Disruption of the ARF–Mdm2–p53 tumor suppressor pathway in Myc-induced lymphomagenesis (original) (raw)
Abstract
Transgenic mice expressing the c-Myc oncogene driven by the immunoglobulin heavy chain enhancer (Eμ) develop B-cell lymphoma and exhibit a mean survival time of approximately 6 months. The protracted latent period before the onset of frank disease likely reflects the ability of c-Myc to induce a p53-dependent apoptotic program that initially protects animals against tumor formation but is disabled when overtly malignant cells emerge. In cultured primary mouse embryo fibroblasts, c-Myc activates the p19ARF–Mdm2–p53 tumor suppressor pathway, enhancing p53-dependent apoptosis but ultimately selecting for surviving immortalized cells that have sustained either p53 mutation or biallelic ARF deletion. Here we report that p53 and ARF also potentiate Myc-induced apoptosis in primary pre-B-cell cultures, and that spontaneous inactivation of the ARF–Mdm2–p53 pathway occurs frequently in tumors arising in Eμ–myc transgenic mice. Many Eμ–myc lymphomas sustained either p53 (28%) or ARF (24%) loss of function, whereas Mdm2 levels were elevated in others. Its overexpression in some tumors lacking p53 function raises the possibility that Mdm2 can contribute to lymphomagenesis by interacting with other targets. Eμ–myc transgenic mice hemizygous for ARF displayed accelerated disease (11-week mean survival), and 80% of these tumors lost the wild-type ARF allele. All ARF-null Eμ–myc mice died of lymphoma within a few weeks of birth. About half of the tumors arising in ARF hemizygous or ARF nullizygous Eμ–myc transgenic mice also overexpressed Mdm2. Therefore, Myc activation strongly selects for spontaneous inactivation of the ARF–Mdm2–p53 pathway in vivo, canceling its protective checkpoint function and accelerating progression to malignancy.
Keywords: Myc, ARF, Mdm2, p53, B-cell lymphoma, apoptosis
Many cancers are characterized by alterations that ultimately lead to constitutive overexpression of the MYC oncogene (for review, see Alitalo et al. 1987). In most somatic cells, c-Myc functions are necessary for the normal rate of progression of quiescent cells into the DNA synthetic (S) phase of the cell cycle (Eilers et al. 1991; Mateyak et al. 1997), and therefore, enforced c-Myc expression can confer an advantage to tumor cells by providing constitutive proliferative signals. However, apart from promoting cell division, activation of c-Myc, particularly under growth-limiting conditions, can initiate an endogenous apoptotic program (Askew et al. 1991; Evan et al. 1992). Therefore, Myc overexpression triggers a potent tumor surveillance response that effectively opposes hyperproliferation by killing those cells in which Myc levels exceed a safe threshold (for review, see Evan et al. 1995; Packham and Cleveland 1995).
An important mediator of Myc-induced apoptosis in mouse embryo fibroblasts (MEFs) is the ARF–Mdm2–p53 tumor suppressor pathway (for review, see Sherr 1998). p19ARF, the product of an alternative open reading frame of the mouse INK4a–ARF locus (Quelle et al. 1995b), stabilizes p53 and induces p53-mediated transcription (Kamijo et al. 1997) by binding to and antagonizing the functions of Mdm 2 (Kamijo et al. 1998; Pomerantz et al. 1998; Stott et al. 1998; Zhang et al. 1998), itself a negative regulator of p53 function (Barak et al. 1993; Wu et al. 1993) (Fig. 1). Myc activation in MEFs rapidly elevates the levels of p19ARF and then p53, thereby accelerating replicative crisis by inducing apoptosis (Zindy et al. 1998). Conversely, MEFs lacking either ARF or p53 function are relatively resistant to Myc-induced apoptosis. MEFs that survive Myc-induced killing generally exhibit either p53 mutation or, more rarely, biallelic ARF loss. One or the other event normally accompanies the establishment of MEF clones capable of continuous cell growth (Harvey and Levine 1991; Harvey et al. 1993b; Zindy et al. 1997; Kamijo et al. 1997), thereby facilitating Myc's ability to immortalize cells and to function as a more potent growth promoter in established cell lines. The general concept is that ARF is activated by hyperproliferative signals from oncogenes such as Myc (Zindy et al. 1998), E1A (De Stanchina et al. 1998), E2F-1 (Bates et al. 1998), mutated Ras (Palmero et al. 1998), and v-Abl (Radfar et al. 1998). By opposing Mdm2 function, ARF can facilitate p53-dependent cell cycle arrest or apoptosis depending on the biologic context (Sherr 1998). Consistent with this notion, ARF loss has been reasoned to attenuate apoptosis in developing mouse lenses lacking the retinoblastoma (Rb) protein and expressing deregulated E2F (Pomerantz et al. 1998) and occurs during v-Abl-induced immortalization of pre-B cells in vitro (Radfar et al. 1998). Although ARF depends on p53 function to induce cell cycle arrest in MEFs (Kamijo et al. 1997), the available data do not preclude the possibility that ARF or Mdm2 might interact with targets other than p53, particularly in other cell lineages (Fig. 1).
Figure 1.

ARF–Mdm2–p53 circuitry. Myc rapidly induces p19ARF expression, but without evidence that Myc transactivates the ARF promoter, its action may be indirect. p19ARF binds directly with Mdm2 to neutralize its function and triggers p53-dependent transcription. In turn, Mdm2 is a p53-responsive gene (pathway A) whose activation cancels the p53 response. Through ill-defined mechanisms, p53 regulates negatively both ARF (pathway B) and Myc (pathway C). However, it is conceivable that the feedback loops affecting ARF and Myc are one and the same.
Regulation of the ARF–Mdm2–p53 pathway is complex, due to several types of feedback control (Fig. 1). The p19ARF protein binds to Mdm2 and can sequester it in the nucleolus (Weber et al. 1999). Coordinately, ARF induces p53 stabilization and activation within the nucleoplasm, leading to the induction of a battery of p53-responsive genes, Mdm2 among them. In turn, Mdm2 acts to antagonize p53 transcription and stabilization and therefore terminates the p53 response (Fig. 1, pathway A). Specifically, Mdm2 binding interferes with p53-dependent transcription (Momand et al. 1992; Chen et al. 1993; Oliner et al. 1993), catalyzes p53 ubiquitination (Honda et al. 1997; Honda and Yasuda 1999), and facilitates p53 shuttling from the nucleus to the cytoplasm, where p53 undergoes degradation in 26S proteasomes (Roth et al. 1998). ARF antagonizes both Mdm2-mediated ubiquitination of p53 (Honda and Yasuda 1999) and Mdm2 shuttling (Tao and Levine 1999; Zhang and Xiong 1999). Although not yet understood mechanistically, p53 negatively regulates the expression of ARF (Fig. 1, pathway B) (Stott et al. 1998) and c-Myc (pathway C) (Levy et al. 1993; Frazier et al. 1998), thereby providing additional avenues for feedback control. In cells lacking p53, p19ARF levels increase significantly due to interruption of the p53–ARF feedback loop; conversely, the enforced ectopic expression of p53 in such cells can restore ARF to normal levels (Kamijo et al. 1998; Stott et al. 1998).
An attractive in vivo model for identifying potential effectors of c-Myc-induced pathways that promote transformation or apoptosis is the Eμ–myc transgenic mouse in which c-Myc is overexpressed in B-cell progenitors under control of the immunoglobulin heavy chain enhancer. After a protracted subclinical course, Eμ–myc transgenic mice develop clonal pre-B and B-cell lymphomas (Adams et al. 1985), similar to those observed in human Burkitt's lymphomas bearing a translocated (t8;14) c-MYC allele (Alitalo et al. 1987). These animals display a marked polyclonal proliferation of large B cells in bone marrow and peripheral blood soon after birth, but the increased rates of cell division are initially offset by a high apoptotic index (Jacobsen et al. 1994). Secondary, and largely unknown, genetic changes occur in these cells that culminate in the expansion of lethal clonal pre-B and B-cell lymphomas (Adams et al. 1985). Notably, crossing Eμ–myc mice with either Eμ–bcl-2 transgenics (Strasser et al. 1990), or p53+/− mice (Hsu et al. 1995; Schmitt et al. 1999), markedly accelerates the course of lymphoma development, suggesting that interference with apoptotic pathways rescues Myc-overexpressing cells from death and enables the growth-promoting functions of Myc to predominate. Here we report that c-Myc-induced lymphomagenesis in Eμ–myc transgenic mice selectively inactivates ARF or p53 in the majority of tumors. ARF deletion was observed as frequently as p53 mutation, and disabling either gene markedly accelerated the rate of tumor development in vivo. Complex patterns of Mdm2 overexpression observed in these lymphomas suggest that Mdm2 also contributes to disease, possibly in both a p53-dependent and independent manner.
Results
ARF and p53 are mediators of c-Myc induced apoptosis in primary pre-B cells
Specific effectors of Myc-induced apoptosis are in part dependent on cell type (Packham and Cleveland 1995; Sakamuro et al. 1995), and this is further complicated by the fact that many previous studies have been performed in immortal cells that lack functional p53 or ARF. To assess the contribution of the ARF–Mdm2–p53 pathway to c-Myc-induced apoptosis in B lymphocytes, we used primary interleukin-7 (IL-7)-dependent pre-B-cell cultures (Whitlock and Witte 1987) derived from bone marrow cells taken from juvenile wild-type, _ARF_−/−, _p53_−/−, and ARF–p53 doubly nullizygous mice. Cells lacking ARF contain an intact INK4a locus and express p16INK4a at normal levels (Kamijo et al. 1997). Wild-type pre-B cells can be grown continuously under these conditions for ∼2 months before reaching replicative senescence. In contrast, polyclonal pre-B cells cultured from the bone marrow of ARF null mice can be maintained in culture for many more months, and perhaps indefinitely (D.L. Randle, C.M. Eischen, M.F. Roussel, and C.J. Sherr, unpubl.). Flow cytometric analyses using markers that distinguish pro-B, pre-B, and immature B cells demonstrated that p53 or ARF loss did not influence the phenotype of cultured pre-B cells. Irrespective of genotype, the cells were >95% CD24+, CD43−, CD19+, and cell surface IgM−, as is characteristic of pre-B cells.
After 1.5–3 weeks of culture on IL-7-producing feeder cells, pre-B cells were infected with a recombinant retrovirus encoding a tamoxifen (TM)-inducible estrogen receptor (ER) module fused to c-Myc (Myc-ERTM) (Eilers et al. 1991; Littlewood et al. 1995) along with the gene encoding green fluorescence protein (GFP) in cis. Even in the absence of 4-hydroxytamoxifen (4-HT), acute infection of wild-type IL-7-dependent pre-B cells with the Myc-ERTM retrovirus led to the death of a significant fraction of the population, whereas ARF null and p53 null cells appeared to be relatively resistant (see below). The efficiency of infection of these cells varied from 15%–65% in different experiments, but cell sorting for green fluorescence yielded >98% GFP-positive cells that were expanded and expressed equivalent levels of Myc-ERTM protein independent of their genetic background (Fig. 2A, top). p53 protein was detected in both wild-type and ARF null cells; p19ARF was barely detectable in infected wild-type pre-B cells but its levels were markedly elevated in p53 null cells (with disrupted feedback loops; see Fig. 1). It should be noted that in general p19ARF is not an abundant protein, although its levels tend to increase as cells are passaged in culture and approach replicative senescence (Zindy et al. 1998). Reevaluation of phenotypes of GFP sorted and expanded Myc-ERTM-infected cells demonstrated that expression of the Myc-ERTM gene was associated with maturation of a proportion of the cells (<20%) to a cell surface IgM-positive phenotype. However, there were again no appreciable differences in the phenotypes of Myc-ERTM-infected wild-type versus infected _ARF_−/−, _p53_−/−, or ARF–p53 double null B cells, allowing a direct comparison of the effects of ARF or p53 status on the Myc apoptotic program.
Figure 2.
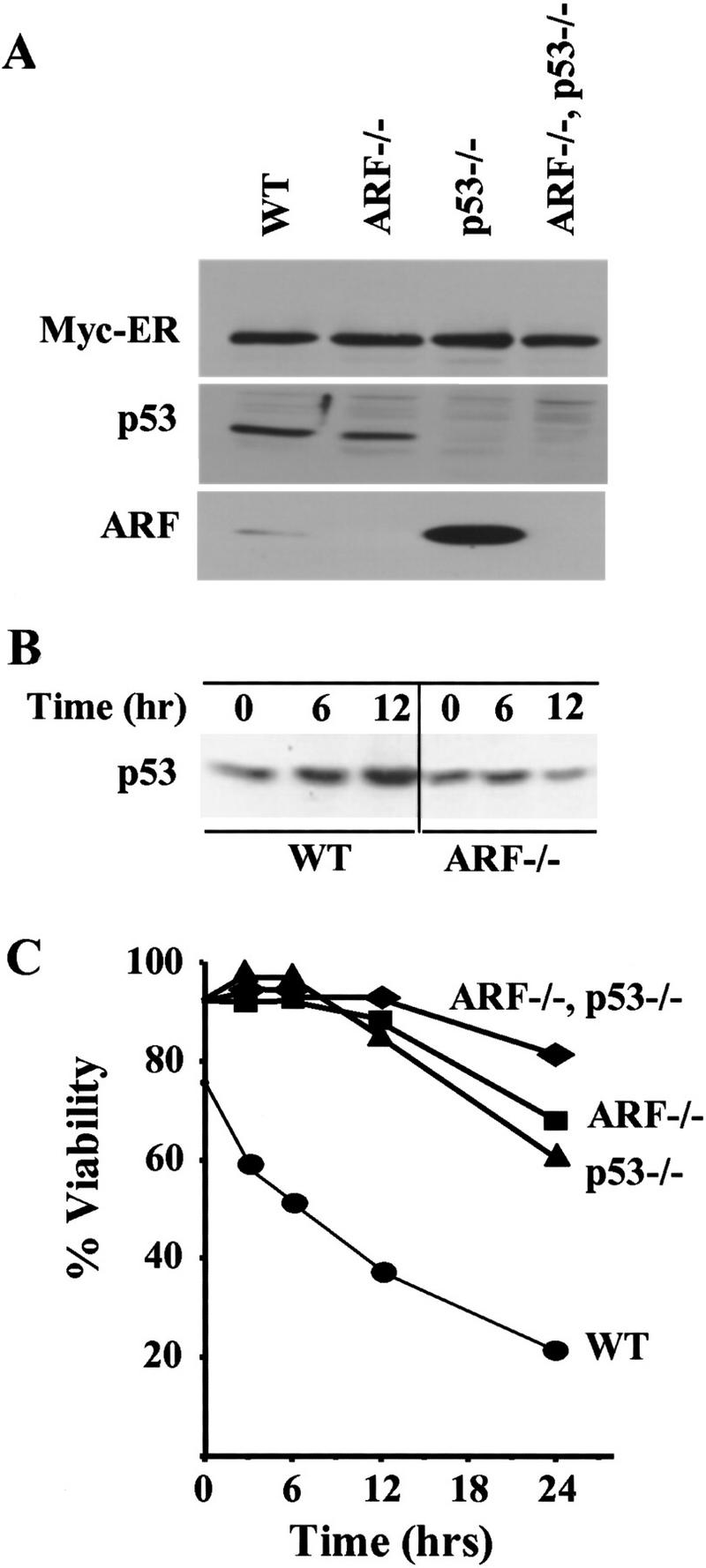
ARF and p53 mediate c-Myc-induced apoptosis in primary B cells. (A) Levels of Myc–ERTM, p53, and ARF in wild-type, _ARF_−/−, _p53_−/−, and ARF–p53 doubly null pre-B cells infected with a retrovirus encoding Myc–ERTM–GFP. Infected pre-B cells sorted for green fluorescence were expanded, lysed, and equal quantities of protein were assessed by immunoblotting with antibodies specific for each protein. (B) Kinetics of p53 induction in response to 4-HT in the same experiment shown in C. p53 was detected by immunoblotting as in A. No signals were detected in lysates of cells lacking p53 (negative data not shown). (C) Steady-state levels of apoptosis in the indicated primary pre-B cells at 14 days after infection are indicated at the 0 hr time point. 4-HT was added to the indicated primary pre-B cell cultures to activate Myc–ERTM, and their viability was determined at intervals thereafter by trypan blue dye exclusion and confirmed by analysis of subdiploid DNA content or staining with Hoescht 33342. The data are representative of three independent experiments.
When maintained in culture, the sorted GFP-positive, Myc–ER-positive population of wild-type cells maintained a three- to fourfold higher apoptotic index (∼25%) than infected cells lacking ARF, p53, or both (Fig. 2C, uninduced cells at time 0; other data not shown). In contrast, the rates of spontaneous apoptosis in sorted wild-type pre-B cells infected with a control GFP-containing vector were negligible. To assess the biologic consequences of Myc activation, 4-HT was added to the cultures. p53 was induced in a time-dependent manner in wild-type cells, whereas ARF null cells exhibited no appreciable response (Fig. 2B). Despite the fact that wild-type pre-B cells were maintained in medium containing IL-7 and serum, Myc activation led to their rapid death. Within 12 hr of 4-HT treatment, >60% of these cells were dead (Fig. 2C), and by 72 hr, very few remained alive (not shown). Cell deaths were apoptotic as judged by morphology, Hoescht 33342 staining, and by propidium iodide staining for subdiploid DNA content. In contrast, ARF null and p53 null Myc-ERTM-infected cultures were more resistant to the effects of Myc activation, as judged by their reduced rates of death after exposure to 4-HT (Fig. 2C). In several such experiments, we observed that ARF–p53 double-null cells were reproducibly more resistant to Myc-induced apoptosis than cells lacking either ARF or p53, and with extended time in the presence of 4-HT, surviving cells emerged more rapidly. These synergistic effects of ARF and p53 loss in pre-B cells were unexpected, as in MEFs, all previously documented effects of ARF expression appeared to be nullified in cells lacking p53 function (Sherr 1998).
The ARF–Mdm2–p53 pathway is frequently inactivated in Myc-induced tumors
Programmed overexpression of c-Myc in the B-cell compartment of Eμ–myc transgenic mice leads to the development of clonal pre-B and B-cell lymphomas (Adams et al. 1985). Generally, tumors do not arise in these mice until they are 6–8 months of age after a prolonged precancerous phase in which increased proliferative rates of B cells in the periphery and bone marrow are offset by a high apoptotic index (Jacobsen et al. 1994). We harvested bone marrow from Eμ–myc transgenic mice at 4 months of age before disease was manifest and cultured these cells in IL-7-conditioned medium. In parallel, we established bone marrow-derived pre-B cell cultures from nontransgenic sex- and age-matched wild-type, ARF_−/−, p53_−/−, and ARF–p53 double-null mice. Although both ARF null and p53 null animals are also prone to tumor development, mice used in these studies lacked signs of disease. During the first 10 days after explantation, most bone marrow cells are not supported by IL-7, and only pre-B cells are maintained and eventually grow out. Emerging proliferating pre-B cells from ARF null and p53 null mice grew at somewhat faster rates than cells from wild-type bone marrow (Fig. 3A). ARF–p53 double-null cells proliferated even more rapidly, again pointing to unanticipated cumulative effects of ARF and p53 loss on cell growth (see above). In contrast, Eμ–_myc_-derived pre-B cell cultures could not be expanded within the same time frame (Fig. 3A), due to an extremely high rate of spontaneous apoptosis (data not shown). Although the overall levels of p53 were roughly equivalent in wild-type versus Eμ–_myc transgenic pre-B cells, all Eμ–_myc_-derived cultures expressed elevated levels of p19ARF (Fig. 3B). Thus, the high rates of spontaneous death of Eμ–_myc transgenic B cells correlated with ARF activation.
Figure 3.
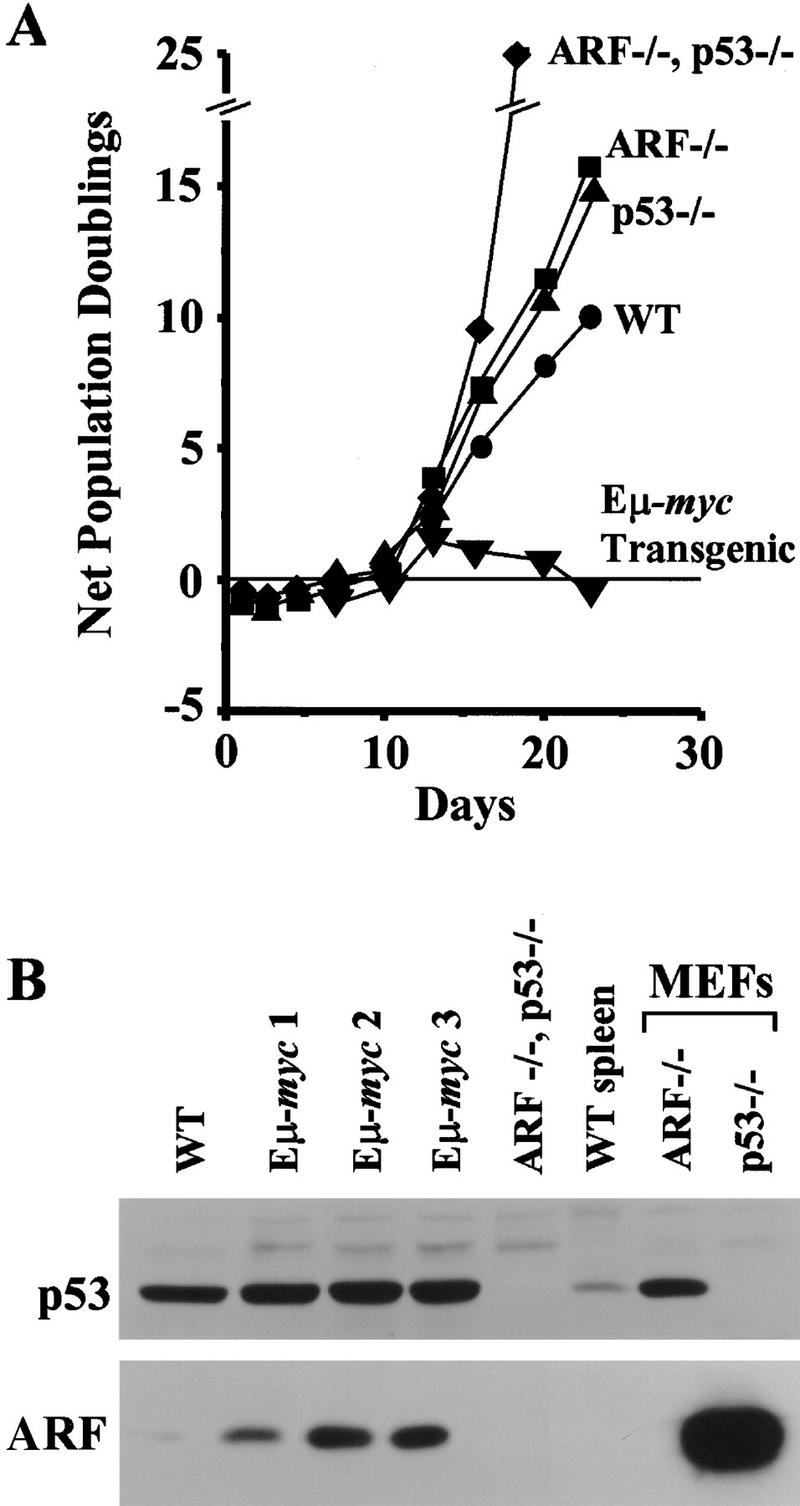
The ARF/p53 checkpoint is constitutively active in Eμ–myc transgenic bone marrow. (A) Growth curves of bone marrow cells explanted into pre-B cell culture medium containing IL-7. (B) Expression of p53 and p19ARF in explanted bone marrow cells at day 9 of culture. Pre-B cells were grown from the bone marrow of age- and sex-matched animals, including one nontransgenic wild-type (WT) mouse, three Eμ–myc transgenic mice, and from an animal lacking both ARF and p53. Spleen extract from a wild-type mouse and MEFs of the indicated genotypes were used as controls. Proteins were detected by immunoblotting as in Fig. 2.
The development of lymphoma in Eμ–myc mice is markedly accelerated to a mean of 2–3 months when Eμ–myc transgenics are crossed to hemizygous p53+/− mice; moreover, many of the tumors arising in these animals have nonfunctional p53 (Hsu et al. 1995; 6 of 11 tumors in their report). However, these studies failed to establish whether mutation of p53 is a natural consequence of Myc-induced tumorigenesis. Therefore, we evaluated ARF and p53 status in tumors arising in Eμ–myc transgenic mice. Direct loss of p53 function can occur through either deletion or point mutation, but the latter is predominant as missense mutation of only one allele is sufficient to generate high levels of expression of dominant-negative mutant p53 (Ko and Prives 1996; Levine 1997). Mutant forms of p53 accumulate to supra-physiologic levels because they are transcriptionally inactive and do not induce Mdm2 to trigger their own destruction (see Fig. 1) (Haupt et al. 1997; Kubbutat et al. 1997). Immunoblotting analyses demonstrated high levels of p53 in 6 of 25 Eμ–_myc_-induced tumors (Fig. 4A, top). Sequencing of the RT–PCR p53 cDNA products amplified from RNA of these tumors revealed missense mutations of p53, whereas p53 was wild type in randomly selected tumors expressing lower levels of p53. Interestingly, three of these mutations occurred at p53 codons that are among the most frequent to undergo mutation in human tumors [mouse Arg-246 (human 249) to Gln; mouse Arg-270 (human 273) to Cys; mouse Arg-279 (human 282) to Leu] (Ko and Prives 1996). Using Southern blotting analysis, we scored p53 deletion in only one tumor (CR320; Fig. 4B, lane 13), indicating that, as expected, p53 point mutations were more frequent than biallelic deletion. Hence, p53 was disabled in a significant fraction (28%) of Myc-induced B-cell lymphomas (summarized in Table 1).
Figure 4.


p53, ARF, and Mdm2 expression and genotypic analysis of ARF and p53 in Eμ–myc lymphomas. (A) Levels of p53 (top), p19ARF (middle), and Mdm2 (bottom) in extracts of tumors from Eμ–myc transgenic mice were assessed by immunoblotting with antibodies specific for each protein. Extracts from _ARF_−/− and _p53_−/− MEFs (right two lanes) were used as controls. The asterisk in the left margin defines the position of a background band detected with Mdm2 C-18 antibody. Arrows indicate the locations of p53, p19ARF, and p92, p90, and p85 Mdm2 isoforms. (B) Southern blot analysis for genomic _Afl_II and _Bam_HI restriction fragments containing ARF exon1β (top) and p53 exons 2–10 (bottom), respectively. Tail DNAs extracted from ARF+/+, ARF+/−, and _ARF_−/− animals were used as controls (top, three right lanes) and indicate the positions of _Afl_II restriction fragments containing (7.8 kb) or lacking (6.0 kb) ARF exon1β.
Table 1.
_ARF–Mdm2–p53 protein expression in lymphomas arising in Eμ–_myc transgenic mice
| Tumor | p53 | p19ARF | Mdm2 |
|---|---|---|---|
| p53 inactivation | |||
| CR20 | mutant | overexpressed | low |
| CR102 | mutant | overexpressed | undetected |
| CR203 | mutant | overexpressed | overexpressed |
| CR246 | mutant | overexpressed | undetected |
| CR320 | absent (deleted) | overexpressed | overexpressed |
| CR413 | mutant | overexpressed | low |
| DF103 | mutant | overexpressed | undetected |
| ARF inactivation | |||
| CR14 | undetected | absent (deleted) | undetected |
| CR73 | undetected | absent (deleted) | overexpressed |
| CR105 | undetected | absent (deleted) | low |
| CR205 | undetected | absent (deleted) | overexpressed |
| CR303 | undetected | absent (deleted) | undetected |
| CR335 | undetected | absent (deleted) | undetected |
| p53 wild type, ARF overexpressed | |||
| CR135 | undetected | overexpressed | undetected |
| CR156 | undetected | overexpressed | overexpressed |
| CR325 | undetected | overexpressed | overexpressed |
| Mdm2 overexpression only | |||
| CR17 | undetected | undetected | overexpressed |
| CR230 | low | undetected | modestly increased |
| DF195 | low | undetected | overexpressed |
| DF795 | low | undetected | modestly increased |
| No detectable alteration | |||
| CR71 | low | undetected | undetected |
| CR107 | low | undetected | undetected |
| CR289 | low | undetected | low |
| CR332 | low | undetected | low |
| DF296 | undetected | undetected | undetected |
Inactivation of the p53 pathway can also occur with ARF loss, which appears to require deletion of both ARF alleles (Kamijo et al. 1997). In MEFs that escape senescence, p53 missense mutations are favored over ARF loss by a frequency of ∼4:1, so we might have expected that ARF loss would have occurred only rarely in Eμ–_myc_-induced tumors. However, by Southern blot analyses using an exon1β _ARF_-specific probe, biallelic deletion of ARF occurred with a frequency comparable to p53 mutation (5 of 20 tumors shown in Fig. 4B, lanes 2,4,6,11,19; 6 of 25 tumors overall, Table 1). To further characterize the extent of the deletions at the INK4a–ARF loci, we also probed Southern blots with probes specific for INK4a exon1α and performed immunoblotting with antibodies specific for p16INK4a, an inhibitor of cyclin D-dependent kinases (Serrano et al. 1993). As expected from their linkage in the genome, ARF and INK4a were codeleted, and p16INK4a was no longer expressed (data not shown). However, several lines of evidence suggest that INK4a loss does not contribute to disease progression in this system (see below).
In all six tumors that expressed high levels of mutant p53 and in the one (Fig. 4A, CR320, lane 14) that had sustained p53 deletion, high levels of p19ARF were detected by immunoblotting (Fig 4A, middle, lanes, 2,7,9,11,14,16,22; Table 1), consistent with disruption of the p53–ARF feedback loop (Fig. 1). However, high levels of p19ARF were also detected in three additional tumors (Fig. 4A, middle; lanes 20,21,24), implying that p53 might have been inactivated through some other mechanism. As Mdm2 is a likely culprit, its expression was examined by immunoblotting (Fig. 4A, bottom). Two of the three tumors with wild-type p53 and elevated levels of p19ARF (lanes 21 and 24) also expressed relatively high amounts of p92, p90, or p85 Mdm2 isoforms, all of which are capable of binding to and inactivating p53 (Olson et al. 1993). Tumor CR135 (lane 20) was a sole exception, representing a case in which an elevated level of p19ARF could not be rationalized by Mdm2 overexpression or p53 loss.
Compared to ARF and p53 status, the patterns of Mdm2 overexpression in Myc-induced lymphomas were complex and fit no simple pattern. Elevated Mdm2 levels were observed in two tumors with either mutant (CR203; Fig. 4A, lane 22) or deleted (CR320; lane 14) p53, and in two tumors lacking ARF (CR73, lane 4 and CR205, lane 6) (summarized in Table 1). Two tumors with no obvious perturbations in p53 or ARF function (CR17, lane 17 and DF195, lane 10) also exhibited high levels of Mdm2, and two others (CR230, lane 13 and DF795, lane 18) expressed minimally increased levels. In principle, the latter cases could conceivably connote p53 loss of function, but we would have expected p19ARF levels to be elevated under such circumstances (see Fig. 1) and they were not. These and other data (see Discussion) raise the possibility that, in some circumstances, Mdm2 might target effectors other than p53 to promote transformation. In no tumor was overexpression of Mdm2 associated with gene amplification (data not shown). Five remaining tumors had no obvious abnormalities (Table 1). Therefore, mechanisms other than p53 mutation or deletion, ARF loss, or Mdm2 overexpression may have contributed to disease in at least 20% of the Eμ–_myc_-induced tumors.
ARF loss accelerates Myc-induced lymphomagenesis
Deletion of p53 in mice generally leads to aggressive T-cell lymphomas that kill the mice by 6 months of age (Donehower et al. 1992; Harvey et al. 1993a; Jacks et al. 1994). In contrast, tumor prone ARF null mice have a mean life expectancy of ∼9 months, and are predisposed to development of sarcomas and carcinomas later in life (Kamijo et al. 1999a). Because B-cell lymphoma in ARF_−/− mice is very rare, we could directly test the contribution of ARF loss to the development of Myc-induced lymphoma. Congenic C57Bl/6 Eμ–_myc transgenic mice were mated to C57Bl/6 × 129svj ARF_−/− mice, and F1 offspring were interbred to obtain ARF+/+, ARF+/−, and ARF_−/− Eμ–_myc transgenic mice. Littermates were followed in parallel for disease development. As expected, Eμ–_myc transgenics that were wildtype for ARF displayed a mortality curve typical for these mice (Fig. 5) (Adams et al. 1985). In contrast, Eμ–myc transgenics that were heterozygous for ARF displayed a greatly accelerated course of disease, with a mean mortality of 11 weeks (Fig. 5). The more rapidly arising tumors in ARF+/− Eμ–myc transgenic mice were phenotypically indistinguishable from those occurring in ARF+/+ Eμ–myc transgenic animals (i.e., pre-B and B-cell lymphoma; data not shown).
Figure 5.

Myc-induced tumorigenesis is accelerated by ARF loss. The genotypes of the mice are indicated next to the survival curves and the number of mice in each group are denoted by the n values. Thin vertical lines indicate ages of surviving mice. The average life spans of ARF+/+ Eμ–myc and ARF+/− Eμ–myc mice were 30 and 11 weeks, respectively. ARF_−/− Eμ–_myc animals were underrepresented because a fraction died soon after birth. Of those that survived weaning and were followed prospectively, their average survival was less than 7 weeks. Lymphoma was documented in all the animals.
Elevated levels of p53 (diagnostic of p53 mutants) were not detected in any of the tumors arising in ARF+/− Eμ–myc transgenic mice, and none deleted the gene. Strikingly, Southern blotting demonstrated that 10 of 13 of these tumors had sustained deletions of the remaining wild-type ARF allele (Fig. 6A). Because the mutant ARF allele containing intact INK4a coding sequences was retained in these tumors, p16INK4a expression was maintained (data not shown), strongly suggesting that loss of ARF but not INK4a is critically important for lymphoma development in Eμ–myc transgenic mice. In agreement with this concept, the rate of tumor formation in Rb+/− Eμ–myc transgenic mice is not significantly accelerated, and the resulting lymphomas retain the wild-type Rb allele (Schmitt et al. 1999). Nor did we observe changes in expression of D-type cyclins or CDK4 in these tumors (data not shown), implying that the p16INK4a–cyclin D/CDK4–Rb pathway was not disrupted.
Figure 6.

ARF deletion and Mdm2 expression in tumors arising in ARF+/− and ARF_−/− Eμ–_myc mice. (A) Southern blot analysis of pre-B and B-cell tumors. Genomic DNAs digested with _Afl_II were hybridized with a probe that detects a 7.8-kb fragment containing ARF exon1β and a mutant 6.0-kb fragment in which the exon was disrupted. The positions of fragments derived from the wild-type and mutant alleles are indicated at the left. Tail DNAs from wild-type, ARF+/−, or ARF null mice (lanes 14_–_16) were run as controls. (B) Tumors from the same mice shown in A (with corresponding lane numbers) were analyzed for Mdm2 expression as in Fig. 4. Immunoblots were probed with Mdm2-specific antibody C-18 to detect p92, p90, and p85 isoforms. (C) Tumors from ARF_−/− Eμ–_myc animals were analyzed for Mdm2 expression as in B. Extracts of MEFs lacking both p53 and Mdm2 were used as a negative control.
Immunoblotting also demonstrated that Mdm2 was highly expressed in 7 of 13 tumors arising in ARF+/− Eμ–myc transgenics (Fig. 6B), including two of three tumors that retained the wild-type ARF allele (DF841, lane 6 and DF851, lane 7). However, overexpression of Mdm2 in the subset of tumors that lacked ARF function again raised the possibility that the genes in the ARF–Mdm2–p53 pathway may contribute independently to disease progression.
A significant fraction of ARF_−/− Eμ–_myc transgenic animals died soon after birth. Litters were normal in size and timed matings failed to reveal gross tumor development in utero, as previously described for Eμ–myc/Eμ–pim-1 double-transgenic mice (Verbeek et al. 1991). Of those ARF_−/− Eμ–_myc transgenic animals that survived and were observed beyond weaning, all died of lymphoma by 8 weeks of age (see Fig. 5). Tumors arising in ARF_−/− Eμ–_myc transgenic mice were phenotypically indistinguishable from those emerging in ARF+/− or wild-type Eμ–myc transgenics. Gross anatomic and histopathological studies demonstrated obvious tumor development in ARF null Eμ–myc transgenic mice that were sacrificed at only 2–4 weeks of age. These malignancies presented as highly aggressive lympholeukemias with high white blood cell counts (>50,000 lymphoblasts per mm3) and metastases to liver and lung. The spleens from ARF nullizygous Eμ–myc transgenics were at least twice the size of ARF+/+ Eμ–myc littermates and exhibited architectural disruption with obliteration of lymphoid follicles due to massive infiltration by tumor cells. High levels of Mdm2 as detected by direct immunoblots were observed in half of the _ARF_−/− tumors (Fig. 6C), again demonstrating the potential for cooperation between Mdm2 overexpression and ARF loss during Myc-induced lymphomagenesis.
Discussion
Induction of p19ARF by Myc in MEFs interferes with Mdm2 function and enhances the execution of an apoptotic program that depends at least in part on p53. Whereas the elevated expression of ARF in MEFs leads to p53-dependent cell cycle arrest, Myc overexpression redirects these cells to commit suicide. The apoptotic response to Myc in MEFs is highly dose dependent and is greatly exacerbated by depriving the cells of serum (Evan et al. 1992). Myc may ‘prime’ cells to die, for example, by triggering the release of cytochrome C from mitochondria (Juin et al. 1999). Because very high levels of Myc can kill p53 null cells, p53-dependent signaling is not obligatory for Myc-induced apoptosis. However, MEFs that lose p19ARF or p53 function are highly resistant to Myc-induced apoptosis (Zindy et al. 1998), so that Myc's ability to trigger cell death likely reflects functional cooperation between ARF–Mdm2–p53-dependent and independent signaling pathways. Further complexity derives from the fact that Myc stimulates cell proliferation through ill-defined mechanisms. Hence, although the ARF-dependent apoptotic response protects cells from Myc-induced hyperproliferation, in the absence of ARF or p53 function, this checkpoint is corrupted and growth promotion by Myc predominates. Do similar events occur in vivo?
ARF and p53 loss occur frequently in Myc-induced lymphoma
A clear prediction is that Myc-induced tumors might sustain lesions that disable the ARF–Mdm2–p53 pathway or that otherwise counter Myc-induced apoptosis. Previous studies with Eμ–myc transgenic mice support this concept in the sense that when such animals were bred to lack p53 function or to overexpress Bcl-2, apoptosis was diminished in proliferating B cell progenitors and the appearance of pre-B and B cell lymphomas was significantly accelerated (Strasser et al. 1990; Hsu et al. 1995). Although many genes can synergize with Myc in inducing disease (for review, see Adams and Cory 1992; Jonkers and Berns 1996), to date the spontaneously occurring genetic events that are responsible for converting the precancerous phase of lymphoma development to overt malignancy have not been cataloged. Our studies now provide direct evidence that ARF and p53 loss of function contribute to about half of the Myc-induced lymphomas arising in these animals and further suggest that Mdm2 overexpression may also abrogate p53 function in additional cases.
Of 25 lymphomas arising in Eμ–myc transgenic animals, seven lost p53 function (six by mutation and one by biallelic deletion), whereas six other tumors sustained biallelic ARF deletions (summarized in Table 1). The idea that loss of ARF or p53 function interferes with Myc-induced apoptosis is supported by several observations. First, Myc overexpression rapidly induced apoptosis in primary cultured pre-B cells, even when the cells were maintained in medium containing serum and IL-7. Conditional activation of Myc in this setting led to p53 accumulation and killed the vast majority of cells within 24 hr. In contrast, pre-B cells lacking either ARF or p53 function were highly resistant to Myc-induced apoptosis, and p53 induction was attenuated in ARF null pre-B cells. Second, when bone marrow cells were explanted directly from Eμ–myc transgenic mice and cultured under similar conditions, the emerging pre-B cells expressed significantly elevated levels of p19ARF, exhibited an unusually high apoptotic index, and unlike normal pre-B cells, could not be expanded in culture. Conversely, pre-B-cell cultures established from bone marrow of ARF null or p53 null mice expanded at faster rates than wild-type cells. Hsu and co-workers (1995) observed p53 loss of heterozygosity in lymphomas arising in Eμ–myc, p53+/− animals, but they concluded that changes in apoptotic index did not account for the reduced tumor latency. This has been challenged by recent work in which significant apoptotic defects were observed by TUNEL assays in Eμ–myc induced tumors lacking either p53 or ARF function, whereas the mitotic indices and S-phase fractions of all lymphomas analyzed were similar (Schmitt et al. 1999).
Although ARF null mice are highly tumor prone and die of cancers with a mean latency of ∼9 months, the appearance of B cell lymphomas is rare and most of the animals die of fibrosarcoma, T-cell lymphoma, carcinomas, and tumors of the central nervous system (Kamijo et al. 1999a). However, ARF+/− Eμ–myc mice developed pre-B and B-cell lymphomas at a much faster rate than their ARF+/+ Eμ–myc littermates, with ∼80% of the tumors having segregated the residual wild-type ARF allele. Equally notable, many ARF_−/− Eμ–_myc mice died soon after birth, and those that survived had clear evidence of aggressive disseminated lympholeukemia by 2–4 weeks of age and died with extensive tumor burdens within 8 weeks of birth. The strong selection for loss of ARF function in ARF+/− Eμ–myc mice and the extremely rapid pace of disease development ARF_−/− Eμ–_myc animals argue that ARF is a potent antagonist of Myc-induced oncogenesis in vivo.
Cooperativity between ARF, Mdm2, and p53
If ARF, Mdm2, and p53 function epistatically to attenuate Myc-induced apoptosis in vivo, we might naively expect that loss of ARF or p53, or overexpression of Mdm2, would lead to similar effects so that only one of the three genes would be targeted in any one tumor. However, this need not be the case. Indeed, tumors arising spontaneously in ARF null mice can acquire p53 mutations, implying that the loss of p53 can further accelerate tumor progression (Kamijo et al. 1997). Unlike ARF, which appears to respond selectively to hyperproliferative signals, p53 is also induced by other forms of stress, including DNA damage by X-rays, ultraviolet light, cytotoxic drugs, and changes in oxygen tension (Levine 1997; Giaccia and Kastan 1998; Prives 1998). Because ARF is not strictly required for the p53-dependent checkpoint response to DNA damage (Kamijo et al. 1997; Stott et al. 1998), p53 loss should have more profound effects on cells than ARF disruption. Moreover, although ARF or p53 inactivation in MEFs (and apparently in mouse pre-B cells; C.M. Eischen et al., unpubl.) enables them to bypass replicative senescence, cultured p53 null cells rapidly increase their ploidy (Fukusawa et al. 1996; Levine 1997), whereas ARF null cells maintain a diploid DNA content throughout many more passages (Zindy et al. 1997). The latter differences pertain as well to ARF null versus p53 null Eμ–_myc_-induced lymphomas (Schmitt et al. 1998; C.M. Eischen et al., unpubl.), implying that these two genes exert differential effects on chromosomal stability. Functional interactions between DNA damage-induced and ARF-dependent p53 signaling pathways have also been documented (De Stanchina et al. 1998; Kamijo et al. 1999b). Although p53 mutations were not observed in Eμ–_myc_-induced lymphomas that sustained ARF loss, two Eμ–_myc_-induced tumors lacking ARF (CR73 and CR205, Table 1) and half of the ARF null tumors arising in ARF+/− and _ARF_−/− animals overexpressed Mdm2, raising the possibility that p53 might be nonfunctional in this setting.
Because p53 provides mechanistically ill-defined feedback controls that negatively regulate ARF (Fig. 1), p53 loss is generally accompanied by significant p19ARF overexpression. This can be readily appreciated at the protein level, particularly in cases where mutant p53 and p19ARF accumulate together. High levels of p19ARF expression are also normally observed when Mdm2 is amplified or when both alleles of p53 are deleted, and in the latter circumstances, p19ARF expression can be suppressed by reintroduction of wild-type p53 into the cells (Kamijo et al. 1998; Stott et al. 1998). Consistent with previous findings, all Myc-induced lymphomas that had sustained p53 mutation or deletion exhibited greatly elevated levels of p19ARF. Under these circumstances then, gross ARF overexpression is generally indicative of p53 loss of function. Three tumors that lacked evidence of p53 mutation expressed very high levels of p19ARF, and two of these overexpressed Mdm2 (CR156 and CR325, Table 1). Yet, one such tumor (CR135) showed no evidence of Mdm2 overexpression implying that additional mechanisms affecting ARF accumulation, possibly involving unidentified genes in the p53–ARF feedback control pathway, might also contribute.
One gene whose overexpression has been demonstrated previously to collaborate with Eμ–myc in retrovirally accelerated lymphomagenesis is Bmi-1 (Haupt et al. 1991; van Lohuizen et al. 1991; Alkema et al. 1997). The Bmi-1 gene encodes a Polycomb group repressor that dampens expression of both p16INK4a and p19ARF. Bmi-1 null mice exhibit severe defects in lymphoid and neurologic development, but these are rescued in double null animals lacking both Bmi-1 and INK4a–ARF (Jacobs et al. 1999). This suggests in turn that collaboration between Bmi-1 and myc in lymphomagenesis depends on INK4a–ARF repression. Our studies imply that the relevant focus of Bmi-1 repression in this context is ARF and not INK4a, given that ARF null tumors arising in Eμ–myc, ARF+/− animals retained p16INK4a function. Moreover, when Eμ–myc transgenic mice were crossed with Rb+/− animals, the rate of tumor formation was not greatly accelerated, and lymphomas that arose retained the wild-type Rb allele (Schmitt et al. 1999). Thus, the p16–cyclin D/cdk4–Rb pathway does not strongly contribute to tumorigenesis in this system. However, the rate of tumor formation in Eμ–myc, INK4a–ARF+/− mice [mean survival 7 weeks, in two independent studies (Jacobs et al. 1999)] although likely to reflect founder effect differences between _INK4a–ARF_−/− and _ARF_−/− strains, leaves open the possibility that p16 loss contributes in a more subtle manner.
Although the above results are consistent with the possible coinvolvement of p53 and ARF in Myc-induced lymphomagenesis, less parsimonious interpretations are plausible. In MEFs, the ability of p19ARF to induce arrest strictly depends on p53, and ARF loss is seemingly without effect in p53 null cells. Similarly, the loss of Mdm2 does not further alter the growth characteristics of p53_-deficient MEFs (McMasters et al. 1996). However, such data do not preclude the possibility that ARF can interact with targets other than Mdm2, or that the ARF–Mdm2 protein complex might affect the ability of Mdm2 to interact functionally with other proteins, particularly in other cell types. In this respect, it seems pertinent that ARF–p53 double-null pre-B cells were somewhat more resistant to Myc-induced apoptosis and proliferated more rapidly in culture than cells lacking p53 or ARF alone. If the effects of ARF were exerted strictly through p53, such results should not be obtained. An Mdm2 transgene can contribute to mammary tumor formation in p53 null mice (Lundgren et al. 1997). Moreover, Mdm2 has been reported to bind to several other protein targets including other p53 family members (Zeng et al. 1999), the p300/CBP transcriptional coactivators (Grossman et al. 1998), the E2F–DP1 family of transcription factors (Martin et al. 1995), and the Rb protein (Xiao et al. 1995; Hsieh et al. 1999). We noted Mdm2 overexpression in two tumors that either expressed mutant p53 or that completely lacked the gene (CR203 and CR320, Table 1). In tumors in which Mdm2 was overexpressed without concomitant ARF overexpression (CR17 and DF195, Table 1), there was no correlative evidence for p53 loss of function. Together, these data point to the possibility that an interaction of Mdm2 with targets other than p53 may also contribute to lymphomas in Eμ–_myc transgenic animals.
In conclusion, we have documented genetic disruption of ARF or p53 in 13 of 25 lymphomas induced by the Eμ–myc transgene and have observed effects of Mdm2 on p19ARF expression, presumably by disruption of p53 feedback control, in two additional cases. ARF and p53 loss of function occurred in a mutually exclusive manner in this series of tumors. In accord with the idea that Myc overexpression can select for the emergence of ARF null tumor cells, Eμ–_myc_-induced tumors arose much more rapidly in both ARF+/− and _ARF_−/− animals, and in the former case, the wild-type ARF allele was frequently deleted. These observations lend strong support to the concept that ARF loss attenuates Myc-induced apoptosis in vivo, enabling tumors to emerge more rapidly. Unexpectedly, Mdm2 was overexpressed in a significant fraction of ARF null tumors. Moreover, the combined loss of ARF and p53 in primary pre-B cells conferred a greater growth and survival advantage than the loss of either gene alone. Such results underscore the potential for collaboration between genes in the ARF–Mdm2–p53 pathway. In two tumors, overexpression of Mdm2 was observed in p53-negative cells, suggesting that ARF–Mdm2 signaling may target genes other than p53 during Myc-induced tumorigenesis, a possibility that merits future scrutiny. Finally, the fact that ∼20% of the analyzed tumors lacked detectable perturbations in ARF, p53, or Mdm2 implies that other genes act in concert with these three in Myc-induced B cell neoplasia.
Materials and methods
Interbreeding of mice, tumor surveillance, and histopathology
ARF null mice (C57Bl/6 × 129/svj) (Kamijo et al. 1997, 1999a) were interbred with Eμ–myc transgenic mice (Adams et al. 1985; Sidman et al. 1988) [inbred C57Bl/6 strain; kindly provided by Dr. Alan Harris (Walter and Eliza Hall Institute, Melbourne, Australia) and Dr. Charles Sidman (University of Cincinnati, OH)]. F1 offspring were intercrossed to obtain ARF+/+, ARF+/−, and ARF_−/− Eμ–_myc transgenic littermates. p53 null mice purchased from the Jackson Laboratories were kindly provided by Dr. Gerard Grosveld [St. Jude Children's Research Hospital (SJCRH)]. Dr. Gerard Zambetti (SJCRH) provided ARF–p53 double-null mice. Eμ–myc transgenic animals were observed daily for signs of morbidity and tumor development. Tumors that arose were harvested immediately after humane sacrifice of animals and were snap frozen in liquid nitrogen. Pieces of tumor tissue were extracted for analysis of DNA, RNA, and protein. Histopathology of formalin-fixed lymph nodes, spleens, and other organs manifesting gross tumor involvement was determined after hematoxylin and eosin staining. Blood smears were stained with Wright–Giemsa.
Cell culture and virus infection
Primary pre-B cell cultures were generated as previously described (Borzillo and Sherr 1989). Briefly, bone marrow was harvested from 6-week-old ARF null, p53 null, and ARF–p53 double null mice, and from wild-type littermate controls. After hypotonic lysis of red blood cells, bone marrow cells were plated in a pre-B-cell culture system (Whitlock and Witte 1987) containing RPMI 1640 medium supplemented with 10% fetal calf serum, 55 μm 2-mercaptoethanol, 2 mm glutamine, penicillin, and streptomycin (GIBCO, Grand Island NY). Cultures of primary pre-B cells were initiated using NIH-3T3 feeder cells engineered to secrete IL-7 and then expanded without feeder layers using 10% IL-7-conditioned medium. After 1.5–3 weeks in culture, the surviving cells were immunophenotyped by flow cytometry and determined to be >95% positive pre-B cells (i.e., CD19+, CD43−, CD24+, IgM−, and negative for T-cell- or myeloid/macrophage-specific markers).
The _myc_-ERTM cDNA provided by Drs. Dean Felsher and J. Michael Bishop (University of California, San Francisco) was subcloned into the MSCV–IRES–GFP virus obtained from Dr. Robert Hawley (Red Cross, New York, NY) (Hawley et al. 1994). The chimeric form of Myc is retained in the cytoplasm complexed to hsp90, but after the addition of the estrogen agonist 4-HT, the chimeric protein is released, relocates to the nucleus, and activates transcription (Littlewood et al. 1995). MSCV–_myc_–ERTM–IRES–GFP and control MSCV–IRES–GFP virus were produced by cotransfection of 293T cells with vector and helper virus plasmids (Roussel et al. 1995). Viruses were harvested at intervals, pooled, filtered, and used to infect naive primary pre-B cell cultures in the presence of 8 μg/ml polybrene. Infected GFP-positive pre-B cells were sorted by FACS and expanded in liquid culture in IL-7-containing growth medium. The efficiency of infection ranged from 15%–65% using this protocol. Expression of the Myc–ERTM fusion protein was confirmed by immunoblotting and was similar in wild-type, ARF null, p53 null, and ARF–p53 double-null pre-B cells (Fig. 2).
Viability and apoptosis quantification
The viability of each of the Myc–ERTM–GFP infected pre-B cell cultures was determined by trypan blue dye exclusion at specific intervals after the addition of 1 μm 4-HT (Sigma, St. Louis, MO). Apoptosis was determined by propidium iodide staining and quantitation of fragmented DNA (subG1) and verified both morphologically and by Hoescht 33342 staining followed by fluorescent microscopy.
Immunoblotting
Whole cell protein extracts from primary pre-B cells or pre-B and B-cell tumors from Eμ–myc transgenic mice were isolated as previously described (Kamijo et al. 1998). Briefly, cells were sonicated 2 × 7 sec after addition of ice-cold lysis buffer [50 mm HEPES (pH 7.5), 150 mm NaCl, 1 mm EDTA, 2.5 mm EGTA, 0.1% Tween 20, 1 mm PMSF, 0.4 U/ml aprotinin, 1 mm NaF, 10 mm β-glycerophosphate, 0.1 mm Na orthovanadate, and 10 μg/ml leupeptin]. Undissolved material was sedimented in a microfuge (4°C, 7 min, 14,000 rpm), and protein in the supernatant was quantified. Protein (100–150 μg/lane) was electrophoretically separated in 7.5% or 10% polyacrylamide gels containing sodium dodecyl sulfate (SDS-PAGE). Proteins were transferred to nitrocellulose membranes (Protran, Schleicher & Schuell, Dassel, Germany) and blotted with antibodies specific for the mouse p19ARF (Quelle et al. 1995b) or p16INK4a (Quelle et al. 1995a) carboxyl termini, p53 (Ab-7, Calbiochem, La Jolla, CA), Mdm2 (C-18, Santa Cruz, Inc., Santa Cruz, CA), and c-Myc (06-340, Upstate Biotechnology, NY).
Southern blotting
Genomic DNA was isolated from mouse tails and tumors arising in ARF+/+ Eμ–myc or ARF+/− Eμ–myc transgenic mice. Equal amounts of genomic DNA were digested with _Afl_II, _Bam_HI, or _Eco_RI, electrophoretically separated in agarose gels, and transferred to nitrocellulose membranes. The cDNAs coding for ARF (exon 1β), p16INK4a (exon 1α), p53 (exons 2–10), and Mdm2 (Fakharzadeh et al. 1991) were used to probe the indicated DNAs.
Acknowledgments
We thank Scott Lowe for helpful discussions throughout the course of this work and both Scott Lowe and Maarten van Lohuizen for communicating data before publication. We thank Alan Harris and Charles Sidman for kindly providing breeders for Eμ–myc transgenic mice; Gerard Zambetti for ARF–p53 double null mice; Charles Sawyers and Robert Hawley for retroviral vectors; Dean Felsher and J. Michael Bishop for Myc–ER cDNA; David Baltimore for 293T cells; Richard Cross and Minas Paktinat for assistance with FACS analyses; David Randle for help growing pre-B cells and for IL7-conditioned medium; James Downing for assistance with histopathology; and the staff of our Animal Resources Center for care and monitoring animals. We also appreciate the excellent technical support of Chunying Yang, Zhen Lu, Esther Van de Kamp, Rose Mathew, and Elsie White, and many helpful suggestions from other colleagues, particularly Frederique Zindy and Gerard Zambetti. This work was supported in part by National Institutes of Health (NIH) grants DK-44158 (J.L.C.), CA-71907 and CA-56819 (M.F.R), Cancer Center core grant CA-21765, NIH postdoctoral research grant CA09346 (C.M.E) and by the American Lebanese Syrian Associated Charities (ALSAC) of St. Jude Children's Research Hospital. C.J.S. is an investigator and J.D.W. is a Research Associate of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked ‘advertisement’ in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL john.cleveland@stjude.org; FAX (901) 525-8025.
References
- Adams JM, Cory S. Oncogene cooperation in leukaemogenesis. Cancer Surveys. 1992;15:119–141. [PubMed] [Google Scholar]
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Alitalo K, Koskinen P, Makela TP, Saksela K, Sistonen L, Winqvist R. Myc oncogenes: Activation and amplification. Biochim Biophys Acta. 1987;907:1–32. doi: 10.1016/0304-419x(87)90016-3. [DOI] [PubMed] [Google Scholar]
- Alkema MJ, Jacobs H, van Lohuizen M, Berns A. Perturbation of B and T cell development and predisposition to lymphomagenesis in Eμ-bmi-1 transgenic mice require the Bmi-1 RING finger. Oncogene. 1997;15:899–910. doi: 10.1038/sj.onc.1201262. [DOI] [PubMed] [Google Scholar]
- Askew DS, Ashmun RA, Simmons BC, Cleveland JL. Constitutive c-myc expression in an IL3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene. 1991;6:1915–1922. [PubMed] [Google Scholar]
- Barak Y, Juven T, Haffner R, Oren M. Mdm2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–468. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates S, Phillips AC, Clarke P, Stott F, Peters G, Ludwig RL, Vousden KH. E2F-1 regulation of p14ARF links pRB and p53. Nature. 1998;395:124–125. doi: 10.1038/25867. [DOI] [PubMed] [Google Scholar]
- Borzillo GV, Sherr CJ. Early pre-B-cell transformation induced by the v-fms oncogene in long-term mouse bone marrow cultures. Mol Cell Biol. 1989;9:3973–3981. doi: 10.1128/mcb.9.9.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993;13:4107–4114. doi: 10.1128/mcb.13.7.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stanchina E, McCurrach ME, Zindy F, Shieh S-Y, Ferbeyre G, Samuelson AV, Prives C, Roussel MF, Sherr CJ, Lowe SW. E1A signaling to p53 involves the p19ARF tumor suppressor. Genes & Dev. 1998;12:2434–2442. doi: 10.1101/gad.12.15.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Eilers M, Schirm S, Bishop JM. The Myc protein activates transcription of the α-prothymosin gene. EMBO J. 1991;10:133–141. doi: 10.1002/j.1460-2075.1991.tb07929.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Water CM, Penn LZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- Evan GI, Brown L, Whyte M, Harrington E. Apoptosis and the cell cycle. Curr Opin Cell Biol. 1995;7:825–834. doi: 10.1016/0955-0674(95)80066-2. [DOI] [PubMed] [Google Scholar]
- Fakharzadeh SS, Trusko SP, George DL. Tumorigenic potential associated with enhanced expression of a gene that is amplified in a mouse tumor cell line. EMBO J. 1991;10:1565–1569. doi: 10.1002/j.1460-2075.1991.tb07676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier MW, He X, Wang J, Gu Z, Cleveland JL, Zambetti GP. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol Cell Biol. 1998;18:3735–3743. doi: 10.1128/mcb.18.7.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukusawa K, Choi T, Kuriyama R, Rulong S, Vande Woude GF. Abnormal centrosome amplification in the absence of p53. Science. 1996;271:1744–1747. doi: 10.1126/science.271.5256.1744. [DOI] [PubMed] [Google Scholar]
- Giaccia AJ, Kastan MB. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes & Dev. 1998;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- Grossman S, Perez R, Kung AL, Joseph M, Mansur C, Xiao ZX, Kumar S, Howley PM, Livingston DM. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell. 1998;2:405–415. doi: 10.1016/s1097-2765(00)80140-9. [DOI] [PubMed] [Google Scholar]
- Harvey DM, Levine AJ. p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes & Dev. 1991;5:2375–2385. doi: 10.1101/gad.5.12b.2375. [DOI] [PubMed] [Google Scholar]
- Harvey M, McArthur MJ, Montgomery CA, Jr, Butel JA, Bradley A, Donehower LA. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat Genet. 1993a;5:225–229. doi: 10.1038/ng1193-225. [DOI] [PubMed] [Google Scholar]
- Harvey M, Sands AT, Weiss RS, Hegi ME, Wiseman RW, Pantazis P, Giovanella BC, Tainsky MA, Bradley A, Donehower LA. In vitro growth characteristics of embryo fibroblasts isolated from p53-deficient mice. Oncogene. 1993b;8:2457–2467. [PubMed] [Google Scholar]
- Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM. Novel zinc finger gene implicated as myc collaborator by retovirally accelerated lymphomagenesis in Eμ–myc transgenic mice. Cell. 1991;65:753–763. doi: 10.1016/0092-8674(91)90383-a. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Hawley RG, Lieu FH, Fong AZ, Hawley TS. Versatile retroviral vectors for potential use in gene therapy. Gene Ther. 1994;1:136–138. [PubMed] [Google Scholar]
- Honda R, Yasuda H. Association of p19ARF with Mdm2 inhibits ubiquitin ligase activity of MDM2 for tumor suppressor p53. EMBO J. 1999;18:22–27. doi: 10.1093/emboj/18.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Hsieh J-K, Chan FSG, O'Connor DJ, Mittnacht S, Zhong S, Lu X. RB regulates the stability of the apoptotic function of p53 via MDM2. Mol Cell. 1999;3:181–193. doi: 10.1016/s1097-2765(00)80309-3. [DOI] [PubMed] [Google Scholar]
- Hsu B, Marin MC, El-Naggar AK, Stephens LC, Brisbay S, McDonnell TJ. Evidence that c-myc mediated apoptosis does not require wild-type p53 during lymphomagenesis. Oncogene. 1995;11:175–179. [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- Jacobs JJL, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- Jacobs, J.J.L., B. Scheijen, J.-W. Voncken, K. Kieboom, A. Berns, and M. van Lohuizen. 1999. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc induced apoptosis via Ink4a/ARF. Genes & Dev. (This issue). [DOI] [PMC free article] [PubMed]
- Jacobsen KA, Prasad VS, Sidman CL, Osmond DG. Apoptosis and macrophage-mediated deletion of precursor B cells in the bone marrow of Eμ–myc transgenic mice. Blood. 1994;84:2784–2794. [PubMed] [Google Scholar]
- Jonkers J, Berns A. Retroviral insertional mutagenesis as a strategy to identify cancer genes. Biochim Biophys Acta. 1996;1287:29–57. doi: 10.1016/0304-419x(95)00020-g. [DOI] [PubMed] [Google Scholar]
- Juin P, Hueber A-O, Littlewood T, Evan G. c-myc induced sensitization to apoptosis is mediated through cytochrome c release. Genes & Dev. 1999;13:1367–1381. doi: 10.1101/gad.13.11.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, Grosveld G, Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci. 1998;95:8292–8297. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999a;59:2217–2222. [PubMed] [Google Scholar]
- Kamijo T, van de Kamp E, Chong MJ, Zindy F, Diehl AJ, Sherr CJ, McKinnon P. Loss of the ARF tumor suppressor reverses premature replicative arrest but not radiation hypersensitivity arising from disabled Atm function. Cancer Res. 1999b;59:2464–2469. [PubMed] [Google Scholar]
- Ko LJ, Prives C. p53: Puzzle and paradigm. Genes & Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Levy N, Yonish-Rouach E, Oren M, Kimchi A. Complementation by wild-type p53 of interleukin-6 effects on M1 cells: Induction of cell cycle exit and cooperativity with c-myc suppression. Mol Cell Biol. 1993;13:7942–7952. doi: 10.1128/mcb.13.12.7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified estrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgren K, Montes de Oca Luna R, McNeill YB, Emerick EP, Spencer B, Barfield CR, Lozano G, Rosenberg MP, Finlay CA. Targeted expression of MDM2 uncouples S phase from mitosis and inhibits mammary gland development independent of p53. Genes & Dev. 1997;11:714–725. doi: 10.1101/gad.11.6.714. [DOI] [PubMed] [Google Scholar]
- Martin K, Trouche D, Hagemeier C, Sorensen TS, LaThangue NB, Kouzarides T. Stimulation of E2F1/DP1 transcriptional activity by mdm2 oncoprotein. Nature. 1995;375:691–694. doi: 10.1038/375691a0. [DOI] [PubMed] [Google Scholar]
- Mateyak MK, Obaya AJ, Adachi S, Sedivy JM. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 1997;8:1039–1048. [PubMed] [Google Scholar]
- McMasters KM, Montes de Oca Luna R, Pena JR, Lozano G. Mdm2 deletion does not alter growth characteristics of p53-deficient embryo fibroblasts. Oncogene. 1996;13:1731–1736. [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–860. doi: 10.1038/362857a0. [DOI] [PubMed] [Google Scholar]
- Olson DC, Marechal V, Momand J, Chen J, Romocki C, Levine AJ. Identification and characterization of multiple mdm-2 protein and mdm-2-p53 protein complexes. Oncogene. 1993;8:2353–2360. [PubMed] [Google Scholar]
- Packham G, Cleveland JL. c-Myc and apoptosis. Biochim Biophys Acta Rev Cancer. 1995;1242:11–29. doi: 10.1016/0304-419x(94)00015-t. [DOI] [PubMed] [Google Scholar]
- Palmero I, Pantoja C, Serrano M. p19ARF links the tumour suppressor p53 to ras. Nature. 1998;395:125–126. doi: 10.1038/25870. [DOI] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liégeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee H-W, Cordon-Cardo C, DePinho R. The Ink4a tumor suppressor gene product, p19ARF, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- Prives C. Signaling to p53: Breaking the MDM2-p53 circuit. Cell. 1998;95:5–8. doi: 10.1016/s0092-8674(00)81774-2. [DOI] [PubMed] [Google Scholar]
- Quelle DE, Ashmun RA, Hannon GJ, Rehberger PA, Trono D, Richter H, Walker C, Beach D, Sherr CJ, Serrano M. Cloning and characterization of murine p16INK4a and p15INK4b genes. Oncogene. 1995a;11:635–645. [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995b;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Radfar A, Unnikrishnan I, Lee H-W, DePinho RA, Rosenberg N. p19ARF induces p53-dependent apoptosis during Abelson virus-mediated pre-B cell transformation. Proc Natl Acad Sci. 1998;95:13194–13199. doi: 10.1073/pnas.95.22.13194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J, Dobbelstein M, Freedman D, Shenk T, Levine AJ. Nucleocytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J. 1998;17:554–564. doi: 10.1093/emboj/17.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel MF, Theodoras AM, Pagano M, Sherr CJ. Rescue of defective mitogenic signaling by D-type cyclins. Proc Natl Acad Sci. 1995;92:6837–6841. doi: 10.1073/pnas.92.15.6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamuro D, Eviner V, Elliott KJ, Showe L, White E, Prendergast GC. c-Myc induces apoptosis in epithelial cells by both p53-dependent and p53-independent mechanisms. Oncogene. 1995;11:2411–2418. [PubMed] [Google Scholar]
- Schmitt, C.A., M.E. McCurrach, E. de Stanchina, and S.W. Lowe. 1999. Impact of Ink4a/ARF and p53 mutations during lymphoma development and therapy. Genes & Dev. (This issue).
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Tumor surveillance via the ARF–p53 pathway. Genes & Dev. 1998;12:2984–2991. doi: 10.1101/gad.12.19.2984. [DOI] [PubMed] [Google Scholar]
- Sidman CL, Marshall JD, Harris AW. Genetic studies on Eμ–myc transgenic mice. Curr Top Microbiol Immunol. 1988;141:94–99. doi: 10.1007/978-3-642-74006-0_13. [DOI] [PubMed] [Google Scholar]
- Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, Palmero I, Ryan K, Hara E, Vousden KH, Peters G. The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998;17:5001–5014. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- Tao W, Levine AJ. p19ARF stabilizes p53 by blocking nucleo-cytoplasmic shuttling of Mdm2. Proc Natl Acad Sci. 1999;96:6937–6941. doi: 10.1073/pnas.96.12.6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, Van der Gulden H, Berns A. Identification of cooperating oncogenes in Eμ–myc transgenic mice by provirus tagging. Cell. 1991;65:737–752. doi: 10.1016/0092-8674(91)90382-9. [DOI] [PubMed] [Google Scholar]
- Verbeek S, van Lohuizen M, van der Valk M, Domen J, Kraal G, Berns A. Mice bearing the Eμ–myc and Eμ–pim-1 transgenes develop pre-B-cell leukemia prenatally. Mol Cell Biol. 1991;11:1176–1179. doi: 10.1128/mcb.11.2.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- Whitlock CA, Witte ON. Long-term culture of murine bone marrow precursors of B lymphocytes. Methods Enzymol. 1987;150:275–286. doi: 10.1016/0076-6879(87)50085-4. [DOI] [PubMed] [Google Scholar]
- Wu X, Bayle JH, Olson D, Levine AJ. The p53–mdm-2 autoregulatory feedback loop. Genes & Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- Xiao ZX, Chen J, Levine A, Modjtahedi N, Xing J, Sellers WR, Livingston DM. Interactions between the retinoblastoma protein and the oncoprotein mdm2. Nature. 1995;375:694–697. doi: 10.1038/375694a0. [DOI] [PubMed] [Google Scholar]
- Zeng X, Chen L, Jost CA, Maya R, Keller D, Wang X, Kaelin WG, Jr, Oren M, Chen J, Lu H. MDM2 suppresses p73 function without promoting p73 degradation. Mol Cell Biol. 1999;19:3257–3266. doi: 10.1128/mcb.19.5.3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y. Mutations in human ARF exon 2 disrupt its nucleolar localization and impair its ability to block nuclear export of MDM2 and p53. Mol Cell. 1999;3:579–591. doi: 10.1016/s1097-2765(00)80351-2. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF–INK4a locus deletion impairs both the Rb and p53 tumor suppressor pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- Zindy F, Quelle DE, Roussel MF, Sherr CJ. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15:203–211. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle D, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes & Dev. 1998;12:2424–2433. doi: 10.1101/gad.12.15.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]