Mutation of the mouse Rad17 gene leads to embryonic lethality and reveals a role in DNA damage-dependent recombination (original) (raw)
Abstract
Genetic defects in DNA repair mechanisms and cell cycle checkpoint (CCC) genes result in increased genomic instability and cancer predisposition. Discovery of mammalian homologs of yeast CCC genes suggests conservation of checkpoint mechanisms between yeast and mammals. However, the role of many CCC genes in higher eukaryotes remains elusive. Here, we report that targeted deletion of an N-terminal part of mRad17, the mouse homolog of the Schizosaccharomyces pombe Rad17 checkpoint clamp-loader component, resulted in embryonic lethality during early/mid-gestation. In contrast to mouse embryos, embryonic stem (ES) cells, isolated from mRad17 5_′Δ/5_′Δ embryos, produced truncated mRad17 and were viable. These cells displayed hypersensitivity to various DNA-damaging agents. Surprisingly, mRad17 5_′Δ/5_′Δ ES cells were able to arrest cell cycle progression upon induction of DNA damage. However, they displayed impaired homologous recombination as evidenced by a strongly reduced gene targeting efficiency. In addition to a possible role in DNA damage-induced CCC, based on sequence homology, our results indicate that mRad17 has a function in DNA damage-dependent recombination that may be responsible for the sensitivity to DNA-damaging agents.
Keywords: cell cycle checkpoint, DNA repair, DNA replication, genomic instability
Introduction
Accurate transmission of genetic information from a dividing cell to its daughters is essential for the survival of organisms. To minimize the number of irreversible mutations, extreme accuracy in DNA replication and equal distribution of chromosomes between daughter cells are critical. In addition, maintaining genome stability requires accurate repair of physiologically and environmentally induced DNA damage. Throughout the cell cycle, DNA integrity is monitored by checkpoint pathways, which arrest cell cycle progression upon the presence of DNA damage or unreplicated DNA (Hartwell and Weinert, 1989; Zhou and Elledge, 2000). This arrest in cell cycle progression, often referred to as DNA damage-induced cell cycle checkpoint (CCC), provides a window of opportunity for DNA damage repair or causes irreversible withdrawal from the cell cycle. Loss of these CCCs can result in genomic instability, which is linked to an increase in cancer susceptibility.
The importance of DNA damage-induced CCC is illustrated by the human disorder ataxia telangiectasia (AT), caused by mutations in the ATM gene (Savitsky et al, 1995) and characterized by increased sensitivity to DNA damage, high frequency of chromosomal aberrations and elevated risk of cancer. Most notably, ATM shares significant homology with Rad3 from the fission yeast Schizosaccharomyces pombe and with TEL1 from the budding yeast Saccharomyces cerevisiae (Savitsky et al, 1995). These sequence similarities and those of other CCC genes between the two distantly related yeast species and the identification of mammalian homologs strongly suggest that CCC mechanisms are highly conserved among eukaryotic cells.
In response to DNA damage, different putative CCC pathways can be identified. The mitotic cell cycle can be inhibited before commitment to S phase DNA replication at the G1/S transition (G1/S checkpoint; Siede et al, 1993), during S phase (intra-S checkpoint; Paulovich and Hartwell, 1995) and upon entry into mitosis from G2 (G2/M checkpoint; Hartwell and Weinert, 1989). In recent years, a variety of genes have been identified that, when defective, lead to hypersensitivity to agents causing DNA damage or replication stalling, such as UV light, γ-radiation and hydroxyurea (HU), and to disturbed cell cycle response upon genome damage. In S. pombe, the products of Rad1, Rad3, Rad9, Rad17, Rad26 and Hus1 have been identified as essential components of DNA damage-induced CCC pathways. Mutants of these checkpoint genes fail to undergo a dose-dependent cell cycle arrest in response to DNA damage or incomplete DNA replication. In S. cerevisiae, a similar group of checkpoint genes RAD9, RAD17, RAD24, TEL1, MEC1, MEC3 and DDC1 is found to be required for DNA damage-induced mitotic cell cycle arrest.
In S. pombe, Rad17 is indispensable for S phase and G2/M arrest in response to both DNA damage and incomplete DNA replication. The structural counterpart in S. cerevisiae is RAD24. In both human and mouse, homologs of the Rad17 gene, hRAD17 and mRad17, respectively, have been identified and characterized (Bluyssen et al, 1999; von Deimling et al, 1999). Homology is also detected between the Rad17 homologs and proteins of mammalian replication factor C (RFC). RFC consists of five structurally related subunits (RFC1–5; Lee et al, 1991), which all contain intrinsic DNA-dependent ATPase activity and together act as a ‘clamp loader' for proliferating cell nuclear antigen (PCNA). PCNA operates as a sliding clamp complex along DNA and is involved in recruiting replicative DNA polymerases δ and ɛ onto primed DNA templates (Hubscher et al, 2002). A recent study has demonstrated that Rad17 associates specifically with members 2–5 of the RFC complex (Green et al, 2000; Ellison and Stillman, 2003), suggesting that Rad17 and RFC1 are competing for interaction with the other four RFC subunits. The homology of Rad17 with RFC and the interaction with the PCNA-like Rad9–Rad1–Hus1 (9-1-1) complex suggests a role for Rad17 in the recruitment of this complex to DNA lesions in a manner similar to PCNA loading by RFC (Zou et al, 2002; Ellison and Stillman, 2003). Consistent with this notion, the 9-1-1 complex is less extractable from the nucleus upon DNA damage induction, suggesting a DNA damage-induced association to chromatin stimulated by Rad17 (Burtelow et al, 2000). Hus1 is phosphorylated after the induction of DNA damage and this modification requires other CCC Rad proteins, including the ATM/ATR-related kinase Rad3 (Kostrub et al, 1998). In S. pombe, Rad3 phosphorylates its tightly bound partner Rad26 after ionizing radiation treatment, independent of Rad17 and the 9-1-1 complex (Edwards et al, 1999), suggesting that the Rad3–Rad26 complex might directly respond to certain types of DNA damage. The recent identification of ATRIP (ATR-interacting protein), the human homolog of Rad26, suggests a similar, Rad17- and 9-1-1 complex-independent, mechanism for detection of certain types of DNA lesions in human cells (Cortez et al, 2001). However, the phosphorylation of Rad17 by ATR, both in different complexes, is stimulated by Hus1 (Zou et al, 2002). These data indicate that the 9-1-1 complex recruited by Rad17 upon DNA damage induction enables ATR to recognize its substrates on the chromatin and thereby combine two independent sensory pathways to activate the DNA-damage response fully.
Although our understanding of DNA damage-induced CCC mechanisms is improving, many aspects of CCC function in mammals remain elusive. To determine the role of mRad17 in mouse development and mouse CCC function, we produced a targeted disruption of mRad17. Here we report that deletion of mRad17 leads to death in early- to mid-gestation embryos. Although no damage-induced CCC defects could be detected, our analyses of viable mRad17 5_′Δ/5_′Δ embryonic stem (ES) cells, producing truncated mRad17, reveal the involvement of mRad17 in DNA damage-dependent recombination.
Results
Deletion of mRad17 results in embryonic lethality during early/mid-gestation
For targeting the mRad17 gene, we constructed a targeting vector with a 110 bp deletion in exon 3. Due to this deletion, the reading frame is shifted, resulting in the formation of three immediate stop codons in the new reading frame. A neomycin selection cassette, driven by the PGK promoter, was cloned in antisense orientation in intron 2 (Figure 1A). Because the region deleted in the targeting vector is present in all different splice variants of mRad17 and the frameshift results in the presence of multiple stop codons, we anticipated that homologous integration of this targeting vector would result in an mRad17 null allele. The linearized targeting construct was electroporated into E14 ES cells and, after G418 selection, resistant clones were screened by DNA blotting of _Pst_I-digested genomic DNA (Figure 1B). Three clones that had undergone homologous recombination events were obtained out of 240 clones tested. These clones were karyotyped and further analyzed for correct 3′ integration of the targeting vector by PCR (data not shown). Two of the clones, F17 and F24, were injected into C57bl/6 blastocysts to produce chimeric mice that transmitted the targeted mRad17 allele through the germ line. No differences between the two independent mouse lines were detected in the experiments described below, indicating that the resulting mouse mutant represents a ‘bona fide' outcome of the genetic modification. Male chimeras were bred to C57bl/6 females to produce mRad17 _+/5_′Δ heterozygous offspring. Heterozygous mRad17 _+/5_′Δ mice were normal and fertile, and no aberrant phenotype has been observed for up to 1 year of age. However, when heterozygous mRad17 _+/5_′Δ mice were intercrossed to produce homozygous mutant mRad17 mice, mRad17 5_′Δ/5_′Δ mice were absent from the large number of offspring analyzed (Table I). Both mRad17 _+/5_′Δ and wild-type mice were present, suggesting that the homozygous mutant mRad17 results in embryonic lethality. As mRad17 _+/5_′Δ pups were born in normal numbers and showed no abnormal phenotype, a dominant-negative effect of the targeted allele is not likely.
Figure 1.
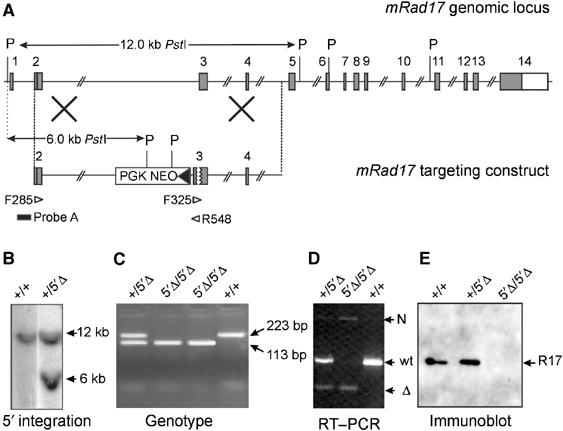
Generation of mRad17 mutant mice. (A) Targeting vector design and screening strategy. Numbered gray boxes represent exons. The positions of _Pst_I (P) restriction sites, size of the _Pst_I restriction fragments and the position of probe A used for DNA blot screening of digested ES cell DNA are indicated. The open arrowheads indicate the position and direction of primers used for PCR. (B) DNA blot analysis of _Pst_I-digested ES cell DNA using probe A. The positions of wild type (+;12 kb) and 5′Δ (6 kb) are indicated. (C) Analysis of genotype by PCR using primers F325 and R548. The positions of wt (223 bp) and 5′Δ (113 bp) bands are indicated. (D) RT–PCR on E8.5 embryos using a poly-dT primer and primers F285 and R548. The positions of ‘wt', ‘Δ' and ‘N' bands are indicated. (E) Immunoblot analysis of E8.5 embryos. The position of mRad17 protein is indicated. Each lane contained half of the total E8.5 embryo lysate.
Table 1.
Genotyping of offspring and embryos from matings of heterozygous mRad17+/5′Δ mice
| Stage (E) | Genotype | Total | ||
|---|---|---|---|---|
| +/+ | +/5′Δ | 5′Δ/5′Δ | ||
| 3.5 | 4 | 12 | 5 | 21 |
| 6.5 | 4 | 4 | 4 | 12 |
| 7.5 | 1 | 8 | 6 | 15 |
| 8.5 | 7 | 41 | 18 (4A)a | 66 |
| 9.5 | 6 | 19 | 9 (3A) | 34 |
| 10.5 | 14 (1A) | 24 (3A) | 17 (6A) | 55+2NDb |
| 11.5 | 7 | 6 (1A) | 8 (5A) | 21 |
| 12.5 | 2 | 5 | 1 (1A) | 8 |
| 13.5 | 4 | 9 | 0 | 13+7ND |
| 15.5 | 3 | 4 | 0 | 7+2ND |
| Total early/mid-embryos | 52 (1.9%)c | 132 (3%) | 68 (23.5%) | 252 |
| (expected) | (63)d | (126) | (63) | |
| Offspring (F2) | 56 | 86 | 0 | 142 |
| (expected) | (47) | (94) | (47) | |
| For each developmental stage, defined by embryonic day postconception (E), the number of embryos/offspring of the indicated genotype is listed. | ||||
| aAbnormal embryo, defined by small size, morphological defects, resorptions or developmental delay. The number in parentheses is included in the total number of the specific genotype and in the total number of embryos analyzed. | ||||
| bND indicates not determined, due to the high level of resorption. | ||||
| cThe percentage of abnormal embryos. | ||||
| dThe expected number of embryos based on Mendelian inheritance. |
Transcription of the targeted mRad17 allele was examined using RT–PCR on RNA isolated from E8.5 embryos, using primers F285 in exon 2 and R548 in exon 3 (Figure 1A). The RT–PCR experiment revealed the presence of three differently sized mRad17 mRNAs, designated ‘wt', ‘Δ' and ‘N', respectively, in wild-type, mRad17 _+/5_′Δ or mRad17 5_′Δ/5_′Δ embryos (Figure 1D). Sequence analysis of the three bands showed that the ‘wt' band, present in the +/+ and +/5′Δ lanes, was derived from the wild-type mRad17 allele, whereas the smaller ‘Δ' band, present in lower quantities in the +/5′Δ and 5′Δ/5′Δ lanes, was derived from the targeted mRad17 allele (data not shown). Sequencing confirmed the reading frameshift and the presence of three immediate stop codons in the ‘Δ' RT–PCR product. The third, weak ‘N' band in the +/5′Δ and 5′Δ/5′Δ lanes contained sequences of the disrupted mRad17 allele and part of the inverted Neo gene with stop codons in all reading frames (data not shown). When mRad17 protein expression was investigated on immunoblot, no mRad17 could be detected in lysates of mRad17 5_′Δ/5_′Δ embryos (Figure 1E), supporting our prediction that the targeted disruption would result in an mRad17 null mutant allele.
To characterize the timing and nature of the embryonic lethality caused by the absence of mRad17, we analyzed early/mid-gestation embryos from heterozygous mRad17 _+/5_′Δ matings. Up to E8.5, no obvious differences were detected between wild-type, mRad17 _+/5_′Δ and mRad17 5_′Δ/5_′Δ embryos (Table I; Figure 2A and B; data not shown). At E8.5, the majority of the _mRad17_-deficient embryos appeared normal. Only a small fraction of the homozygous mRad17 5_′Δ/5_′Δ embryos showed growth retardation, morphological defects or had an inviable appearance (Table I; Figure 2C and D). At days E9.5 and E10.5, a larger proportion of the mRad17 5_′Δ/5_′Δ embryos showed morphological abnormalities, but still most embryos were essentially normal (Table I; Figure 2E and H). At E11.5, the majority showed numerous and heterogeneous morphological abnormalities. The most striking were growth retardation, reduction in the number of somites, bleedings in rhomencephalon, mesencephalon and prosencephalon, failure to close the head folds, delays and abnormalities in the development of the neural tube and pharyngeal arches, bleedings and morphological abnormalities of heart and liver (Table I; Figure 2I and J). Viable _mRad17_-deficient embryos were not detected at later stages of gestation (Table I). In conclusion, deletion of mRad17 results in a wide range and extensive heterogeneity of defects over a relative long period of embryonic development.
Figure 2.
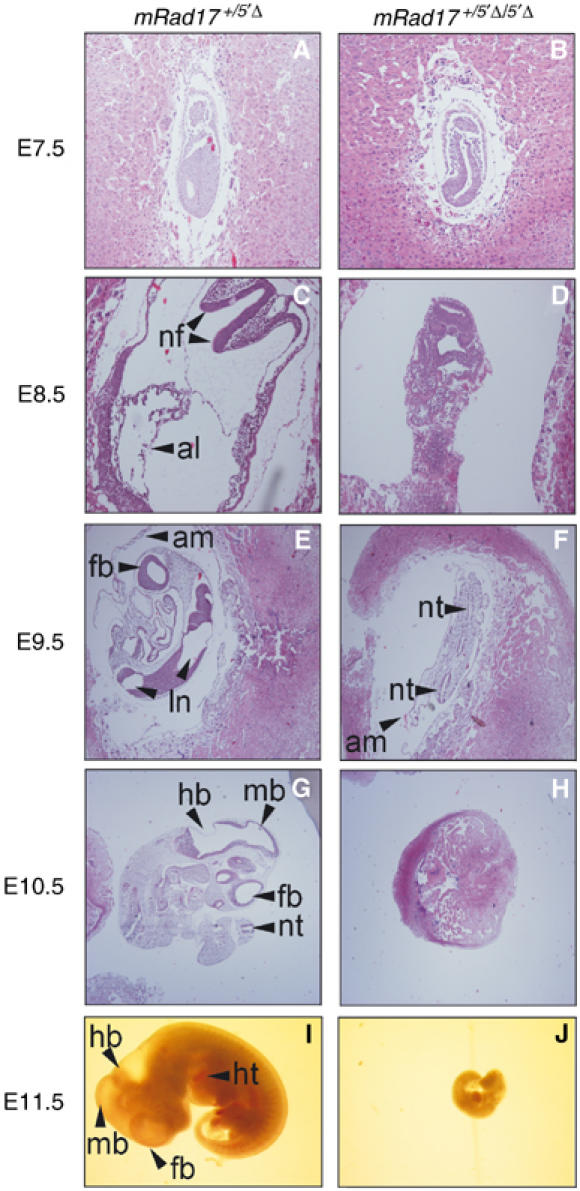
Development of mRad17 _+/5_′Δ and mRad17 5_′Δ/5_′Δ embryos. Examples of hematoxylin–eosin-stained sections (A–H) and whole embryos (I–J). Original magnifications are × 100 for (A–D) and × 40 for (E–J). Genotypes of embryos are given at the top. Developmental stage, in embryonic days (E), is indicated on the left. al: allantois; am: amnion; nf: neural fold (head fold); nt: neural tube; ln: lumen neural tube; ht: hart; fb: forebrain; mb: midbrain; hb: hindbrain.
Generation of mRad17-deficient mouse cell lines
The results described above suggest that mRad17 5_′Δ/5_′Δ embryos are viable up to E8.5 of gestation. Metaphase spreads from such E8.5 embryos did not show obvious genomic instability and no endoreplication (Table II). This is in contrast to Rad17 flox/− human colon epithelial cells, which exhibited acute chromosomal aberrations and underwent endoreplication at high rate upon Rad17 deletion (Wang et al, 2003). Therefore, we attempted to derive mouse embryonic fibroblasts (MEFs) from E8.5 embryos. Out of 40 embryos isolated from mRad17 _+/5_′Δ heterozygous matings, three mRad17 5_′Δ/5_′Δ primary MEF cultures were obtained. However, in contrast to wild-type and mRad17 _+/5_′Δ MEFs, which did not show culture abnormalities, all homozygous mRad17 5_′Δ/5_′Δ MEFs grew poorly and became senescent around passage 5, from which they failed to recover, even when grown under low (3%) oxygen conditions (data not shown).
Table 2.
Karyotype of cells from embryos
| Genotype | No. of metaphases counted | Loss of 1–2 chrom. | 40 chrom. | Gain of 1–2 chrom. | Tetraploid | NDa |
|---|---|---|---|---|---|---|
| Wild type | 15 | 5 | 9 | 0 | 0 | 1 |
| 15 | 2 | 11 | 0 | 0 | 2 | |
| 15 | 2 | 9 | 0 | 2 | 2 | |
| 15 | 2 | 13 | 0 | 0 | 0 | |
| 5′Δ/+ | 16 | 2 | 13 | 0 | 0 | 1 |
| 15 | 0 | 0 | 0 | 14 | 1 | |
| 15 | 3 | 10 | 0 | 0 | 2 | |
| 15 | 0 | 11 | 0 | 0 | 4 | |
| 15 | 2 | 11 | 0 | 0 | 2 | |
| 15 | 3 | 8 | 0 | 1 | 3 | |
| 15 | 1 | 13 | 0 | 0 | 1 | |
| 15 | 0 | 10 | 0 | 4 | 1 | |
| 5′Δ/5′Δ | 16 | 2 | 7 | 0 | 0 | 7 |
| 14 | 1 | 10 | 1 | 1 | 1 | |
| aND indicates nondeterminable. |
Next we tried to generate ES cells from E3.5 blastocysts of mRad17 _+/5_′Δ heterozygous matings. In contrast to the MEFs, we succeeded in establishing multiple ES cell lines. An mRad17 _+/5_′Δ, three mRad17 5_′Δ/5_′Δ together with a C57bl/6 wild-type control ES cell line were used for further experiments (Figure 3A). When mRad17 protein expression was investigated on immunoblots using an mRad17 antibody, unexpectedly, a smaller protein band could be detected in mRad17 _+/5_′Δ and mRad17 5_′Δ/5_′Δ ES whole-cell extracts (WCE; Figure 3B). This is in contrast to mRad17 5_′Δ/5_′Δ MEFs, in which no mRad17 could be detected (data not shown). Usage of a second, independent polyclonal mRad17 antibody confirmed these results (data not shown). Skipping of the natural start codon and usage of a second ATG in exon 3, which is out of frame in the natural mRad17 sequence, could lead to production of truncated mRad17 protein because the 129 bp deletion and the addition of 19 bp cloning sequence results in a frameshift. Due to this frameshift, usage of the second ATG results in the loss of 78 N-terminal amino acids of the regular mRad17 frame and the addition of 16 N-terminal nonsense amino acids (Figure 3C). The wild-type and 5′Δ-mRad17 were expressed to a similar level, since no difference in the amount of the proteins was detected in WCE (Figure 3B). However, immunoblotting of subcellular fractions revealed that, compared to wild-type mRad17, less of the 5′Δ-mRad17 protein was present in the nucleus. This is evident from a reduction in the one-to-one ratio of the 5′Δ-mRad17 to wild-type mRad17 in the WCE compared to the nuclear fraction. This is observed when comparing extracts from wild-type cells with extracts from mRad17 5_′Δ/5_′Δ cells and when directly comparing the amounts of wild-type and 5′Δ-mRad17 protein within extracts from mRad17 _+/5_′Δ cells. Importantly, the truncation of the mRad17 protein resulted in less efficient association of the protein with chromatin (Figure 3B). Using standard cell culture conditions, both mRad17 5_′Δ/5_′Δ and mRad17 _+/5_′Δ ES cell lines showed growth characteristics comparable to wild-type ES cells and no genomic instability was detected between early and late passages (Table III).
Figure 3.
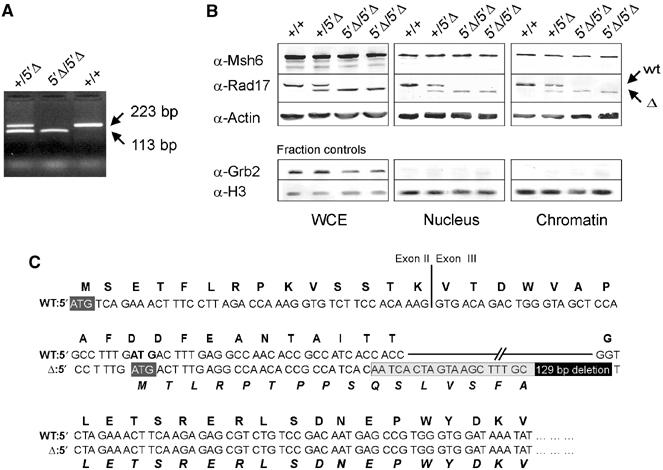
Generation of mRad17 mutant ES cell lines. (A, B) Genotypes are indicated above the lanes. (A) PCR analysis of genotypes using primers F325 and R548 (see Figure 1A). The positions of wild-type (223 bp) and targeted (113 bp) mRad17 bands are indicated. (B) Immunoblot analysis on ES cell lysates and cell fractions. The positions of wild-type mRad17 (+) and targeted mRad17 (Δ) are indicated. Actin and Msh6 were used as loading controls. Histone 3 (H3) and Grb2 were used as subcellular fractionation controls. (C) Sequence analysis of ‘wt' and ‘Δ' RT–PCR fragments. Bold capitals represent mRad17 amino acids, capitals represent nucleotides and bold italic capitals represent mutant mRad17 amino acids. II and III indicate exons 2 and 3, respectively. Dark gray boxes indicate translation start sites. In the ‘Δ' sequence, the positions of the deletion (black box) and additional sequence due to cloning (light gray box) are indicated.
Table 3.
Karyotype of ES cells
| Genotype | Passage no. | No. of metaphases counted | 39 chrom. | 40 chrom. | 41 chrom. | |||
|---|---|---|---|---|---|---|---|---|
| — | Markera | — | Markera | — | Markera | |||
| Wild type | 30 | 15 | 1 | 1 | 13 | 0 | 0 | 0 |
| 53 | 15 | 0 | 1 | 14 | 0 | 0 | 0 | |
| 5′Δ/5′Δ | 17 | 15 | 0 | 0 | 0 | 13 | 2 | 0 |
| 43 | 15 | 0 | 0 | 0 | 14 | 0 | 1 | |
| aMetacentric chromosome. |
mRad175′Δ/5′Δ ES cells are sensitive to _γ_-radiation, UV light, mitomycin C, illudin-S, methyl methane sulfonate and hydroxyurea
S. pombe Rad17 and S. cerevisiae RAD24 mutants are characterized by hypersensitivity to a wide variety of DNA-damaging agents. We therefore investigated the sensitivity of mRad17 5_′Δ/5_′Δ, mRad17 _+/5_′Δ and wild-type ES cells after exposure to γ-rays by determining their colony forming ability. Upon γ-irradiation, mRad17 5_′Δ/5_′Δ ES cell lines were found to be two- to three-fold more sensitive than wild-type ES cells. As a positive control for irradiation, γ-irradiation-sensitive ES cells deficient for the DNA repair protein Rad54 were used (Figure 4A; Essers et al, 1997).
Figure 4.
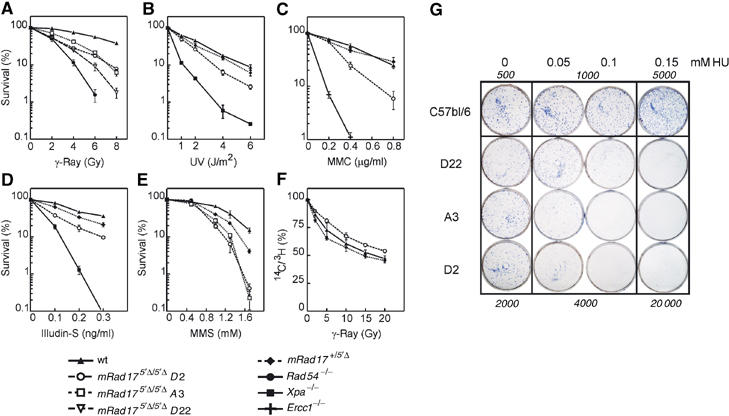
Effect of DNA-damaging agents on survival of mRad17 5_′Δ/5_′Δ, mRad17 _+/5_′Δ and wild-type ES cells. (A) Colony survival assay on ES cells after γ-irradiation, (B) UV irradiation, (C) MMC exposure, (D) illudin-S exposure and (E) MMS exposure. (F) RDS analysis on ES cells. (G) Colony survival assay after HU exposure. The numbers of seeded cells are indicated in italics.
To investigate whether mRad17 5_′Δ/5_′Δ causes a general sensitivity to genotoxic stress, we tested cell survival after treatment with other DNA-damaging agents, producing different types of DNA lesions that are repaired by different pathways. mRad17 5_′Δ/5_′Δ ES cells appeared ∼1.5-fold more sensitive to UV light (Figure 4B). However, they are not as UV light sensitive as Xpa −/− ES cells, which are deficient in nucleotide excision repair. In addition, mRad17 5_′Δ/5_′Δ ES cells displayed ∼2-fold hypersensitivity to the DNA interstrand crosslinks-inducing agent mitomycin C (MMC), which is not as high as crosslink repair-defective Ercc1 −/− ES cells (Figure 4C; Niedernhofer et al, 2001). Moreover, mRad17 5_′Δ/5_′Δ ES cells were ∼2.5-fold more sensitive than wild-type ES cells but less sensitive than Xpa −/− ES cells to illudin-S, which induces lesions presumed to be removed by transcription-coupled repair and which are also detected by the DNA replication machinery (Figure 4D; Jaspers et al, 2002). Methyl methane sulfonate (MMS) exposure, inducing DNA lesions repaired by recombination and DNA translesion synthesis, revealed an ∼1.5- to 2-fold increased sensitivity of mRad17 5_′Δ/5_′Δ ES cells compared to wild-type ES cells, whereas mRad17 _+/5_′Δ ES cells showed an intermediate sensitivity (Figure 4E). Exposure to the replication inhibitor HU showed a very high sensitivity of the mRad17 5_′Δ/5_′Δ ES cells compared to wild-type ES cells (Figure 4G). The observation that mRad17 5_′Δ/5_′Δ ES cells are sensitive to agents causing a variety of different DNA lesions or replication stalling suggests that mRad17 is involved in a DNA damage processing or repair step that is shared between a number of distinct DNA repair or processing pathways, or that it harbors a defect in one or more CCCs.
Effect of mRad175′Δ/5′Δ mutation on DNA damage-induced cell cycle arrest
We subsequently investigated whether γ-ray sensitivity of the mRad17 5_′Δ/5_′Δ ES cells was due to a defect in the intra-S checkpoint. Surprisingly, we found that mRad17 5_′Δ/5_′Δ ES cells did not display radiation-resistant DNA synthesis (RDS; Figure 4F). Because RDS is a hallmark of cells known to be defective in the induction of intra-S checkpoints, the results suggest that, in ES cells, deletion of the 5′ end of mRad17 has no detectable effect on this checkpoint or, alternatively, that mRad17 is not essential for it.
The absence of an overt RDS in mRad17 5_′Δ/5_′Δ ES cells prompted us to analyze other CCCs. Asynchronously growing ES cultures were exposed to various DNA-damaging agents and at different time points DNA content was ascertained by propidium iodide staining and analyzed by flow cytometry. The responses to γ-irradiation and to MMC are presented in Figure 5. Wild-type ES cells responded initially (6 h) by accumulation in S phase and G2 leading to a depletion of cells in the G1 pool, followed by a strong increase in cells in the G2 fraction (at 12 h) and in the case of γ-rays normalization of the G1 pool at 24 h. At the dose of MMC used, cells accumulated in G2; however, at a lower dose (0.2 μg/ml), a similar recovery of the G1 fraction was noted as observed for the γ-ray response (data not shown). Similar findings were obtained with different doses of both DNA-damaging agents. All three independently isolated mRad17 5_′Δ/5_′Δ ES cell lines behaved identical in these experiments (data not shown). Importantly, mRad17 _+/5_′Δ as well as mRad17 5_′Δ/5_′Δ ES cells behaved indistinguishable from the wild-type ES cells. Even in experiments using a phosphospecific histone 3 antibody, as described by Xu et al (2002), no differences in damage-induced CCC could be detected between wild-type, mRad17 _+/5_′Δ and mRad17 5_′Δ/5_′Δ ES cells (data not shown). We also investigated the effect of 5′Δ-mRad17 on the apoptotic response upon DNA damage, but did not find any differences compared to wild-type ES cells (data not shown). We conclude that, at least in ES cells, deletion of the 5′ end of mRad17 has no detectable effect on DNA damage-induced CCC nor on apoptosis.
Figure 5.
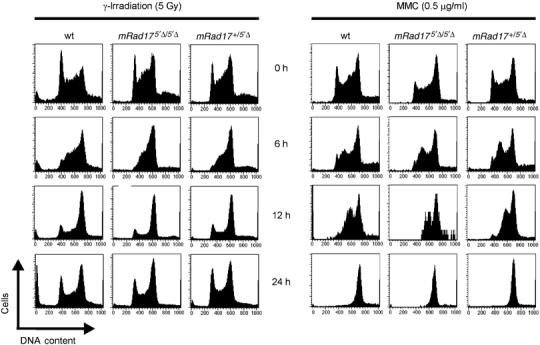
Fluorescent activated cell sorting (FACS) analysis of the effect of DNA damage on cell cycle progression of ES cells. Shown on the _x_-axis is the DNA content of the cells (stained with propidium iodide) and on the _y_-axis the number of cells. The left panel shows the effect after γ-irradiation, while the right panel displays the effect after MMC exposure. Genotypes of ES cells are shown on top. Time (h) is shown between the two panels.
Effect of mRad175′Δ/5′Δ on homologous recombination
Since lack of wild-type mRad17 expression does not lead to impairment of intra-S and G2/M cell cycle arrests, nor increased apoptosis upon exposure to DNA-damaging agents, we investigated whether there are differences in genomic stability after acquiring DNA damage between wild-type, mRad17 _+/5_′Δ and mRad17 5_′Δ/5_′Δ ES cells. Therefore, we analyzed the ability of the cells to mediate sister chromatid exchanges (SCEs). No difference was observed in the level of spontaneous and MMC-induced SCE between wild-type, mRad17 _+/5_′Δ and mRad17 5_′Δ/5_′Δ ES cells (data not shown). These results indicate that mRad17 5_′Δ/5_′Δ ES cells have a normal capacity to perform SCE as observed cytologically. However, compared to wild-type and mRad17 _+/5_′Δ ES cells, only a low percentage of mRad17 5_′Δ/5_′Δ ES cells were in second division after MMC treatment (data not shown). This indicates that these cells have a reduced capacity to repair or otherwise properly respond to interstrand DNA crosslinks.
The SCE analyses presented above suggest that homologous recombination between sister chromatids is unaffected in mRad17 5_′Δ/5_′Δ ES cells. A similar observation has been made for mutants in the structure-specific endonuclease Ercc1/Xpf (Niedernhofer et al, 2001). For Ercc1 −/− ES cells, it has been demonstrated that although homologous recombination between identical DNAs, as measured by SCE, is unaffected, the cells are defective in homologous gene replacement. The difference between homologous recombination between sister chromatids and homologous gene replacement is that the former recombination occurs between identical sequences while in the latter the homology is interrupted by nonidentical sequences, for example, the selectable marker. We investigated the possibility that mRad17 is also involved in the homologous recombination subpathway that mediates homologous gene replacement. The capacity of wild-type and mutant cells to conduct gene targeting was assessed using a green fluorescent protein (GFP)-based gene targeting assay. ES cells were electroporated with a construct designed to introduce a GFP tag in the mouse Rad54 locus. This targeting construct is referred to as 11.1hRad54GFP knock-in construct (Figure 6A; Abraham et al, 2003). Targeted integration of the construct within the Rad54 locus results in expression of GFP-tagged mRad54 from the endogenous mRad54 promoter. Homologous integration can be scored in individual cells as green fluorescence, using FACS analysis, as established by DNA blot analysis of genomic DNA of individually sorted and expanded green fluorescent cells (data not shown). Random integration of the Rad54-GFP construct resulted in nonfluorescent cells.
Figure 6.
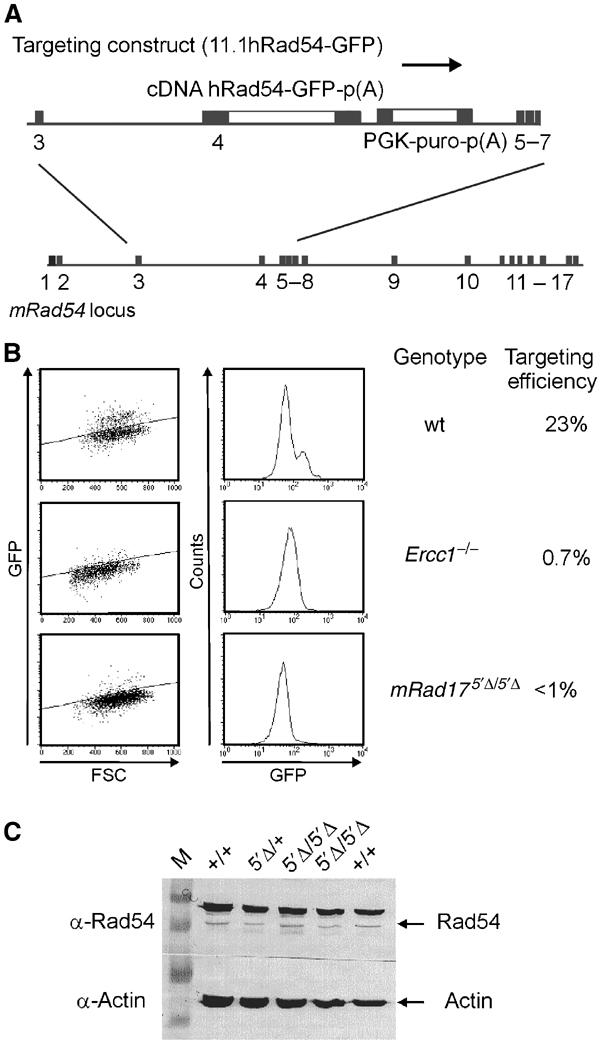
Targeted gene replacement is severely reduced in mRad17 5_′Δ/5_′Δ cells. (A) Schematic overview of the Rad54 targeting strategy. The Rad54 targeting construct (11.1hRad54-GFP) and the mRad54 gene locus are shown. Targeted gene replacement by the Rad54-GFP construct results in GFP expression, while random integration results in nonfluorescent cells. (B) (left) Cells of the indicated genotype were electroporated with the Rad54-GFP knock-in construct and 1 × 105 cells were analyzed by flow cytometry. Shown are green fluorescence (GFP) versus forward scatter (FSC) plots. Fluorescent cells appear above and nonfluorescent cells below the diagonal line (left panels). To determine the percentage of gene targeting efficiency, results were also plotted in a fluorescence (GFP) histogram (right panels). In wild-type ES cells, two peaks are visible, representing non-GFP- and GFP-expressing cells. Ercc1 −/− and mRad17 5_′Δ/5_′Δ cells show only one peak representing nonfluorescent cells. (C) Immunoblot using anti-Rad54, showing that mRad54 expression is not affected by the mRad17 genotype (indicated on top). Actin was used as a loading control. The upper band in the Rad54 blot is a nonspecific band (Essers et al, 1997).
Using this assay, transfection of the Rad54-GFP construct into the wild-type ES cells resulted in targeting efficiencies of ∼23% (Figure 6B). Similar targeting efficiencies were obtained with ES cells from both 129Sv and C57bl/6 genetic background, indicating that the genetic background does not influence the efficiency of targeted gene replacement at the mRad54 locus (data not shown). The targeting efficiency is similar to that determined by DNA blotting analysis (Niedernhofer et al, 2001). As a negative control, we used Ercc1 −/− ES cells that are completely deficient in targeted gene replacement (Niedernhofer et al, 2001). Transfection of the Rad54-GFP construct to Ercc1 −/− ES cells resulted in 0.7% green fluorescent cells (Figure 6B), which is similar to the background level in this assay. We then examined the capability of mRad17 5_′Δ/5_′Δ ES for targeted gene replacement. In experiments using the three independent mRad17 5_′Δ/5_′Δ ES cells with the Rad54-GFP knock-in construct, the percentage of green fluorescent cells never exceeded background levels (Figure 6B). Rad54 expression is not affected by the Rad17 mutation, as shown in the immunoblot (Figure 6C). Together, these data show that mRad17 plays an important role in recombination.
Discussion
S. pombe Rad17 is one of the components of the cellular machinery that responds to DNA damage and stalled DNA replication. In this study, we examined the role of the mouse homolog of Rad17 by analyzing the consequences of a targeted deletion of mRad17. Our findings reveal an important role for mRad17 in cell growth and/or differentiation at an early stage of embryonic development, as mRad17-deficient embryos die during early/mid-gestation. In contrast, S. pombe Rad17 and S. cerevisiae RAD24 mutants are viable under normal growth conditions (al-Khodairy and Carr, 1992). This may indicate that mRad17 has acquired functions not mediated by S. pombe Rad17 or S. cerevisiae RAD24. However, in view of the strong sequence conservation of the Rad17 protein between yeast and mammals and the notion that it is part of a larger complex, this option may not seem very likely. A more plausible explanation is that fundamental physiological differences between yeast and mammalian cells increase the requirement for mRad17. During early embryogenesis, cells are constantly under mitotic stress as divisions in some regions of the embryo can occur as rapidly as every 2–3 h. Under these circumstances, correct ordering of cell cycle events and fast repair of DNA damage or resolution of replication stalling are crucial. Failures, caused by mitotic stress, may lead to random drop out of cells in early embryogenesis. This may lead to heterogeneous effects later in development and explains the observed heterogeneity in time and cause of embryonic lethality. In this respect, it is not surprising that targeted disruption of other mouse cell cycle genes, including Atr (Brown and Baltimore, 2000; de Klein et al, 2000), Hus1 (Weiss et al, 2000) and mRad1 (A Maas, unpublished results) also results in heterogeneity in early embryonic lethality. Conditional gene targeting therefore should give more insight into the role of mRad17 in adult tissue.
mRad17 5_′Δ/5_′Δ MEFs isolated from E8.5 embryos failed to grow beyond passage 5. This is consistent with RAD17 flox/− human HCT116 colon epithelial cells, of which >90% were unable to form colonies upon Ad-Cre infection and subsequent loss of Rad17 (Wang et al, 2003). However, in cells freshly isolated from mRad17 5_′Δ/5_′Δ embryos, we failed to observe detectable levels of chromosomal instability and endoreplication, as present in RAD17 flox/− epithelial cells (Wang et al, 2003). In the established Ad-Cre-infected RAD17 flox/− epithelial cells, inefficient apoptotic responses or DNA repair may result in chromosomal aberrations and endoreplication. Possibly, a more efficient apoptotic response in primary mRad17 5_′Δ/5_′Δ cells prevents the detection of chromosomal instability and endoreplication. Alternatively, the difference in the site of mutation in the mRad17 protein may be responsible for this phenotypic difference.
In contrast to more differentiated cells, as shown here, and to Rad17 flox/− human epithelial cells (Wang et al, 2003), mRad17 5_′Δ/5_′Δ ES cells appeared viable. Unexpectedly and in contrast to other more differentiated cell types, a shorter truncated mRad17 protein is produced in mRad17 5_′Δ/5_′Δ ES cells. The molecular mechanism for the observed discrepancy of truncated mRad17 expression between ES cells and somatic cells is unclear at this point, but might involve differences in translation initiation and/or protein stability between these cell types.
Although a mutant form of mRad17 is expressed in these ES cells, they show hypersensitivity to different types of DNA damage induced by γ-irradiation, UV light, MMC, MMS and illudin-S and replication stalling induced by HU treatment. Thus, like Rad17 in S. pombe, mouse Rad17 is involved in cellular responses to genotoxic stress. However, in striking contrast to S. pombe Rad17 and S. cerevisiae RAD24 mutants, cell cycle arrest during S phase and the G2/M transition upon induction of DNA damage is not affected in mRad17 5_′Δ/5_′Δ ES cells, expressing the shorter mRad17 mutant form (Weinert et al, 1994; Lydall and Weinert, 1997). In addition, our results with mRad17 also differ from those obtained with other mammalian cell lines defective in CCC genes. For example, ATM and members of the DNA repair complex Rad50/Mre11/Nbs1 influence cell cycle progression. Cell lines with mutations in these components display RDS (de Jager and Kanaar, 2002; Falck et al, 2002). DNA synthesis in mRad17 5_′Δ/5_′Δ ES cells is not significantly different after γ-irradiation treatment compared to wild-type and mRad17 _+/5_′Δ ES cells. The broad-spectrum DNA damage sensitivity combined with apparently normal DNA damage signaling and CCC exhibited by the mRad17 5_′Δ/5_′Δ ES cells indicates a defect in DNA damage processing, likely in relation to replication of a damaged DNA template.
Our results in mouse ES cells lead us to consider that mRad17 may be involved in both CCC and DNA repair processes. This is not unprecedented because a similar dual function is found for the Rad50/Mre11/Nbs1 complex, which also has both DNA repair and CCC functions (Bender et al, 2002; Falck et al, 2002). It is possible that ES cells require only the contribution of mRad17 to DNA repair, while in more differentiated cells both functions of mRad17 are required, making it essential for cellular viability. This might also explain the fact that, in contrast to mRad17 5_′Δ/5_′Δ ES cells, mRad17 5_′Δ/5_′Δ MEFs fail to grow and become senescent. In contrast to MEFs, ES cells do not arrest at the G1/S transition (Aladjem et al, 1998; Schmidt-Kastner et al, 1998). DNA damage can trigger G1/S cell cycle arrest in mRad17 5_′Δ/5_′Δ MEFs, which could lead to irreversible withdrawal from cell cycling. However, this cannot be the sole explanation as the ES cell CCCs for intra-S and G2 do not appear to be abnormal, except when the initial DNA damage signaling in ES cells is principally different for all CCCs when compared to other cell types. In this respect, it is interesting that deletion of p21 can rescue poor growth of other cell cycle protein-deficient MEFs, including ATM_−/−_ (Wang et al, 1997; Xu et al, 1998) and Hus1_−/−_ cells (Weiss et al, 2000). However, a more plausible explanation is that the reduced amount of 5′Δ-mRad17 compared to wild-type mRad17, present in the nucleus and bound to chromatin, may be sufficient for mRad17 5_′Δ/5_′Δ ES cell viability but not for DNA repair functions.
mRad17 5_′Δ/5_′Δ ES cells are severely impaired in homologous gene targeting and show hypersensitivity to a variety of DNA-damaging agents. One conclusion from these findings is that the hypersensitive phenotype apparent in these mRad17 5_′Δ/5_′Δ ES cells is not due to a compromised CCC function but is primarily derived from the defect in homologous recombination, most likely dependent on DNA damage. This opens the possibility that the hypersensitivity of other CCC genes is also caused by inefficient handling of DNA lesions. A common intermediate in homologous recombination and DNA damage repair is a single-stranded DNA gap. It is possible that mRad17 has a role in restoration of these single-stranded DNA gaps to duplex DNA, which requires the action of a DNA polymerase (Figure 7; Venclovas and Thelen, 2000; Ellison and Stillman, 2003). A docking site for DNA polymerase δ and ɛ on the chromatin is provided by PCNA, which is loaded by RFC1–5 (Hubscher et al, 2002). Similarly, mRad17 can bind to the four small subunits of RFC (mRad17/RFC2–5; Green et al, 2000; Ellison and Stillman, 2003). mRad17/RFC2–5 may have a higher affinity for nicked or gapped DNA, resulting in the loading of the 9-1-1 trimer (Rauen et al, 2000). Rad9, Rad1 and Hus1 may be involved in further processing or stabilization of the DNA repair intermediate structures and/or provide a platform for other DNA repair enzymes, including DNA lesion bypass polymerases (Ellison and Stillman, 2003). CCC may be mediated by the mammalian homologs for the S. pombe Rad3/Rad26 complex, ATR/ATRIP (Cortez et al, 2001), binding to the damaged DNA independently of Rad17.
Figure 7.
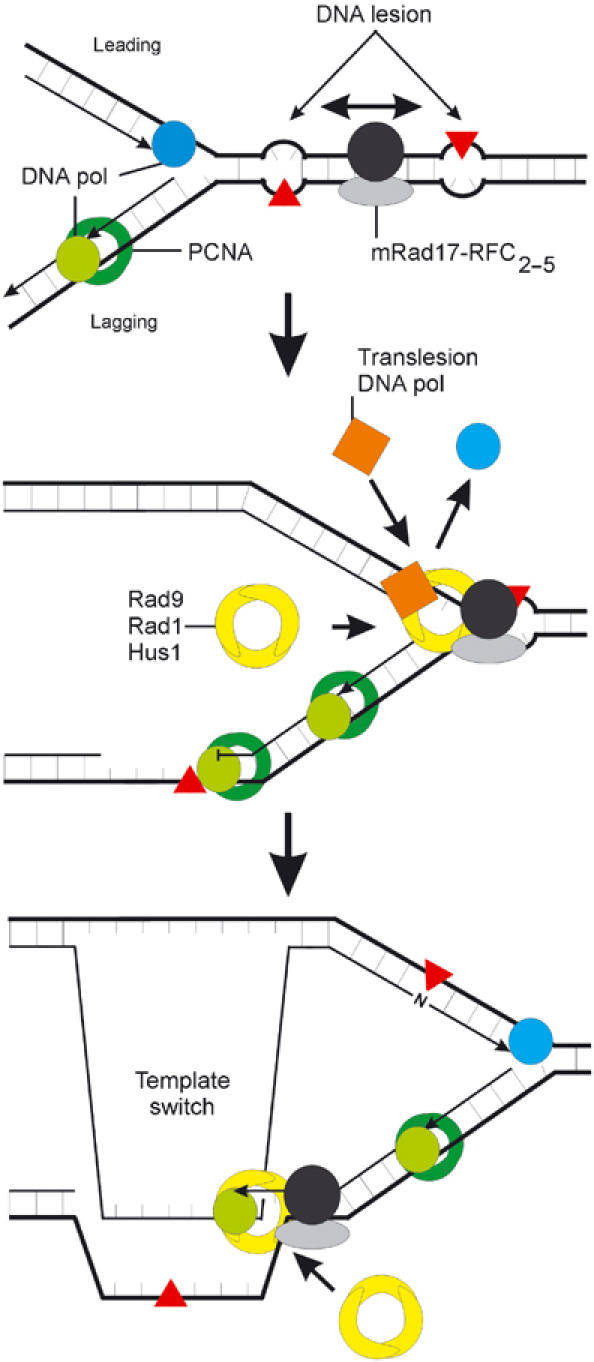
Schematic model of the role of mRad17 in DNA damage. In response to DNA damage, RFC1 from the RFC1–5 complex is replaced by mRad17 to form a Rad17–RFC2–5 complex. If this complex has a higher affinity for damaged or gapped DNA than RFC1–5, then this could result in the preferential loading of the Rad9–Rad1–Hus1 heterotrimer onto the site of damage compared to PCNA. Phosphorylation of mRad17, by ATR/ATRIP and stimulated by Hus1, may result in dissociation of Rad17–RFC2–5 from Rad9–Rad1–Hus1, which then forms a docking site for translesion bypass DNA polymerases or may result in a template switch. In this model, CCC is not necessarily mediated by mRad17 but by other protein complexes, possibly ATR/ATRIP, binding to the site of damage independently of mRad17.
Materials and methods
Construction of the mRad17 targeting vector
To construct an mRad17 targeting vector, an mRad17 genomic 4.8 kb _Xba_I fragment was used as the 3′ homologous fragment. This fragment, which contains 38 bp of the end of exon 3, was subcloned into the _Xba_I site of pGEM-9Zf (Promega), excised as an _Spe_I_–Sal_I fragment and cloned into _Spe_I_–Sal_I sites of pGem-5Zf (Promega). For the 5′ homologous fragment, a 5 kb PCR fragment, generated with primers F285 (5′-GTCAGAAACTTTCCTTAGACCAAAGGTGTC-3′) and R380 (5′-GTGATGGCGGTGTTGGC CTCAAAGTC-3′) and containing 55 bp of the beginning of exon 3, was cloned into pGem T easy (Promega). A pgk-Neo selection cassette was cloned in antisense orientation into a unique _Ava_I site in intron 2, approximately 200 bp 5′ of exon 3. The insert was then excised from the pGem T easy vector using a partial _Spe_I–_Sac_II digest and cloned into _Sac_II_–Spe_I sites of pGem-5Zf, which already contained the 3′ homologous fragment. In this way, a 110 bp deletion of exon 3 was generated. The targeting vector was linearized using a unique _Pme_I site that was generated 5′ of the insert.
ES cell culture, gene targeting and generation of mRad17 mutant mice
E14 ES cells (subclone IB10) were cultured in BRL-conditioned medium supplemented with 1000 U/ml leukemia inhibitory factor. A 20 μg portion of the _Pme_I-linearized targeting vector was electroporated into approximately 107 ES cells in 500 μl. Selection with 0.2 μg/ml G418 was started 24 h after electroporation. After 8–10 days, G418-resistant clones were isolated. Screening for homologous recombinants was performed using DNA blot analyses of _Pst_I-digested DNA with a 350 bp 5′ external probe. Correct integration of the 3′ end of the targeting construct was verified by PCR using a neomycin-specific primer (FNeo: 5′-AACCTGCGTGCAATCCAT-3′) and a primer outside the targeting vector in exon 5 (R348: 5′-GGTCGTTGTCTTTCCACAT CCAGG-3′). Out of 240 G418-resistant clones, three ES clones had a correctly targeted mRad17 allele. Two out of the three correctly targeted ES clones (designated F17 and F24) were injected into blastocysts of C57Bl/6 mice and transplanted into B10/CBA foster mothers. Chimeric offspring was mated to C57bl/6 females and resulted in mice heterozygous for the targeted mRad17 allele. These mice were further intercrossed, resulting in mice with a mixed 129Sv/C57bl/6 genetic background. Germline transmission and genotyping of offspring were carried out by PCR. Primers F325 (5′-GTGACAGACTGGGTAGCTCCAGCC-3′) and R548 (5′-GGCTCATTGTCGGACAGACGCT CTC-3′) encompassing the deletion in exon 3 were used.
Cell fractionation and immunoblotting
Cell fractionation was performed as described (Zou et al, 2002). For immunoblotting, α-Rad17 H-300 (Santa Cruz), α-histone H3 (Upstate), α-actin (Chemicon Int.), α-Grb2, α-Msh6 (Becton Dickinson) and α-hRad54 (Essers et al, 1997) were used.
Colony survival curves
Sensitivity of ES cells to increasing doses of DNA-damaging agents was determined as described previously (Essers et al, 1997). Briefly, cells were plated in 6 cm dishes, at various dilutions. After 12–16 h, cells were irradiated with a single dose in the range of 0–10 Gy using a 137Cs source or UV (0–8 J/m2) or treated for 1 h with MMC (0–0.8 μg/ml), illudin-S (0–0.3 ng/ml) or MMS (0–1.7 mM). Cells were grown for 4–6 days, fixed, stained and colonies were counted. For the HU colony survival assay, 500–20 000 ES cells were plated in a 6 cm dish 4 h prior to the addition of HU. After 5–7 days, cells were fixed and stained. All experiments were performed in triplicate.
Flow cytometric analysis
Confluent ES cells (60–70%) on 10 cm culture dishes were either irradiated with a single dose of γ-radiation (5 or 10 Gy) using a 137Cs source or treated for 1 h with MMC (0.2 or 0.5 μg/ml). At different time points after treatment, cells were collected and fixed with 70% ethanol. After a minimum of 2 h on ice, cells were washed with phosphate buffered saline (PBS) and resuspended in 400 μl PBS containing 0.1% Triton X-100, 0.1 mg/ml propidium iodide and 0.1 mg/ml RNAse. Cells were incubated overnight and analyzed on a Facscan (Becton Dickinson).
Transfections and recombination assays
To quantitate the efficiency of homologous gene targeting, ES cells were transfected with a Rad54-GFP knock-in construct containing a puromycin selectable marker (Abraham et al, 2003). At 1 week after puromycin selection, plates were trypsinized, resuspended to single-cell suspensions in PBS and fixed with 1% paraformaldehyde. After permeabilization with 0.1% Triton X-100 and RNAse treatment, cells were analyzed in a Becton Dickinson FacsCalibur on a green fluorescence (FL1) versus forward scatter (FSC-H) plot (Figure 6). GFP-positive and -negative cells appeared in separate populations, above and below the diagonal line, respectively (Figure 6). Results were also plotted in a fluorescence (GFP) histogram (Figure 6). The expression level of mRad54 was analyzed using α-hRad54 antibodies (Essers et al, 1997).
Acknowledgments
This work was supported by grants from the Dutch Cancer Society (EMCR 2002-2073), HFSP (RG 0245/1999M), AICR (99-111), EU (RTN2-2001-00276) and the Netherlands Organization for Scientific Research (900-99-003). We thank Y Verhoeven, DC van Gent, P Molenbeek and D Zondervan for assistance.
References
- Abraham J, Lemmers B, Hande MP, Moynahan ME, Chahwan C, Ciccia A, Essers J, Hanada K, Chahwan R, Khaw AK, McPherson P, Shehabeldin A, Laister R, Arrowsmith C, Kanaar R, West SC, Jasin M, Hakem R (2003) Eme1 is involved in DNA damage processing and maintenance of genomic stability in mammalian cells. EMBO J 22: 6137–6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aladjem MI, Spike BT, Rodewald LW, Hope TJ, Klemm M, Jaenisch R, Wahl GM (1998) ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr Biol 8: 145–155 [DOI] [PubMed] [Google Scholar]
- al-Khodairy F, Carr AM (1992) DNA repair mutants defining G2 checkpoint pathways in Schizosaccharomyces pombe. EMBO J 11: 1343–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender CF, Sikes ML, Sullivan R, Huye LE, Le Beau MM, Roth DB, Mirzoeva OK, Oltz EM, Petrini JH (2002) Cancer predisposition and hematopoietic failure in Rad50(S/S) mice. Genes Dev 16: 2237–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluyssen HA, Naus NC, van Os RI, Jaspers I, Hoeijmakers JH, de Klein A (1999) Human and mouse homologs of the Schizosaccharomyces pombe rad17+ cell cycle checkpoint control gene. Genomics 55: 219–228 [DOI] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D (2000) ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 14: 397–402 [PMC free article] [PubMed] [Google Scholar]
- Burtelow MA, Kaufmann SH, Karnitz LM (2000) Retention of the human Rad9 checkpoint complex in extraction-resistant nuclear complexes after DNA damage. J Biol Chem 275: 26343–26348 [DOI] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ (2001) ATR and ATRIP: partners in checkpoint signaling. Science 294: 1713–1716 [DOI] [PubMed] [Google Scholar]
- de Jager M, Kanaar R (2002) Genome instability and Rad50(S): subtle yet severe. Genes Dev 16: 2173–2178 [DOI] [PubMed] [Google Scholar]
- de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH (2000) Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 10: 479–482 [DOI] [PubMed] [Google Scholar]
- Edwards RJ, Bentley NJ, Carr AM (1999) A Rad3–Rad26 complex responds to DNA damage independently of other checkpoint proteins. Nat Cell Biol 1: 393–398 [DOI] [PubMed] [Google Scholar]
- Ellison V, Stillman B (2003) Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol 1: E33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers J, Hendriks RW, Swagemakers SM, Troelstra C, de Wit J, Bootsma D, Hoeijmakers JH, Kanaar R (1997) Disruption of mouse RAD54 reduces ionizing radiation resistance and homologous recombination. Cell 89: 195–204 [DOI] [PubMed] [Google Scholar]
- Falck J, Petrini JH, Williams BR, Lukas J, Bartek J (2002) The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet 30: 290–294 [DOI] [PubMed] [Google Scholar]
- Green CM, Erdjument-Bromage H, Tempst P, Lowndes NF (2000) A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr Biol 10: 39–42 [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA (1989) Checkpoints: controls that ensure the order of cell cycle events. Science 246: 629–634 [DOI] [PubMed] [Google Scholar]
- Hubscher U, Maga G, Spadari S (2002) Eukaryotic DNA polymerases. Annu Rev Biochem 71: 133–163 [DOI] [PubMed] [Google Scholar]
- Jaspers NG, Raams A, Kelner MJ, Ng JM, Yamashita YM, Takeda S, McMorris TC, Hoeijmakers JH (2002) Anti-tumour compounds illudin S and Irofulven induce DNA lesions ignored by global repair and exclusively processed by transcription- and replication-coupled repair pathways. DNA Repair (Amst) 1: 1027–1038 [DOI] [PubMed] [Google Scholar]
- Kostrub CF, Knudsen K, Subramani S, Enoch T (1998) Hus1p, a conserved fission yeast checkpoint protein, interacts with Rad1p and is phosphorylated in response to DNA damage. EMBO J 17: 2055–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Kwong AD, Pan ZQ, Hurwitz J (1991) Studies on the activator 1 protein complex, an accessory factor for proliferating cell nuclear antigen-dependent DNA polymerase delta. J Biol Chem 266: 594–602 [PubMed] [Google Scholar]
- Lydall D, Weinert T (1997) G2/M checkpoint genes of Saccharomyces cerevisiae: further evidence for roles in DNA replication and/or repair. Mol Gen Genet 256: 638–651 [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Essers J, Weeda G, Beverloo B, de Wit J, Muijtjens M, Odijk H, Hoeijmakers JH, Kanaar R (2001) The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. EMBO J 20: 6540–6549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulovich AG, Hartwell LH (1995) A checkpoint regulates the rate of progression through S phase in S. cerevisiae in response to DNA damage. Cell 82: 841–847 [DOI] [PubMed] [Google Scholar]
- Rauen M, Burtelow MA, Dufault VM, Karnitz LM (2000) The human checkpoint protein hRad17 interacts with the PCNA-like proteins hRad1, hHus1, and hRad9. J Biol Chem 275: 29767–29771 [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali S, Simmons A, Clines G, Sartiel A, Gatti R, Chessa L, Sanal O, Lavin M, Jaspers N, Taylor A, Arlett C, Miki T, Weissman S, Lovett M, Collins F, Shiloh Y (1995) A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268: 1749–1753 [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner PK, Jardine K, Cormier M, McBurney MW (1998) Absence of p53-dependent cell cycle regulation in pluripotent mouse cell lines. Oncogene 16: 3003–3011 [DOI] [PubMed] [Google Scholar]
- Siede W, Friedberg AS, Friedberg EC (1993) RAD9-dependent G1 arrest defines a second checkpoint for damaged DNA in the cell cycle of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 90: 7985–7989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venclovas C, Thelen MP (2000) Structure-based predictions of Rad1, Rad9, Hus1 and Rad17 participation in sliding clamp and clamp-loading complexes. Nucleic Acids Res 28: 2481–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Deimling F, Scharf JM, Liehr T, Rothe M, Kelter AR, Albers P, Dietrich WF, Kunkel LM, Wernert N, Wirth B (1999) Human and mouse RAD17 genes: identification, localization, genomic structure and histological expression pattern in normal testis and seminoma. Hum Genet 105: 17–27 [DOI] [PubMed] [Google Scholar]
- Wang X, Zou L, Zheng H, Wei Q, Elledge SJ, Li L (2003) Genomic instability and endoreduplication triggered by RAD17 deletion. Genes Dev 17: 965–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YA, Elson A, Leder P (1997) Loss of p21 increases sensitivity to ionizing radiation and delays the onset of lymphoma in atm-deficient mice. Proc Natl Acad Sci USA 94: 14590–14595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert TA, Kiser GL, Hartwell LH (1994) Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev 8: 652–665 [DOI] [PubMed] [Google Scholar]
- Weiss RS, Enoch T, Leder P (2000) Inactivation of mouse Hus1 results in genomic instability and impaired responses to genotoxic stress. Genes Dev 14: 1886–1898 [PMC free article] [PubMed] [Google Scholar]
- Xu B, Kim ST, Lim DS, Kastan MB (2002) Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell Biol 22: 1049–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Yang EM, Brugarolas J, Jacks T, Baltimore D (1998) Involvement of p53 and p21 in cellular defects and tumorigenesis in Atm−/− mice. Mol Cell Biol 18: 4385–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439 [DOI] [PubMed] [Google Scholar]
- Zou L, Cortez D, Elledge SJ (2002) Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev 16: 198–208 [DOI] [PMC free article] [PubMed] [Google Scholar]