for 24 h. Right: western blot documenting the expression of E2F2-ER upon retroviral infection. (B) FACScan profile showing the DNA content of serum-starved p27–/– E2F2-ER cells before (left) and after (right) addition of 4-OHT for 24 h. (C) FSC profiles of p27–/– E2F2-ER cells under the same conditions as (B).
The data show that activation of Myc promotes cell growth in the absence of activation of cyclin E-dependent kinase and of E2F-dependent transcription, and that the induction of E2F-dependent transcription does not account for the growth-promoting effect of Myc. Taken together, the data show that the activation of cyclin E-dependent kinase, of E2F-dependent transcription and the stimulation of cell growth are genetically separable functions of Myc.
Discussion
While the role of c-myc as a regulator of cell proliferation is well established, the mechanisms by which Myc protein promotes G1 progression are far from clear. In particular, specific roles for Myc in the control of E2F-dependent transcription, in activation of cyclin E–cdk2 kinase activity and in cell growth have been suggested, raising the question as to whether any one of these roles is upstream of other effects during Myc-stimulated G1 progression.
A previous approach to address this issue made use of centrifugal elutriation of RAT1 MycER cells growing exponentially in the presence of serum growth factors (Pusch et al., 1997). Under these conditions, activation of Myc does not affect the rate of G1 progression and cell proliferation. However, activation of Myc leads to a premature activation of cyclin E–cdk2 kinase and of E2F-dependent transcription in small cells early after exit from mitosis. Since activation of Myc does not affect the rate of proliferation in these cells, Myc’s stimulatory effect on both cyclin E kinase and on E2F-dependent transcription is not linked to any effect on cell growth.
We have now used a genetic strategy to further dissect signalling pathways downstream of Myc. We show that Myc is unable to regulate cyclin E–cdk2 kinase activity in p27–/– cells. In serum-starved p27–/– cells, cyclin E-dependent kinase activity is limited by association with p130 (Coats et al., 1999), and cyclin E–p130 complexes are stable after activation of Myc; hence, there is no activation of cyclin E-dependent kinase. The amounts of p130 are limiting and are insufficient to inhibit both cyclin E–cdk2 and newly synthesized cyclin A–cdk2 complexes. Therefore, the increased synthesis of cyclin A results in a preferential activation of cyclin A–cdk2 complexes in p27–/– cells.
Loss of p27 does not impair the ability of Myc to induce E2F-dependent transcription, G1 progression and cell growth, demonstrating that upregulation of cyclin E–cdk2 kinase activity is not sufficient to explain the ability of Myc to stimulate E2F-dependent transcription and cell growth. Ectopic expression of constitutive or inducible alleles of E2F2 induces E2F-dependent transcription and S-phase entry, but not cell growth. In contrast, activation of Myc stimulates cell growth even in the presence of deregulated E2F-dependent transcription. Therefore, neither induction of cyclin E–cdk2 kinase activity nor stimulation of E2F-dependent transcription accounts for the ability of Myc to stimulate cell growth. Taken together with previous data, the data strongly suggest that regulation of kinase activity, E2F-dependent transcription and growth are distinct downstream signalling pathways of Myc that are regulated by distinct (sets of) target genes.
Several lines of evidence support this conclusion. First, induction of c-myc in a B-cell line that harbours a tetracyclin-regulated allele of c-myc (Schuhmacher et al., 1999) in the absence of growth factors promotes cell growth, but not cell proliferation; in contrast, activation of c-myc in the presence of growth factors promotes cell proliferation. Although the molecular basis for the difference remains unclear, the data support the view that Myc’s ability to promote cell growth can be separated from its ability to promote cell cycle progression. Similarly, B cells expressing an Ig-Myc transgene are enlarged during all phases of the cell cycle, demonstrating that Myc can enhance growth independently of its effects on cell proliferation (Iritani and Eisenman, 1999).
Secondly, the growing list of putative target genes of Myc includes genes directly involved in the control of cyclin E–cdk2 kinase: cdc25A, cyclin D2 and cdk4 (Galaktionov et al., 1996; Bouchard et al., 1999; Perez-Roger et al., 1999; Coller et al., 2000; Hermeking et al., 2000), and of E2F-dependent transcription: E2F2 (Sears et al., 1997, 1999). In all cases, direct binding sites for Myc–Max protein complexes have been identified in the promoter or enhancer elements of these genes. For both cyclin D2 and cdc25A, activation of transcription upon induction of Myc is resistant to the addition of cycloheximide, demonstrating that the activation does not depend on prior induction of protein synthesis and cell growth. Conversely, several putative target genes of Myc are involved in metabolic pathways and several, including eIF4, have been directly implicated in the control of translation and cell growth (Johnston et al., 1998; Dang, 1999; Coller et al., 2000; Greasley et al., 2000; O’Hagan et al., 2000a). One likely conclusion from these studies is that the capacity of Myc to promote cell proliferation results from coordinate changes in expression of a potentially large number of target genes.
Thirdly, the present study points to a high degree of conservation between the function of c-myc and Drosophila (d)Myc genes (Johnston et al., 1999). Studies on dMyc mutant cells in the wing disc have shown that dMyc, like cyclin E, stimulates progression through the G1 phase. In contrast to cyclin E and E2F, however, dMyc also promotes cell growth, not only cell cycle progression, showing that dMyc, like c-myc, has a growth-promoting activity distinct from its ability to induce cyclin E kinase activity.
We note that in wing discs, dMyc is effective at promoting the G1–S progression but poorly effective at promoting progression through the G2–M phases of the cell cycle and, therefore, fails to enhance the rate of cell cycle progression. In contrast, dE2F promotes both transitions by enhancing both cyclin E and string (cdc25) expression (Neufeld et al., 1998). Similar to dMyc, activation of c-Myc effectively promotes progression from G1 to S phase in the mouse cell lines studied here, but fails to promote G2–M progression effectively in the absence of serum growth factors. It appears, therefore, that neither c-myc nor dMyc is immediately involved in stimulating progression through the G2 phase of the cell cycle.
Finally, we would like to point out that we have used a gain-of-function approach, mimicking the situation found in tumours, where Myc is frequently overexpressed. Experiments using c–_myc_–/– fibroblasts or dominant-negative alleles of c-myc suggest that endogenous Myc protein has a rate-limiting function in cell cycle progression, cyclin E–cdk2 regulation, E2F-dependent transcription and cell growth, suggesting that our findings extrapolate to the functions of endogenous Myc (Berns et al., 1997; Mateyak et al., 1997, 1999; Santoni-Rugiu et al., 2000). Elimination of p27 enhances the rate of proliferation of c-_myc_–/– MEFs, suggesting that the function of Myc in cyclin E–cdk2 kinase regulation is rate-limiting in these cells (O’Hagan et al, 2000b). However, as long as the mechanisms by which Myc controls proliferation are not fully resolved, it remains a formal possibility that overexpressed and endogenous Myc proteins carry out different functions in cellular metabolism.
Materials and methods
Cell culture and retroviruses
Primary MEFs were isolated from 9.5 day embryos as described before (Haas et al., 1997) and genotyped by PCR using primers specific for the wild-type and knock-out alleles of p27 (Fero et al., 1996). MEFs were maintained in Dulbecco’s modified Eagle’s medium containing 10% FCS, 2 mM glutamine, 100 µg/ml streptomycin and 100 U/ml penicillin. Cells were infected at passage 3 or used directly to generate established cell lines according to a 3T3 protocol (3 × 105 cells transferred at 3 day intervals).
Recombinant retroviruses were generated by transient transfection of Phoenix packaging cells (Grignani et al., 1998). The pbabe-puro-Myc, pbabe-puro-MycER™ and pbabe-puro-E2F2-ER vectors have been described (Littlewood et al., 1995; Bouchard et al., 1999; Vigo et al., 1999). To express rat cyclin D2 or human E2F2 (gift from Willi Krek), full-length cDNAs were amplified by PCR and inserted into the pbabe-phleo vector (Morgenstern and Land, 1990). Selection was carried out using 40 µg/ml Zeocin™ (Cayla). Rosocovitine was obtained from Calbiochem, adriamycin from Pharmacia and Upjohn, and aphidicolin from Sigma.
Cell cycle analysis
Western blot analysis and immunoprecipitations of cyclin–cdk complexes were performed as described (Steiner et al., 1995; Bouchard et al., 1999). Immune depletion of cdk2 complexes was performed by three consecutive rounds of depletions with protein-G-coupled α-cdk2 antibodies (M2; Santa Cruz). The following antibodies were used for western blotting: cyclin D2, DCS-3 (Lukas et al., 1995); p19ARF (Abcam); p53, CM-5 (Novocastra); cyclin A, C-19 (western blot) and H-432 (IP); cyclin E, M-20; cdk2, M2; E2F2, C-20; p130, C-20 (all Santa Cruz). p130 was depleted using a mixture of C-20 (Santa Cruz) and R27020 (Transduction Laboratories) antibodies.
BrdU labelling was carried out with cells grown on coverslips. Before fixation, cells were incubated with 50 µM BrdU for 1 h. BrdU-positive cells were counted by fluorescence microscopy after staining with an α-BrdU monoclonal antibody (RPN202; Amersham). cdk assays were carried out as described previously (Steiner et al., 1995) using histone H1 as substrate and the following antibodies: cyclin E, M-20; cyclin A, H432; cdk2, M2 (all Santa Cruz). Antibodies directed against cyclin E2 were a kind gift from Bruno Amati. FACScan analysis was performed using a FACScalibur instrument (Becton-Dickinson). The ‘Modfit’ integrating software package (Verity Software House) was used for cell cycle analysis. ‘Cellquest’ was used for FSC analysis.
RT–PCR assays of E2F target genes was carried out exactly as described in Vigo et al., (1999), except that the primers were adapted to represent the mouse cDNA sequences.
RNA from cell pellets was prepared using the RNeasy kit (Qiagen).
The conditions for microinjections have been described (Rudolph et al., 1996); the pCMVRb and pCMVRbΔCDK vectors were gifts of Jiri Bartek (Lukas et al., 1997).
Supplementary data
Supplementary data for this paper are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We would like to thank N.Liu and Rolf Müller for help with E2F-bandshift experiments. We thank Myung-Ho Lee, Bruno Amati, Willi Krek, Jiri Bartek, Kristian Helin and Roger Watson for reagents, and Bruno Amati and members of the laboratory for stimulating discussions. This work was supported by the Hess program of the Deutsche Forschungsgemeinschaft, a grant from the Human Frontiers of Science Organisation and a Biomed 2 grant from the European community.
References
- Alessi F., Quarta,S., Savio,M., Riva,F., Rossi,L., Stivala,L.A., Scovassi,A.I., Meijer,L. and Prosperi,E. (1998) The cyclin-dependent kinase inhibitors olomoucine and roscovitine arrest human fibroblasts in G1 phase by specific inhibition of CDK2 kinase activity. Exp. Cell Res., 245, 8–18. [DOI] [PubMed] [Google Scholar]
- Amati B., Alevizopoulos,K. and Vlach,J. (1998) Myc and the cell cycle. Front. Biosci., 15, D250–D268. [DOI] [PubMed] [Google Scholar]
- Berns K., Hijmans,E.M. and Bernards,R. (1997) Repression of c-Myc responsive genes in cycling cells causes G1 arrest through reduction of cyclin E/CDK2 kinase activity. Oncogene, 15, 1347–1356. [DOI] [PubMed] [Google Scholar]
- Bouchard C. et al. (1999) Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J., 18, 5321–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castano E., Kleyner,Y. and Dynlacht,B.D. (1998) Dual cyclin-binding domains are required for p107 to function as a kinase inhibitor. Mol. Cell. Biol., 18, 5380–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coats S., Flanagan,W.M., Nourse,J. and Roberts,J.M. (1996) Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science, 272, 877–880. [DOI] [PubMed] [Google Scholar]
- Coats S., Whyte,P., Fero,M.L., Lacy,S., Chung,G., Randel,E., Firpo,E. and Roberts,J.M. (1999) A new pathway for mitogen-dependent cdk2 regulation uncovered in p27Kip1-deficient cells. Curr. Biol., 9, 163–173. [DOI] [PubMed] [Google Scholar]
- Coller H.A., Grandori,C., Tamayo,P., Colbert,T., Lander,E.S., Eisenman,R.N. and Golub,T.R. (2000) Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signalling and adhesion. Proc. Natl Acad. Sci. USA, 97, 3260–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang C.V. (1999) c-Myc target genes involved in cell growth, apoptosis and metabolism. Mol. Cell. Biol., 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M., Picard,D., Yamamoto,K. and Bishop,J.M. (1989) Chimaeras between the MYC oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature, 340, 66–68. [DOI] [PubMed] [Google Scholar]
- Eilers M., Schirm,S. and Bishop,J.M. (1991) The MYC protein activates transcription of the α-prothymosin gene. EMBO J., 10, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fero M.L. et al. (1996) A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis and female sterility in p27Kip1-deficient mice. Cell, 85, 733–744. [DOI] [PubMed] [Google Scholar]
- Galaktionov K., Chen,X. and Beach,D. (1996) Cdc25 cell-cycle phosphatase as a target of c-myc. Nature, 382, 511–517. [DOI] [PubMed] [Google Scholar]
- Greasley P.J., Bonnard,C. and Amati,B. (2000) Myc induces the nucleolin and BN51 genes: possible implications in ribosome biogenesis. Nucleic Acids Res., 28, 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grignani F., Kinsella,T., Mencarelli,A., Valtieri,M., Riganelli,D., Lanfrancone,L., Peschle,C., Nolan,G.P. and Pelicci,P.G. (1998) High-efficiency gene transfer and selection of human hematopoietic progenitor cells with a hybrid EBV/retroviral vector expressing the green fluorescence protein. Cancer Res., 58, 14–19. [PubMed] [Google Scholar]
- Gudas J.M., Payton,M., Thukral,S., Chen,E., Bass,M., Robinson,M.O. and Coats,S. (1999) Cyclin E2, a novel G1 cyclin that binds Cdk2 and is aberrantly expressed in human cancers. Mol. Cell. Biol., 19, 612–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas K., Staller,P., Geisen,C., Bartek,J., Eilers,M. and Möröy,T. (1997) Mutual requirement of CDK4 and Myc in malignant transformation: evidence for cyclin D1/CDK4 and p16INK4A as upstream regulators of Myc. Oncogene, 15, 179–192. [DOI] [PubMed] [Google Scholar]
- He T.C., Sparks,A.B., Rago,C., Hermeking,H., Zawel,L., da Costa,L.T., Morin,P.J., Vogelstein,B. and Kinzler,K.W. (1998) Identification of c-MYC as a target of the APC pathway. Science, 281, 1509–1512. [DOI] [PubMed] [Google Scholar]
- Hermeking H. et al. (2000) Identification of CDK4 as a target of c-MYC. Proc. Natl Acad. Sci. USA, 97, 2229–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iritani B.M. and Eisenman,R.N. (1999) c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc. Natl Acad. Sci. USA, 96, 13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen-Dürr P., Meichle,A., Steiner,P., Pagano,M., Finke,K., Botz,J., Wessbecher,J., Draetta,G. and Eilers,M. (1993) Differential modulation of cyclin gene expression by MYC. Proc. Natl Acad. Sci. USA, 90, 3685–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston K.A., Polymenis,M., Wang,S., Branda,J. and Schmidt,E.V. (1998) Novel regulatory factors interacting with the promoter of the gene encoding the mRNA cap binding protein (eIF4E) and their function in growth regulation. Mol. Cell. Biol., 18, 5621–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston L.A., Prober,D.A., Edgar,B.A., Eisenman,R.N. and Gallant,P. (1999) Drosophila myc regulates cellular growth during development. Cell, 98, 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauper N., Beck,A.R., Cariou,S., Richman,L., Hofmann,K., Reith,W., Slingerland,J.M. and Amati,B. (1998) Cyclin E2: a novel CDK2 partner in the late G1 and S phases of the mammalian cell cycle. Oncogene, 17, 2637–2643. [DOI] [PubMed] [Google Scholar]
- Leone G., DeGregori,J., Sears,R., Jakoi,L. and Nevins,J.R. (1997) Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature, 387, 422–426. [DOI] [PubMed] [Google Scholar]
- Leone G., DeGregori,J., Jakoi,L., Cook,J.G. and Nevins,J.R. (1999) Collaborative role of E2F transcriptional activity and G1 cyclin dependent kinase activity in the induction of S phase. Proc. Natl Acad. Sci. USA, 96, 6626–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood T.D., Hancock,D.C., Danielian,P.S., Parker,M.G. and Evan,G.I. (1995) A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res., 23, 1686–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J., Bartkova,J., Welcker,M., Petersen,O.W., Peters,G., Strauss,M. and Bartek,J. (1995) Cyclin D2 is a moderately oscillating nucleoprotein required for G1 phase progression in specific cell types. Oncogene, 10, 2125–2134. [PubMed] [Google Scholar]
- Lukas J., Herzinger,T., Hansen,K., Moroni,M.C., Resnitzky,D., Helin,K., Reed,S.I. and Bartek,J. (1997) Cyclin E-induced S phase without activation of the pRb/E2F pathway. Genes Dev., 11, 1479–1492. [DOI] [PubMed] [Google Scholar]
- Mateyak M.K., Obaya,A.J., Adachi,S. and Sedivy,J.M. (1997) Pheno types of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ., 8, 1039–1048. [PubMed] [Google Scholar]
- Mateyak M.K., Obaya,A.J. and Sedivy,J.M. (1999) c-Myc regulates cyclin D-cdk4 and -cdk6 activity but affects cell cycle progression at multiple independent points. Mol. Cell. Biol., 19, 4672–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern J.P. and Land,H. (1990) A series of mammalian expression vectors and characterisation of their expression of a reporter gene in stably and transiently transfected cells. Nucleic Acids Res., 18, 1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller D., Bouchard,C., Rudolph,B., Steiner,P., Stuckmann,I., Saffrich,R., Ansorge,W., Huttner,W. and Eilers,M. (1997) Cdk2-dependent phosphorylation of p27 facilitates its Myc-induced release from cyclin E/cdk2 complexes. Oncogene, 15, 2561–2576. [DOI] [PubMed] [Google Scholar]
- Neufeld T.P., de la Cruz,A.F., Johnston,L.A. and Edgar,B.A. (1998) Coordination of growth and cell division in the Drosophila wing. Cell, 93, 1183–1193. [DOI] [PubMed] [Google Scholar]
- Obaya A.J., Mateyak,M.K. and Sedivy,J.M. (1999) Mysterious liaisons: the relationship between c-Myc and the cell cycle. Oncogene, 18, 2934–2941. [DOI] [PubMed] [Google Scholar]
- O’Hagan R.C. et al. (2000a) Gene-target recognition among members of the myc superfamily and implications for oncogenesis. Nature Genet., 24, 113–119. [DOI] [PubMed] [Google Scholar]
- O’Hagan R.C., Ohh,M., David,G., Moreno de Alboran,I., Alt,F.W., Kaelin,W.G. and DePinho,R. (2000b) Myc-enhanced expression of Cul1 promotes ubiquitin-dependent proteolysis and cell cycle progression. Genes Dev., 14, 2185–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Roger I., Kim,S.H., Griffiths,B., Sewing,A. and Land,H. (1999) Cyclins D1 and D2 mediate Myc-induced proliferation via sequestration of p27Kip1 and p21Cip1. EMBO J., 18, 5310–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon R.Y.C., Toyoshima,H. and Hunter,T. (1995) Redistribution of the CDK inhibitor p27 between different cyclin–CDK complexes in the mouse fibroblast cell cycle and in cells arrested with lovastatin or ultraviolet irradiation. Mol. Biol. Cell, 6, 1197–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch O., Bernaschek,G., Eilers,M. and Hengstschlager,M. (1997) Activation of c-Myc uncouples DNA replication from activation of G1-cyclin-dependent kinases. Oncogene, 15, 649–656. [DOI] [PubMed] [Google Scholar]
- Rosenwald I.B., Rhoads,D.B., Callanan,L.D., Isselbacher,K.J. and Schmidt,E.V. (1993) Increased expression of eukaryotic translation initiation factors eIF-4E and eIF-2α in response to growth induction by c-myc. Proc. Natl Acad. Sci. USA, 90, 6175–6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph B., Zwicker,J., Saffrich,R., Henglein,B., Müller,R., Ansorge,W. and Eilers,M. (1996) Activation of cyclin dependent kinases by Myc mediates transcriptional activation of cyclin A, but not apoptosis. EMBO J., 15, 3065–3076. [PMC free article] [PubMed] [Google Scholar]
- Santoni-Rugiu E., Falck,J., Mailand,N., Bartek,J. and Lukas,J. (2000) Involvement of Myc activity in a G1/S-promoting mechanism parallel to the pRb/E2F pathway. Mol. Cell. Biol., 20, 3497–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmacher M., Staege,M.S., Pajic,A., Polack,A., Weidle,U.H., Bornkamm,G.W., Eick,D. and Kohlhuber,F. (1999) Control of cell growth by c-Myc in the absence of cell division. Curr. Biol., 9, 1255–1258. [DOI] [PubMed] [Google Scholar]
- Sears R., Ohtani,K. and Nevins,J.R. (1997) Identification of positively and negatively acting elements regulating expression of the E2F2 gene in response to cell growth signals. Mol. Cell. Biol., 17, 5227–5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears R., Leone,G., DeGregori,J. and Nevins,J.R. (1999) Ras enhances Myc protein stability. Mol. Cell, 3, 169–179. [DOI] [PubMed] [Google Scholar]
- Steiner P., Philipp,A., Lukas,J., Godden-Kent,D., Pagano,M., Mittnacht,S., Bartek,J. and Eilers,M. (1995) Identification of a Myc-dependent step during the formation of active G1 cyclin/cdk complexes. EMBO J., 14, 4814–4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigo E., Muller,H., Prosperini,E., Hateboer,G., Cartwright,P., Moroni,M.C. and Helin,K. (1999) CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol. Cell. Biol., 19, 6379–6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlach J., Hennecke,S., Alevizopoulos,K., Conti,D. and Amati,B. (1996) Growth arrest by the cyclin-dependent kinase inhibitor p27Kip1 is abrogated by c-Myc. EMBO J., 15, 6595–6604. [PMC free article] [PubMed] [Google Scholar]
- Warner B.J., Blain,S.W., Seoane,J. and Massague,J. (1999) Myc downregulation by transforming growth factor β required for activation of the p15Ink4b G1 arrest pathway. Mol. Cell. Biol., 19, 5913–5922. [DOI] [PMC free article] [PubMed] [Google Scholar]