QIIME allows analysis of high-throughput community sequencing data (original) (raw)
. Author manuscript; available in PMC: 2011 Aug 16.
Published in final edited form as: Nat Methods. 2010 Apr 11;7(5):335–336. doi: 10.1038/nmeth.f.303
To the Editor
High-throughput sequencing is revolutionizing microbial ecology studies. Efforts like the Human Microbiome Projects1 and the US National Ecological Observatory Network2 are helping us to understand the role of microbial diversity in habitats within our own bodies and throughout the planet.
Pyrosequencing using error-correcting, sample-specific barcodes allows hundreds of communities to be analyzed simultaneously in multiplex3. Integrating information from thousands of samples, including those obtained from time series, can reveal large-scale patterns that were inaccessible with lower-throughput sequencing methods. However, a major barrier to achieving such insights has been the lack of software that can handle these increasingly massive datasets. Although tools exist to perform library demultiplexing and taxonomy assignment4,5, tools for downstream analyses are scarce.
Here we describe ‘quantitative insights into microbial ecology’ (QIIME; prounounced ‘chime’), an open-source software pipeline built using the PyCogent toolkit6, to address the problem of taking sequencing data from raw sequences to interpretation and database deposition. QIIME, available at http://qiime.sourceforge.net/, supports a wide range of microbial community analyses and visualizations that have been central to several recent high-profile studies, including network analysis, histograms of within- or between-sample diversity and analysis of whether ‘core’ sets of organisms are consistently represented in certain habitats. QIIME also provides graphical displays that allow users to interact with the data. Our implementation is highly modular and makes extensive use of unit testing to ensure the accuracy of results. This modularity allows alternative components for functionalities such as choosing operational taxonomic units (OTUs), sequence alignment, inferring phylogenetic trees and phylogenetic and taxon-based analysis of diversity within and between samples (including incorporation of third-party applications for many steps) to be easily integrated and benchmarked against one another (Supplementary Fig. 1).
We applied the QIIME workflow to a combined analysis of previously collected data (see Supplementary Discussion) for distal gut bacterial communities from conventionally raised mice, adult human monozygotic and dizygotic twins and their mothers, and a time series study of adult germ-free mice after they received human fecal microbiota (Fig. 1, Supplementary Table 1 and Supplementary Discussion). This analysis combined ten full 454 FLX runs and one partial run, totalling 3.8 million bacterial 16S rRNA sequences from previously published studies, including reads from different regions of the 16S rRNA gene.
Figure 1.
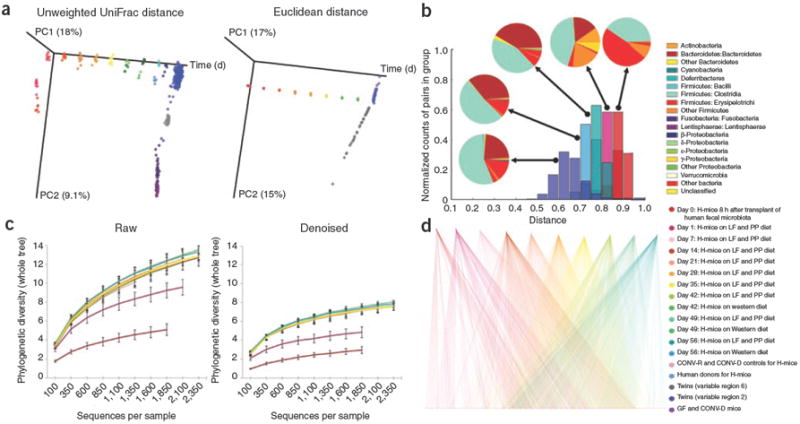
QIIME analyses of the distal gut microbiotas of conventionally raised and conventionalized mice, gnotobiotic mice colonized with a human fecal gut microbiota (H-mice), and human adult mono- and dizygotic twins. (a) Principal coordinates analysis plots for mice, H-mice and twins. Colors correspond to separate samples by species and time point, and are consistent throughout the panels. (b) Unweighted UniFrac distance histograms between the data for fecal microbiota of human twins; human donors for the H-mice study; day 56 post-transplant H-mice on a low-fat (LF) and plant polysaccharide–rich (PP) diet; day 1 H-mice (LF and PP diet); and day 0 H-mice. Taxonomic classifications are presented at the class level. (c) Alpha diversity rarefaction plots of phylogenetic diversity for the H-mice samples. (d) OTU network connectivity of H-mice time series data. CONV-D, conventionalized mice; CONV-R, conventionally raised mice; and GF, germ-free mice.
QIIME is thus a robust platform for combining heterogeneous experimental datasets and for rapidly obtaining new insights about various microbial communities. Because QIIME scales to millions of sequences and can be used on platforms from laptops to high-performance computing clusters, we expect it to keep pace with advances in sequencing technology and to facilitate characterization of microbial community patterns ranging from normal variations to pathological disturbances in many human, animal and other environmental ecosystems.
Supplementary Material
Supp
Acknowledgments
We thank our collaborators for their helpful suggestions on features, documentation and the manuscript, and our funding agencies for their commitment to open-source software. This work was supported in part by Howard Hughes Medical Institute and grants from the Crohn’s and Colitis Foundation of America, the German Academic Exchange Service, the Bill and Melinda Gates Foundation, the Colorado Center for Biofuels and Biorefining and the US National Institutes of Health (DK78669, GM65103, GM8759, HG4872 and its ARRA supplement, HG4866, DK83981 and LM9451).
Footnotes
COMPETING FINANCIAL INTERESTS The authors declare competing financial interests: details accompany the full-text HTML version of the paper at http://www.nature.com/naturemethods/.
Note: Supplementary information is available on the Nature Methods website.
References
- 1.National Institutes of Health Human Microbiome Project Working Group et al. Genome Res. 2009;19:2317–2323. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hopkin M. Nature. 2006;444:420–421. doi: 10.1038/444420a. [DOI] [PubMed] [Google Scholar]
- 3.Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Nat Methods. 2008;5:235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cole JR, et al. Nucleic Acids Res. 2009;37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schloss PD, et al. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knight R, et al. Genome Biol. 2007;8:R171. doi: 10.1186/gb-2007-8-8-r171. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp