Structural basis of long-term potentiation in single dendritic spines (original) (raw)
. Author manuscript; available in PMC: 2014 Sep 9.
Published in final edited form as: Nature. 2004 Jun 9;429(6993):761–766. doi: 10.1038/nature02617
Abstract
Dendritic spines of pyramidal neurons in the cerebral cortex undergo activity-dependent structural remodelling1–5 that has been proposed to be a cellular basis of learning and memory6. How structural remodelling supports synaptic plasticity4,5, such as long-term potentiation7, and whether such plasticity is input- specific at the level of the individual spine has remained unknown. We investigated the structural basis of long-term potentiation using two-photon photolysis of caged glutamate at single spines of hippocampal CA1 pyramidal neurons8. Here we show that repetitive quantum-like photorelease (uncaging) of glutamate induces a rapid and selective enlargement of stimulated spines that is transient in large mushroom spines but persistent in small spines. Spine enlargement is associated with an increase in AMPA-receptor-mediated currents at the stimulated synapse and is dependent on NMDA receptors, calmodulin and actin polymerization. Long-lasting spine enlargement also requires Ca/calmodulin-dependent protein kinase II. Our results thus indicate that spines individually follow Hebb’s postulate for learning. They further suggest that small spines are preferential sites for long-term potentiation induction, whereas large spines might represent physical traces of long-term memory.
We investigated the structural plasticity of individually identified spines on proximal apical dendrites of CA1 pyramidal neurons in rat hippocampal slice preparations, using two-photon uncaging (at 720nm) of a caged glutamate compound (MNI (4-methoxy-7-nitroindolinyl)-glutamate)8. Neurons within the hippocampal slices were transfected biolistically with an expression vector for enhanced green fluorescent protein (eGFP) and were imaged with a second two-photon laser at 910 nm. Two-photon uncaging allows the release of glutamate within a small focal volume8, so that activation of glutamate receptors is limited to a region within ca. 1 μm of the uncaging beam. The power of the laser was set at ca. 5mW, which induced currents through AMPA (α-amino-3- hydroxy-5-methyl-4-isoxazole propionic acid)-sensitive receptors with amplitudes in the range of miniature excitatory postsynaptic currents (see Methods). Repetitive stimulation of NMDA (N-methyl-D-aspartate)-sensitive receptors at a frequency of 1 Hz for 1 min was achieved using an external solution lacking Mg.
This stimulation procedure induced marked increases (of .50%) in spine fluorescence in most spines examined (38 out of 40 spines, Fig. 1a, b; Supplementary Movie 1). The fluorescence profile (Fig. 1e) of the spine shown in Fig. 1a was indicative of a 28% increase in spine-head diameter at 81 min after stimulation, which would account for the 111% increase in fluorescence at this time (Fig. 1b). The maximal increase in spine-head volume (ΔV) was 203 +/− 37% (mean +/− s.e.m., n = 40) (Fig. 1f) and was apparent 1–5 min after stimulation. Such spine enlargement did not occur in the absence of MNI-glutamate. Furthermore, it was either transient, with spines recovering their original volume, or persistent for periods of at least 100 min. The latter fate was observed in 30% of stimulated spines and increases of more than 50% were detected for 70–100 min after stimulation. The spine enlargement was detected within 60s of stimulation, being first apparent within 10s of initiation of the stimulus (Fig. 1g; for such rapid imaging, the fluorescence sample had to be limited to a single plane through the centre of the spine head). The enlargement was selective towards stimulated spines (Fig. 1a, b, h); only 9% of neighbouring spines (mean distance from stimulated spines, 1.2mm; mean ΔV = 8 +/− 8%; n = 34) showed enlargement (of >50%) and only 3% showed long-lasting enlargement.
Figure 1.

Spine-head enlargement induced by repetitive uncaging of MNI-glutamate. a, Time-lapse images of dendritic spines on a hippocampal CA1 pyramidal neuron expressing eGFP. The arrowhead indicates the spot of two-photon uncaging of MNI-glutamate, which was achieved by stimulation for 1 min at 1 Hz, beginning at time zero in a bathing solution lacking Mg. Images were stacked along the z axis. Scale bar, 1 μm. b, Time course of spine-head volume, estimated from stacked images, of the stimulated (black circles) and neighbouring (green diamonds) spines shown in a. c, Time-lapse images of a spine on a dendrite that was affected by electrical stimulation of presynaptic fibres at 2 Hz for 1 min; d, time course of the head volume. e, Fluorescence intensity along a line crossing the centre of the stimulated spine head shown in a at −3 min (black circles) and 81 min (red circles). Smooth black and red lines represent the predictions made for the z-stacked images of spheres with diameters of 0.46 and 0.59 μm, respectively (see Supplementary Methods). The dashed line is scaled from the smooth black one. f, h, Averaged time courses of spine-head volume for two-photon uncaging of MNI-glutamate (circles, n = 40), neighbouring spines (green diamonds in h, n = 34), 2-Hz electrical stimulation in the absence of Mg (blue open triangles, n = 19) or 100 Hz electrical stimulation in the presence of Mg (blue closed triangles, n = 14). Error bars represent +/− s.e.m. g, Initial time course of spine fluorescence. The intensity in a single plane drawn through the centre of the spines was sampled; the average for 11 spines is shown. Dotted lines show time courses corresponding to the mean +/− s.d., and the horizontal bar indicates the period of repetitive uncaging.
Similar spine enlargement was also induced by repetitive stimulation of Schaffer collateral fibres, either at low frequency (2 Hz, 1 min) in the absence of Mg (Fig. 1c, d, f; blue open triangles) or at high frequency (100 Hz for 1 s, twice with a 20-s interval) in the presence of Mg (Fig. 1f, blue closed triangles). With such stimulation of presynaptic fibres, however, it was not possible to ascertain whether the enlarged spines corresponded to the stimulated spines. A fundamental advantage of our optical approach is that it allows the location of stimulation to be related directly to the site of spine enlargement. Given that stimulation of Schaffer collaterals and focal glutamate application induced similar structural changes in dendritic spines, we conclude that uncaging of glutamate mimics synaptic stimulation with a resolution at the level of the individual spine.
Enlargement (ΔV > 50%) was detected in 100% of small spines (volume < 0.1 μm3, n = 20) and 90% of large spines (volume > 0.1 μm3, n = 20) after stimulation by uncaging of glutamate (Fig. 2a–c). The amplitude of transient enlargement (ΔVtransient) (Fig. 2c) did not differ significantly between small and large spines (Fig. 2d). In contrast, long-lasting enlargement (ΔVlong) was markedly dependent on original spine size (Supplementary Fig. 2b); the mean value of ΔVlong was thus greater for small spines than for large ones (Fig. 2d), and long-lasting enlargement (ΔVlong > 50%) was observed in 55% and 5% of small and large spines, respectively (see Supplementary Information). The high-frequency electrical stimulation (Fig. 1f, 100 Hz) consistently induced long-lasting enlargement (>50%) in only small spines (volume < 0.1 μm3, n = 5).
Figure 2.

Properties of the spine-head enlargement. a, c, Averaged time course (+/− s.e.m.) of spine-head volume for large (a) and small (c) spines with an initial head volume of > 0.1 μm3 (n = 20) and < 0.1 μm3 (n = 20), respectively. ΔV transient represents the difference in spine-head volume between 1–5 and 70–100 min after stimulation, whereas ΔV long represents that between 70–100 min after and 0–20 min before stimulation. b, Example of an enlargement of a large spine. The arrowhead indicates the point of two-photon uncaging of MNI-glutamate. d, Mean values of ΔV transient and DV long for small and large spines (n = 20 each). e, Effects of 50 μM AP5 or 4 μM KN62 on the averaged time course (+/− s.e.m.) of spine-head volume for small spines. f, Pharmacological properties of the transient and long-lasting phases of head enlargement in small spines. Antagonists examined were 1 mM MCPG (n = 15), 50 μM AP5 (n = 7), 20 μM W7 (n = 11), 4 μM KN62 (n = 12) and 20 nM (n = 12) or 100 nM latrunculin A (n = 7). *P < 0.05, **P < 0.01 (Mann–Whitney U test). Errors bars in d and f show s.e.m.
The long-lasting spine enlargement showed pharmacological properties similar to those of the induction of long-term potentiation (LTP)9,10. Spine enlargement was thus prevented by the NMDA receptor antagonist, D(−)-2-amino-5-phosphonovaleric acid (AP5), (Fig. 2e, f) but unaffected by the metabotropic glutamate receptor blocker, (S)-alpha-methyl-4-carboxyphenylglycine (MCPG) (Fig. 2f), indicating that it was dependent on the activation of NMDA receptors. The structural plasticity was also blocked by the calmodulin inhibitors, W7 (N-(6-amino-hexyl)-5- chloro-1-naphthalene-sulphonamide hydrochloride) (Fig. 2f) and calmidazolium (data not shown), as well as by latrunculin A (100 nM) (Fig. 2f), an inhibitor of actin polymerization, implicating Ca- and calmodulin-dependent actin reorganization in this process. An inhibitor of Ca/calmodulin-dependent protein kinase II, KN62, blocked the long-lasting (Fig. 2e, f) but not the transient spine enlargement. We also found that a low concentration of latrunculin A (20nM) selectively affected the long-lasting enlargement, consistent with the previous observation that a low concentration of this agent blocked long-term but not early potentiation11, and suggesting that distinct cytoskeletal organizations may underlie transient and long-lasting spine enlargements.
We next investigated expression of postsynaptic LTP during spine structural plasticity using three-dimensional mapping of AMPA receptor-mediated currents. The currents were mapped using two-photon uncaging of MNI-glutamate8, rather than by iontophoretic glutamate application12. Pyramidal neurons expressing eGFP were subjected to whole-cell clamping in the perforated-patch mode to prevent washout of intracellular proteins13. Paired stimulation— consisting of postsynaptic depolarization to 0 mV and repetitive glutamate application to a single spine at 2 Hz for 60 s in a solution containing a physiological concentration (1mM) of Mg— induced structural plasticity (Fig. 3a) similar to that evoked by uncaging of MNI-glutamate in a solution lacking Mg (Fig. 1a). Because, in the presence of Mg, spine enlargement was observed only when depolarization was paired with glutamate application, we were able to obtain AMPA current maps8 (holding potential, −60 mV) both before and after the induction of structural plasticity without influencing plasticity itself. AMPA currents were markedly potentiated by paired stimulation at sites restricted to the stimulated spine (Fig. 3a, b). The potentiation was rapid, being detected as early as 3 min after stimulation (designated ‘early potentiation’; Fig. 3a). Furthermore, current potentiation (>30% increase) persisted for at least 30 min in 44% of small spines (Fig. 4b; n = 9). The AMPA currents were always detected at the same locations in the spines, possibly reflecting the postsynaptic density, before and after stimulation (Fig. 3). Long-lasting potentiation of AMPA currents is therefore identical to LTP in that it probably occurs at the postsynaptic density, is induced by glutamate application14 and is blocked by AP5 (see below).
Figure 3.

Colocalization of enlargement of spine heads and potentiation of AMPA- receptor-mediated currents. a, b, Examples of small spines that showed transient (a) or long-lasting (b) enlargement (upper panels) and potentiation of AMPA currents (lower panels). Three-dimensional mapping of AMPA currents was performed in neurons that were clamped in the perforated-patch mode. The amplitude of AMPA currents is pseudocolour coded and stacked along the z axis by the maximum-intensity method. The neurons were depolarized to 0 mV and the small spines, indicated by the arrowheads, were stimulated by two-photon uncaging of MNI-glutamate at 2 Hz between times 0 and 60 s. White lines in the lower panels indicate contours of dendrites. Scale bars, 1 μm. c, d, Examples of small (c) and large (d) spines whose heads failed to enlarge in response to paired stimulation.
Figure 4.
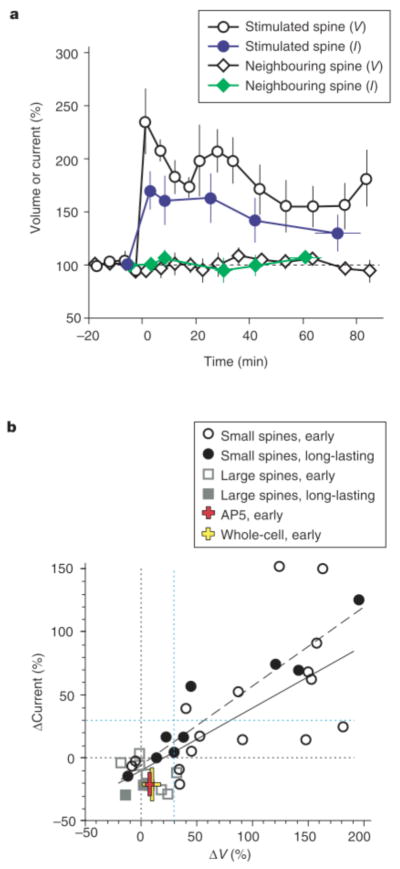
Relationship between spine-head enlargement and potentiation of AMPA- receptor-mediated currents. a, Time courses (+/− s.e.m.) of spine-head volume (V, open symbols) and maximal AMPA currents (I, filled symbols) normalized to the original values. The data were derived from all the small spines (n = 9) that showed enlargement immediately after pairing stimulation and for which it was possible to obtain current maps more than 30 min after paired stimulation in the perforated-patch recording mode. Circles and diamonds represent stimulated and neighbouring spines, respectively. b, Relationships between normalized amplitudes of head enlargement (ΔV) and potentiation of AMPA currents (ΔCurrent) in the early phase (1–5 min after stimulation) and long- lasting phase (30–120 min) in small and large spines. The continuous and dashed lines are regression lines for the early and long-lasting phases, respectively; correlation coefficients are 0.72 (n = 24, P < 0.0001) and 0.95 (n = 9, P < 0.0001), respectively. The red and yellow crosses show data obtained in the presence of AP5 (n = 5) or with whole-cell recording (n = 7), respectively. Black and blue dotted lines are 0% and 30% levels, respectively.
Figure 4 summarizes the results from many dual imaging/mapping experiments. We found that potentiation of AMPA-receptor-mediated currents at small spines was strongly correlated with enlargement of the same spine head. The following findings indicate this correlation: (1) early potentiation was detected only at spines that underwent enlargement (Fig. 4b, open circles), being apparent in seven of the 14 spines that showed enlargement (of > 30%; we set the criterion level based on two s.d. values of spontaneous fluctuation; see Supplementary Information) but in neither of the two spines that did not (< 30% increase in volume) (Fig. 3c, d). (2) LTP was induced in four of the five spines that showed long-lasting enlargement but in none of the four spines that did not (Fig. 4b, filled symbols). (3) The magnitude of potentiation correlated well with that of early (ΔVtransient + ΔVlong) (Fig. 4b, solid line) or long-lasting (Fig. 4b, dashed line) spine enlargement. (4) Both current potentiation and morphological enlargement were detected only in stimulated spines, not in neighbouring spines (Figs 3a, b and 4a). (5) Neither current potentiation nor spine enlargement was detected in cells in which the induction of LTP was prevented by AP5. Paired stimulation in the presence of AP5 did not substantially alter spine volume (increase of only 8 +/− 4%, n = 5) but did induce a slight decrease (of 21 +/− 9%, n = 5) in the amplitude of AMPA currents in the early phase (Fig. 4b, red cross) that was probably due to a side effect of repetitive optical illumination (similar reductions were also induced in the following two conditions). (6) In neurons subjected to whole-cell dialysis, we detected little structural plasticity (ΔV = 9 +/− 8%, n = 7) and a slight decrease in AMPA currents (of 21 +/− 13%) (Fig. 4b, yellow cross); the absence of spine enlargement and LTP was probably due to washout of the responsible molecules15. Large spines showed little enlargement when cells were clamped in the perforated-patch mode (Figs 3d and 4b, squares in Fig. 4b; n = 8; see Supplementary Information), and a slight reduction in the AMPA current was detected (Fig. 4b).
We have shown that synaptic potentiation is closely related to enlargement of spine heads in terms of pharmacology, amplitude, time course and spatial localization within the dendrite. Spine enlargement was induced with little time delay, as has been reported for LTP16. In contrast, new filopodia or spines require at least 20 min to emerge from dendrites after the induction of LTP4,5 and therefore cannot explain the rapid onset of LTP. Under our experimental conditions, in which only one spine is stimulated, the generation of new spines was not observed. The growth of new spines might thus require the stimulation of many synapses and contribute to a later phase of LTP. The expression of AMPA receptors in spines is dependent on F-actin11,17 and scaffolding proteins18,19. Spine enlargement, which is also dependent on actin polymerization, might therefore promote the accumulation of AMPA receptors. However, the precise molecular mechanisms of F-actin reorganization20,21 and AMPA receptor expression18,19,22,23 remain to be clarified.
With the use of location-specific two-photon photolysis of caged glutamate to stimulate single synapses, we have shown that both structural and functional plasticity can be induced at the level of the individual spine. Our results thus indicate that a hebbian mechanism24 can operate individually in single spines of hippocampal CA1 pyramidal neurons as has been suggested25 but not proved26. We also found that the preferential sites of LTP induction are small spines, which express a small number of AMPA receptors8. Small spines might thus correspond to the postsynaptic structures of so-called silent synapses27–29. Given that large spines have been shown to express AMPA receptors abundantly8 and to be stable for months in the mouse cerebral cortex in vivo2,3, and given that we have now shown that small spines can be converted into larger spines, we suggest that spines act as memory units, with large spines being the physical traces of long-term memory. Consistent with this idea, we found that large spines are resistant to LTP, which is critical for information storage. The memory stored in large spines might thus be protected from further potentiation caused by its readout and new memory formation30. Further clarification of the structural bases of synaptic potentiation and depression should give greater insight into the operation of memory units in the brain.
Methods
Preparation of slice cultures
Hippocampal slices with a thickness of 350 μm were prepared from 6–8-day-old Sprague– Dawley rats. The slices were mounted on 0.4-mm culture inserts (Millipore) and incubated at 35 °C under 5% CO2 in a medium comprising 50% MEM (Invitrogen), 25% Hanks’ balanced salt solution (Invitrogen), 25% horse serum (Nichirei) and 6.5 g 121 glucose. After 5–6 days in vitro, slices were transfected using a gene gun system (PDS-1,000; Bio-Rad), with a vector containing eGFP complementary DNA under the control of the chicken b-actin gene promoter and cytomegalovirus enhancer. Two to five days after transfection, each slice was transferred to a recording chamber and superfused at room temperature (23–25 °C) with a solution that contained 125 mM NaCl, 2.5 mM KCl, 3 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3 and 20 mM glucose, and which was bubbled with 95% O2 and 5% CO2. The bathing solution also contained 1 μM tetrodotoxin, 50 μM picrotoxin and 200 μM Trolox (Aldrich). For electrical stimulation, 0.1-ms voltage pulses were delivered either with a glass pipette in the absence of Mg, or with a bipolar tungsten electrode in the presence of Mg. In experiments with the antagonists AP5, MCPG (Tocris), W7 (Calbiochem), KN62 (Seikagaku) and latrunculin A (Molecular Probes), slices were superfused with bathing solution containing each drug at least 30 min before two-photon uncaging of caged glutamate. The experiments were approved by the animal experiment committee of the National Institute for Physiological Sciences.
Two-photon excitation imaging and uncaging
Time-lapse two-photon imaging of dendritic spines was performed with an upright microscope (BX50WI; Olympus) equipped with a water immersion objective lens (LUMPlanFI/IR 60x, numerical aperture of 0.9; Olympus) and with Fluoview software (Olympus). Two mode-locked fs-pulse Ti:sapphire lasers (Spectra Physics) set at wavelengths of 720 and 910 nm were connected to the laser-scanning microscope through two independent scan heads. For imaging of dendritic spines we used the laser set at a wavelength of 910 nm, with which the point-spread function of the focal volume was estimated using 0.1-μm fluorescent beads as 0.44 μm (full-width at half-maximum) laterally and 2.1 μm axially. For three-dimensional reconstruction, 13–17 xy images separated by 0.5 μm were stacked by summation of fluorescence values at each pixel, thus the enlargements appear as an increase in diameter and intensity. Immediately before two-photon uncaging, one image was acquired at the 720-nm wavelength for precise adjustment of the point of photolysis; MNI-glutamate (12 mM) was then applied locally from a glass pipette positioned close to the selected dendrite. Repetitive (1 or 2 Hz) photolysis of MNI-glutamate was performed on the specimen at a 720-nm wavelength with a pulse-train duration of 0.6 ms and a power of 5 mW. A self-made program based on LabView (National Instruments) controlled a galvano-scanner driver and a mechanical shutter (Uniblitz).
Electrophysiology
For perforated-patch recordings, 1 mM MgCl2 was added to the extracellular solution and the concentration of CaCl2 was increased to 4 mM. The patch-clamp electrodes (open-tip resistance, 1.5–2.5 MQ) were filled with a solution containing 136.5 mM potassium gluconate, 17.5 mM KCl, 9 mM NaCl, 1 mM MgCl2, 0.2 mM EGTA (tetrapotassium salt), 10 mM Hepes–KOH (pH 7.2) and 0.3 mg/mL amphotericin B. Neurons were voltage-clamped at −60 mV and the currents were low-pass filtered at 2 kHz and sampled at 10 kHz. Series resistance was 32 +/− 8.1 MΩ (mean +/− s.d.) during mapping of AMPA receptors. Amplitudes of AMPA currents were 1–25 pA, (measured with perforated patch-clamp experiments), which were within the range of miniature excitatory postsynaptic currents8. For such mapping, a pseudorandom sequence of scanning pixels in a region of interest was constructed to maintain the distance between two successive pixels at greater than 2.5–5 mm and AMPA currents were sampled at an interval of 100 ms. For three-dimensional mapping of AMPA currents, xy scanning of a region of interest comprising 16 × 16 pixels (1 pixel = 0.33 mm) was performed at 4–6 different z axis planes, each separated by 1 mm. Peak amplitudes of AMPA currents were assigned to each pixel, linear-interpolated8, stacked along the z axis by the maximum-intensity method and displayed with a pseudocolour coding (Fig. 3a).
Supplementary Material
Supplement-movie
Supplement-text
Acknowledgments
We thank P. Haydon for reading the manuscript, N. Takahashi and T. Kise for technical assistance and Y. Yanagawa and M. Okabe for the eGFP construct. This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (H.K. and M.M.) and by the Human Frontier Science Program Organization (G.C.R.E.-D. and H.K.), the NIH (G.C.R.E.-D. and H.K.), the NSF (G.C.R.E.-D) and the McKnight Endowment Fund for Neuroscience (G.C.R.E.-D).
Footnotes
Competing interests statement The authors declare that they have no competing financial interests.
References
- 1.Van Harreveld A, Fifkova E. Swelling of dendritic spines in the fascia dentata after stimulation of the perforant fibers as a mechanism of post-tetanic potentiation. Exp Neurol. 1975;49:736–749. doi: 10.1016/0014-4886(75)90055-2. [DOI] [PubMed] [Google Scholar]
- 2.Trachtenberg JT, et al. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- 3.Grutzendler J, Kasthuri N, Gan WB. Long-term dendritic spine stability in the adult cortex. Nature. 2002;420:812–816. doi: 10.1038/nature01276. [DOI] [PubMed] [Google Scholar]
- 4.Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- 5.Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- 6.Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]
- 7.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuzaki M, et al. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nature Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malenka RC, et al. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- 10.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 11.Krucker T, Siggins GR, Halpain S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci USA. 2000;97:6856–6861. doi: 10.1073/pnas.100139797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies SN, Lester RA, Reymann KG, Collingridge GL. Temporally distinct pre- and post-synaptic mechanisms maintain long-term potentiation. Nature. 1989;338:500–503. doi: 10.1038/338500a0. [DOI] [PubMed] [Google Scholar]
- 13.Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cormier RJ, Mauk MD, Kelly PT. Glutamate iontophoresis induces long-term potentiation in the absence of evoked presynaptic activity. Neuron. 1993;10:907–919. doi: 10.1016/0896-6273(93)90206-7. [DOI] [PubMed] [Google Scholar]
- 15.Kato K, Clifford DB, Zorumski CF. Long-term potentiation during whole-cell recording in rat hippocampal slices. Neuroscience. 1993;53:39–47. doi: 10.1016/0306-4522(93)90282-k. [DOI] [PubMed] [Google Scholar]
- 16.Gustafsson B, Wigstrom H. Long-term potentiation in the hippocampal CA1 region: its induction and early temporal development. Prog Brain Res. 1990;83:223–232. doi: 10.1016/s0079-6123(08)61252-2. [DOI] [PubMed] [Google Scholar]
- 17.Kim CH, Lisman JE. A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci. 1999;19:4314–4324. doi: 10.1523/JNEUROSCI.19-11-04314.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schnell E, et al. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rumbaugh G, Sia GM, Garner CC, Huganir RL. Synapse-associated protein-97 isoform-specific regulation of surface AMPA receptors and synaptic function in cultured neurons. J Neurosci. 2003;23:4567–4576. doi: 10.1523/JNEUROSCI.23-11-04567.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 21.Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M. Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron. 2001;31:289–303. doi: 10.1016/s0896-6273(01)00355-5. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi Y, et al. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- 23.Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nature Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 24.Hebb DO. The Organization of Behavior. Wiley; New York: 1949. [DOI] [PubMed] [Google Scholar]
- 25.Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- 26.Engert F, Bonhoeffer T. Synapse specificity of long-term potentiation breaks down at shortdistances. Nature. 1997;388:279–284. doi: 10.1038/40870. [DOI] [PubMed] [Google Scholar]
- 27.Durand GM, Kovalchuk Y, Konnerth A. Long-term potentiation and functional synapse induction in developing hippocampus. Nature. 1996;381:71–75. doi: 10.1038/381071a0. [DOI] [PubMed] [Google Scholar]
- 28.Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- 29.Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 30.Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure–stability–function relationships of dendritic spines. Trends Neurosci. 2003;26:360–368. doi: 10.1016/S0166-2236(03)00162-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement-movie
Supplement-text