Comparative genomics of the classical Bordetella subspecies: the evolution and exchange of virulence-associated diversity amongst closely related pathogens (original) (raw)
Abstract
Background
The classical Bordetella subspecies are phylogenetically closely related, yet differ in some of the most interesting and important characteristics of pathogens, such as host range, virulence and persistence. The compelling picture from previous comparisons of the three sequenced genomes was of genome degradation, with substantial loss of genome content (up to 24%) associated with adaptation to humans.
Results
For a more comprehensive picture of lineage evolution, we employed comparative genomic and phylogenomic analyses using seven additional diverse, newly sequenced Bordetella isolates. Genome-wide single nucleotide polymorphism (SNP) analysis supports a reevaluation of the phylogenetic relationships between the classical Bordetella subspecies, and suggests a closer link between ovine and human B. parapertussis lineages than has been previously proposed. Comparative analyses of genome content revealed that only 50% of the pan-genome is conserved in all strains, reflecting substantial diversity of genome content in these closely related pathogens that may relate to their different host ranges, virulence and persistence characteristics. Strikingly, these analyses suggest possible horizontal gene transfer (HGT) events in multiple loci encoding virulence factors, including O-antigen and pertussis toxin (Ptx). Segments of the pertussis toxin locus (ptx) and its secretion system locus (ptl) appear to have been acquired by the classical Bordetella subspecies and are divergent in different lineages, suggesting functional divergence in the classical Bordetellae.
Conclusions
Together, these observations, especially in key virulence factors, reveal that multiple mechanisms, such as point mutations, gain or loss of genes, as well as HGTs, contribute to the substantial phenotypic diversity of these versatile subspecies in various hosts.
Keywords: Genome, Bordetella, SNP, Pan-genome, Virulence, Evolution, Horizontal gene transfer, Host adaptation, Pertussis toxin
Background
The genus Bordetella contains nine designated species, three of which are so closely related that they are considered subspecies and are referred to as the “classical _Bordetella_”; B. pertussis, B. parapertussis and _B. bronchiseptica_[1]. These three present an outstanding experimental system because, although they are highly similar at the DNA sequence level, they vary in critical aspects of bacterial pathogenesis, including host specificity, severity of diseases, and duration of infections (acute versus chronic) [2]. For example, B. bronchiseptica causes infections ranging from lethal pneumonia to asymptomatic respiratory carriage [2] and chronically colonizes the respiratory tracts of various mammalian hosts, with some lineages primarily isolated from humans [3]. B. pertussis and B. parapertussis hu , which are thought to have evolved independently from a _B. bronchiseptica_-like progenitor [4], are causative agents of whooping cough in humans. Another distinct lineage only isolated from sheep has been designated B. parapertussis _ov_[5]. The very close evolutionary relationship between these subspecies and the diversity of their interactions with various hosts provides an opportunity to explore the evolutionary genetic basis for changes in important pathogen characteristics.
The prior description of a single genome of each of the classical Bordetella subspecies reported an extensive number of gene inactivations, deletions and genomic rearrangements, as associated with host-adaptation of other pathogens, such as _Yersinia pestis_[2]. However, unlike Y. pestis and E. coli, there is limited evidence of gene acquisition amongst the Bordetella species. Microarray-based comparative genome hybridization (CGH) has suggested that the more distantly related B. holmesii may have obtained some genes from B. pertussis, but did not reveal evidence of gene acquisition or exchange within the classical Bordetellae [6]. We have previously reported evidence that the distributions of subsets of the O-antigen encoding genes correlate with O-antigen type, but not with MLST-based phylogeny of B. bronchiseptica strains [7]. However, genomic-signatures associated with horizontal gene transfer (HGT) events have not been described for the classical Bordetellae. Furthermore, the genomic plasticity of these pathogens, which is related to novel gene acquisition [8], has not been investigated. Understanding the pan-genome of the classical Bordetellae and identifying genes that may have been acquired at important points in their natural evolutionary history will provide insight into the potential role of HGTs in their past and ongoing evolution.
In this study, we sequenced seven divergent classical Bordetella isolates, including five B. bronchiseptica complex I (non-human) and IV (human) strains, one B. parapertussis ov (sheep) strain, and one B. pertussis (human) strain. Phylogenetic trees of the classical Bordetellae based on genome-wide single nucleotide polymorphism (SNP) data provide a more detailed and robust view of their genetic relationship, as well as some new insights. For example, a shared evolutionary history for B. pertussis and B. bronchiseptica complex IV strains, and a closer relationship between B. parapertussis isolates of ovine and human sources. Genome content analyses revealed that only approximately 50% of genes are shared by all strains in the core genome consisting of 2,857 gene families. Variably present genes encode a range of functions, including many potential virulence factors, in a pan-genome of at least 5,558 gene families. Notably, mathematical analysis of these genomes suggests that the classical Bordetellae pan-genome is “open” with limited gene acquisition. Additional evidence of HGT events are identified within the loci of genes required for Pertussis Toxin (Ptx) assembly and secretion, suggesting that HGT, followed by divergent evolution, contribute to strain diversity and host adaptation of the classical Bordetella subspecies.
Results
Sequencing of seven new classical Bordetella genomes
In 2003, Parkhill et al. published the first comparative genomic analysis of B. pertussis, B. parapertussis, and _B. bronchiseptica_[2]. Since then, _B. petrii_[9], _B. avium_[3], and seven B. pertussis strains [10,11] have been sequenced. Here, we sequenced seven divergent classical Bordetella strains, including five B. bronchiseptica strains isolated from both human and non-human hosts (strains 253, 1289, MO149, Bbr77, and D445), one B. parapertussis ovine strain (Bpp5), and one B. pertussis strain (18323) to comprehensively examine the evolutionary relationships of the classical Bordetellae (Table 1).
Table 1.
Summary of sequenced strain information
| Strain | ST | Species | Host | Location | Year | Complex | Reference | Note |
|---|---|---|---|---|---|---|---|---|
| RB50 | 12 | B. bronchiseptica | Rabbit | USA | Unknown | I | [2] | Published |
| Tohama I | 1 | B. pertussis | Human | Japan | 1954 | II | [2] | Published |
| CS | 1 | B. pertussis | Human | China | 1951 | II | [11] | Published |
| B0558 | 2 | B. pertussis | Human | Netherlands | 1949 | II | [10] | Published |
| B1193 | 1 | B. pertussis | Human | Netherlands | 1950 | II | [10] | Published |
| B1831 | 2 | B. pertussis | Human | Netherlands | 1999 | II | [10] | Published |
| B1834 | 2 | B. pertussis | Human | Netherlands | 1999 | II | [10] | Published |
| B1917 | 2 | B. pertussis | Human | Netherlands | 2000 | II | [10] | Published |
| B1920 | 2 | B. pertussis | Human | Netherlands | 2000 | II | [10] | Published |
| 12822 | 19 | B. parapertussis hu | Human | Germany | Unknown | III | [2] | Published |
| 1289 | 32 | B. bronchiseptica | Monkey | South America | Unknown | I | [12] | This study |
| 253 | 27 | B. bronchiseptica | Dog | USA | Unknown | I | [13] | This study |
| MO149 | 15 | B. bronchiseptica | Human | USA | Unknown | IV | [4] | This study |
| D445 | 17 | B. bronchiseptica | Human | USA | Unknown | IV | [4] | This study |
| Bbr77 | 18 | B. bronchiseptica | Human | Germany | Unknown | IV | [4] | This study |
| 18323 | 24 | B. pertussis | Human | USA | 1946 | II | [14] | This study |
| Bpp5 | 16 | B. parapertussis ov | Sheep | New Zealand | Unknown | I | [15] | This study |
B. bronchiseptica strain 1289 was sequenced and assembled at the Pennsylvania State University using Illumina [16], 454 with GSFLX Titanium chemistry [17,18], and optical maps [19] from OpGen (Gaithersburg, MD). The other six strains were sequenced at the Sanger Institute. The genomes of Bpp5, 253, MO149, and 18323 were sequenced to approximately 6 to 9-fold coverage with ABI3730 sequencers [20]. The genomes of D445 and Bbr77 were sequenced on Illumina [16] and 454 [17,18]. Read assembly resulted in finished genomes of MO149, Bpp5, 1289 and 18323, 4 contigs in 253, 11 scaffolds in D445, and 16 scaffolds in Bbr77. The sum of the contig or scaffold lengths ranged from 4.1 Mb to 5.2 Mb, which is comparable to genome sizes of previously sequenced strains.
These new genomes and previously sequenced genomes share similar characteristics, such as G + C content, number of rRNA operons, or tRNAs (Table 2). The B. bronchiseptica strains have the largest genomes (~5.3 MB), followed by B. parapertussis strains (~4.8 MB) and B. pertussis strains (~4.1 MB), as previously observed [2]. This work confirmed the presence of different Insertion Sequence (IS) elements in different species [5]. Overall, even though these strains have shown diverse phenotypes in the mouse model of infection [2,4,12-15] and were isolated from different hosts, they appear to share similar genomic organization and characteristics.
Table 2.
Summary of sequenced genome annotation information
| Species | B. bronchiseptica | B. parapertussis | B. pertussis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Strain Name | RB50 | 1289 | 253 | MO149 | Bbr77 | D445 | 12822 | Bpp5 | Bpp5 plasmid | Tohama I | 18323 | CS |
| Contig (C) / Scaffold (S) | 1 | 1 | 4 C | 1 | 16 S | 11 S | 1 | 1 | 1 | 1 | 1 | 1 |
| Genome Size (bp) | 5339179 | 5208522 | 5264383 | 5091817 | 5115717 | 5243194 | 4773551 | 4887379 | 12195 | 4086189 | 4043846 | 4124236 |
| Gaps (Ns) | 0 | 628 | 2 | 0 | 146101 | 251924 | 0 | 0 | 0 | 0 | 0 | 0 |
| G + C content (%) | 68.49 | 68.65 | 68.64 | 68.86 | 68.51 | 68.23 | 68.43 | 68.15 | 61.39 | 68.12 | 68.11 | 68.1 |
| Predicted CDSs | 5009 | 4785 | 4845 | 4669 | 4667 | 4775 | 4402 | 4558 | 15 | 3806 | 3746 | 3822 |
| Pseudogenes | 12 | 39 | 60 | 44 | 76 | 256 | 217 | 389 | 0 | 359 | 369 | 357 |
| CDSs with Ns | 0 | 0 | 0 | 0 | 320 | 473 | 0 | 0 | 0 | 0 | 0 | 0 |
| Average Gene Size (bp) | 983 | 1000 | 989 | 1002 | 974 | 953 | 1000 | 986 | 534 | 983 | 986 | 991 |
| % of CDSs | 92.23 | 91.9 | 91.04 | 91.88 | 91.51 | 91.21 | 92.23 | 92.16 | 65.6 | 91.62 | 91.38 | 91.84 |
| rRNA operons | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 0 | 3 | 3 | 3 |
| tRNA | 55 | 54 | 54 | 54 | 54 | 54 | 54 | 54 | 0 | 51 | 51 | 51 |
| IS481 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 230 | 239 | 236 |
| IS1001 | 0 | 0 | 0 | 0 | 0 | 0 | 22 | 27 | 0 | 0 | 0 | 0 |
| IS1002 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | 0 | 0 | 5 | 7 | 4 |
| IS1663 | 0 | 0 | 0 | 7 | 0 | 9 | 0 | 0 | 0 | 17 | 18 | 18 |
B. pertussis strains (Tohama I, 18323, and CS) contained no novel genes except transposases or insertion elements, confirming that B. pertussis has evolved from a _B. bronchiseptica_-like ancestor by genome decay (Figure 1). While we did not observe many genome rearrangements between Tohama I and CS, extensive genome rearrangement distinguishes Tohama I and 18323 (unpublished). More phage-related and membrane protein encoding genes are present in B. bronchiseptica non-human isolates (RB50, 253, and 1289), while more transposases are present in B. bronchiseptica human-isolated strains (D445, Bbr77, and MO149). Notably, a locus novel to the classical Bordetellae was observed in both D445 (D445_1578 – D445_1707) and Bbr77 (Bb77_4113 - Bbr77_4216) strains. This locus is also present in B. petrii and includes a Type IV secretion system-like locus, which is being further investigated (unpublished). B. parapertussis ov Bpp5 strain has about 150 strain-specific gene families comprised mostly of hypothetical proteins, phage-related proteins, and putative secreted proteins. Comparing the two lineages designated B. parapertussis, there are about 300 gene families that are present in either B. parapertussis hu or B. parapertussis ov, but not both. Most of these gene families are shared with B. bronchiseptica strain RB50 and include exported proteins, hypothetical proteins, and membrane proteins. While B. parapertussis hu 12822 is missing the locus encoding a putative type II secretion system, B. parapertussis ov strain Bpp5 is missing the locus encoding flagella. Furthermore, Bpp5 has a unique 12 KB plasmid containing genes predicted to encode proteins involved in partition, replication, plasmid conjugal transfer, mobilization and transcriptional regulation. We have not observed this plasmid in any other classical Bordetella strains.
Figure 1.

Comparative genome content of eleven classical Bordetellae strains. The outermost circle indicates the predicted functional categories of each gene families in the pan-genome. Species-specific gene families are individually blocked with black line (B. bronchiseptica, B. parapertussis, and B. pertussis). Internal circles indicate the presence (solid color) or absence (unfilled) of each gene family in each strain examined. Circles from outer to inner are started with B. bronchiseptica strains followed by B. parapertussis strains and B. pertussis strains. Gene families that are shared in all strains are in the region A, while gene families that are not conserved in all strains are in the region B, C, and D. Region C indicates the phage encoding gene families that are only present in RB50. This figure was created using the Circos software [21].
The classical Bordetella pan-genome and core genome
To determine the global gene repertoire (pan-genome) and the universally shared “core” genome, we identified orthologous gene families via OrthoMCL [22,23] using all of the published Bordetella genomes that have manually curated annotations (RB50, Tohama I, CS, 12822, B. petrii, and B. avium). The pan-genome of these strains has 8,425 gene families, consisting of 1,778 core gene families and 6,647 gene families that are missing in at least one strain (see Additional file 1). While four Bordetella species remain to be sequenced and others are not finished or adequately annotated, these results do provide solid, yet preliminary, insight into the global gene repertoire of Bordetella. Throughout the remainder of this study, all investigations will focus on the evolutionary relationship of the classical Bordetellae. The classical Bordetella pan-genome includes 5,558 gene families, consisting of 2,857 core gene families (Region A in Figure 1) and 2,701 non-core gene families (Regions B, C and D in Figure 1) (see Additional file 2). The core gene families are mainly involved in energy metabolism, central metabolism and information transfer (Figure 2A). The most abundant gene family groups in these genomes encode surface proteins and 40% of these belong to the non-core genome. Variability in these proteins, which are likely surface exposed and antigenic, would be expected to affect host immune recognition [24].
Figure 2.
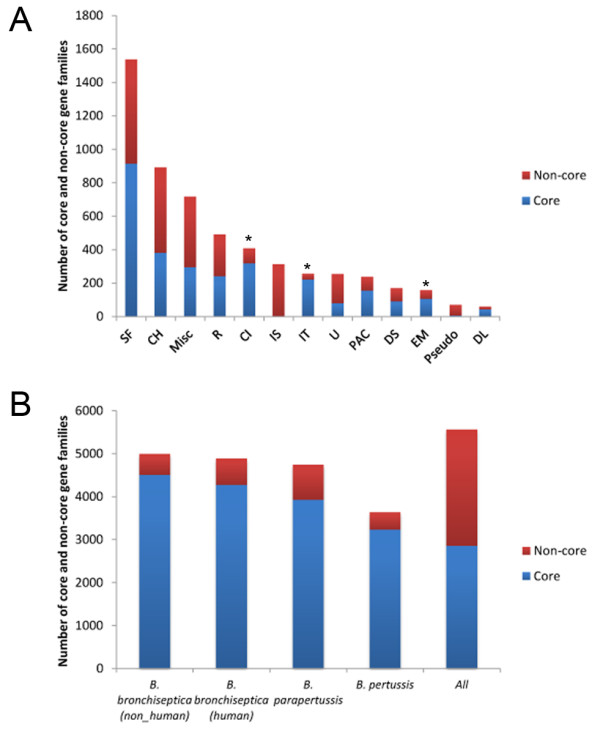
Core and non-core genome of eleven classical Bordetellae pan-genome. The number of core and non-core gene families in each predicted functional category of the classical Bordetellae pan-genome (A) and each species pan-genome (B) are summarized (B. bronchiseptica non-human isolates: RB50, 253 and 1289, B. bronchiseptica human isolates: MO149, D445, and Bbr77, B. parapertussis strains: 12822 and Bpp5, and B. pertussis strains: Tohama I, CS and 18323). Functional categories are SF: surface proteins, CH: conserved hypothetical proteins, Misc: miscellaneous information, R: regulators, CI: central/intermediary metabolism, IS: phage/insertion sequence (IS) elements, IT: information transfer proteins, U: unknown proteins, PAC: pathogenicity/adaptation/chaperones, DS: degradation of small molecules, EM: energy metabolism, Pseudo: pseudogenes and DL: degradation of large molecules. * indicates the gene families that more than 80% of gene families are conserved in all strains.
When bacteria specialize to an ecological niche, this adaptation is often accompanied by a loss of genes that are no longer required or beneficial in the new niche [25]. This concept can explain the large-scale genome loss exhibited by B. pertussis and B. parapertussis, which appear to have evolved from a zoonotic generalist into specialized human pathogens [2]. If other lineages had adapted to particular environments (i.e. subsets of hosts), we would expect those lineages to lose genes not required in that environment, but retain a set of shared core genes required for their shared characteristics, such as the ability to infect the respiratory tract of their particular host. B. bronchiseptica complex I non-human isolates (RB50, 253, and 1289; sharing 4,509 gene families) and complex IV human isolates (MO149, Bbr77 and D445; sharing 4,271 gene families) strains have larger core genomes, than B. pertussis strains (Tohama I, 18323, and CS; sharing 3,235 gene families) (Figure 2B) [4]. In fact, the core genome of all available classical Bordetella strains excluding B. pertussis isolates (3,652 gene families) was larger than that of B. pertussis strains, suggesting that this lineage has selectively lost genes/functions that are retained in all others. The non-core genome (2,701 gene families not present in all strains) contains a large variety of gene families and many phage-encoding genes, and likely contributes to the phenotypic variation of this group.
Open pan-genome with limited gene acquisition
To evaluate and compare the total gene pool of all classical Bordetella subspecies (i.e. all eleven sequenced strains), compared to that of all strains excluding B. pertussis strains, and that of B. bronchiseptica strains alone (RB50, 253, 1289, MO149, Bbr77, and D445), we employed the prediction method described by Tettelin _et al._[26]. Based on the number of novel gene families per additional genome analyzed, all of the groups (the classical Bordetella subspecies, all the strains excluding B. pertussis strains, and B. bronchiseptica strains only) appeared to have what Tettlin et al. described as open but “slowly closing” pan-genomes [27] (Figure 3A). This indicates that more sequenced genomes have the potential to provide additional genomic diversity but that beyond a couple of hundred sequenced genomes novel genes are less likely to be discovered.
Figure 3.
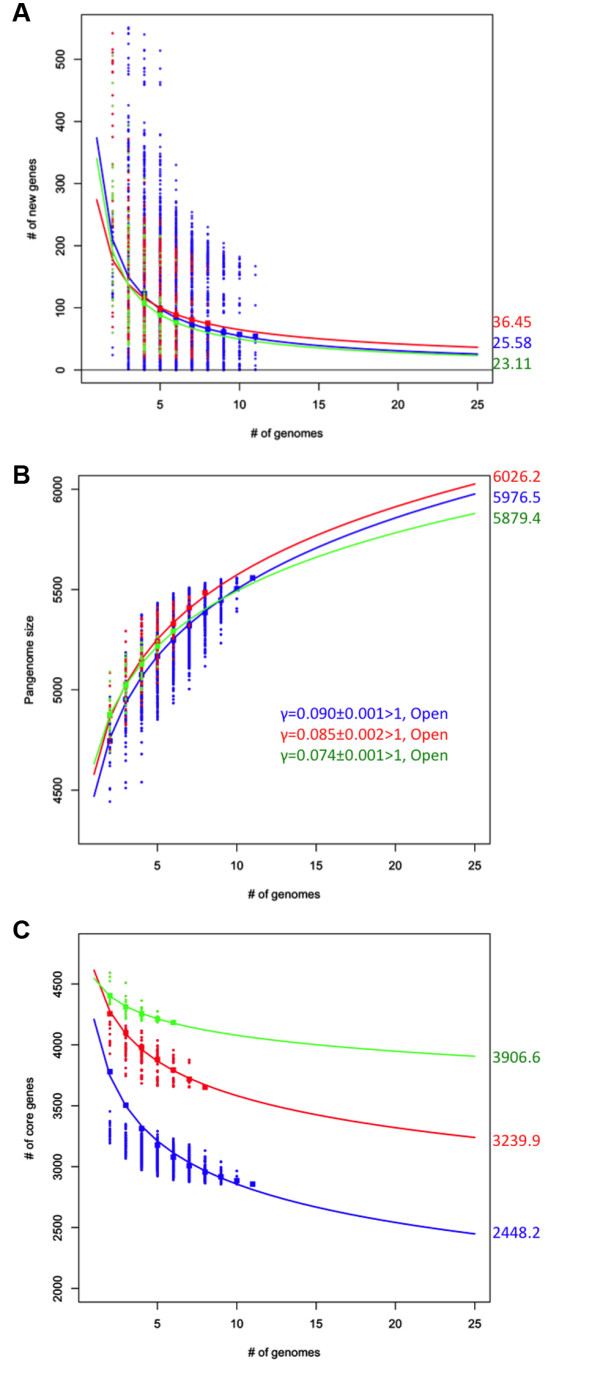
Mathematical estimation of size of novel gene families, pan-genome, and core gene families. The number of novel gene families (A), pan-genome size (B), and the number of core gene families (C) were estimated for the classical Bordetellae (blue: all eleven genomes), B. bronchiseptica strains only (green: RB50, 253, 1289, MO149, D445, and Bbr77), and all the strains except B. pertussis strains (red: RB50, 253, 1289, MO149, D445, Bbr77, 12822, and Bpp5). If n genomes are selected from 11, there are 11!/ [(n-1)!*(11-n)!] possible combinations. Each possible combination is plotted as a point, and the line is fitted to the power law model adapted from the methods of Tetellin et al. [26]. γ in the power law model for the pan-genome size estimation was reported for each group. The numbers shown on the right side of each graph are the number of expected novel gene families, pan-genome size, and core gene families with 25 genomes.
A complementary approach to assess gene acquisition was used to calculate the increase in pan-genome size with each additional genome sequenced (Figure 3B). This expected pan-genome size (n) upon sequencing an Nth genome was modeled by the power law function n = κN γ, where an open pan-genome has a γ value greater than zero and less than one, with lower values signifying a more closed genome with fewer acquired genes. The γ value for the classical Bordetella subspecies (0.090) is lower than that for Bacillus cereus (0.43) [28], indicating that the pan-genome of the classical Bordetellae is open but grows more slowly than that of B. cereus with each additional genome sequenced. With 25 additional genomes, the pan-genome of the classical Bordetella subspecies will reach approximately 6,000 gene families, while the pan-genome of the group excluding B. pertussis strains is estimated to contain 6,026 gene families. Furthermore, since a smaller core genome size reflects smaller sets of shared functional genes, the small size of the core genome containing B. pertussis strains is additional evidence that this lineage has lost the functions encoded by approximately 800 genes (Figure 3C). This further supports the previous hypothesis that B. pertussis strains have adapted to their niche by genome reduction [2] and that there is a correlation between the pan/core genome size and the host range.
Variations in virulence factors amongst the classical Bordetellae
To examine the possible basis for differences in virulence phenotypes amongst strains/lineages, we compared the presence/absence of factors thought to be involved in interactions with the host, loosely referred to as “virulence factors”, including filamentous hemagglutinin (FHA), fimbriae (Fims), pertactin (PRN), tracheal colonization factor (TcfA), adenylate cyclase/hemolysin (ACT), dermonecrotic toxin (Dnt), pertussis toxin (Ptx), Bordetella resistance to killing (BrkA) protein [29], O-antigen [30], Type III secretion system (TTSS) [31], and Type VI secretion system (T6SS) [32]. Only genes encoding FHA, PRN, and TTSS locus are conserved in all strains (Figure 4). B. bronchiseptica complex IV strains (D445, Bbr77, and MO149) have the most divergent fha locus (as low as 92% identity) compared to RB50, while B. pertussis strains have the most divergent fim loci (as low as 84% identity) with gene deletions and multiple internal sequence variations. Although tcfA varies in all strains (as low as 84% identity), genes encoding PRN segregate into clear distinctive groups between B. bronchiseptica complex I/B. parapertussis strains and B. bronchiseptica complex IV/B. pertussis strains, as previously described [4]. Together, the presence, absence or divergence of these virulence factors do not always follow the phylogenetic relationships, suggesting some other processes, in addition to sequence divergence by accumulation of point mutations, may contribute to variation of virulence factor genes.
Figure 4.

Diversity in virulence factor genes/loci among the classical Bordetellae compared to RB50. The heatmap was generated based on nucleotide percentage identity compared to RB50 for each gene/loci. Absence of a certain gene and presence of a pseudogene are highlighted with white and sky blue color with #, respectively. * indicates the missing nucleotides due to the draft status of the genome. The gene content tree, the dendrogram of hierarchical clustering of the complete pan-genome matrix, was superimposed on top of the heatmap. Manhattan distances, linkage method, and 1,000 bootstrap replicates were used for the clustering.
In a prior analysis of the hypovirulent B. bronchiseptica strain 253, we observed that it does not contain the whole ACT locus [13]. Notably, strains 1289, D445, Bbr77, MO149, and 18323 all contain a frameshift mutation within cyaB; however, each strain remains beta-hemolytic and grows within the murine respiratory tracts (unpublished). Therefore, the functional effects of this mutation on ACT secretion and hemolysis appear less dramatic than that of the mutation in strain 253. Strikingly, strains Bbr77 and MO149 do not contain the entire ptx locus and the other human isolate (D445) has a divergent ptx locus (as low as 88% identity) compared to RB50. dnt is divergent (as low as 93% identity) or missing in complex IV strains compared to RB50. One of the complement resistance factors, brkA, is very similar (>97% identity) in all strains except that it is a pseudogene in 12822 and RB50. Combined, these data reveal a discord between the phylogenetic relatedness of strains and the variation in their individual virulence genes.
In addition to small loci (<10 kb) responsible for encoding adhesins and toxins, we compared large loci (>10 kb) containing contiguous genes with highly coordinated functions. The T3SS locus is very similar (>98% identity) among all strains, with the notable exception being a pseudogene present only in B. parapertussis hu strain 12822 [12,33]. However, other loci were much more variable. Consistent with previous reports [2,4,7], the genetic makeup of the O-antigen locus (0-26 kb) widely fluctuated among strains, with the entire locus missing in all three B. pertussis stains. In strains in which they are present, these genes share as little as 90% identity with their apparent orthologs. In some cases, the locus includes completely different sets of genes. Two pseudogenes are present within the O-antigen locus in Bpp5, suggesting relatively recent loss of function. Similarly, a locus encoding a putative T6SS shows a high degree of variations. For example, B. pertussis strains and D445 are missing parts of the T6SS locus, while both human and ovine B. parapertussis strains either have pseudogenes or are missing subsets of genes within this locus. A predicted pseudogene is also present in this locus of B. bronchiseptica strain 1289. Together, these data reveal the intriguing tendency of virulence loci to be lost or divergent in the human isolates (B. bronchiseptica complex IV strains, B. parapertussis hu strains_,_ and B. pertussis strains), possibly signifying differential roles in different hosts [34].
Phylogenomic analysis
The observation that there are discrepancies between the phylogenetic relationships of sets of genes suggests that a small set of genes may be limited in its ability to accurately represent phylogenetic relationships among these strains [35]. Thus, to more accurately and robustly evaluate the genetic relationships between Bordetella isolates, we built a phylogenetic tree with all the genomic content, using a previously defined pan-genome matrix in our analysis. This matrix was constructed with genomes and gene families as columns and rows, respectively. Each cell in the matrix presents 1 or 0, depending on the presence or absence of the gene family in each genome. By generating a gene content tree based on this matrix, excluding unique gene families in each strain, we observed many similarities and some notable differences as compared to earlier studies that only used a small set of housekeeping genes. B. pertussis strains form a separate lineage that is isolated from the other two species (Figure 4). This clustering trend can be explained by the drastic genome reduction (absence of genes) in B. pertussis strains. However, B. parapertussis strains are closely related to B. bronchiseptica strains_,_ reflecting a relatively large set of shared core genes. Notably, the B. parapertussis ov strain (Bpp5) clustered together with B. parapertussis hu strain 12822, consistent with the species designation but different from the previous minimum spanning tree based on MLST [4], which clustered Bpp5 within B. bronchiseptica complex I strains and separated 12822 from B. bronchiseptica strains.
To include all the additional information contained in the individual SNPs, a phylogenetic tree was generated based on genome-wide SNP candidate sites against the reference genome (RB50) with B. petrii as an outgroup (Figure 5). Similar to the previously published MLST minimum spanning tree, this SNP-based tree suggests that B. pertussis is closely related to B. bronchiseptica complex IV strains [4]. Moreover, building the phylogenetic tree using genome-wide SNPs supports two important genetic relationships that previous phylogenetic analyses did not reveal. First, B. pertussis and B. bronchiseptica complex IV appear to share a more recent common ancestor distinct from that shared by B. parapertussis and B. bronchiseptica complex I strains. Second, B. parapertussis ov strain Bpp5 is more closely related to B. parapertussis hu strain 12822 than to B. bronchiseptica complex I strains. The agreement on the closer relationship between the two B. parapertussis clades of both the SNP-based and gene content-based trees suggests that the more distant relationship observed in the previously published MLST-based tree is likely due to the limited amount of information in the seven housekeeping genes that were used.
Figure 5.

Maximum likelihood phylogenetic tree of eleven classical Bordetellae with genome-wide SNP sites. The phylogenetic tree was reconstructed with genome-wide SNP sites based on B. bronchiseptica RB50.
SNP densities across each genome, compared to the RB50 genome, differ between strains. B. bronchiseptica complex IV strains (MO149, D445, and Bbr77) have the highest overall density of SNP sites (~12 SNPs/1,000 bp), followed by B. pertussis strains (~9 SNPs/1,000 bp), B. bronchiseptica complex I strain 253 (~7 SNPs/1,000 bp), B. parapertussis strains (~4 SNPs/1,000 bp), and complex I strain 1289 (~2 SNPs/1,000 bp), reflecting their overall relatedness to RB50. Of all SNPs, about 61% are synonymous (average dS = 0.0170), while ~26% are non-synonymous (average dN = 0.0023), reflecting the overall pressure of purifying selection. ~12% of SNPs were found within intergenic regions, and approximately 1% are in pseudogenes. Genes encoding phage-related, hypothetical proteins and some virulence factors, including ptx, contained amongst the highest SNP densities (see Additional file 3). One explanation for this observation could be positive selection, for example via the proposed vaccine-driven selection for antigenic variation of genes encoding vaccine components [37,38]. However, dN/dS ratios are lower than one in most of these genes, indicating overall negative (purifying) rather than positive (diversifying) selection. These results do not support the view that the normal rate of SNP generation, followed by positive selection for variation, resulted in this diversity, and raise the possibility that other mechanisms of diversity generation, such as insertions or deletions, HGT and functional divergence, occurred within these discrete loci.
Horizontal gene transfer and divergent evolution of the classical Bordetellae
To more closely examine the divergence of the ptx/ptl loci, we compared the percent sequence similarity of these genes and their flanking genes in all sequenced strains to that of Tohama I (Figure 6). The ptx/ptl loci of B. pertussis 18323 and CS and B. parapertussis 12822 and Bpp5 are closely related to that of Tohama I (>98% and >95% identity, respectively), while those of B. bronchiseptica non-human strains are much more divergent (as low as 87% identity). Conversely, B. bronchiseptica human isolates, which are much more closely related based on overall SNPs (Figure 5), have either low percentage similarity to Tohama I (D445; as low as 82% identity) or do not contain the entire ptx/ptl loci (Bbr77 and MO149) (Figure 6). There were ten or less SNPs distinguishing the ptx/ptl loci of seven recently published B. pertussis genomes (see Additional file 4). The relatively high SNP densities of the ptx/ptl loci do not appear to be due to positive selection because pair-wise dN/dS ratios are below one, indicating overall purifying selection (see Additional file 5).
Figure 6.

Percent sequence similarity of ptx/ptl locus with flanking genes against Tohama I. Percent sequence similarity of the ptx/ptl locus and flanking genes based on Tohama I was plotted between 80% and 100% using zPicture [36]. Intergenic regions, coding regions, and a tRNA were highlighted with red, blue, and green, respectively.
An alternative explanation for the high SNP density in the ptx/ptl loci could be the introduction of variation via horizontal gene transfer (HGT). Gerlach et al. previously speculated that the ptx/ptl loci had features of pathogenicity islands (PAIs) based only on the clusters of virulence genes in these loci, the presence of a tRNA (known to be associated with HGT and often a DNA integration target site [39]), and the absence of these loci in some strains [40]. Using the entire sequences of all eleven classical Bordetella strains, we analyzed each genome with Alien_hunter [41], which detects horizontally transferred genome segments. Nine to eighteen percent of each genome was identified as containing potential HGT candidates based on atypical genomic composition (see Additional file 6). The loci encoding phage-related proteins, alcaligin biosynthesis proteins, cytochrome ubiquinol oxidase, NADH-ubiquinone oxidoreductase, ATP synthase and many adhesins were identified as HGT candidates. Among the candidates were a number of genes required for assembly of O-antigen, consistent with our prior prediction of HGT within this locus [7,42,43]. Notably, Alien_hunter also predicted that the ptx locus and a part of its associated secretion system (ptl) locus were acquired by HGT.
To further investigate possible HGT of ptx/ptl genes, phylogenetic trees were generated based on individual genes within the ptx/ptl loci; these gene trees (Figure 7 and see Additional file 7) were then compared to the genome-wide SNP tree (Figure 5) to identify incongruence in the tree topology. The majority of the ptx/ptl gene trees are similar to the genome-wide SNP tree in that each strain is clustered based on their species designation, such as B. pertussis, B. parapertussis, and B. bronchiseptica non-human strains. However, the B. bronchiseptica human isolate D445 locus is located on a long branch separate from all the other strains, implying that the ptx/ptl loci in D445 has an evolutionary history different from other Bordetella species. Additionally, B. pertussis clades and B. parapertussis clades clustered together in the individual gene trees unlike in the genome-wide SNP tree. These results suggest HGT being a source of diversity within this locus, although they do not rule out other possibilities.
Figure 7.

The phylogenetic tree of the ptx/ptl locus. The tree was reconstructed by maximum likelihood methods with 1,000 bootstrap replicates based on the ptx/ptl locus sequences. B1920 represents B1834 and Tohama I represents CS because they have the identical sequences.
In addition to the higher overall SNP density, the distribution of SNPs across the ptx/ptl loci appears to be non-random, with areas of low density (consistent with the flanking genes and much of the genome) and areas of much higher density. For example, B. bronchiseptica complex IV strains have accumulated the highest density of SNPs (D445) or have lost the entire locus (MO149 and Bbr77), suggesting a change in the requirement for Ptx in this lineage. However, much of the ptl locus of the non-human B. bronchiseptica strains (RB50, 1289, and 253) have low SNP density, while the ptx locus, most notably the ptxC gene, has much higher SNP density (Figure 6). Interestingly, the large majority of these SNPs across the locus result in silent (synonymous) mutations (dN/dS < 0.6), suggesting the high SNP density is not due to positive selection for variations. It is also interesting to note that sequence similarity differs between the ptx locus (average 96% identity) and the ptl locus (average 98% identity) in all the strains except strain D445, suggesting a different evolutionary history for the ptx and ptl loci. Together, the ptx/ptl loci appear to have been horizontally transferred to the classical Bordetellae and have diverged in different lineages.
Discussion
The recent availability of classical Bordetella genomes gives us insight into the evolutionary changes involved in their divergence, including extensive genome decay, rearrangements and, for the first time, specific sequence data revealing evidence of limited gene acquisition [2,5]. Our mathematical prediction suggests that we will need over a hundred genomes to comprehensively describe a classical Bordetella pan-genome, although these numbers may change when more strains are added to this analysis (Figure 3). The core genome of the classical Bordetellae represents only a small fraction (~50%) of the pan-genome, and many genes in the non-core genome are likely to contribute to the diverse characteristics of these strains (Figures 1 and 2). Substantial variation is observed in virulence factor genes in particular, with human isolates appearing to have more inactivated genes than non-human isolates (Figure 4). Some of these changes are similar in strains with a shared evolutionary history, reflected by the genome-wide SNP-based phylogenetic tree. This tree is similar to the previously constructed MLST minimum spanning tree [4] with two important differences. The first is that the branching pattern of the genome-wide SNP tree links human and ovine strains of B. parapertussis, which can explain their shared characteristics that led to their similar species designation. This common branch suggests some divergence from B. bronchiseptica complex I prior to the divergence of these two lineages. Second, a more recent last common ancestor was shared by B. pertussis and B. bronchiseptica complex IV, making them more closely related to each other than to the other lineages (Figures 5). The resolution of our genome-wide SNP tree also provides a firm foundation for the comparisons with trees built based on individual genes, supporting or refuting the possibility of HGT candidates identified by Alien_hunter (Figures 6 and 7).
Species with limited diversity, such as Chlamydia trachomatis and Bacillus anthracis, share ~90% of their closed pan-genomes [44,45], while species with open pan-genomes, such as E. coli, can share as little as 20% of their pan-genomes, with nearly 80 new genes discovered for each additional sequenced genome [46]. Like Streptococcus pneumoniae, B. bronchiseptica has an open pan-genome, but the number of novel gene families is likely to decrease more rapidly with additional genomes sequenced, suggesting an intermediate category between open and closed pan-genome (Figure 3) [27]. Pan-genome size often reflects host range and gene pool availabilities. For example, a human pathogen restricted to human oral-nasal mucosa, Streptococcus pyogenes, has a smaller pan-genome than that of a zoonotic pathogen capable of infecting multiple organs, _Streptococcus agalactiae_[47]. Although we could not use our computational analysis to estimate the pan-genome sizes of B. parapertussis and B. pertussis due to limited number of fully annotated genomes (Figure 3), we speculate that the pan-genome of these two species would be smaller than that of B. bronchiseptica due to restricted host range, consistent with our core and pan-genome analysis using eleven genomes (Figure 2). This suggests that the larger pan-genome of B. bronchiseptica may be essential for survival in a broader niche of multiple host species (Figures 1 and 2). Furthermore, a similar analysis comparing two B. bronchiseptica complexes (I versus IV) with more strains may elucidate how some B. bronchiseptica isolates that are more closely related to B. pertussis share the propensity for isolation from human hosts.
Many known virulence factor genes or loci are not conserved in all the classical Bordetella strains, and the presence, absence, or inactivation of these virulence factors does not always follow the phylogenetic species relationships (Figures 4 and 5). Our study suggests that limited HGTs between foreign species and the classical Bordetellae have occurred, resulting in an open pan-genome with limited growth. Although we cannot determine the precise origin of horizontally transferred loci due to limited availability of other closely related Bordetella genomes, the atypical genomic composition detected by Alien_hunter, being flanked by a tRNA, as well as high SNP densities but low dN/dS ratios, and incongruent phylogenetic trees together provide evidence of HGT within the ptx/ptl loci (Figures 6 and 7). Expression of Ptx has not yet been observed in B. bronchiseptica or B. parapertussis strains; however, the low dN/dS ratio reflects purifying selection, indicating there are conditions under which these loci must be expressed. The facts that the entire operon is conserved in both B. bronchiseptica and _B. parapertussis_[48], and there is non-random distribution of SNP accumulations in different genes of this locus further support this view. Ptx is known for multiple biological activities, including both enzymatic activity on a range of different G proteins [49] and the ability to bind to various mammalian cells [50,51]. The ability to vary these activities to confront different hosts could be important to the broad host range of these organisms. In this light, the large number of amino acid substitutions in ptxC is intriguing. ptxB and ptxC are highly homologous and appear to have arisen via gene duplication. Since the proteins they encode are known to contain regions that determine binding specificity [50,51] and interact with extracellular receptors [52], the greater variation in these genes could contribute to adaptation to new hosts (see Additional file 8). Possible partial redundancy between the two gene products could allow diversifying without the cost of deleterious mutations necessarily resulting in complete loss of Ptx activity [48]. Despite their close relationship to B. pertussis, B. bronchiseptica complex IV strains have ptx/ptl loci that are either highly divergent (D445) or completely lost (MO149 and Bbr77), supporting the immune-mediated competition [53] or change of the requirement for the ptx/ptl loci in these strains. The extensive variation in SNP densities within the ptx locus, and the substantially different SNP densities of the ptx and ptl loci suggest that there are more complexities to the evolution of these lineages than can be explained by simple descent with variation and positive/negative selection.
Conclusions
The current comparative genomic analysis of multiple classical Bordetella subspecies has revealed a complex and flexible pan-genome with limited introduction of new genetic material. Genome-wide SNP-based phylogenetic trees of the classical Bordetellae provide a robust model of their genetic relationship against which to measure the relative evolutionary pressures on the various factors that might affect their success differently in various environments. Evolution of virulence-associated genes appears to occur via mechanisms that include the random SNP accumulations as well as more directed mechanisms, such as HGTs, that can explain high SNP densities. Both of these or potentially more interesting novel mechanisms are likely to contribute to the substantial phenotypic diversity amongst the classical Bordetella subspecies. The importance of these organisms as pathogens of humans and other mammals as well as their recent evolutionary changes in critical pathogen characteristics raise the significance of understanding the genesis and effects of these diversity-generating mechanisms.
Materials and methods
Sequencing and assembly of genomes
The genomes of Bpp5 (accession number HE965803, plasmid accession number HE965804), 253 (accession number HE965806), MO149 (accession number HE965807), also known as D444, and 18323 (accession number HE965805) were sequenced to approximately 9-fold coverage (or 6-fold for 18323) by shotgun sequencing from two genomic shotgun libraries, pMAQ1Sac_BstXI (with insert sizes of 6-9 kb) and pOTWI2 (with insert sizes of 4-5 kb; 5-6 kb), that were sequenced using big-dye terminator chemistry on ABI3730 automated sequencers [20]. End sequences from large insert fosmid libraries in pCC1FOS with an insert size of 38-42 kb were used as a scaffold. This generated approximately 0.2- to 0.4-fold coverage. The assembly was generated using phrap2gap. All repeat regions and gaps were bridged by read-pairs or end-sequenced polymerase chain reaction (PCR) products that were again sequenced with big dye terminator chemistry on ABI3730 capillary sequencers. The sequences were manipulated to the ‘Finished’ standard for Bpp5, MO149, and 18323 or to the ‘Improved High Quality Draft’ standard for 253 [54]. The genomes of D445 (accession number HE983627) and Bbr77 (accession number HE983628) were sequenced on both the Illumina GAII platform (54 bp paired end library) and the Roche 454 machine using GSFLX Titanium chemistry (3 kb insert paired end library) [16]. The Illumina data were assembled using Velvet [55] at a k-mer value of 39 or 31, respectively. This assembly was then shredded and combined with 454 data using Newbler [17,18].
The genome of 1289 (accession number HE983626) was sequenced using the Roche 454 GSFLX sequencing platform [12,56] and assembled using Newbler [17,18], which resulted in 32-fold coverage and 133 contigs. The order of these contigs was determined by using RB50 as a reference genome. Gaps were closed by using sequences from PCR amplification of gap regions generated using big dye terminator chemistry on an ABI3730 capillary sequencer, and were assembled using PHRED, PHRAP, and CONSED [57-59], sequences generated from a 76 bp paired end library using a GAIIX [16] (resulting in 188-fold coverage), and sequences generated from a 3 kb insert paired end library using a GSFLX platform [17,18]. These long-tag paired end reads and optical maps [19] (OpGen, Gaithersburg, MD) were used as scaffolds to identify and correct misassemblies and verify the final assembly. SNP sequencing errors were corrected by mapping the sequences generated by the GAIIX to the final genome assembly using inGAP [60]. The final draft of the 1289 genome consists of seven ordered contigs, covering >99.99% of the genome.
Annotation of genomes
Both RAST [61] and an automated annotation transfer tool at the Sanger Institute (unpublished) were used to annotate the finished or draft genomes of MO149, 1289, 253, D445, Bbr77, Bpp5, and 18323. The annotation tools were also used to annotate the ptx/ptl loci of six B. pertussis genomes (B0558, B1193, B1831, B1834, B1917, and B1920). We used RAST results for the novel regions that were not automatically transferred from the reference genomes, RB50, 12822, and Tohama I, based on their species classification. Each novel gene prediction was also curated with BLAST [62] and FASTA [63] results, and Pfam [64] and Prosite [65] were used to identify protein motifs. Transmembrane domains, signal sequences, and rRNA genes were identified with TMHMM [66], SignalP [67], and BLASTN [62], respectively. ISFinder was also used to detect Insertion Sequence (IS) elements in the genomes [68]. Manual curation was done with these novel regions using Artemis [69] and Artemis Comparison Tool (ACT) [70].
Pan-genome analysis
Coding sequences were extracted from the eleven classical Bordetella genomes as well as B. petrii and B. avium genomes, and orthologous gene families were determined using OrthoMCL [22,23], which defines putative pairs of orthologs based on reciprocal all-against-all BLASTP [62] with a cutoff E-value of 10-5, over 70% length coverage, and at least 70% identity. The orthologs are then clustered using Markov cluster algorithm. When gene families were not clustered and had no BLAST hits, they were considered strain specific gene families. The results of OrthoMCL were converted to a pan-genome matrix profile for further analysis. A pan-genome matrix was constructed with each column as a genome and each row as a gene family. Cell (i,j) in the matrix is 1 when gene family “i” is present in genome “j”, or 0 when gene family is absent. A graphical representation of the classical Bordetellae pan-genome (Figure 1) was created using the Circos software [21].
The sequential inclusion of up to eleven strains (eleven columns in a pan-genome matrix) was simulated in all possible combinations (N = 11!/ [(n-1)!*(11-n)!]). The number of new gene families, the core genome, and pan-genome size were estimated using methods adapted from Tettelin _et al._[26]. The R [71] function nls was used for non-linear least squares regression on the mean of the new gene families, core gene families, and pan-genome distributions. The number of expected new gene families and core gene families (n) determined by sequencing an Nth genome was modeled by the power law function n = _κN_-α, and the pan-genome size (n) by a power law n = κN γ. For estimation of novel gene families, the functions were fit to the mean values for all N ≥ 4, when there are more than 6 genomes to avoid the left side bias that could skew the results.
Gene content tree
Hierarchical clustering was performed for the complete pan-genome matrix using pvclust in R [71], modified from Lukjancenko _et al._[72]. The dendrogram was generated by hierarchical clustering with complete linkage method and Manhattan distances using R [71]. Bootstrap values were computed by resampling the rows of the matrix 1,000 times.
Virulence factor comparison
Genes and the loci that encode the known virulence factors, filamentous haemagglutinin (FHA), fimbriae (Fims), pertactin (PRN), tracheal colonization factor (TcfA), invasive adenylate cyclase/haemolysin (ACT), dermonecrotic toxin (Dnt), pertussis toxin (Ptx), Bordetella resistance to killing (BrkA) [29], O-antigen [30], Type III secretion system (TTSS) [31], and Type VI secretion system (T6SS) [32], were compared among the eleven genomes via ACT [70]. Percent sequence similarity was calculated based on RB50 sequences with BLASTN [62], and genes that either contain a frame-shift mutation or an in-frame stop codon, or that are absent were highlighted with different colors in the heatmap that was generated by R [71]. The phylogenetic tree (Gene content tree) based on the presence and absence of each gene family in the pan-genome, excluding strain-specific genes, was superimposed on the heatmap.
SNP analysis
All seventeen (B. bronchiseptica strains: 253, 1289, D445, Bbr77, and MO149, B. parapertussis strains: 12822 and Bpp5, B. pertussis strains: Tohama I, 18323, CS, B0558, B1193, B1831, B1834, B1917, and B1920, and B. petrii strain DSM 12804) genomic sequences were randomly shredded into 54 bp long reads and mapped onto the reference genome (RB50), using Ssaha v2.2.1 [73]. High quality candidate SNPs were identified using ssaha_pileup, and 128,752 SNP sites were identified in at least one strain based on RB50. Phylogenetic trees were constructed with RAxML v7.0.4 [74] for all SNP sites in the reference genome, using a General Time Reversible (GTR) model with a gamma correction for among site rate variation and ten random starting trees [75].
dN/dS Analysis
The available Bordetella genomes were aligned onto the reference genome (RB50) using MAUVE aligner with default parameters [76]. Orthologous sequences were extracted from the alignments using gene coordinates defined in RB50 and processed to cover the entire coding regions of each individual genome, excluding pseudogenes. The nucleotide sequence alignments were later refined using their corresponding amino acid sequence alignments. dN and dS values were computed using PAML package [77] with the Nei-Gojobori method [78].
HGT Detection
Putative horizontally acquired regions in the eleven classical Bordetella strains were predicted by Alien_hunter, which is able to detect genomic regions that may originate from foreign sources using Interpolated Variable Order Motifs (IVOMs) [41]. For the phylogenetic tree comparison, multiple alignments of individual genes and the entire ptx/ptl loci were generated by the MEGA5 software [79], and maximum likelihood trees were constructed with a Tamura-Nei model and 1,000 bootstrap replicates.
Abbreviations
SNP: Single nucleotide polymorphism; HGT: Horizontal gene transfer; CGH: Comparative genome hybridization; MLST: Multi-locus sequence typing; PCR: Polymerase chain reaction; IS: Insertion sequence; GTR: General time reversible; IVOM: Interpolated variable order motifs; PAI: Pathogenicity island; Ptx: Pertussis toxin.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
J. Park, YZ and AMB conceived and designed the research, performed the research, analyzed data, and wrote the manuscript; ETH conceived and designed the research, analyzed data, and wrote the manuscript; J. Parkhill and JFM conceived and designed the research; SCS conceived and designed the research as well as analyzed the data; UA and ML conceived and designed the research, analyzed the data, and contributed materials and reagents; MS analyzed the data and contributed analysis tools; XZ and SB performed the research. All authors read and approved the final manuscript.
Supplementary Material
Additional file 1
Comparative genome content of thirteen Bordetella strains. Circles indicate the presence (solid color) or absence (unfilled) of each gene family in each strain examined. Circles from outer to inner are started with B. petrii strain followed by B. avium strain and the pan-genome of the classical Bordetella strains. Then, individual circle of B. bronchiseptica, B. parapertussis and B. pertussis strains were shown. This figure was created using the Circos software [21].
Additional file 2
Lists of gene families that are present in the pan-genome of the classical Bordetella strains. This table summarized the gene family numbers, representative gene name, predicted function of encoding proteins, presence (1) or absence (0) of each gene family in individual genome, and the sum of each row for a sorting purpose.
Additional file 3
The number of SNPs in the genes and dN/dS ratios of genes that have high number of SNPs. This table summarized the start and end position of genes in RB50, gene name, strand, gene length, predicted function of encoding proteins, number of SNPs among the classical Bordetellae based on RB50, and number of SNPs per 100 base pair. dN, dS, and dN/dS ratio are included only for fifty genes that have high number of SNPs. The mean SNPs per 100 bp for the whole genome was 2.24. When the orthologs are pseudogenes, we did not calculate dN/dS ratios, indicating NA in the columns.
Additional file 4
Percent sequence similarity of B. pertussis ptx/ptl loci with flanking genes against Tohama I. Percent sequence similarity of B. pertussis ptx/ptl loci and flanking genes compared to Tohama I was plotted between 95% and 100% using zPicture [36]. Intergenic regions, coding regions, and a tRNA were highlighted with red, blue, and green, respectively.
Additional file 5
Pair-wise dN/dS ratios for individual gene in the classical Bordetellae ptx/ptl locus. dN/dS ratios of ten classical Bordetellae were plotted for individual gene in the ptx/ptl locus. B. pertussis strain Tohama I represents another B. pertussis strain CS as well because they share identical nucleotide sequences in the ptx/ptl loci. Each figure represents ptx/ptl genes (A), ptx genes (B), ptxA (C), ptxB (D), ptxD (E), ptxE (F), ptxC (G), ptl genes (H), ptlA (I), ptlB (J), ptlC (K), ptlD (L), ptlI (M), ptlE (N), ptlF (O), ptlG (P), and ptlH (Q).
Additional file 6
Genome-wide horizontal gene transfer candidates for the classical Bordetellae. The position of the candidates is plotted in each genome (Bpp5 (A), 12822 (B), Tohama I (C), CS (D), 18323 (E), RB50 (F), 1289 (G), 253 (H), MO149 (I), Bbr77 (J), and D445 (K)) and the height represents the score of Alien_hunter. Red line represents the threshold for each genome.
Additional file 7
Phylogenetic trees for the classical Bordetellae ptx/ptl locus. Phylogenetic trees were constructed by maximum likelihood method with 1,000 bootstrap replicates with individual gene sequences in the locus, or the entire ptx or ptl locus. Each figure represents the entire ptx locus (A), ptxA (B), ptxB (C), ptxD (D), ptxE (E), ptxC (F), ptl locus (G), ptlA (H), ptlB (I), ptlC (J), ptlD (K), ptlI (L), ptlE (M), ptlF (N), ptlG (O), and ptlH (P).
Additional file 8
Multiple sequence alignments of the classical Bordetellae ptxB and ptxC. Multiple amino acid sequence alignments of the classical Bordetellae ptxB and ptxC were presented in this figure.
Contributor Information
Jihye Park, Email: jup139@psu.edu.
Ying Zhang, Email: zhan2142@umn.edu.
Anne M Buboltz, Email: anne.buboltz@gmail.com.
Xuqing Zhang, Email: Xuqing_Zhang@hms.harvard.edu.
Stephan C Schuster, Email: scs@bx.psu.edu.
Umesh Ahuja, Email: uahuja@ucla.edu.
Minghsun Liu, Email: mliu@alum.mit.edu.
Jeff F Miller, Email: jfmiller@ucla.edu.
Mohammed Sebaihia, Email: ms5@sanger.ac.uk.
Stephen D Bentley, Email: sdb@sanger.ac.uk.
Julian Parkhill, Email: parkhill@sanger.ac.uk.
Eric T Harvill, Email: Harvill@psu.edu.
Acknowledgements
We acknowledge all members of the Harvill Lab for discussion of the manuscript, especially Sara E. Hester and Laura S. Weyrich for their critical review of the manuscript. We also thank the Sanger Institute Core Informatics and Sequencing Facilities, particularly Michael A. Quail, Scott Thurston, Heidi Hauser, Andries van Tonder, Nicola Corton, Danielle Walker, and Karen Brooks. Additionally, we are grateful to Drs. Fangqing Zhao, Ji Qi and Bodo Linz for their bioinformatic guidance and comparative microbial genomics expertise, and to Dr. Daniela Drautz and Lynn Tomsho for sequencing that occurred at Pennsylvania State University. This work was supported by the Wellcome Trust (Grant number 098051), NIH (5R01GM083113), and by the Agriculture and Food Research Initiative Competitive Grants Program Grant no. 2010-65110-20488 from the USDA National Institute of Food and Agriculture.
References
- van der Zee A, Groenendijk H, Peeters M, Mooi FR. The differentiation of Bordetella parapertussis and Bordetella bronchiseptica from humans and animals as determined by DNA polymorphism mediated by two different insertion sequence elements suggests their phylogenetic relationship. Int J Syst Bacteriol. 1996;46:640–647. doi: 10.1099/00207713-46-3-640. [DOI] [PubMed] [Google Scholar]
- Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, Holden MTG, Churcher CM, Bentley SD, Mungall KL, Cerdeño-Tárraga AM, Temple L, James K, Harris B, Quail MA, Achtman M, Atkin R, Baker S, Basham D, Bason N, Cherevach I, Chillingworth T, Collins M, Cronin A, Davis P, Doggett J, Feltwell T, Goble A, Hamlin N, Hauser H, Holroyd S, Jagels K, Leather S, Moule S, Norberczak H, O’Neil S, Ormond D, Price C, Rabbinowitsch E, Rutter S, Sanders M, Saunders D, Seeger K, Sharp S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Unwin L, Whitehead S, Barrell BG, Maskell DJ. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis, and Bordetella bronchiseptica. Nat Genet. 2003;35:32–40. doi: 10.1038/ng1227. [DOI] [PubMed] [Google Scholar]
- Sebaihia M, Preston A, Maskell DJ, Kuzmiak H, Connell TD, King ND, Orndorff PE, Miyamoto DM, Thomson NR, Harris D, Goble A, Lord A, Murphy L, Quail MA, Rutter S, Squares R, Squares S, Woodward J, Parkhill J, Temple LM. Comparison of the genome sequence of the poultry pathogen Bordetella avium with those of B. bronchiseptica, B. pertussis, and B. parapertussis reveals extensive diversity in surface structures associated with host interaction. J Bacteriol. 2006;188:6002–6015. doi: 10.1128/JB.01927-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diavatopoulos DA, Cummings CA, Schouls LM, Brinig MM, Relman DA, Mooi FR. Bordetella pertussis, the causative agent of whooping cough, evolved from a distinct, human-associated lineage of B. bronchiseptica. PLoS Pathog. 2005;1:e45. doi: 10.1371/journal.ppat.0010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings CA, Brinig MM, Lepp PW, van de Pas S, Relman DA. Bordetella species are distinguished by patterns of substantial gene loss and host adaptation. J Bacteriol. 2004;186:1484–1492. doi: 10.1128/JB.186.5.1484-1492.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diavatopoulos DA, Cummings CA, van der Heide HGJ, van Gent M, Liew S, Relman DA, Mooi FR. Characterization of a Highly Conserved Island in the Otherwise Divergent Bordetella holmesii and Bordetella pertussis Genomes. J Bacteriol. 2006;188:8385–8394. doi: 10.1128/JB.01081-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buboltz AM, Nicholson TL, Karanikas AT, Preston A, Harvill ET. Evidence for Horizontal Gene Transfer of Two Antigenically Distinct O Antigens in Bordetella bronchiseptica. Infect Immun. 2009;77:3249–3257. doi: 10.1128/IAI.01448-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The bacterial pan-genome: a new paradigm in microbiology. http://revistes.iec.cat/index.php/IM/article/view/11019. [DOI] [PubMed]
- Gross R, Guzman CA, Sebaihia M, dos Santos VAPM, Pieper DH, Koebnik R, Lechner M, Bartels D, Buhrmester J, Choudhuri JV, Ebensen T, Gaigalat L, Herrmann S, Khachane AN, Larisch C, Link S, Linke B, Meyer F, Mormann S, Nakunst D, Rückert C, Schneiker-Bekel S, Schulze K, Vorhölter F-J, Yevsa T, Engle JT, Goldman WE, Pühler A, Göbel UB, Goesmann A, Blöcker H, Kaiser O, Martinez-Arias R. The missing link: Bordetella petrii is endowed with both the metabolic versatility of environmental bacteria and virulence traits of pathogenic Bordetellae. BMC Genomics. 2008;9:449. doi: 10.1186/1471-2164-9-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bart MJ, van Gent M, van der Heide HG, Boekhorst J, Hermans P, Parkhill J, Mooi FR. Comparative genomics of prevaccination and modern Bordetella pertussis strains. BMC Genomics. 2010;11:627. doi: 10.1186/1471-2164-11-627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Xu Y, Zhou Z, Wang S, Yang R, Wang J, Wang L. Complete Genome Sequence of Bordetella Pertussis CS, a Chinese Pertussis Vaccine Strain. J Bacteriol. 2011;193:4017–4018. doi: 10.1128/JB.05184-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buboltz AM, Nicholson TL, Weyrich LS, Harvill ET. Role of the type III secretion system in a hypervirulent lineage of Bordetella bronchiseptica. Infect Immun. 2009;77:3969–3977. doi: 10.1128/IAI.01362-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buboltz AM, Nicholson TL, Parette MR, Hester SE, Parkhill J, Harvill ET. Replacement of adenylate cyclase toxin in a lineage of Bordetella bronchiseptica. J Bacteriol. 2008;190:5502–5511. doi: 10.1128/JB.00226-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebo P, Glaser P, Sakamoto H, Ullmann A. High-level synthesis of active adenylate cyclase toxin of Bordetella pertussis in a reconstructed Escherichia coli system. Gene. 1991;104:19–24. doi: 10.1016/0378-1119(91)90459-O. [DOI] [PubMed] [Google Scholar]
- Brinig MM, Register KB, Ackermann MR, Relman DA. Genomic features of Bordetella parapertussis clades with distinct host species specificity. Genome Biol. 2006;7:R81. doi: 10.1186/gb-2006-7-9-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER. Next-Generation DNA Sequencing Methods. Annu. Rev. Genom. Human Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen Y-J, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim J-B, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvie T, Harkins T. De novo assembly and genomic structural variation analysis with genome sequencer FLX 3K long-tag paired end reads. Biotechniques. 2008;44:829–831. doi: 10.2144/000112894. [DOI] [PubMed] [Google Scholar]
- Latreille P, Norton S, Goldman BS, Henkhaus J, Miller N, Barbazuk B, Bode HB, Darby C, Du Z, Forst S, Gaudriault S, Goodner B, Goodrich-Blair H, Slater S. Optical mapping as a routine tool for bacterial genome sequence finishing. BMC Genomics. 2007;8:321. doi: 10.1186/1471-2164-8-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applied Biosystems. 3730/3730xl DNA Analyzers User Guide. 2007.
- Krzywinski MI, Schein JE, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Mackey AJ, Stoeckert CJ Jr, Roos DS. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006;34:D363–D368. doi: 10.1093/nar/gkj123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley S. Sequencing the species pan-genome. Nat Rev Micro. 2009;7:258–259. doi: 10.1038/nrmicro2123. [DOI] [PubMed] [Google Scholar]
- Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MTG, Prentice MB, Sebaihia M, James KD, Churcher C, Mungall KL, Baker S, Basham D, Bentley SD, Brooks K, Cerdeno-Tarraga AM, Chillingworth T, Cronin A, Davies RM, Davis P, Dougan G, Feltwell T, Hamlin N, Holroyd S, Jagels K, Karlyshev AV, Leather S, Moule S, Oyston PCF, Quail M, Rutherford K, Simmonds M, Skelton J, Stevens K, Whitehead S, Barrell BG. Genome sequence of Yersinia pestis, the causative agent of plague. Nature. 2001;413:523–527. doi: 10.1038/35097083. [DOI] [PubMed] [Google Scholar]
- Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N. Margarit y Ros I et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome. Proc Natl Acad Sci USA. 2005;102:13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tettelin H. SYBIL:STREPNEUMO:PANGENOME. 2008.
- Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. 2008;11:472–477. doi: 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Smith AM, Guzmán CA, Walker MJ. The virulence factors of Bordetella pertussis: a matter of control. FEMS Microbiol Rev. 2001;25:309–333. doi: 10.1111/j.1574-6976.2001.tb00580.x. [DOI] [PubMed] [Google Scholar]
- Preston A, Maskell D. The molecular genetics and role in infection of lipopolysaccharide biosynthesis in the Bordetellae. J Endotoxin Res. 2001;7:251–261. [PubMed] [Google Scholar]
- Stockbauer KE, Foreman-Wykert AK, Miller JF. Bordetella type III secretion induces caspase 1-independent necrosis. Cell Microbiol. 2003;5:123–132. doi: 10.1046/j.1462-5822.2003.00260.x. [DOI] [PubMed] [Google Scholar]
- Weyrich LS, Rolin OY, Muse SJ, Park J, Spidale N, Kennett MJ, Hester SE, Chen C, Dudley EG, Harvill ET. A Type VI Secretion System Encoding Locus Is Required for Bordetella bronchiseptica Immunomodulation and Persistence In Vivo. PLoS ONE. 2012;7:e45892. doi: 10.1371/journal.pone.0045892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuk MH, Harvill ET, Miller JF. The BvgAS virulence control system regulates type III secretion in Bordetella bronchiseptica. Mol Microbiol. 1998;28:945–959. doi: 10.1046/j.1365-2958.1998.00850.x. [DOI] [PubMed] [Google Scholar]
- Baumler AJ, Tsolis RM, Ficht TA, Adams LG. Evolution of Host Adaptation in Salmonella enterica. Infect Immun. 1998;66:4579–4587. doi: 10.1128/iai.66.10.4579-4587.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, Douady CJ. Horizontal gene transfer and phylogenetics. Curr Opin Microbiol. 2003;6:498–505. doi: 10.1016/j.mib.2003.09.008. [DOI] [PubMed] [Google Scholar]
- Ovcharenko I, Loots GG, Hardison RC, Miller W, Stubbs L. zPicture: dynamic alignment and visualization tool for analyzing conservation profiles. Genome Res. 2004;14:472–477. doi: 10.1101/gr.2129504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooi FR, van Oirschot H, Heuvelman K, van der Heide HGJ, Gaastra W, Willems RJL. Polymorphism in the Bordetella pertussis Virulence Factors P.69/Pertactin and Pertussis Toxin in The Netherlands: Temporal Trends and Evidence for Vaccine-Driven Evolution. Infect Immun. 1998;66:670–675. doi: 10.1128/iai.66.2.670-675.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Octavia S, Maharjan RP, Sintchenko V, Stevenson G, Reeves PR, Gilbert GL, Lan R. Insight into Evolution of Bordetella pertussis from Comparative Genomic Analysis: Evidence of Vaccine-Driven Selection. Mol Biol Evol. 2011;28:707–715. doi: 10.1093/molbev/msq245. [DOI] [PubMed] [Google Scholar]
- Schubert S, Darlu P, Clermont O, Wieser A, Magistro G, Hoffmann C, Weinert K, Tenaillon O, Matic I, Denamur E. Role of Intraspecies Recombination in the Spread of Pathogenicity Islands within the Escherichia coli Species. PLoS Pathog. 2009;5:e1000257. doi: 10.1371/journal.ppat.1000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach G, von Wintzingerode F, Middendorf B, Gross R. Evolutionary trends in the genus Bordetella. Microbes Infect. 2001;3:61–72. doi: 10.1016/S1286-4579(00)01353-8. [DOI] [PubMed] [Google Scholar]
- Vernikos GS, Parkhill J. Interpolated variable order motifs for identification of horizontally acquired DNA: revisiting the Salmonella pathogenicity islands. Bioinformatics. 2006;22:2196–2203. doi: 10.1093/bioinformatics/btl369. [DOI] [PubMed] [Google Scholar]
- Wildschutte H, Preheim SP, Hernandez Y, Polz MF. O‐antigen diversity and lateral transfer of the wbe region among Vibrio splendidus isolates. Environ Microbiol. 2010;12:2977–2987. doi: 10.1111/j.1462-2920.2010.02274.x. [DOI] [PubMed] [Google Scholar]
- Iguchi A, Ooka T, Ogura Y, Nakayama K, Frankel G, Hayashi T. Asadulghani. Genomic comparison of the O-antigen biosynthesis gene clusters of Escherichia coli O55 strains belonging to three distinct lineages. Microbiology. 2008;154:559–570. doi: 10.1099/mic.0.2007/013334-0. [DOI] [PubMed] [Google Scholar]
- Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev. 2005;15:589–594. doi: 10.1016/j.gde.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Joseph SJ, Didelot X, Gandhi K, Dean D, Read TD. Interplay of recombination and selection in the genomes of Chlamydia trachomatis. Biol Direct. 2011;6:28. doi: 10.1186/1745-6150-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snipen L, Almøy T, Ussery DW. Microbial comparative pan-genomics using binomial mixture models. BMC Genomics. 2009;10:385. doi: 10.1186/1471-2164-10-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefébure T, Stanhope MJ. Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol. 2007;8:R71. doi: 10.1186/gb-2007-8-5-r71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausman SZ, Cherry JD, Heininger U, von König CHW, Burns DL. Analysis of proteins encoded by the ptx and ptl genes of Bordetella bronchiseptica and Bordetella parapertussis. Infect Immun. 1996;64:4020–4026. doi: 10.1128/iai.64.10.4020-4026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro-Neto FA, Rodbell M. Pertussis toxin induces structural changes in G alpha proteins independently of ADP-ribosylation. Proc Natl Acad Sci USA. 1989;86:2577–2581. doi: 10.1073/pnas.86.8.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saukkonen K, Burnette WN, Mar VL, Masure HR, Tuomanen EI. Pertussis toxin has eukaryotic-like carbohydrate recognition domains. Proc Natl Acad Sci USA. 1992;89:118–122. doi: 10.1073/pnas.89.1.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witvliet MH, Burns DL, Brennan MJ, Poolman JT, Manclark CR. Binding of pertussis toxin to eucaryotic cells and glycoproteins. Infect Immun. 1989;57:3324–3330. doi: 10.1128/iai.57.11.3324-3330.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider OD, Weiss AA, Miller WE. Pertussis Toxin Signals Through the TCR to Initiate Cross-Desensitization of the Chemokine Receptor CXCR4. J Immunol. 2009;182:5730–5739. doi: 10.4049/jimmunol.0803114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørnstad ON, Harvill ET. Evolution and emergence of Bordetella in humans. Trends Microbiol. 2005;13:355–359. doi: 10.1016/j.tim.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Chain PSG, Grafham DV, Fulton RS, Fitzgerald MG, Hostetler J, Muzny D, Ali J, Birren B, Bruce DC, Buhay C, Cole JR, Ding Y, Dugan S, Field D, Garrity GM, Gibbs R, Graves T, Han CS, Harrison SH, Highlander S, Hugenholtz P, Khouri HM, Kodira CD, Kolker E, Kyrpides NC, Lang D, Lapidus A, Malfatti SA, Markowitz V, Metha T. et al. Genomics. Genome project standards in a new era of sequencing. Science. 2009;326:236–237. doi: 10.1126/science.1180614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche Applied Science. Genome Sequencer FLX Data Analysis Software Manual. 2007.
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: a graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Qi J, Zhao F, Buboltz A, Schuster SC. inGAP: an integrated next-generation genome analysis pipeline. Bioinformatics. 2010;26:127–129. doi: 10.1093/bioinformatics/btp615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer EL, Eddy SR, Durbin R. Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins. 1997;28:405–420. doi: 10.1002/(SICI)1097-0134(199707)28:3<405::AID-PROT10>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- de Castro E, Sigrist CJA, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 2006;34:W362–W365. doi: 10.1093/nar/gkl124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- Carver TJ, Rutherford KM, Berriman M, Rajandream M-A, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- R Development Core Team: R. A language and environment for statistical computing. Vienna, Austria; 2008. (R Foundation for Statistical Cmputing). [Google Scholar]
- Lukjancenko O, Wassenaar TM, Ussery DW. Comparison of 61 sequenced Escherichia coli genomes. Microb Ecol. 2010;60:708–720. doi: 10.1007/s00248-010-9717-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Z, Cox AJ, Mullikin JC. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–1729. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Harris SR, Feil EJ, Holden MTG, Quail MA, Nickerson EK, Chantratita N, Gardete S, Tavares A, Day N, Lindsay JA, Edgeworth JD, de Lencastre H, Parkhill J, Peacock SJ, Bentley SD. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327:469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1
Comparative genome content of thirteen Bordetella strains. Circles indicate the presence (solid color) or absence (unfilled) of each gene family in each strain examined. Circles from outer to inner are started with B. petrii strain followed by B. avium strain and the pan-genome of the classical Bordetella strains. Then, individual circle of B. bronchiseptica, B. parapertussis and B. pertussis strains were shown. This figure was created using the Circos software [21].
Additional file 2
Lists of gene families that are present in the pan-genome of the classical Bordetella strains. This table summarized the gene family numbers, representative gene name, predicted function of encoding proteins, presence (1) or absence (0) of each gene family in individual genome, and the sum of each row for a sorting purpose.
Additional file 3
The number of SNPs in the genes and dN/dS ratios of genes that have high number of SNPs. This table summarized the start and end position of genes in RB50, gene name, strand, gene length, predicted function of encoding proteins, number of SNPs among the classical Bordetellae based on RB50, and number of SNPs per 100 base pair. dN, dS, and dN/dS ratio are included only for fifty genes that have high number of SNPs. The mean SNPs per 100 bp for the whole genome was 2.24. When the orthologs are pseudogenes, we did not calculate dN/dS ratios, indicating NA in the columns.
Additional file 4
Percent sequence similarity of B. pertussis ptx/ptl loci with flanking genes against Tohama I. Percent sequence similarity of B. pertussis ptx/ptl loci and flanking genes compared to Tohama I was plotted between 95% and 100% using zPicture [36]. Intergenic regions, coding regions, and a tRNA were highlighted with red, blue, and green, respectively.
Additional file 5
Pair-wise dN/dS ratios for individual gene in the classical Bordetellae ptx/ptl locus. dN/dS ratios of ten classical Bordetellae were plotted for individual gene in the ptx/ptl locus. B. pertussis strain Tohama I represents another B. pertussis strain CS as well because they share identical nucleotide sequences in the ptx/ptl loci. Each figure represents ptx/ptl genes (A), ptx genes (B), ptxA (C), ptxB (D), ptxD (E), ptxE (F), ptxC (G), ptl genes (H), ptlA (I), ptlB (J), ptlC (K), ptlD (L), ptlI (M), ptlE (N), ptlF (O), ptlG (P), and ptlH (Q).
Additional file 6
Genome-wide horizontal gene transfer candidates for the classical Bordetellae. The position of the candidates is plotted in each genome (Bpp5 (A), 12822 (B), Tohama I (C), CS (D), 18323 (E), RB50 (F), 1289 (G), 253 (H), MO149 (I), Bbr77 (J), and D445 (K)) and the height represents the score of Alien_hunter. Red line represents the threshold for each genome.
Additional file 7
Phylogenetic trees for the classical Bordetellae ptx/ptl locus. Phylogenetic trees were constructed by maximum likelihood method with 1,000 bootstrap replicates with individual gene sequences in the locus, or the entire ptx or ptl locus. Each figure represents the entire ptx locus (A), ptxA (B), ptxB (C), ptxD (D), ptxE (E), ptxC (F), ptl locus (G), ptlA (H), ptlB (I), ptlC (J), ptlD (K), ptlI (L), ptlE (M), ptlF (N), ptlG (O), and ptlH (P).
Additional file 8
Multiple sequence alignments of the classical Bordetellae ptxB and ptxC. Multiple amino acid sequence alignments of the classical Bordetellae ptxB and ptxC were presented in this figure.