Hedgehog Antagonist RENKCTD11 Regulates Proliferation and Apoptosis of Developing Granule Cell Progenitors (original) (raw)
Abstract
During the early development of the cerebellum, a burst of granule cell progenitor (GCP) proliferation occurs in the outer external granule layer (EGL), which is sustained mainly by Purkinje cell-derived Sonic Hedgehog (Shh). Shh response is interrupted once GCPs move into the inner EGL, where granule progenitors withdraw proliferation and start differentiating and migrating toward the internal granule layer (IGL). Failure to interrupt Shh signals results in uncoordinated proliferation and differentiation of GCPs and eventually leads to malignancy (i.e., medulloblastoma). The Shh inhibitory mechanisms that are responsible for GCP growth arrest and differentiation remain unclear. Here we report that REN, a putative tumor suppressor frequently deleted in human medulloblastoma, is expressed to a higher extent in nonproliferating inner EGL and IGL granule cells than in highly proliferating outer EGL cells. Accordingly, upregulated REN expression occurs along GCP differentiation in vitro, and, in turn, REN overexpression promotes growth arrest and increases the proportion of p27/Kip1+ GCPs. REN also impairs both Gli2-dependent gene transcription and Shh-enhanced expression of the target Gli1 mRNA, thus antagonizing the Shh-induced effects on the proliferation and differentiation of cultured GCPs. Conversely, REN functional knock-down impairs Hedgehog antagonism and differentiation and sustains the proliferation of GCPs. Finally, REN enhances caspase-3 activation and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling apoptotic GCP numbers; therefore, the pattern of REN expression, its activity, and its antagonism on the Hedgehog pathway suggest that this gene may represent a restraint of Shh signaling at the outer to inner EGL GCP transitions. Medulloblastoma-associated REN loss of function might withdraw such a limiting signal for immature cell expansion, thus favoring tumorigenesis.
Keywords: cerebellar granule progenitors, Hedgehog, Gli, tumor suppressor, apoptosis, medulloblastoma
Introduction
Medulloblastoma, the most common and aggressive brain malignancy in childhood, is believed to arise from cerebellar granule cell progenitors (GCPs) in which the differentiation program is altered by molecular events that lead to uncontrolled growth (Wechsler-Reya and Scott, 2001; Ruiz i Altaba et al., 2002). During development, GCPs migrate from the rhombic lip over the outer layer of the cerebellar surface [external granule layer (EGL)] (Hatten and Heintz, 1995; Wang and Zoghbi, 2001); after an initial burst of cell proliferation, they start a differentiation program by exiting the cell cycle and moving into the inner EGL. They migrate farther inward into the internal granule layer (IGL), where differentiated granule neurons reside (Hatten and Heintz, 1995; Wang and Zoghbi, 2001). Sonic Hedgehog (Shh) is the most potent mitogen of GCPs (Dahmane and Ruiz i Altaba, 1999; Wechsler-Reya and Scott, 1999; Wallace, 1999), whereas what causes GCPs to stop dividing in the inner EGL is not well understood. Importantly, failure to contain the proliferation of developing GCPs is involved in tumorigenesis, because constitutive activation of the Hedgehog pathway is responsible for medulloblastoma development (for review, see Wetmore, 2003; De Smaele et al., 2004).
We have shown recently that chromosome 17p deletion, the most frequent genetic lesion and poor prognosis marker in human medulloblastoma (Lamont et al., 2004), leads to the loss of RENKCTD11, a novel putative tumor suppressor that inhibits medulloblastoma growth by antagonizing Hedgehog signaling (Di Marcotullio et al., 2004). REN expression is enhanced by epidermal growth factor (EGF) (Gallo et al., 2002), which regulates cerebellar development, when EGF receptor is expressed in the inner EGL and is related to postmitotic events (migration and differentiation) (Carrasco et al., 2003). Retinoic acid and nerve growth factor (NGF), which promote neuronal differentiation and growth arrest, also upregulate REN (Gallo et al., 2002). Consistently, REN expression is initially detected in the neural fold epithelium during gastrulation and subsequently throughout the embryonal neural tube and the encephalic neuroepithelium (Gallo et al., 2002). A role for REN in neurogenesis is suggested by its ability to promote neuroblastoma and striatal cell differentiation and proneural neurogenin-1 and neurogenic differentiation (NeuroD) expression (Gallo et al., 2002).
The impairment of REN tumor suppression function, which occurs in human medulloblastoma (Di Marcotullio et al., 2004), raises the question of the role of this gene in the development of cerebellar GCPs. We show here that REN expression is restricted to low-proliferating inner EGL GCPs and granule neurons of the IGL during development. By overexpression and functional knock-down studies, we show that REN promotes growth arrest, differentiation, and apoptosis, and it antagonizes Hedgehog signaling in GCPs, suggesting that it is required for withdrawing GCP expansion at the outer to inner EGL transition. Loss of REN therefore may release a restraint of the Hedgehog pathway, favoring tumorigenesis.
Materials and Methods
Animals and materials. CD-1 mice were obtained from Charles River Laboratories (Wilmington, MA). Recombinant mouse Shh C-terminal peptide was from R & D Systems (Minneapolis, MN). All other chemicals were from Sigma (St. Louis, MO), unless indicated otherwise.
In situ hybridization. In situ hybridization on paraffin sections was performed as described previously (Sassoon and Rosenthal, 1993). Brains of postnatal day 8 (P8) mice were rapidly dissected after cervical dislocation and then immediately fixed by immersion into 4% paraformaldehyde. Brains of P21 mice were perfused with 4% paraformaldehyde. Dissected brains were postfixed by immersion into 4% paraformaldehyde overnight, dehydrated, and embedded in Paraplast Plus Tissue Embedding Medium (Società Italiana Chimici, Rome, Italy); next, serial 9-μm-thick sections were prepared. mRNA transcripts were detected with digoxigenin-labeled 780 bp riboprobes as described by Gallo et al. (2002). The probe that was used revealed reproducible hybridization patterns when in the antisense orientation and no signal when in the sense orientation.
Immunohistochemistry. For immunohistochemistry, 6-μm-thick sections were prepared from Formalin-fixed and paraffin-embedded tissues. Sections were subjected to microwaves for 15 min at a high energy setting in 0.1 m sodium citrate buffer, pH 6.0. After antigen retrieval, endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 10 min, and nonspecific binding was prevented by incubation with 5% normal swine serum (Denmark code X0901; Dako Cytomation, Glostrop, Denmark) in Large Volume UltrAb Diluent (Lab Vision, Fremont, CA) for 1 h at room temperature. Sections were subsequently incubated either with rabbit monoclonal anti-Ki67 (clone SP6; Lab Vision; 1:100 dilution in Large Volume UltrAb Diluent) for 2 h at room temperature or with 1 μg/ml affinity-purified rabbit polyclonal anti-REN (Gallo et al., 2002) overnight at 4°C. Controls included replacement of the primary antibody with normal serum. The slides were subsequently washed and incubated with swine anti-rabbit Ig biotin (code 0353; Dako Cytomation) for 30 min. For Gli1 immunostaining, sections were fixed, blocked with normal serum, and then incubated overnight at 4°C with a 1:200 dilution of either rabbit polyclonal anti-Gli1 H300 (20687; Santa Cruz Biotechnology, Santa Cruz, CA) or rabbit serum as a negative control. After two buffer washes, secondary biotinylated anti-rabbit antibody (Vector Laboratories, Burlingame, CA) was applied for an overnight incubation at 4°C. Binding of antibodies was detected with the biotin–avidin-based Vectastain Elite ABC reagent (Vector Laboratories), according to the manufacturer's protocol, and with UltraVision Detection System DAB Substrate (TA-125H; Lab Vision). Sections were counterstained with hematoxylin.
Cell culture and constructs. Cerebellar GCPs were prepared from 4- to 7-d-old mice according to established protocols (Wechsler-Reya and Scott, 1999). Briefly, cerebella were removed aseptically, cut into small pieces, and incubated at room temperature for 15 min in digestion buffer [Dulbecco's PBS (Invitrogen, Gaithersburg, MD) with 0.1% trypsin, 0.2% EDTA, and 100 μg/ml DNase]. Tissues were then triturated with fire-polished Pasteur pipettes to obtain a single-cell suspension. Cells were centrifuged, resuspended in Neurobasal medium supplemented with B27, penicillin–streptomycin, and l-glutamine (2 mm) (Invitrogen) and plated at a density of 8 × 105 cells/cm2 on tissue-culture dishes or eight-well Lab-Tek chamber slides (Permanox slide; Nunc, Naperville, IL) coated with 1 mg/ml poly-l-lysine.
GCPs were transfected with Lipofectamine 2000 (Invitrogen) at the same time that the cells were plated. Transfection efficiency was ∼15% of the GCP cell population. Luciferase activity was assayed with a dualluciferase assay system (Promega, Madison, WI) 40 h after transfection with 0.7 μg of total plasmid DNA per 48-well plate [including 100 ng of luciferase reporter and _Renilla_-expressing vector pRL-TK (Promega)]. REN-encoding plasmids (pCDNA-RENmyc, pCDNA-ΔPOZ-REN-myc, and pEGFP-REN) have been described previously (Gallo et al., 2002; Di Marcotullio et al., 2004). pCXN2-REN-AS has been described previously (Gallo et al., 2002) and was obtained by cloning a 5′ fragment of mouse REN cDNA (1–2015 bp) into pCXN2 vector in an antisense orientation. GLI-RE-Luc (containing 12 repeated Gli consensus sequences in a TK minimal promoter vector) and pcDNA-HIS-hGLI2 (encoding Gli2 cDNA) have been described previously (Di Marcotullio et al., 2004).
Recombinant adenovirus infection. The mouse REN coding region sequence was cloned at the _Bgl_II–_Xho_I sites into pAdTrack-cytomegalovirus, which also expresses green fluorescent protein (GFP). Recombinant adenoviruses were prepared with the pAdEasy system as described by He et al. (1998). Recombinant viruses were obtained by transfection of human embryonic kidney 293 cells, and the viruses were amplified and purified to ∼1011 particles per milliliter. REN or control recombinant adenoviruses were added at a multiplicity of infection of 50, 250, and 500 pfu per cell-to-cell suspension immediately before plating. Infected GCPs were harvested 40 h after infection.
Immunofluorescence. GCPs were cultured in Lab-Tek chamber slides, fixed in 4% paraformaldehyde for 20 min at room temperature, incubated in 0.2% Triton X-100 to permeabilize cells, and then incubated in blocking buffer (PBS with 3% BSA). The primary antibodies were as follows: mouse monoclonal antibody (MAB) βIII-tubulin (βIII-Tub), neuronal-specific nuclear protein (NeuN) [MAB1637 and MAB377 (Chemicon, Temecula, CA) and p27/Kip1 (K25020; Transduction Laboratories, Lexington, KY)], rabbit polyclonal anti-ACTIVE caspase-3 [G748 (Promega) or #9661 (Cell Signaling Technology, Beverly, MA)], anti-Myc Tag (06–549; Upstate Biotechnology, Lake Placid, NY), affinity-purified rabbit polyclonal anti-REN (Gallo et al., 2002), and anti-GFP (8334; Santa Cruz Biotechnology). The secondary antibodies were FITC-conjugated anti-rabbit (F1262; Sigma), Texas Red-conjugated anti-mouse (Jackson ImmunoResearch, West Grove, PA), and Alexa Fluor 594-conjugated anti-rabbit (A11037; Invitrogen, Eugene, OR).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. At 48 and 72 h after transfection with test plasmids, apoptosis was detected by terminal deoxynucleotidyl transferase-mediated UTP nick end labeling (TUNEL) assay with the In Situ Cell Death Detection Kit (Roche, Welwyn Garden City, UK), according to the manufacturer's instructions.
Cell proliferation assay. Cell proliferation was evaluated by bromodeoxyuridine (BrdU) labeling assay (Roche) at different times after transfection with test plasmids. GCPs were fixed after BrdU incorporation and permeabilized as described above. After the primary and secondary labeling with anti-GFP or anti-Myc Tag, cells were refixed for 10 min with 4% paraformaldehyde in PBS at room temperature and treated with 2N HCl to denature DNA; BrdU detection was performed according to the manufacturer's instructions.
Western blot analysis. Cell lysates were prepared as described previously (Gallo et al., 2002), separated by a 12% SDS-PAGE, and transferred to a nitrocellulose membrane. Immunoblotting was performed with a rabbit polyclonal antibody against caspase-3 (#9662; Cell Signaling Technology) or anti-Myc Tag. HRP-conjugated secondary antibody anti-rabbit IgG (Santa Cruz Biotechnology) was used, and immunoreactive bands were visualized by enhanced chemiluminescence (Pierce, Rockford, IL).
mRNA expression analysis. Total RNA was isolated with the RNeasy Mini Kit (Qiagen, Hilden, Germany). One microgram of DNase-treated total RNA was reverse transcribed with Superscript II reverse transcriptase and random hexamers (Invitrogen). The PCR cycles for semiquantitative PCR of the α6 subunit of GABA receptor (GABRA6) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were designed to maintain the amplification in the exponential phase. Mouse REN mRNA expression was analyzed by real-time quantitative PCR (RT-Q-PCR) (Di Marcotullio et al., 2004). The primer sequences used were as follows: GABRA6 sense primer, 5′-ctggctcttcattattctatggc-3′; GABRA6 antisense primer, 5′-ctctcatcagtccaagtcatctc-3′; GAPDH sense primer, 5′-caccatggagaaggccgggg-3′; GAPDH antisense primer, 5′-gacggacacattgggggtag-3′; REN sense primer, 5′-gggtacggagaaactgccct-3′; REN antisense primer, 5′-gggccggatctggtagaag-3′; and probe FAM-conjugated, 5′-cttaaggcagaggctg-3′. 18S and hypoxanthine-guanine phosphoribosyl transferase (HPRT) endogenous controls are commercial TaqMan assay reagents (Applied Biosystems, Foster City, CA).
Statistical analysis. Results are expressed as means ± SD from an appropriate number of experiments as indicated in the figure legends. Each experiment was considered to be a sample size of 1 and was represented by an individual primary culture of GCPs obtained from a pool of 10–16 mice. At least triplicate assays per treatment group were performed in each experiment. Statistical differences were analyzed with the Mann–Whitney U test for nonparametric values with StatView 4.1 software (Abacus Concepts, Berkeley, CA). Values of p < 0.05 were considered statistically significant.
Results
REN is expressed in low-proliferating inner EGL GCPs and IGL cerebellar granule cells
REN expression is significantly reduced in medulloblastoma cells with respect to normal cerebellar tissue as a consequence of allelic deletion and gene silencing (Di Marcotullio et al., 2004). Because medulloblastoma cells are believed to arise from highly proliferating cerebellar GCPs with developmental programs that have been interrupted at an early stage in which cells are located in the outer EGL (Ruiz i Altaba et al., 2002), our observations suggest that REN expression might be physiologically upregulated in association with reduced cell proliferation and increased differentiation along the outer to inner EGL transition, a condition that might be lost in medulloblastoma. To test this hypothesis, we first examined REN mRNA expression by in situ hybridization in immature (P8) and differentiated (P21) neonatal mouse cerebella (Fig. 1_A_). At P4–P8, most GCPs are at their early developmental stage and located in the EGL. GCPs subsequently migrate away from this position (until P21) and generate the differentiated neurons in the IGL. As shown by in situ hybridization analysis, REN mRNA appears to be expressed mostly in granule cells in both the EGL and the IGL (Fig. 1_A_). An enhanced hybridization signal was observed at the bottom layer of the EGL (inner layer), with mRNA-positive cells deepening into the underlying molecular and Purkinje cell layers (PCLs) toward the IGL (Fig. 1_A_). REN mRNA expression was also detected in Purkinje cells (Fig. 1_A_).
Figure 1.

REN expression during cerebellar development. A, In situ hybridization in P8 mice cerebella (left) reveals REN expression (arrowheads) in the EGL and to a higher extent in cells going through the PCL (arrow) into the newly forming IGL, whereas at P21 (right), mRNA-positive cells are in the IGL. B, C, Immunohistochemical staining of REN and Ki67 proteins in P4 (B, left) and P8 (B, right, C, left) mice cerebella shows REN immunoreactivity in the inner zone of the EGL and in granule cells migrating toward the IGL, whereas Ki67 immunoreactivity is in the outer EGL progenitors. At P21, REN-positive cells are located in IGL granule cells (C, right).
To evaluate REN protein expression during development, we performed immunohistochemical staining with an antibody raised against REN (Gallo et al., 2002). In P4 and P8 cerebella, REN immunoreactivity, which was inhibited by a competing REN peptide (data not shown), was localized in granule and Purkinje cells (Fig. 1_B,C_). Strikingly, most REN-positive cells are located in the inner zone of the EGL and in granule cells migrating throughout the molecular layer toward the IGL (Fig. 1_B_, left panel, P4, and right panel, P8). Consistent with these observations, staining with REN antibody was also detected in differentiated IGL granule cells in P8 (Fig. 1_C_, left panel) and P21 (Fig. 1_C_, right panel) cerebella. We also stained P4–P8 cerebella with an antibody against Ki67, a marker of cell proliferation. In contrast to REN localization, Ki67-positive proliferating cells are located mostly in the outer EGL, with no staining in the inner EGL (Fig. 1_B_); thus, REN and Ki67 staining appear to be mutually exclusive in developing cerebellar granule cells.
Together, these observations suggest that REN is preferentially expressed as soon as granule cells stop proliferating and start the differentiation and migration processes toward the IGL, where REN expression is maintained.
Upregulation of REN expression in cerebellar GCPs along differentiation in culture
The preferential expression of REN in low-proliferating GCPs suggests that REN expression might be upregulated along the differentiation process occurring during the transition from EGL to IGL; therefore, we isolated GCPs from the cerebella of P4 mice, allowed them to differentiate in vitro, and monitored REN expression. GCPs cultured for 8 d stopped proliferating (data not shown) (Miyazawa et al., 2000) and significantly differentiated into mature granule cells displaying long neurite outgrowth expressing βIII-Tub (Fig. 2_A_, bottom panel). Concomitantly, the mRNA levels for GABRA6 were strongly increased (Fig. 2_A_, top panel). In a parallel manner, REN mRNA levels increased significantly (p < 0.01) in GCPs cultured for 8 d in vitro (DIV8), as long as the growth arrest and differentiation processes occurred, thus mimicking the modulation of REN expression in the EGL and IGL in vivo (Fig. 2_B_). Similar results were obtained with P7-derived GCPs (data not shown).
Figure 2.
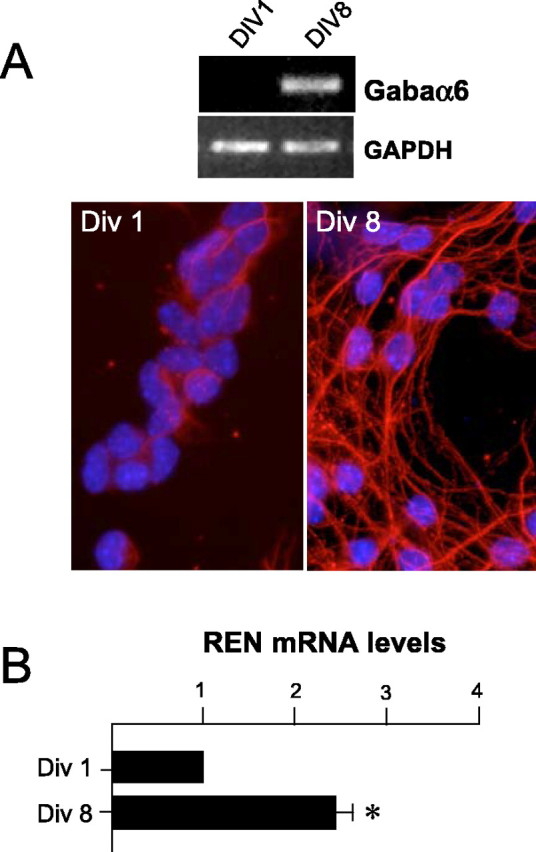
Upregulation of REN expression in cultured cerebellar GCPs along differentiation. A, Top, RT-PCR of GABRA6 mRNA expression in GCPs cultured for 1 d (DIV1) or 8 d (DIV8) versus GAPDH expression, as a loading control. Bottom, Immunofluorescence with anti-βIII-Tub antibody (red) and Hoechst staining (blue) of DIV1- and DIV8-cultured GCPs. B, RT-Q-PCR of REN expression in DIV1- and DIV8-cultured GCPs. Results are expressed as arbitrary units (mean ± SD from 6 experiments; *p < 0.01) normalized to endogenous controls (18S and HPRT RNA).
REN inhibits proliferation and enhances p27/Kip1 expression in cultured cerebellar GCPs
The association of upregulated REN expression with decreased proliferation of GCPs suggests that REN might play a role in slowing GCP growth. To test this hypothesis, we overexpressed REN in P4 GCPs by transfecting expression vectors encoding REN cDNA. REN-transfected GCPs displayed a significantly (p < 0.01) reduced incorporation of BrdU when compared with control cells transfected with a GFP-expressing vector (Fig. 3_A_). Similarly, a dose-dependent inhibition of GCP proliferation was also observed in cultured GCPs infected with an adenovirus encoding REN with respect to a GFP-expressing adenovirus as a control (Fig. 3_A_).
Figure 3.

REN promotes growth arrest and p27/Kip1 expression in cultured cerebellar GCPs. A, BrdU incorporation, evaluated by immunofluorescence (mean ± SD from 3 experiments; *p < 0.01) in GCPs isolated from cerebella of P4 mice, 40 h after transfection with pEGFP-REN (REN, black box) or control pEGFP (GFP, white box) expression vectors or infection with increasing amounts (50, 250, and 500 pfu per cell) of REN-expressing (Ad-REN) or GFP-expressing (Ad-C) adenovirus. B, Percentage (mean ± SD from 3 experiments; *p < 0.01) of p27/Kip1-expressing GCPs, isolated from cerebella of P4 mice, 40 h after transfection with pEGFP-REN or pEGFP vector, evaluated by immunofluorescence.
p27/Kip1 has been reported previously to play a critical role in regulating GCP proliferation (Miyazawa et al., 2000); therefore, we tested whether REN was able to modulate p27 expression in GCPs in culture. A significant (p < 0.01) increase in the percentage of cells expressing p27 was detected after transfection of REN vector with respect to GFP-transfected GCPs (Fig. 3_B_), suggesting that p27 might be involved in the inhibition of GCP proliferation induced by REN.
REN is an antagonist of Hedgehog activity in cultured cerebellar GCPs
A major enhancer of GCP proliferation has been reported to be Shh, which triggers Gli2 and Gli3 transcription factors, in turn promoting the transcription of downstream target genes (i.e., Gli1) (Dahmane and Ruiz i Altaba, 1999; Wallace, 1999; Wechsler-Reya and Scott, 1999; Ruiz i Altaba et al., 2002). We found Gli1 expression restricted to high-proliferating outer EGL GCPs, whereas it was absent in the inner EGL (Fig. 4_A_), thus inversely mimicking in vivo REN expression; therefore, these observations suggest that REN might target the Shh-induced GCP growth. To test this hypothesis, we first investigated the effect of REN-expressing vectors on the Gli2-dependent activation of a luciferase reporter driven by a Gli-responsive element, transfected into cultured GCPs. Figure 4_B_ shows that REN overexpression significantly (p < 0.01) inhibits the luciferase transcription induced by Gli2. Likewise, adenovirus-mediated overexpression of REN was able to reduce mRNA levels of the target gene Gli1 in response to Shh treatment of cultured GCPs (Fig. 4_C_).
Figure 4.

REN is an antagonist of Hedgehog activity in cultured cerebellar GCPs. A, Immunohistochemistry of Gli1 in P8 mouse cerebella reveals Gli1 expression in the outer granule cells of the EGL. A higher magnification of the EGL is shown in the bottom panel. B, REN inhibits the activation of GLI-RE-Luc induced by Gli2. pCDNA-HIS-hGLI2 and REN expression vectors in combination with each other or with empty vectors were transfected into P4-cultured GCPs, and after 40 h, luciferase activity was evaluated relative to Renilla activity. Values indicated are the means ±SD from three experiments (*p < 0.01). C, REN suppresses the induction of the target gene Gli1 in response to Shh treatment of cultured GCPs. An RT-Q-PCR of Gli1 expression in P4 GCPs 40 h after infection with REN- or GFP-expressing adenovirus (500 pfu per cell) in the absence or presence of treatment with Shh (3μg/ml) was performed. Results are expressed as arbitrary units (mean ± SD from 3 experiments; *p < 0.01) normalized to endogenous controls (18S and HPRT RNA).
REN was also able to overcome the mitogenic effect of Shh, because REN overexpression significantly (p < 0.01) reduced GCP proliferation in the presence of Shh added to the culture (Fig. 5_A_). A region spanning amino acids 18–80 [named the poxvirus and zinc finger (POZ) domain] has been shown previously to be responsible for the inhibitory activity on both proliferation and Gli function in human medulloblastoma cells (Di Marcotullio et al., 2004), making it the strongest candidate to mediate the growth-suppressing activity on GCPs. We therefore transfected a REN mutant deleted of the POZ domain (ΔPOZ) into GCPs and compared its activity with that of wild-type REN and GFP expression vectors (Fig. 5_B_, top panel). ΔPOZ was much less active in inhibiting BrdU incorporation in GCPs when compared with wild-type REN, in either the absence or presence of added exogenous Shh (Fig. 5_B_, bottom panel).
Figure 5.

REN antagonizes Shh-induced proliferation of cultured cerebellar GCPs. BrdU incorporation was evaluated by immunofluorescence (mean ± SD from 3 experiments; *p < 0.01) in Shh-treated (3 μg/ml) or untreated P4GCPs, 40 h after transfection with pEGFP-REN, or pEGFP control vector (A) and either REN wild-type (pcDNA-REN-myc) or deletion mutant (pcDNA-ΔPOZ-REN-myc) (B). REN WT-transfected cells display significantly lower levels of BrdU-positive cells compared with REN ΔPOZ-transfected cells (mean ± SD from 3 experiments; *p < 0.01). Protein levels of transfected wild-type or mutant REN were revealed by Western blot with anti-Myc Tag antibody (B, top). WT, Wild type.
These observations suggest that REN is an antagonist of Hedgehog signaling in cerebellar GCPs and that it may be able to suppress a number of Hedgehog-dependent responses during cerebellar development, including mitogenesis.
REN enhances the differentiation of cultured GCPs and antagonizes the ability of Hedgehog to maintain undifferentiated cells
GCPs undergo spontaneous neuronal differentiation during culture, a process that is hampered by Shh treatment (Dahmane and Ruiz i Altaba, 1999; Wallace, 1999; Wechsler-Reya and Scott, 1999; Solecki et al., 2001). REN overexpression in cultured GCPs enhanced the differentiation process in the absence of Shh treatment. In fact, REN-expressing GCPs showed an increased proportion of cells positive for the neural differentiation marker NeuN (Fig. 6_A_). Interestingly, REN was able to antagonize the inhibitory effect of Shh on GCP differentiation in culture. Indeed, REN-positive GCPs kept expressing NeuN, despite the presence of Shh, which instead significantly (p < 0.05) reduced the percentage of NeuN-positive GFP-transfected cells (Fig. 6_A_). REN-induced NeuN-positive cells also displayed morphological features of differentiated cells, as indicated by considerable neurite outgrowth observed with respect to GFP-transfected cells (Fig. 6_B_); therefore, REN not only antagonizes Shh-enhanced GCP proliferation but also hampers the ability of this growth factor to maintain undifferentiated cells.
Figure 6.
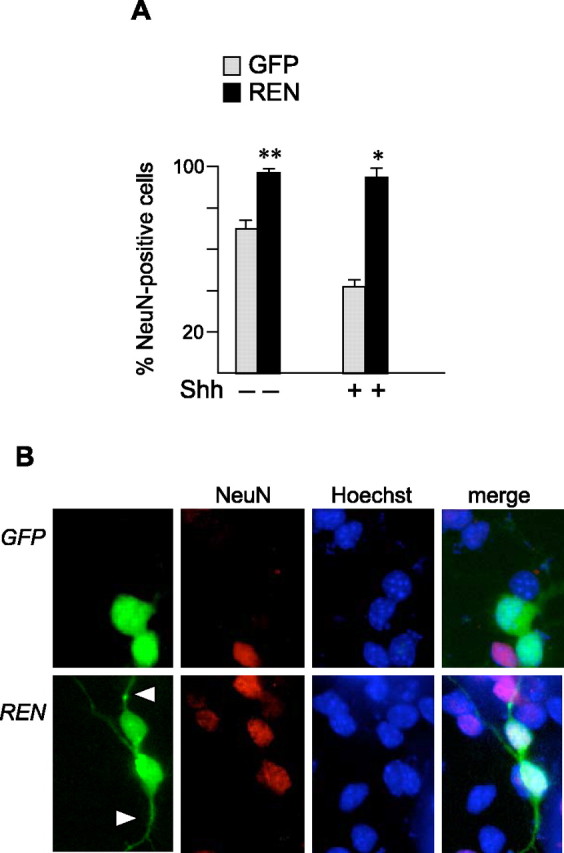
REN enhances the differentiation of cultured GCPs and antagonizes the ability of Hedgehog to maintain undifferentiated cells. A, NeuN expression in Shh-treated (3 μg/ml) or untreated P4 GCP cells 40 h after transfection with pEGFP-REN or pEGFP. The percentage (mean ± SD from 3 experiments; *p < 0.01; **p < 0.05) of NeuN-incorporating cells in the REN+ or GFP+ population was determined by coimmunostaining with anti-NeuN and anti-GFP antibodies. B, Coimmunofluorescence of NeuN (red) and REN or GFP (green) in either pEGFP-REN- or pEGFP-REN-transfected and Shh-treated GCPs, 40 h after transfection. Nuclei were visualized by Hoechst staining (blue). Neurite outgrowth in REN-transfected cells is indicated by arrowheads.
REN loss of function enhances Hedgehog signaling and proliferation and impairs differentiation in cultured GCPs
The ability of REN to antagonize the Hedgehog pathway suggests that loss of function of this gene might enhance Hedgehog signaling. We have reported previously that a vector encoding an antisense RNA against REN transcript (pCXN2-REN-AS) is effective in abrogating endogenous REN protein levels, thereby impairing the activation of a number of REN-dependent cell responses (i.e., the expression of NeuroD) (Gallo et al., 2002).
We confirmed the abrogation of REN expression by pCXN2-REN-AS in GCP cells, as measured by immunofluorescence staining (Fig. 7_A_). We also observed a significant (p < 0.01) increase in Gli-dependent activation of luciferase activity driven by a Gli-responsive element in REN-AS with respect to mock-transfected GCPs (Fig. 7_B_), suggesting that REN loss of function relieves a restraint of Hedgehog signaling.
Figure 7.

Abrogation of REN expression impairs Hedgehog antagonism and differentiation and increases proliferation of cultured GCPs. A, Antisense REN inhibits REN expression. P4 GCPs were transfected with either pCXN2-REN-AS (REN-AS) or pCXN2 (control) together with a 10-fold lower amount of pEGFP to monitor transfected cells. Coimmunofluorescence staining of endogenous REN protein (red; stained with affinity-purified rabbit polyclonal antibody) and GFP(green) is indicated. Nuclei were visualized by Hoechststaining(blue). Arrowheads indicate either REN-positive (control) or REN-negative (REN-AS) cells. B, REN loss of function enhances Hedgehog signaling. P4 GCPs were transfected with pCXN2-REN-AS (REN-AS) or pCXN2 (control) together with either Gli1 or empty vector and Gli-RE-luciferase reporter. Luciferase activity (evaluated as described in Fig. 4 B; mean ± SD from 3 experiments; *p < 0.01) is indicated after subtraction of basal values observed in cells transfected with pCXN2/Gli-RE-luciferase alone. C, D, REN loss of function enhances cell proliferation and impairs NeuN expression. BrdU incorporation (C) and NeuN expression (D) were evaluated 60 and 40 h, respectively, after transfection of either pCXN2-REN-AS (REN-AS) or pCXN2 (control) into P4 GCPs. A 10-fold lower amount of pEGFP was also cotransfected to monitor transfected cells. Values indicated are the means ± SD from three experiments (*p < 0.01).
To investigate whether abrogation of REN expression was also able to affect the GCP cell proliferation rate, we transfected GCPs isolated from P4 mice with either pCXN2-REN-AS or control vector pCXN2 (mock). We observed that antisense RNA-mediated REN functional knock-down was able to sustain the GCP proliferation rate, because a twofold increase in the fraction of GCP cells that were still proliferating was detected 60 h after transfection with REN-AS with respect to mock-transfected cells (Fig. 7_C_).
Finally, we also unveiled the ability of the functional knock-down of REN to affect differentiation of cultured GCPs. Indeed, we observed a reduction in GCPs expressing NeuN in REN-AS-transfected GCP cells compared with mock controls (Fig. 7_D_). Overall, these findings suggest that REN expression is required to antagonize Hedgehog activity and to promote growth arrest and differentiation of GCPs in culture.
REN enhances the apoptosis of cultured cerebellar GCPs
Apoptosis has been described in early phases of neuroblast development when it plays a crucial role in the control of the overall number of differentiating cells by limiting neurons produced in excess (Miyazawa et al., 2000; for review, see Yeo and Gautier, 2004); therefore, loss of apoptotic function during this developmental phase is suggested to favor malignant transformation. The tumor-suppressor function that we have described previously for REN led us to further investigate whether the overall growth-inhibitory activity of REN might also be associated with the ability to regulate GCP apoptosis. To test this hypothesis, we transfected either P4 or P7 GCPs with REN-expressing vectors and assessed cell apoptosis by monitoring pycnotic nuclei by Hoechst staining and TUNEL. Overexpression of REN was able to enhance apoptosis of both P4 and P7 GCPs, because a higher percentage of REN-positive cells displayed picnic nuclei (data not shown) and TUNEL staining after 48 h and, more significantly (p < 0.01), 72 h of transfection, with respect to GFP-transfected cells (Fig. 8_A_).
Figure 8.
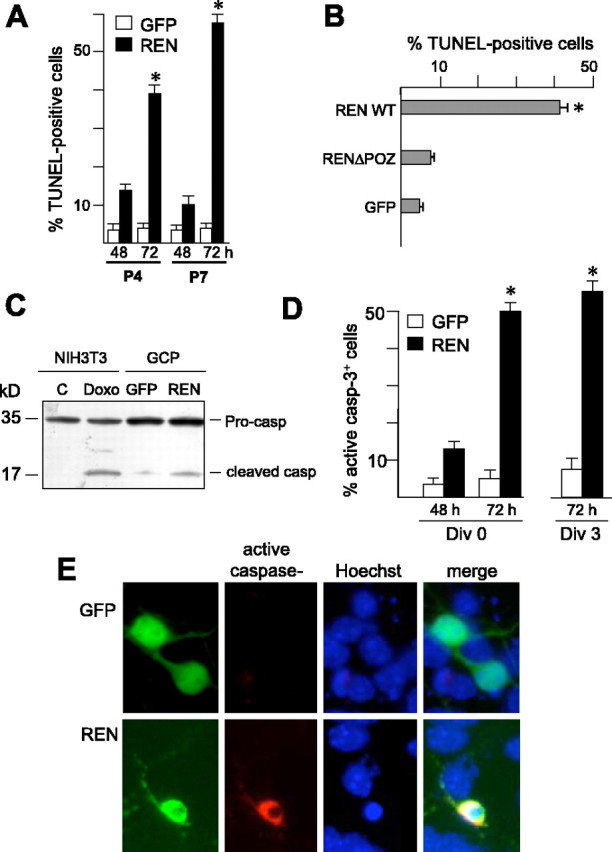
REN enhances apoptosis of cultured cerebellar GCPs. The percentages of TUNEL-positive (mean ± SD from 3 experiments; *p < 0.01) cultured GCPs from P4 or P7 mice cerebella transfected with pEGFP-REN or pEGFP control vector (A, 48 and 72 h after transfection) or with wild-type (pcDNA-REN-myc) or deletion mutant REN (pcDNA-ΔPOZ-RENmyc) (B, 72 h after transfection) are shown. The percentage of TUNEL-positive cells in the REN+ or GFP+ population was determined by coimmunostaining with anti-myc or anti-GFP antibodies and TUNEL. C, Western blot analysis of caspase-3 in GCPs 48 h after transfection with REN or GFP vectors. Uncleaved pro-caspase (35 kDa) and cleaved caspase active fragment (17 kDa) are indicated. Doxorubicin-treated NIH-3T3 cells are also shown, as a control of cleaved caspase-3 generation. D, Active (cleaved) caspase-3 expression in P7 GCPs 48 and 72 h after transfection with pEGFP-REN or pEGFP on the first day (DIV0) or third day (DIV3) of culture, detected by coimmunostaining with anti-GFP and anti-active-caspase-3 antibodies (mean ± SD from 3 experiments; *p < 0.01). E, Coimmunofluorescence of active caspase-3 (red) and REN or GFP (green) expression in either pEGFP-REN or pEGFP DIV0-transfected GCPs. Nuclei were visualized by Hoechst staining (blue).
To study whether the growth-inhibitory and proapoptotic activities of REN were mediated by overlapping domains in the protein, we assessed the effect of the REN mutant deleted of the POZ domain (ΔPOZ), which was shown previously to be devoid of growth-suppressing properties (Fig. 5_B_). ΔPOZ was unable to increase the percentage of TUNEL-positive apoptotic GCPs, being almost indistinguishable from control GFP expression vector, whereas wild-type REN was very active in promoting cell apoptosis (Fig. 8_B_). These findings suggest that the growth-inhibitory and proapoptotic activities of REN in GCPs overlap and map to the POZ domain.
To investigate whether REN-induced cell death was a consequence of the activation of the caspase cascade, we evaluated the cleavage of pro-caspase-3 generating an active caspase-3 fragment as an effector of the apoptotic process. Figure 8_C_ shows that overexpression of REN in cultured GCPs is able to induce the generation of the 17 kDa active fragment from uncleaved caspase-3, whereas no significant cleavage was detected in GFP-transfected control cells. Immunofluorescence staining of transfected GCPs with an antibody against active caspase-3 confirmed the above data, because a significant (p < 0.01) increase in the percentage of active caspase-3-positive cells was detected in GCPs transfected at DIV0 with REN compared with GFP-transfected cells (Fig. 8_D,E_).
Finally, to investigate whether REN induced apoptosis in either proliferating or differentiating cells, we cultured GCPs for 3 d and transfected them with REN at DIV3 to detect active caspase-3 expression. Under these conditions, GCP cells are almost differentiated, and the proliferation rate is very low (∼5%) at the time of triggering apoptotic stimulus by REN. REN overexpression in these cells resulted in a substantial increase in both TUNEL-positive (data not shown) and active caspase-3-positive granule cells (Fig. 8_D_), suggesting that REN is able to induce apoptosis in differentiating nonproliferating cells.
Discussion
The development of the cerebellum is known to be controlled by a coordinated sequence of events that regulate the proliferation, differentiation, and death of GCPs. Cell proliferation is restricted to the outer EGL, under the action of Purkinje cell-secreted Shh, which is the most potent physiological mitogen for GCPs (Dahmane and Ruiz i Altaba, 1999; Wallace, 1999; Wechsler-Reya and Scott, 1999); however, despite the persistent presence of Shh, GCPs exit the cell cycle on their entrance into the inner EGL (Ruiz i Altaba et al., 2002). This raises the question of the identity of the signals responsible for the growth arrest observed at the transition from the outer to inner EGL.
A role for p27, a cyclin-dependent kinase inhibitor, has been suggested, because its expression is upregulated in inner compared with outer EGL GCPs (Miyazawa et al., 2000). Likewise, high p27 expression correlated with decreased cell proliferation in cultured GCPs, and, conversely, inactivation of p27 leads to increased growth of GCPs in cell-culture assays and in vivo (Miyazawa et al., 2000); however, even GCPs from p27–/– mice ultimately exit the cell cycle and differentiate into mature granule cells (Miyazawa et al., 2000), suggesting that additional factors control EGL cell proliferation and differentiation programs.
Cell-cycle machinery has also been reported to be regulated by Shh-induced signals, via the transcriptional activation of a number of genes involved in cell-cycle progression (i.e., cyclin D1, cyclin D2, and N-myc) (Yoon et al., 2002; Oliver et al., 2003). Cyclin D2 is required for GCP proliferation, as indicated by the reduced number of granule cells in cyclin D2-deficient mice (Huard et al., 1999). The presence of a mechanism capable of interrupting Shh signaling during the outer to inner EGL GCP transition is suggested by the absence of expression of the Shh target Gli1 transcription factor that we have observed in the inner EGL, which instead is restricted to high-proliferating outer EGL cells. These findings are in agreement with the pattern of Gli1 expression described recently in Gli1_–_LacZ mice (Corrales et al., 2004). A combination of activation of p27 expression and silencing of Shh–Gli-induced target genes (i.e., cyclin D2) may allow GCPs to exit the cell cycle in the inner EGL; however, how Shh signaling is withdrawn while p27 expression is enhanced needs to be elucidated.
We indicate in this paper that REN is a candidate for playing such a role in EGL development, as suggested by the following evidence. First, REN expression inversely correlates with granule cell proliferation along the transition from outer to inner EGL GCPs and in IGL cells of the developing cerebellum. Such a cell maturation process in vivo is paralleled by the upregulation of REN expression also occurring along GCP differentiation in culture. Second, we report that REN is able to enhance p27 and inhibit Shh-dependent events, thus antagonizing both Shh-induced growth and block of differentiation of cultured GCPs. Finally, an active role for REN is also suggested by the requirement of REN expression to restrain Hedgehog signaling and cell growth and to promote the differentiation of cultured GCPs. These observations strongly support the notion that REN may be involved in the control of the expansion of GCPs during the transition from outer to inner EGL.
Cell death plays a crucial role in maintaining an appropriate number of differentiated neurons during early cerebellar development (for review, see Yeo and Gautier, 2004). Indeed, mice that are deficient in proapoptotic caspase-3 die perinatally with marked hyperplasia, which is attributed to a reduction of apoptosis in neural progenitor cells in different brain regions, including the cerebellum (Kuida et al., 1996; Pompeiano et al., 2000). Apoptotic events are suggested to occur as a consequence of undergoing differentiation (for review, see Hoffman and Liebermann, 1994), implying a role for molecules involved in growth arrest and the execution of the apoptotic processes.
Here we report that such an apoptotic response is also targeted by REN, which triggers caspase-3 activation and increases the number of TUNEL-positive GCP cells. Although the underlying mechanisms need to be investigated further, REN-induced apoptosis might be linked to its activity on cell cycle and differentiation. To this regard, the effect of REN is reminiscent of the apoptosis induced by other proteins involved in early neural cell development such as the basic helix-loop-helix (bHLH) Math1 or its inducer PC3, both of which promote neuronal differentiation and apoptosis in progenitors of the ventricular zone and cerebellar GCPs (Isaka et al., 1999; Canzoniere et al., 2004). Likewise, loss of Hes1, a bHLH factor that negatively regulates differentiation, results in accelerated neuronal differentiation with increased apoptosis (Nakamura et al., 2000). Moreover, the cyclin-dependent kinase inhibitor p27, a promoter of cell-cycle arrest and differentiation, has been reported to also have a proapoptotic function (Wang et al., 1997; for review, see Philipp-Staheli et al., 2001; Coqueret, 2003). A role for Shh signaling in controlling cell-death events has also been suggested by the wide-spread apoptosis observed in neural progenitors after inhibition of Shh activity (Ahlgren and Bronner-Fraser, 1999; Charrier et al., 2001) and by the ability of the Hedgehog receptor Patched to trigger apoptosis in the absence of Shh signal (Thibert et al., 2003). Whether REN-induced apoptosis is related to its ability to regulate both p27 and Hedgehog signaling needs to be investigated further. Also, how REN precisely regulates these events at the molecular level is still unclear. We have shown that the REN region containing a POZ domain is essential for its function (Di Marcotullio et al., 2004; this paper). Because this domain is known to mediate protein–protein interactions, REN could bind to still unidentified proteins or known components of the Hedgehog pathway to induce the observed biological effects.
In summary, we have shown that REN is able to limit the expansion of GCPs (growth arrest and apoptosis) at a critical stage of early cerebellar development, possibly favoring their physiological progression toward terminal differentiation. These observations have important implications with regard to pathological conditions that arise from aberrant development, such as neoplastic transformation. Indeed, the early stage of cerebellar progenitor development appears to be critical for the occurrence of medulloblastoma. This is suggested by the high frequency of medulloblastoma development triggered by defects responsible for inappropriate activation of Hedgehog signaling, which occur in Ptc1–/+ mice (for review, see Wetmore, 2003) and in a consistent number (up to 50%) of human medulloblastomas, as a consequence of either genetic or epigenetic events (for review, see De Smaele et al., 2004; Hallahan et al., 2004). Lack of extinguishing Hedgehog signals at an early developmental stage of cerebellar progenitors is expected to lead to uncontrolled growth and to the impairment of apoptotic and differentiation events, resulting in abnormal granule cell expansion eventually favoring cell transformation. Additionally, the loss of p27, which also plays a critical role in early GCP development (Miyazawa et al., 2000), is involved in medulloblastoma formation, as indicated by the increased susceptibility of tumor incidence in p27–/– mice (Lee et al., 2003).
Interestingly, we have shown previously that 17p deletion, a genetic defect observed most frequently in human medulloblastoma (>50% of cases), leads to the loss of REN, the expression of which is substantially reduced in tumor samples as a consequence of allelic deletion and epigenetic gene silencing (Di Marcotullio et al., 2004). REN behaves as a tumor suppressor in human medulloblastoma by impairing G1/S cell-cycle transition and suppressing tumor growth in vitro and in vivo via antagonism of Hedgehog signaling (Di Marcotullio et al., 2004). As shown here, this antagonism is also present in cerebellar GCPs at early stages of development, providing a possible link between tumorigenic events stemming from aberrant development and tumor progression.
Our observations therefore suggest a model (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) in which the loss of function of REN, as a consequence of cancer-associated genetic–epigenetic events, might relieve a restraint of Hedgehog signaling at the transition from outer to inner EGL GCP and withdraw a limiting signal for immature cell expansion, thus favoring tumorigenic events.
Footnotes
This work was partially supported by the Associazione Italiana per la Ricerca sul Cancro, Telethon Grant GGP04168, the National Research Council, the Ministry of University and Research, and the Ministry of Health and Center of Excellence for Biology and Molecular Medicine. We thank M. Zani, C. Rinaldi, S. Martini, A. Porcellini, A. Corsi, D. Zei, M. Moretti, and F. Lombardi for experimental assistance.
Correspondence should be addressed to Dr. Alberto Gulino, Department of Experimental Medicine and Pathology, University La Sapienza, 324 Viale Regina Elena, 00161 Rome, Italy. E-mail: alberto.gulino@uniroma1.it.
DOI:10.1523/JNEUROSCI.2438-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258338-09$15.00/0
References
- Ahlgren SC, Bronner-Fraser M (1999) Inhibition of sonic hedgehog signaling in vivo results in craniofacial neural crest cell death. Curr Biol 9: 1304–1314. [DOI] [PubMed] [Google Scholar]
- Canzoniere D, Farioli-Vecchioli S, Conti F, Ciotti MT, Tata AM, Augusti-Tocco G, Mattei E, Lakshmana MK, Krizhanovsky V, Reeves SA, Giovannoni R, Castano F, Servadio A, Ben-Arie N, Tirone F (2004) Dual control of neurogenesis by PC3 through cell cycle inhibition and induction of Math1. J Neurosci 24: 3355–3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco E, Blum M, Weickert CS, Casper D (2003) Epidermal growth factor receptor expression is related to post-mitotic events in cerebellar development: regulation by thyroid hormone. Brain Res Dev Brain Res 140: 1–13. [DOI] [PubMed] [Google Scholar]
- Charrier JB, Lapointe F, Le Douarin NM, Teillet MA (2001) Anti-apoptotic role of Sonic hedgehog protein at the early stages of nervous system organogenesis. Development 128: 4011–4020. [DOI] [PubMed] [Google Scholar]
- Coqueret O (2003) New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol 13: 65–70. [DOI] [PubMed] [Google Scholar]
- Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL (2004) Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellar development. Development 131: 5581–5590. [DOI] [PubMed] [Google Scholar]
- Dahmane N, Ruiz i Altaba A (1999) Sonic Hedgehog regulates the growth and patterning of the cerebellum. Development 126: 3089–3100. [DOI] [PubMed] [Google Scholar]
- De Smaele E, Di Marcotullio L, Ferretti E, Screpanti I, Alesse E, Gulino A (2004) Chromosome 17p deletion in human medulloblastoma: a missing checkpoint in the Hedgehog pathway. Cell Cycle 3: 1263–1266. [DOI] [PubMed] [Google Scholar]
- Di Marcotullio L, Ferretti E, De Smaele E, Argenti B, Mincione C, Zazzeroni F, Gallo R, Masuelli L, Napolitano M, Maroder M, Modesti A, Giangaspero F, Screpanti I, Alesse E, Gulino A (2004) RENKCTD11 is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma. Proc Natl Acad Sci USA 101: 10833–10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo R, Zazzeroni F, Alesse E, Mincione C, Borello U, Buanne P, D'Eugenio R, Mackay AR, Argenti B, Gradini R, Russo MA, Maroder M, Cossu G, Frati L, Screpanti I, Gulino A (2002) REN: a novel, developmentally regulated gene that promotes neural cell differentiation. J Cell Biol 158: 731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA, Olson JM (2004) The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res 64: 7794–7800. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Heintz N (1995) Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci 18: 385–408. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman B, Liebermann DA (1994) Molecular controls of apoptosis: differentiation/growth arrest primary response genes, proto-oncogenes, and tumor suppressor genes as positive and negative modulators. Oncogene 9: 1807–1812. [PubMed] [Google Scholar]
- Huard JM, Forster CC, Carter ML, Sicinski P, Ross ME (1999) Cerebellar histogenesis is disturbed in mice lacking cyclin D2. Development 126: 1927–1935. [DOI] [PubMed] [Google Scholar]
- Isaka F, Ishibashi M, Taki W, Hashimoto N, Nakanishi S, Kageyama R (1999) Ectopic expression of the bHLH gene Math1 disturbs neural development. Eur J Neurosci 11: 2582–2588. [DOI] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA (1996) Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384: 368–372. [DOI] [PubMed] [Google Scholar]
- Lamont JM, McManamy CS, Pearson AD, Clifford SC, Ellison DW (2004) Combined histopathological and molecular cytogenetic stratification of medulloblastoma patients. Clin Cancer Res 10: 5482–5493. [DOI] [PubMed] [Google Scholar]
- Lee Y, Miller HL, Jensen P, Hernan R, Connelly M, Wetmore C, Zindy F, Roussel MF, Curran T, Gilbertson RJ, McKinnon PJ (2003) A molecular fingerprint for medulloblastoma. Cancer Res 63: 5428–5437. [PubMed] [Google Scholar]
- Miyazawa K, Himi T, Garcia V, Yamagishi H, Sato S, Ishizaki Y (2000) A role for p27/Kip1 in the control of cerebellar granule cell precursor proliferation. J Neurosci 20: 5756–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Sakakibara S, Miyata T, Ogawa M, Shimazaki T, Weiss S, Kageyama R, Okano H (2000) The bHLH gene Hes1 as a repressor of the neuronal commitment of CNS stem cells. J Neurosci 20: 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, Wickramasinghe R, Scott MP, Wechsler-Reya RJ (2003) Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci USA 100: 7331–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philipp-Staheli J, Payne SR, Kemp CJ (2001) P27/kip1: regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res 264: 148–168. [DOI] [PubMed] [Google Scholar]
- Pompeiano M, Blaschke AJ, Flavell RA, Srinivasan A, Chun J (2000) Decreased apoptosis in proliferative and postmitotic regions of the Caspase 3-deficient embryonic central nervous system. J Comp Neurol 423: 1–12. [PubMed] [Google Scholar]
- Ruiz i Altaba A, Sanchez P, Dahmane N (2002) Gli and Hedgehog in cancer: tumors, embryos and stem cells. Nat Rev Cancer 2: 361–382. [DOI] [PubMed] [Google Scholar]
- Sassoon D, Rosenthal N (1993) Detection of messenger RNA by in situ hybridization. Methods Enzymol 225: 384–404. [DOI] [PubMed] [Google Scholar]
- Solecki DJ, Liu XL, Tomoda T, Fang Y, Hatten ME (2001) Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron 31: 557–568. [DOI] [PubMed] [Google Scholar]
- Thibert C, Teillet MA, Lapointe F, Mazelin L, Le Douarin NM, Mehlen P (2003) Inhibition of neuroepithelial patched-induced apoptosis by sonic hedgehog. Science 301: 843–846. [DOI] [PubMed] [Google Scholar]
- Wallace VA (1999) Purkinje-derived Sonic Hedgehog regulated granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 9: 445–448. [DOI] [PubMed] [Google Scholar]
- Wang VY, Zoghbi HY (2001) Genetic regulation of cerebellar development. Nat Rev Neurosci 2: 484–491. [DOI] [PubMed] [Google Scholar]
- Wang X, Gorospe M, Huang Y, Holbrook NJ (1997) p27/kip1 overexpression causes apoptotic death of mammalian cells. Oncogene 15: 2291–2297. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP (1999) Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 22: 103–114. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP (2001) The developmental biology of brain tumors. Annu Rev Neurosci 24: 385–428. [DOI] [PubMed] [Google Scholar]
- Wetmore C (2003) Sonic hedgehog in normal and neoplastic proliferation: insight gained from human tumors and animal models. Curr Opin Genet Dev 13: 34–42. [DOI] [PubMed] [Google Scholar]
- Yeo W, Gautier J (2004) Early neural cell death: dying to become neurons. Dev Biol 274: 233–244. [DOI] [PubMed] [Google Scholar]
- Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, Jacob H, Walterhouse D, Iannaccone P (2002) Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem 277: 5548–5555. [DOI] [PubMed] [Google Scholar]