Fission Yeast Homologs of Human CENP-B Have Redundant Functions Affecting Cell Growth and Chromosome Segregation (original) (raw)
Abstract
Two functionally important DNA sequence elements in centromeres of the fission yeast Schizosaccharomyces pombe are the centromeric central core and the K-type repeat. Both of these DNA elements show internal functional redundancy that is not correlated with a conserved DNA sequence. Specific, but degenerate, sequences in these elements are bound in vitro by the S. pombe DNA-binding proteins Abp1p (also called Cbp1p) and Cbhp, which are related to the mammalian centromere DNA-binding protein CENP-B. In this study, we determined that Abp1p binds to at least one of its target sequences within S. pombe centromere II central core (cc2) DNA with an affinity (Ks = 7 × 109 M−1) higher than those of other known centromere DNA-binding proteins for their cognate targets. In vivo, epitope-tagged Cbhp associated with centromeric K repeat chromatin, as well as with noncentromeric regions. Like abp1+/cbp1+, we found that cbh+ is not essential in fission yeast, but a strain carrying deletions of both genes (Δ_abp1 Δcbh_) is extremely compromised in growth rate and morphology and missegregates chromosomes at very high frequency. The synergism between the two null mutations suggests that these proteins perform redundant functions in S. pombe chromosome segregation. In vitro assays with cell extracts with these proteins depleted allowed the specific assignments of several binding sites for them within cc2 and the K-type repeat. Redundancy observed at the centromere DNA level appears to be reflected at the protein level, as no single member of the CENP-B-related protein family is essential for proper chromosome segregation in fission yeast. The relevance of these findings to mammalian centromeres is discussed.
The faithful inheritance of a cell's genetic blueprint depends on flawless chromosome segregation at each cell division. Mistakes that result in gain or loss of chromosomes (aneuploidy) are associated with death and disease in humans. Aneuploidy is estimated to be the cause of 25% of spontaneous abortions, is the leading cause of mental retardation, and is associated with certain types of cancers, including most colorectal tumors (9, 26, 36). Proper segregation in mitosis and meiosis is governed by the centromere, a multifunctional region on each chromosome that is the site of sister chromatid cohesion, kinetochore assembly, and spindle attachment and is subject to cell cycle surveillance (reviewed in references 1 and 16) and epigenetic regulation (reviewed in references 13 and 34). Despite the functional similarities of centromeres in various organisms, wide variation is seen in the underlying centromeric DNA sequences that nucleate assembly of the kinetochore. Not surprisingly, the centromere appears to be a target of species divergence.
With the exception of the budding yeasts, which utilize small “point” centromeres (<0.2 kb), other organisms as diverse as fission yeast (Schizosaccharomyces pombe), Drosophila, and humans contain large “regional” centromeres (40 to 5,000 kb) that are characterized by the presence of repeated DNAs (13, 54). Fission yeast employs the smallest regional centromere yet known, which spans 40 to 100 kb and is composed of a limited number of repeated sequences organized into an inverted repeat around a central core region (Fig. 1A). Centromeric DNAs in higher eukaryotes are characterized by satellite DNAs whose repeats are reiterated hundreds to thousands of times. In Drosophila, low-complexity satellite sequences are interspersed with complex transposon sequences, resulting in centromere regions spanning hundreds of kilobases of DNA. Mammalian centromeric DNA is primarily composed of satellite DNAs and a small number of repetitive short and long interspersed retrotransposon and transposon elements, forming centromeres that reach sizes of up to 5,000 kb. Of the various satellite DNA families present in humans, α-satellite (alphoid) DNA is the only member that is found at every human centromere (reviewed in reference 37). Alphoid DNA consists of tandem, divergent 171-bp monomers arranged into higher-order repeats that are chromosome specific (69). These alphoid repeats can be subdivided based on their protein binding characteristics, and the α-I subfamily is highly enriched in protein binding sites. On human chromosome 21, for example, the α-I region extends for ∼1,300 kb and contains, at intervals of approximately 0.4 kb, a 17-bp repeat sequence designated the CENP-B box, the binding site for centromere-binding protein B (18, 30, 31, 40, 41). In contrast, the α-II region of centromere 21 extends ∼1,900 kb and has very few CENP-B boxes. Thus, as a consequence of the heterogeneity of alphoid repeats, the frequency of CENP-B boxes is highly variable among human centromeres.
FIG. 1.

Organization of Abp1p and Cbh1p in vitro binding sites in S. pombe centromeres from strain Sp223. The ovals and triangles represent Abp1p and Cbh1p binding sites, respectively, summarized from this and previous studies (24, 38, 50). (A) The three S. pombe centromeres consist of a limited set of repeated DNA elements that are arranged in an inverted repeat around a central core (cc) region that, in the case of cc2, is unique in the genome, whereas cc1 and cc3 share extensive homology (14, 65). The central core and K-type repeat, which are necessary and sufficient for centromere function, were examined for in vitro protein binding sites by electrophoretic mobility shift assay. The open symbols mark the locations of sites identified in mobility shift assays. The solid symbols correspond to homologous sequences in the other centromeres. The arrows indicate the orientations of the centromeric repeat arrays. (B) Enlarged view of cc2 showing the numbered restriction fragments that were tested in mobility shift assays and the approximate locations of Abp1p binding sites within those fragments that show specific interactions. (C) Electrophoretic mobility shift assays of the central core fragments c2-13 (359 bp), c2-15 (376 bp), and c2-18 (383 bp) indicated in panel B. Five femtomoles of radiolabeled cc2 fragment DNA (∼1 ng) and an excess of poly(dI-dC) nonspecific competitor DNA (1 μg) were mixed in binding buffer with 1.2 μg of protein from a 0.4 M KCl crude extract of chromatin prepared from abp1+ (Sp223) or Δ_abp1_ (SBP070196-F5C) logarithmically growing cultures (24). The black dots to the left of each panel indicate the positions of specific protein-DNA complexes formed. (D) Enlargement of a portion of the cen1 K" repeat, corresponding to the black bar in panel A, showing the location of Abp1p and Cbh1p binding sites. The _Kpn_I-_Kpn_I subfragment is termed the centromere enhancer and is found in all K repeat elements. Within this region, Cbh1p binds specifically to fragment HI (PCR product corresponding to the thin black line).
It has been suggested that in mammals CENP-B organizes centromeric DNA into an ordered structure. This is based on the presence of CENP-B in vivo at most centromeres and on its ability to bind in vitro to DNA as a monomer through an NH2-terminal helix-turn-helix domain, followed by dimerization via COOH-terminal domains to bring together two noncontiguous CENP-B binding sites (32, 48, 72). However, the presence of DNA-bound CENP-B is not diagnostic for an active centromere; CENP-B is found at both loci on a dicentric chromosome in which one centromere is active and the other is epigenetically inactive (18). Moreover, CENP-B is an abundant protein in African green monkey cells but is barely detectable at centromeres in that species (21, 71), and it is not detectable by indirect immunofluorescence on mouse or human Y chromosomes or at “neocentromeres” on human marker chromosomes (17, 68). Importantly, recent studies have shown that CENP-B is not an essential protein in mice (29, 33, 53). Thus, mitosis and meiosis appear to proceed normally in the absence of CENP-B, suggesting either that CENP-B is not absolutely required for these functions or that multiple CENP-B-related proteins may play functionally redundant roles in these processes.
Indeed, at least one CENP-B-like protein of unknown function has been previously described in mice, as have three in humans. These are the mouse and human jerky gene products; human JH8 (GenBank accession no. AF072467), also a _jerky_-like protein; and the transposase encoded by human Tigger transposable elements (35, 66). CENP-B and its relatives show significant homology to bacterial insertion sequence transposases, Drosophila pogo transposase, and other _pogo_-like proteins (35, 59). Mechanistically, transposases are thought to bring together a pair of terminal inverted repeats into a protein-DNA complex and to catalyze DNA cleavage and strand exchange. Although CENP-B proteins from humans, mice, and hamsters contain alterations within an invariant motif associated with the cleavage and strand transfer activities, the structure formed in the initial proposed step of transposition is somewhat similar to that envisioned in the simplest models of higher-order chromatin structure at the centromere.
Two DNA-binding proteins with amino acid homology to CENP-B have been previously described in fission yeast. Abp1p (autonomously replicating sequence [ARS]-binding protein 1) (also called Cbp1p [centromere-binding protein 1]) and Cbhp (CENP-B homolog), which we will hereafter refer to as Abp1p and Cbh1p, show ∼25% identity and ∼50% similarity to CENP-B over their entire lengths (24, 38, 47). Like those of their mammalian counterparts, the NH2-terminal and central domains of these proteins bear significant homology with IS_630_-Tc1 and _pogo_-like transposases. As is true of mammalian CENP-B, the transposase-invariant motif is not conserved in the fission yeast homologs, which probably signifies a lack of transposase activity. Because Abp1p and Cbh1p share considerable homology with CENP-B, they are believed to bind DNA via NH2-terminal domains. In vitro experiments have shown that both fission yeast CENP-B homologs Abp1p and Cbh1p bind A+T-rich DNA sequences derived from S. pombe centromeres, as well as an ARS (24, 38, 47). Importantly, in vitro binding sites for these proteins have been found within the S. pombe centromeric central core and K repeat, DNA elements that together are sufficient for centromere function on a circular minichromosome (6). As is the case in higher eukaryotes, a single conserved primary DNA sequence required for function has not been found at fission yeast centromeres. Instead, in vivo experiments have demonstrated that centromeric sequences within both the central core and the K-type repeat show functional redundancies (6, 50). Consequently, artificial chromosomes with two different DNA sequences derived from two native S. pombe centromeres can confer centromere function on minichromosomes, although in one case the sequences must be epigenetically activated after prolonged propagation in vivo (50, 61). Distinct, but functionally redundant, DNA sequences are thus capable of nucleating kinetochore formation in S. pombe.
Fission yeast centromeric central-core regions, based on their unusual chromatin structure, are probable sites of kinetochore assembly (14). Central-core chromatin is a nuclease-resistant region that exhibits a highly atypical micrococcal nuclease digestion pattern compared to other centromeric sequences or to bulk chromatin (39, 55, 65). This protected pattern is associated with the presence in cis of a portion of the K repeat, termed the centromere enhancer (_Kpn_I-_Kpn_I [Fig. 1D]). The centromere enhancer, although not strictly necessary for centromere function, accelerates the rate at which naked centromeric DNA attains a functional (active) conformation, presumably by recruitment of centromere-binding proteins that efficiently organize the chromatin into a higher-order structure (50).
We present evidence here that CENP-B-related proteins in fission yeast play an important, albeit redundant, role in fission yeast chromosome segregation. We show that epitope-tagged Cbh1p associates with centromeric DNA in vivo but that similar to centromere-binding protein Cbf1p from budding yeast, it also associates with noncentromeric DNA. We find that Cbh1p, like Abp1p, is not an essential protein, although strains carrying null mutations for either abp1+ or cbh1+ lose artificial chromosomes at slightly elevated rates. A strain carrying deletions of both genes, however, is extremely compromised in growth and shows a dramatic increase in chromosome missegregation and aneuploidy. We propose that Abp1p and Cbh1p belong to a family of structural proteins that organize A+T-rich chromatin, such as that found at centromeres, into a higher-order structure. Multiple CENP-B homologs in fission yeast may reflect the redundancy observed within the centromeric central cores and K elements. Thus, the fission yeast centromere, a simple example of a regional centromere, exhibits layers of functional redundancy, making it a useful model with potentially important implications for mammalian systems.
MATERIALS AND METHODS
Strains, plasmids, and media.
The following fission yeast strains were used: Sp223 (h− ade6-216 leu1-32 ura4-294), SBP040398 (h+ leu1-32 ura4-D18), ED666 and SBP060498-21D (both h+ ade6-210 leu1-32 ura4-D18), ED667 and SBP060498-21A (both h− ade6-216 leu1-32 ura4-D18), SBP070196-F5C (h− abp1Δ::ura4+ ade6-216 leu1-32 ura4-D18), SBPD021298 (h+/h− ade6-210/ade6-216 leu1-32/leu1-32 ura4-D18/ura4-D18), SBPD051198 (h+/h− ade6-210/ ade6-216 cbh1+/cbh1Δ::hisG-ura4+-hisG leu1-32/leu1-32 ura4-D18/ura4-D18), SBP060498-21B (h+ ade6-216 cbh1Δ::hisG-ura4+-hisG leu1-32 ura4-D18), SBP060498-21C (h− ade6-210 cbh1Δ::hisG-ura4+-hisG leu1-32 ura4-D18), SBP072198-1A (h+ abp1Δ::ura4+ ade6-216 leu1-32 ura4-D18), SBP072198-1B (h+ ade6-216 leu1-32 ura4-D18), SBP072198-1C (h− abp1Δ::ura4+ ade6-216 cbh1Δ::hisG-ura4+-hisG leu1-32 ura4-D18), SBP072198-1D (h− ade6-216 cbh1Δ::hisG-ura4+-hisG leu1-32 ura4-D18), and SBP071198-2 (h+ cbh1::GFP-pBSIIKS-ura4+-cbh1C leu1-32 ura4-D18). Strain SBPD021298 was isolated from a cross between ED666 and ED667. Strains SBP060498-21A to -D are the four sporulation products from one SBPD051198 tetrad. Strains SBP072198-1A to -D are the meiotic products of one tetrad from a mating between SBP060498-21B and SBP070196-F5C.
The following fission yeast strains carry the 78-kb linear cen1 artificial chromosome pSp(cen1)-7L-sup3E (23): SBP072098-21B3 (h− ade6-704 leu1-32 ura4−/cen1+ sup3E+ ura4+ TRP1 URA3), SBP072098-21B5 (h− ade6-704 cbh1Δ::hisG-ura4+-hisG leu1-32 ura4−/cen1+ sup3E+ ura4+ TRP1 URA3), SBP072098-21B7 (h− ade6-704 cbh1Δ::hisG-ura4+-hisG leu1-32 ura4-294/cen1+ sup3E+ ura4+ TRP1 URA3), SBP082898-4C-204 (h+ abp1Δ::ura4+ ade6-704 cbh1Δ::hisG-ura4+-hisG leu1-32 ura4-294/cen1+ sup3E+ ura4+ TRP1 URA3), and SBP082996/pSp(cen1)-7L-sup3E (abp1Δ::ura4+ ade6-704 leu1-32 ura4-294/cen1+ sup3E+ ura4+ TRP1 URA3). Strains SBP072098-21B3, -21B5, and -21B7 are from a mating between SBP060498-21B and SBP32590/pSp(cen1)-7L-sup3E (h− ade6-704 leu1-32 ura4-294/cen1+ sup3E+ ura4+ TRP1 URA3). Tetrads were dissected using an MSM Systems series 200 microscope and micromanipulator (Singer Instruments, Somerset, United Kingdom). Growth media were as previously described (22, 46). DNA fragments used for gene replacement were introduced into S. pombe by alkali-cation yeast transformation (Bio101, La Jolla, Calif.).
Protein purification and mobility shift assays.
S. pombe whole-cell protein extracts were prepared and mobility shift assays were performed as previously described (24).
Determination of an Abp1p equilibrium binding constant.
An apparent equilibrium binding constant was calculated for Abp1p using a previously described electrophoretic mobility shift method that corrects for nonspecific binding of protein to DNA (3, 4, 7). Increasing amounts of radiolabeled c2-10 DNA fragment (0.04 to 5.0 fmol; ∼0.01 to 1 ng) were mixed with a constant amount of affinity-purified Abp1 protein (1 μl; estimated to be 4 to 8 ng by silver staining) (24) and poly(dI-dC) nonspecific DNA (100 ng). Reaction mixtures (10 μl) were incubated for 20 min at room temperature in binding buffer containing 10 mM HEPES (pH 8.0), 6 mM MgCl2, 2 mM NaF, 120 mM KCl, 0.5 mM dithiothreitol, 0.5 μg of bovine serum albumin, and 10% glycerol. The amounts corresponding to retarded protein-DNA complexes ([CD_s_]) and unbound DNA probe ([_Ds_]) were determined by liquid scintillation counting or with a molecular imager (Bio-Rad Laboratories, Hercules, Calif.). At equilibrium the relationship between free and bound specific probe DNA is given by the following equations:
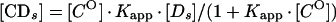 |
1 |
|---|
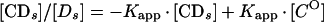 |
2 |
|---|
where [_C_O] is the number of Abp1p DNA-binding sites present. Equation 1 and equation 2 describe a hyperbolic curve (Fig. 2A) and straight line (Fig. 2B), respectively. Equation 2 is solved graphically by plotting [CD_s_]/[_Ds_] versus [CD_s_]. _K_app is given by the slope of the line, and [_C_O] is given by the x intercept.
FIG. 2.

Determination of an equilibrium binding constant for Abp1p to c2-10 DNA. (A) Saturation binding curve using increasing amounts of radiolabeled c2-10 DNA and a constant amount of affinity-purified Abp1p in the presence of nonspecific poly(dI-dC) DNA. The concentration of Abp1p complexed with c2-10 [CD_s_] is plotted versus free c2-10 [_Ds_]. (B) The same data are plotted to graphically solve equation 2 (see Materials and Methods). The slope of the line equals _K_app, and the x intercept equals _C_O, the number of binding sites in the reaction. (C) Correction of _K_app for the contribution made by binding to nonspecific DNA. Binding between constant amounts of affinity-purified Abp1p and radiolabeled c2-10 DNA was competed by increasing amounts of nonspecific poly(dI-dC) DNA. The inverse of _K_app, determined from equation 3, is plotted versus the concentration of nonspecific DNA [_Dn_O] to graphically solve equation 5 for Kn and Ks, the nonspecific and specific binding constants, respectively (3, 7). The y intercept equals 1/Ks, and the slope of the line is equal to Kn/Ks. Thus, Kn is equal to the slope divided by the y intercept.
From a parallel set of reactions with constant amounts of protein (∼4 to 8 ng) and radiolabeled c2-10 DNA fragment (0.63 fmol; ∼0.1 ng), together with increasing amounts of poly(dI-dC) nonspecific DNA (250- to 22,500-fold mass excess over specific DNA), nonspecific and specific equilibrium binding constants, Kn and Ks, were determined using the following relationships (3, 7):
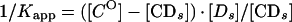 |
3 |
|---|
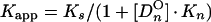 |
4 |
|---|
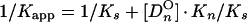 |
5 |
|---|
The value for _C_O, determined in Fig. 2B, was used to calculate 1/_K_app from equation 3. The values for Kn and Ks were determined graphically from equation 5 by plotting 1/_K_app versus [_Dn_O] (Fig. 2C). Kn/Ks is given by the slope of the line, and 1/Ks equals the y intercept. Thus, Kn equals the slope of the line divided by the y intercept.
Gene replacement, green fluorescent protein (GFP) fusion, and other genetic manipulations.
A plasmid bearing the cbh1 cDNA as a _Nde_I-_Sal_I fragment, kindly supplied by J. Hurwitz (Memorial Sloan-Kettering Cancer Center, New York, N.Y.), was used for a one-step gene replacement of the cbh1+ open reading frame with a selectable marker gene (57). The plasmid was digested with _Eco_RI, treated with Klenow fragment, and then digested with _Bam_HI to release a 1.3-kb fragment from the 1.5-kb open reading frame of cbh1+. A 4.3-kb _Bgl_II (blunted with Klenow fragment)-_Bam_HI fragment carrying the hisG-ura4+-hisG cassette from pDM291 (43) was ligated in its place. The altered cbh1 gene was released with _Nde_I-_Sal_I such that it carries 80 bp of homologous sequences that correspond to the first 27 amino acids at the N terminus of Cbh1p and 188 bp that correspond to the last 22 amino acids at the C terminus and sequences downstream of the stop codon. Gene replacement was carried out in the diploid S. pombe strain SBPD021298, and 13 Ura+ transformants were screened by Southern analysis to identify heterozygous diploids. Only one isolate showed the correct structure. It was sporulated, and tetrad analysis was performed as previously described (24) to examine the haploid progeny carrying the cbh1 deletion.
A cbh1-GFP gene fusion was produced in strain SBP071198-2 by integrating a plasmid into the native cbh1+ gene such that cbh1-GFP expression was controlled by the native promoter and wild-type cbh1+ expression was eliminated. Construction of this integration plasmid was carried out in a multistep process. A 0.6-kb fragment (cbh1C) representing the C terminus of Cbh1p was amplified by PCR with a primer that spanned the second from the left _Eco_RI site in cbh1+ (Fig. 3A) and a primer that substituted an _Spe_I site in place of the stop codon. The resulting DNA product was digested with _Eco_RI and _Spe_I, cloned into pBluescript II KS(−) (Stratagene, La Jolla, Calif.), and sequenced to confirm that only the intended base changes were introduced. A pBluescript II KS(−) ura4+-GFP integration vector was constructed by blunt-end ligation of S. pombe ura4+ contained on a 1.8-kb _Hin_dIII-_Hin_dIII fragment into the _Acc_65I site of the polylinker (after treatment of both DNA preparations with Klenow fragment) and by ligation of wild-type GFP on a 0.9-kb _Spe_I-_Sac_II fragment (from pRS316GalGFP; a gift from H. Mosch and S. Kron, Whitehead Institute, Biomedical Research, Cambridge, Mass. [11]) into the corresponding pBluescript polylinker sites. The 0.6-kb _Eco_RI-_Spe_I fragment carrying cbh1C was then cloned upstream of GFP into the _Eco_RI-_Spe_I sites of the integration vector such that cbh1C and GFP were joined in frame at the _Spe_I site. This cbh1C-GFP-ura4+-bearing plasmid was linearized within cbh1 sequences at a unique _Pst_I site 192 bp from the C terminus of Cbh1p and used to transform strain SBP040398 (_ura4_Δ). DNAs from Ura+ transformants were screened by Southern analysis. Five of six isolates showed a hybridization pattern consistent with integration at the native cbh1+ gene. Recombination between cbh1+ and cbh1C-GFP produced cbh1-GFP, whose expression is driven by the native promoter. cbh1-GFP encodes full-length Cbh1p (514 amino acids [aa]) linked by the amino acid sequence TSS to full-length GFP (238 aa), followed by the remainder of the integration vector and a duplication of cbh1C that is not expressed because it lacks a promoter.
FIG. 3.

The CENP-B-related gene cbh1+ is not essential, but deletion of both abp1+ and cbh1+ has a synergistic effect on fission yeast growth. (A) Structures of cbh1+ and the Δ_cbh1_ construct (see Materials and Methods). The solid bars correspond to radiolabeled probes used for panel B. Restriction site abbreviations: B, _Bam_HI; Bg, _Bgl_II; E, _Eco_RI; N, Nhe_I; S, Sal_I. (B) The viability of Δ_cbh1 strains was verified by Southern analysis of the meiotic products (A to D; SBP060498-21) corresponding to one tetrad that resulted from sporulation of a heterozygous (Δ_cbh1/cbh1+) parent (P; SBPD051198). Duplicate aliquots of _Sal_I-digested fission yeast DNA were electrophoresed on a 1% agarose gel, blotted, and probed with a 1.3-kb _Nhe_I-_Bam_HI hisG fragment, which hybridizes to a 6.4-kb _Sal_I fragment (left), or a mixture of the 0.6-kb _Eco_RI-_Bgl_II and 0.7-kb Bgl_II-Bam_HI fragments from cbh1+, which hybridize to a 2.9-kb Sal_I fragment (right). (C) Extract prepared from a Δ_cbh1 strain (SBP060498-21C) lacks HI-binding activity. Binding reactions were performed as described in the legend to Fig. 1. The specificity of Cbh1p for the HI PCR-generated K" fragment (Fig. 1D) is demonstrated in the rightmost lane, where binding to 5 fmol of radiolabeled HI fragment was competed by addition of a 25-fold molar excess of unlabeled HI fragment. (D) Growth of a Δ_cbh1 strain was equivalent to that of the wild type, but a Δ_abp1 Δcbh1 strain grew extremely slowly. The strains (SBP072198-1A to -D) were grown for 9 days at room temperature and photographed.
Immunoprecipitation of formaldehyde-fixed chromatin.
Exponentially growing 100-ml cultures of S. pombe (107 cells/ml) were fixed for 30 min at 32°C by the addition of 10 ml of fixation solution (11% formaldehyde, 0.1 M NaCl, 1 mM NaEDTA, 0.5 mM NaEGTA, 50 mM Tris-HCl, pH 8.0). Formaldehyde-induced cross-linking was quenched for 5 min at room temperature by the addition of glycine to a final concentration of 120 mM. The cells were washed and lysed with glass beads, and the chromatin was sheared to an average length of 0.8 kb as previously described (58). GFP-tagged Cbh1p was incubated overnight at 4°C with 5 μg of affinity-purified polyclonal rabbit antibody against GFP (a gift from Jason Kahana, Ludwig Institute for Cancer Research, University of California, San Diego [73]), followed by capture of the immunocomplexes with protein G-Sepharose beads for 4 h at 4°C. The immunoprecipitates were washed as previously described (28), and bound DNA was released and purified according to standard methods (52) with the following modifications. Sheared lambda DNA (2 μg) was added before cross-linking was reversed, and RNase treatment was delayed until after the cross-links were reversed. Portions of total and immunoprecipitated DNA (1/2,200 and 1/12, respectively, for centromeric K repeat primers and 1/1,100 and 1/6, respectively, for ars3002, leu1, top2, and hypothetical gene SBPC1A4.01 [accession no. AL031174] primers) were used for PCR amplification (25 cycles). The locations of the primers are indicated in Fig. 4A. The PCR products were electrophoresed on 2% NuSieve 3:1 agarose gels (FMC BioProducts, Rockland, Maine), visualized with ethidium bromide, and digitally photographed with an AlphaImager 2000 system (Alpha Innotech, San Leandro, Calif.).
FIG. 4.

GFP-tagged Cbh1p is associated with both centromeric and noncentromeric chromatin in vivo. (A) Schematic of a portion of chromosome II shows the locations that were examined for association with Cbh1p-GFP using a ChIP technique (51, 52). Besides ars3002 and fragment HI of the centromeric K repeat, which were tested as likely cognate targets because they bind Cbh1p in vitro, several other random noncoding and coding sequences were also tested. PCR primer sets are indicated by vertical lines. Fragments AB, HI, and M are located within the centromeric K repeat that is found in 16 to 18 copies in the S. pombe genome. The single-copy genes top2, leu1, and a hypothetical open reading frame (hyp) are located greater than 300 kb from cen2 (Sanger Centre S. pombe Genome Project [http://www.sanger.ac.uk]). The location of ars3002, a telomere-proximal unique sequence on chromosome III, is not shown. Centromeric DNA elements are indicated by solid or patterned boxes. Genes are marked by open, pointed symbols, with the direction of transcription proceeding toward the tip. (B) Coimmunoprecipitation of centromeric K repeat DNA, ars3002 DNA, and random transcribed DNA with Cbh1p-GFP. Cross-linked chromatin from an integrated cbh1-GFP (SBP071198-2) or cbh1+ (SBP040398) strain was immunoprecipitated with antibody (+Ab) against GFP (lanes 1 and 3, respectively) or beads only (−Ab) (lanes 2 and 4, respectively). Total DNA (lanes 5 and 6) and immunoprecipitated DNA were used for PCR with primers specific for the indicated fragments (see Materials and Methods). The corresponding densitometry values for immunoprecipitated DNA normalized to total DNA from cbh1-GFP and cbh1+ strains, respectively, are as follows: AB, 0.707 and 0.192; HI, 0.385 and 0.167; M, 0.391 and 0.147; top2, 0.268 and 0.184; leu1, 0.255 and 0.196; hyp, 0.253 and 0.212; and ars3002, 0.253 and 0.212. The size of each amplified fragment is indicated in base pairs.
Minichromosome stability assay.
The loss of a linear cen1 minichromosome, pSp(cen1)-7L-sup3E, was assayed by the appearance of red colony color following plating on limiting adenine medium Y+4 (0.5% yeast extract, 3% glucose, 2% agar plus 4 supplements [histidine, leucine, lysine, and uracil; 50 μg each/ml]). In cells that retain the sup3E+-bearing minichromosome, the ade6-704 mutation is complemented by expression of an opal suppressor tRNA encoded by sup3E+, yielding white colonies. The minichromosome loss rate is equal to the number of half-red colonies (or greater than half sectored) divided by the total number of colonies, excluding those that are entirely red.
Cell viability and growth rate measurements.
S. pombe cultures were monitored spectrophotometrically (_A_600) over a period of 30 to 60 h until the cultures reached stationary growth. Growth curves were plotted, and the generation time was calculated from the time frame corresponding to exponential growth. Viability tests were conducted by counting cells on a hemacytometer (3 × 106 cells to 2 × 107 cells/ml) and plating serial dilutions of a known number of cells on rich medium. The plates were grown at 32°C for 3 to 14 days, and the viability was calculated by dividing the number of colonies formed by the theoretical number of cells plated.
FACS analysis and microscopy.
The strains were grown to a density of ∼3 × 106 cells/ml in rich medium or for three generations under conditions of nitrogen starvation in synthetic proline medium (19, 44, 56). Approximately 2 × 107 cells were fixed in 5 ml of 70% ethanol overnight at 4°C. The fixed cells were washed with 1 ml of water, treated with 1 ml of RNase A (1 mg/ml) for 3 h at 37°C, digested with 0.2 ml of pepsin (5 mg/ml in 55 mM HCl) for 45 min at 37°C, and then stained overnight at 4°C in 0.5 ml of 10× propidium iodide solution (5 mg of propidium iodide + 1.42 g of MgCl2 · 6H2O dissolved in 100 ml of 180 mM Tris [pH 7.5]–190 mM NaCl buffer). Approximately 2 × 106 cells were diluted in 2 ml of 0.1× propidium iodide solution in Falcon 2054 tubes and shipped overnight for fluorescence-activated cell sorter (FACS) analysis (Cytometry Research Services, La Jolla, Calif.). From each preparation, a monolayer of cells was dried onto polyethylenimine-treated glass slides, coverslips were mounted with 10 μl of Vectashield (H-1000; Vector Laboratories, Inc., Burlingame, Calif.), and the strains were examined by fluorescence microscopy on the rhodamine channel of a Nikon Microphot-SA fluorescence microscope (A. G. Heinze Precision Optics, Irvine, Calif.). The numbers of branched or multiseptated cells, dividing cells with normal or aberrant chromatin masses, interphase cells, and stained cells lacking a discernible chromatin mass were tabulated. Digital images were recorded with Image-1 version 4.0 software (Universal Imaging Corporation, West Chester, Pa.), and photographs were prepared with Adobe Photoshop 5.0.
RESULTS
The S. pombe CENP-B-related protein Abp1p binds in vitro to at least six sites within cc2.
The central core and K repeat, DNA elements that are necessary and sufficient for centromere function in S. pombe, contain several protein binding sites for Abp1p and Cbh1p (24, 38, 50). In addition to a single site for Cbh1p in the centromere enhancer, we have demonstrated previously that Abp1p binds in vitro to three small (<400-bp) fragments in one half of the centromeric central core of chromosome II (cc2), based on electrophoretic mobility shift assays and a supershift assay with antibody directed against Abp1p (Fig. 1A) (24). These fragments carry A+T-rich sequences (73 to 75% A+T) but do not share a conserved primary sequence or palindromic motif. However, these fragments, designated c2-8, c2-10, and c2-11, are not the only locations of DNA-binding sites for Abp1p in cc2 (Fig. 1B). Electrophoretic mobility shift assays were carried out with small DNA fragments (<500 bp) from the other half of cc2 (c2-13 to c2-20) and whole-cell protein extracts enriched for the chromatin fraction from abp1+ and Δ_abp1_ fission yeast strains (24). Specific mobility shifts were observed with fragments c2-13, c2-15, and c2-18, equivalent to those observed for fragments c2-8 or c2-11 (24). With fragment c2-13, a single band was retarded that was not detectable if the protein extract was prepared from a Δ_abp1_ strain, demonstrating that this protein-DNA interaction is Abp1p dependent (Fig. 1C). Similarly, fragments c2-15 and c2-18 with extract from an abp1+ strain each yielded two shifted bands that were not detectable or were greatly reduced with extract from a Δ_abp1_ strain. In these cases, the fastest-migrating band was absent and the slower-migrating band was diminished when Δ_abp1_ extracts were used. Altogether, six in vitro binding sites for Abp1p have now been identified in cc2, excluding any that may be overlooked because of their proximity to restriction fragment ends. Of the nine centromeric DNA fragments that have been identified as in vitro binding sites for Abp1p, five (c2-8, -10, -13, -18, and O [Fig. 1B and D]) show an 11-nucleotide A+T-rich palindromic sequence separated by 3 nucleotides from the sequence CAAT [(A/T)2(A/G)(A/T)2N(A/T)2T(A/T)2N3CA2T]. Only one of these fragments, c1-8, contains a sequence match to the 10-nucleotide Cbh1p consensus binding site previously derived from in vitro DNase I footprinting assays [(C/T)(A/G)ATAT(C/T)(A/G)TA] (38). Thus, Abp1p and Cbh1p appear to recognize distinct binding motifs based on the lack of homology in pairwise comparisons between the two groups of DNA fragments and the lack of competition by a 25-fold molar excess of a fragment from one group in an electrophoretic mobility shift of a fragment from the other group.
Abp1p binds in vitro to cc2 DNA with high affinity.
Because high-affinity in vitro binding may be indicative of an in vivo-relevant association, we quantitatively measured the affinity of Abp1p for a cognate site in cc2 DNA. A bandshift method that accounts for nonspecific binding was chosen to determine an equilibrium binding constant for Abp1p to c2-10 DNA (3, 4, 7). This cc2 fragment exhibits the strongest affinity for Abp1p, based on the proportion of bound to unbound radiolabeled DNA probe. Binding reactions, containing a constant amount of affinity-purified Abp1p and nonspecific poly(dI-dC) DNA mixed with serial dilutions of labeled c2-10 fragment, were electrophoresed, and the amount of protein-DNA complex versus unbound DNA was plotted (Fig. 2A). A prominent fast-migrating complex and a slower-migrating minor complex are visible on the autoradiograph (Fig. 2A, inset). Saturation binding results for the predominant complex are plotted, for which the simplest case, one Abp1p molecule bound to one c2-10 DNA molecule, is assumed. No cooperative binding, as evidenced by a sigmoidal curve, was apparent. When the data are plotted in the equivalent of a Scatchard plot, a line is defined whose slope equals the negative of _K_app and whose x intercept equals _C_O, the total number of binding sites present (Fig. 2B and equation 2). Values for _K_app and _C_O were determined in three separate experiments using a single preparation of affinity-purified Abp1p. The average value for _K_app was 1.1 × 1010 M−1 (range, 0.49 × 1010 to 1.5 × 1010 M−1), and the average for _C_O was 2.1 × 10−10 M (range, 1.3 × 10−10 to 2.5 × 10−10 M), both values showing good reproducibility.
The apparent equilibrium binding constant is influenced by the presence of nonspecific DNA in the binding reactions. To correct for this influence, we ran a parallel set of reactions that contained increasing amounts of nonspecific poly(dI-dC) DNA while the amount of radiolabeled specific c2-10 DNA and Abp1p protein were kept constant. The binding constants for nonspecific (Kn) and specific (Ks) binding were ascertained from a plot of 1/_K_app versus the concentration of nonspecific DNA (_Dn_O) (Fig. 2C). The slope of the line and the y intercept gave Kn/Ks and 1/Ks, respectively. Based on the average of three experiments, Kn and Ks were 2.6 × 104 M−1 (range, 0.56 × 104 to 4.0 × 104 M−1) and 7.4 × 109 M−1 (range, 2.7 × 109 to 11 × 109 M−1), respectively. This value for Ks is an order of magnitude stronger than that obtained for human centromere protein CENP-B and yeast centromere-binding protein Cbf1p to their cognate sites but weaker than the binding constants for some transcription factors (Table 1).
TABLE 1.
Abp1p shows an intermediate binding constant compared to other DNA-binding proteins
| Protein | _K_app (M−1)a | Reference |
|---|---|---|
| Yeast Cbf1p | 3 × 108 b | 4 |
| Human CENP-B | 6 × 108 b | 48 |
| Xenopus 5S rRNA TFIIIA | 1 × 109 | 25 |
| Fission yeast Abp1p | 7 × 109 b | This study |
| Adenovirus major late TF | 1 × 1010 | 12 |
| Yeast transcription factor tau | 5 × 1010 b | 3 |
| Human TFIIIC | 2 × 1011 b | 7 |
When excess affinity-purified Abp1p is used in a mobility shift assay with the c2-10 fragment, a ladder of up to three bands of retarded mobility is seen. In vitro experiments designed to discriminate between additional protein molecules bound to the c2-10 DNA fragment versus protein molecules bridging the same site on separate DNA molecules, the situation observed in vitro with mammalian CENP-B (72), did not detect any evidence for the latter possibility in the case of Abp1p (data not shown). Thus, for all of our calculations we presumed that the fastest-migrating complex represents Abp1p bound at a single high-affinity DNA site and that the slower-migrating bands represent complexes between a second and third Abp1p molecule and a single c2-10 DNA molecule. We previously reported that, using a strain overexpressing Abp1p under control of its native promoter on a multicopy plasmid, we obtained ∼50 to 100 pmol of affinity-purified Abp1p from ∼5 × 1010 cells and recovered 2% of the total binding activity from the preparation (24). This corresponds to a predicted number of 30,000 to 60,000 Abp1p molecules per cell in the overexpressing strain. We also previously showed that Abp1p is enriched ∼2-fold over wild-type levels in whole-cell extracts isolated from this strain and that ∼45% of the binding activity is found in the chromatin fraction from wild-type cells (24). Therefore, we estimate that roughly 6,000 to 13,000 chromatin-associated Abp1p molecules are present per wild-type cell.
Cbh1p, a second member of the CENP-B-related protein family in S. pombe, is not essential for viability.
The fission yeast _cenpB_-homologous gene cbh1+ encodes a 60-kDa putative DNA-binding protein which recognizes a site distinct from that bound by Abp1p (38). To date, a single in vitro binding site for Cbh1p has been found in the HI fragment within the centromere enhancer portion of the S. pombe centromeric K repeat (Fig. 1D), and three sites have been located in ars3002 DNA (38, 50). Contrary to a previous report (38), however, we find that cbh1+ is not an essential gene. In the null strain constructed in this study, the cbh1+ open reading frame between the indicated Eco_RI and Bam_HI sites was removed (Fig. 3A), leaving 27 N-terminal amino acids and 22 C-terminal amino acids encoded by cbh1, separated by a selectable marker cassette carrying ura4+. Following sporulation of a diploid strain that is heterozygous for the null allele (cbh1+/Δ_cbh1), all four spores from a tetrad were viable, and two of the four spores were Ura4+. Southern analysis confirmed that the two Ura4− spores carried a wild-type cbh1+ gene and the two Ura4+ spores carried Δ_cbh1 (Fig. 3B). Although the null construct encodes an altered product that contains a short portion of the amino terminus of cbh1+, extracts made from this strain lacked a protein that was capable of binding to fragment HI from the centromeric K repeat, demonstrating that the electrophoretic mobility shift that we previously reported (6, 50) is indeed Cbh1p dependent (Fig. 3C).
Cbh1p is present at centromeric chromatin in vivo but is not limited to this region.
The association of Cbh1p protein with centromeric and noncentromeric regions was examined by chromatin immunoprecipitation (ChIP) using an S. pombe strain that expresses a GFP-tagged version of Cbh1p as the only form of Cbh1p in the cell (28, 51, 52). Strains expressing recombinant Cbh1p-GFP or native Cbh1p were treated in situ with formaldehyde, a reversible, high-resolution (2-Å) cross-linking agent that preserves protein-protein and protein-nucleic acid interactions in vivo. Cross-linked chromatin from cbh1-GFP and cbh1+ strains was sheared to an average DNA length of 800 bp and immunoprecipitated with antibody against GFP. In order to purify ChIP-enriched DNA, protein-DNA cross-links were reversed by an overnight incubation at 65°C, and samples were treated with RNase and deproteinated. This material was used as a template for PCR with centromeric primers corresponding to fragments AB, HI, and M of the K repeat and noncentromeric primers corresponding to top2, leu1, and an adjacent hypothetical open reading frame, all located greater than 300 kb from cen2 (Fig. 4A). Primers to ars3002, located near one telomere of chromosome III and previously implicated in Cbh1p binding (38), were also employed. With total DNA from either a cbh1-GFP or a cbh1+ strain, all of these primers directed amplification of PCR products of the expected size (Fig. 4B, lanes 5 and 6, respectively). Amplification from immunoprecipitated DNA was dependent on addition of GFP antibody to a cbh1-GFP strain (Fig. 4, lane 1) and was either not detected or barely detected without antibody or with immunoprecipitated DNA from a cbh1+ strain (Fig. 4, lanes 2 to 4). Cbh1p-GFP was detected readily by ChIP at the centromeric K repeat, which contains an in vitro binding site for Cbh1p, and weakly at all of the other regions tested. These results seem to indicate that the Cbh1p binding site is more degenerate than previously determined (38), as only the top2 region contains a match to the consensus site. Nonetheless, Cbh1p, expressed as a GFP fusion protein, interacts with both centromeric and noncentromeric chromatin in vivo.
A Δ_abp1 Δcbh1_ double mutation results in a profound poor-growth phenotype.
In contrast to cellular depletion of Abp1p, which results in cold sensitivity at 17°C, a low growth rate at 32 and 37°C, and a profound defect in events leading to meiosis (24), depletion of Cbh1p has no measurable effect on growth rate or meiotic events. Depletion of either protein, however, subtly elevates mitotic loss of a cen1_-derived artificial chromosome. The growth rates for wild-type and Δ_cbh1 strains are identical at 17, 25, 32, and 37°C, with a generation time of 2 h at 32°C (Fig. 3D and data not shown) in comparison to a 4-h generation time at 32°C for a Δ_abp1_ strain. Among strains carrying a mitotically stable 78-kb linear cen1 minichromosome marked with a selectable gene, a Δ_cbh1_ strain shows a fourfold increase in loss frequency compared to the wild type, similar to the sevenfold increase in loss frequency seen in a Δ_abp1_ strain (Table 2). Progeny carrying both deletions Δ_abp1_ and Δ_cbh1_, however, are very compromised for growth, although the combination is not lethal. A Δ_abp1 Δcbh1_ strain grew extremely slowly at all four temperatures tested (Fig. 3D and data not shown), with an 8-h generation time at 32°C and a defect in cytokinesis. Three independent Δ_abp1 Δcbh1_ isolates were examined and are equivalent in growth rate and morphology. Because of these growth defects, it was not possible to measure a minichromosome loss frequency for a double-deletion strain. Thus, although cbh1+ and abp1+ are not essential genes, they appear to share a related function such that cell growth is severely compromised if both gene products are lacking.
TABLE 2.
Absence of Cbh1p has a subtle effect on the mitotic stability of a 78-kb linear minichromosome
| Strain | Genotype | Chromosome loss per divisiona | Fold increase |
|---|---|---|---|
| SBP072098-21B3 | abp1+ cbh1+ | 5.8 × 10−4 | 1 |
| SBP072098-21B7 | abp1+ cbh1Δ | 2.3 × 10−3 | 4 |
| SBP082996/pSp(cen1)- 7L-sup3E | abp1Δ cbh1+ | 3.9 × 10−3 | 7 |
| SBP082898-4C-204 | abp1Δ cbh1Δ | NAb |
Abnormal morphology and unequal distribution of chromatin in a Δ_abp1 Δcbh1_ strain.
To identify or observe potential morphological and cell cycle distribution defects in a Δ_abp1 Δcbh1_ strain, cells were fixed, stained with propidium iodide, and examined microscopically and by FACS for evidence of gross chromosome missegregation or cell cycle anomalies. When observed by fluorescence microscopy, cultures of wild-type, Δ_abp1_, or Δ_cbh1_ strains appeared similar (Fig. 5A to C). Approximately 20 to 30% of the cells contained a C-shaped chromatin mass typical of interphase nuclei, whereas ∼70% of cells had entered mitosis (Table 3). Altogether, less than 5% of the cells lacked a discernible chromatin mass or showed abnormal cellular morphology. In contrast, nearly 50% of the cells in an exponentially growing Δabp1 Δcbh1 culture appeared anucleate, branched, or multiseptated (Table 3 and Fig. 5F to G). The increased proportion of anucleate cells presumably reflects a corresponding increase in asymmetric chromosome segregation during mitosis in the double-deletion strain. However, branched and multiseptated cells, a defect commonly seen in nutritionally deprived cells (42), may be the result of an extremely low growth rate, leading to an uncoupling of temporal events that coordinate cellular growth and mitosis. In the remaining 51% of Δ_abp1 Δcbh1_ cells, about one-quarter (12%) had typical C-shaped chromatin masses corresponding to interphase nuclei, and three-quarters (39%) had entered mitosis, proportions similar to those observed for a wild-type strain (Table 3). Among the 39% of Δ_abp1 Δcbh1_ cells that had entered mitosis, only ∼70% appeared to segregate chromosomes normally compared to >99% of wild-type or Δ_cbh1_ mitotic cells and >90% of Δ_abp1_ mitotic cells (Table 4). Unequal distribution of chromatin (assessed by focusing through the entire depth of the chromatin mass) was seen in 14 and 4% of Δ_abp1 Δcbh1_ and Δ_abp1_ cells, respectively (Fig. 5J and E), whereas lagging chromosomes (incomplete segregation, with some chromatin left at the midzone) were seen in 6 and 2% of Δ_abp1 Δcbh1_ and Δ_abp1_ cells, respectively (Fig. 5H, I, and D). Asymmetric distribution of chromatin, in which all of the staining material was found in one half of a cell with a septum, was seen in 3% of the Δ_abp1 Δcbh1_ dividing cell population. In general, segregation defects were more pronounced and more varied in Δ_abp1 Δcbh1_ cells than in Δ_abp1_ cells and were not evident or were rare in wild-type and Δ_cbh1_ cells. Overall, ∼30% of normally shaped Δ_abp1 Δcbh1_ dividing cells showed gross chromosome missegregation.
FIG. 5.

Chromosome missegregation and defective cytokinesis in a Δ_abp1 Δcbh1_ strain. Ethanol-fixed, propidium iodide-stained cells were examined by fluorescence microscopy as described in footnote a to Table 3. Wild-type (A) and Δ_cbh1_ (B) cells appeared uniformly normal, whereas Δ_abp1_ (C to E) cells showed rare instances of missegregation. In contrast, Δ_abp1 Δcbh1_ (F to J) cells showed frequent mistakes in chromosome segregation, as well as many cells with branched, multiseptated morphology (magnification, ×120). Cell septa (arrowheads), lagging chromosomes (arrows), and unequal distribution of chromatin masses (asterisks) are indicated. (See Tables 3 and 4 for a summary of the data). All cultures were grown at 32°C.
TABLE 3.
Abnormal morphology and chromosome segregation is evident in Δ_abp1 Δcbh1_ fission yeasta
| Strain description | Branched or multiseptated | Anucleate | Interphase | Mitotic | Total | Viability (%) | Generation time (h) |
|---|---|---|---|---|---|---|---|
| WT | 0 (<0.3) | 14 (4) | 71 (20) | 264 (76) | 349 | 71 | 2 |
| Δcbh1 | 0 (<0.4) | 0 (<0.4) | 81 (31) | 177 (69) | 258 | 81 | 2 |
| Δabp1 | 1 (0.4) | 7 (3) | 70 (29) | 168 (68) | 246 | 47 | 4 |
| Δabp1 Δcbh1 | 39 (30) | 24 (19) | 16 (12) | 50 (39) | 129 | 24b | 8 |
TABLE 4.
Unequal chromosome segregation and other mitotic defects are elevated in Δ_abp1 Δcbh1_ fission yeasta
| Strain genotypeb | No. (%) of cells | ||||||
|---|---|---|---|---|---|---|---|
 |
|||||||
| Normal | Unequal | Lagging | Placement | Asymmetric | “Cut” | Total | |
| WT | 183 (100) | 0 (<0.6) | 0 (<0.6) | 0 (<0.6) | 0 (<0.6) | 0 (<0.6) | 183 |
| Δcbh1 | 123 (100) | 0 (<0.8) | 0 (<0.8) | 0 (<0.8) | 0 (<0.8) | 0 (<0.8) | 124 |
| Δabp1 | 147 (92.5) | 6 (3.8) | 3 (1.9) | 3 (1.9) | 0 (<0.6) | 0 (<0.6) | 159 |
| Δabp1 Δcbh1 | 71 (69.6) | 14 (13.7) | 6 (5.9) | 6 (5.9) | 3 (2.9) | 2 (2.0) | 104 |
Because Cbh1p binds in vivo to the ars3002 sequence and, as previously shown, both Cbh1p and Abp1p bind in vitro to this sequence (38), suggesting they might play a role in DNA replication, we investigated whether strains with these proteins depleted showed any evidence of a cell cycle delay. Fission yeast, a haploid organism, spends most of the cell cycle in G2 with a 2C DNA content. A replication defect, however, is predicted to result in a higher proportion of cells accumulating in G1 with unreplicated DNA. Typically, less than 15% of the fission yeast cell cycle is spent in G1 (45), and a G1 peak is often not observed, as is the case for the wild-type, Δ_abp1_, and Δ_cbh1_ strains examined here (Fig. 6A to C). The predominant peak represents G2 cells with a 2C DNA content, followed by a smaller 4C peak attributable to exponentially dividing cells that initiated a new round of DNA synthesis before completing cytokinesis. In contrast, the Δ_abp1 Δcbh1_ population of cells showed a broad, flattened distribution, with many cells at higher ploidy and a slight increase in cells with a DNA content less than 2C (Fig. 6D). These results are consistent with a defect that results in unequal chromosome segregation, producing cells with both higher and lower ploidy in a Δ_abp1 Δcbh1_ strain. However, in a haploid organism such as S. pombe, loss of a chromosome is lethal, effectively leading to an accumulation of cells only at higher ploidy. Also contributing to the apparent high ploidy in this strain is a cytokinesis defect that results in chains of connected cells. Thus, Abp1p and Cbh1p appear to share some overlapping functions necessary for equal mitotic chromosome segregation. The fact that the Δ_abp1 Δcbh1_ double mutation has a synergistic, but nonlethal, effect on chromosome missegregation hints at the possible existence of additional members of this CENP-B-related protein family in fission yeast.
FIG. 6.

A Δ_abp1 Δcbh1_ double-deletion strain accumulates cells at higher ploidy. DNA content was estimated by FACS analysis. See Materials and Methods for details of the procedure and footnote a to Table 3 for strain names. Peaks representing 1C (nitrogen-starved haploid cells), 2C, and 4C DNA content are indicated. Fluorescence, which corresponds to DNA content, is plotted on an arbitrary scale (x axis) versus relative cell number (y axis). All cultures were grown at 32°C.
DISCUSSION
Like Abp1p, Cbh1p, a second member of an S. pombe family of proteins related to the mammalian alphoid DNA-binding protein CENP-B, has been found to be nonessential. Cells lacking both Abp1p and Cbh1p, however, are synergistically compromised in growth and show abnormal chromatin distribution in ∼30% of the mitoses examined, suggesting that the two proteins perform related functions.
Extracts with Abp1p or Cbh1p depleted have allowed us to identify in vitro binding sites for the two proteins, which bind to distinct sites. Six in vitro binding sites for Abp1p have now been identified within the centromeric central core region of cen2 (cc2) and two each in cc1 and cc3; an additional site is found two to three times in the flanking repeated DNA at each centromere. The results to date are summarized in Fig. 1, but this assignment of binding sites is not exhaustive. In addition to the in vivo binding sites for Cbh1p identified in noncentromeric locations, a single in vitro and several in vivo binding sites for Cbh1p lie within the centromere enhancer portion of the K repeat (_Kpn_I-_Kpn_I [Fig. 1D]), a sequence element that is found in 2 to 12 copies at the S. pombe cen loci. We have previously described a model for centromere activation that invokes protein-mediated looping between the central core and K repeat sequences (14, 39). Human CENP-B is capable of binding to DNA and dimerizing, thereby bringing together noncontiguous stretches of α-satellite DNA (72). Both Abp1p and Cbh1p form dimers or multimers (38, 50). Thus, there is an intriguing possibility that Abp1p and Cbh1p may form homo- or heterodimers, accomplishing a chromatin condensation function similar to that of CENP-B.
Although the Δ_abp1 Δcbh1_ double mutation has a dramatic effect on chromosome segregation, a single deletion of either gene has only a minor effect. This would be expected if the two proteins perform redundant functions. Related to this, we have found a 10-fold discrepancy between the Δ_abp1_ minichromosome loss frequency reported by Halverson et al. (24) (3.8 × 10−2) and measurements conducted here (3.9 × 10−3). There are two possible explanations for this result. First, chromosome missegregation leads over time to an increase in the cell population that is disomic for the minichromosome, which might cause an apparent decrease in the loss frequency. Meiotic analysis of strains exhibiting minichromosome loss frequencies of 10−2 to 10−3 suggests that acquired disomy is a significant factor (J. Irelan and L. Clarke, unpublished observation). Thus, loss frequency data for S. pombe often is an overestimate of mitotic stability, since it is not possible to distinguish between 2+:0− (missegregation) and 1+:0− (loss) events based on the colony color assay described in Materials and Methods. Secondly, chromatin remodeling or adaptation in the absence of Abp1p may contribute to minichromosome stability over time. Evidence for this comes from in vitro experiments. Electrophoretic mobility shift assays with protein extracts made from a Δ_abp1_ strain and several centromeric fragments that predominantly bind to Abp1p lack the major band corresponding to the Abp1p-DNA complex but show the appearance of a new prominent shifted band (50). The appearance of this new complex suggests that binding sites for other proteins may be utilized in vivo in the absence of Abp1p.
The equilibrium binding constant determined here for Abp1p is intermediate compared to other DNA-binding proteins, but it is ∼10-fold higher than that observed for centromere-binding proteins in other organisms (Table 1; yeast Cbf1p and human CENP-B). Abp1p binds to nonspecific DNA with low affinity (3 × 104 M−1), 5 orders of magnitude weaker than its affinity for c2-10 DNA (7 × 109 M−1). The benchmark figure for sequence-specific binding is generally accepted as a difference of 106 between Ks and Kn (15), placing Abp1p near the borderline for a sequence-specific DNA-binding protein. It has not yet been possible to determine a consensus binding site among all nine of the identified Abp1p in vitro binding sites because of their A+T richness and the lengths of the fragments involved, although it seems certain that the binding motif will be degenerate. Budding yeast centromere protein Cbf1p and human CENP-B recognize degenerate consensus sites, and similar to Abp1p, they show Ks values that are 105 and 104 higher, respectively, than their affinities for nonspecific DNA. We were unable to footprint the Abp1p binding site with 1,10-phenanthroline-copper ion nuclease (unpublished observation), which detects minor-groove interactions, perhaps because Abp1p may contact the major groove of DNA.
In S. pombe, ARS elements, ∼0.8-kb A+T-rich sequences that are capable of conferring a high-frequency transformation efficiency on plasmids, are found more frequently throughout centromeric DNA than in bulk genomic sequences. However, physical mapping of replication origins by two-dimensional gel techniques suggests that ARS elements do not function as origins of replication in the central-core portion of the centromere (60). Because Abp1p and Cbh1p also bind to the ars3002 origin of replication, however, we cannot rule out the possibility that the effect of the Δ_abp1 Δcbh1_ double mutation is related to some aspect of DNA replication. There is no apparent increase in cells with a 1C DNA content in a Δ_abp1_ or Δ_cbh1_ strain, and while there is a small increase in a Δ_abp1 Δcbh1_ strain, this can be explained by the increased incidence of unequal and asymmetric segregation in the double-deletion strain. However, we cannot rule out the possibility that this strain is defective in a replication checkpoint and passes through S phase with unreplicated DNA, leading to aberrant chromosome segregation in mitosis.
To date, the Abp1p in vitro binding sites that have been located within centromeres and ars3002 account for only a handful of the estimated 6,000 to 13,000 chromatin-associated Abp1p molecules in an Sp223 cell. If Abp1p dimerizes at each binding site (50), the total number of sites is reduced to 3,000 to 6,500, enough to bind an Abp1p dimer once every 2 to 4 kb on average in the S. pombe genome. Similar calculations for affinity-purified Cbh1p preparations indicate that ∼10-fold less Cbh1p than Abp1p is chromatin associated, enough to bind once every 20 to 40 kb on average (M. Baum, V. Ngan, and L. Clarke, unpublished observations). Abp1p and Cbh1p, respectively, are ∼13- and ∼130-fold less abundant than histone H3, based on the assumption that the total histone protein mass is approximately equal to the mass of the S. pombe genome (67). The abundance of Abp1p suggests that it may be a generalized chromatin condensation protein, perhaps binding to A+T-rich sequences not only at centromere regions but also at replication origins and matrix attachment regions (8, 62–64). ChIP experiments (51, 52) with antibody directed against a Cbh1p-GFP fusion protein cross-linked in vivo to its cognate sites showed reproducible responses with all of the DNA fragments tested and did not indicate that centromeric sequences were enriched substantially compared to noncentromeric DNA for binding to this protein. Because Abp1p and Cbh1p appear to differ in relative abundance and to recognize different binding motifs, it is likely that they differ somewhat in chromatin localization, which might account for the disparity in growth phenotypes and mating ability observed for Δ_abp1_ and Δ_cbh1_ strains that exhibit very similar mitotic loss frequencies. This pleiotropic effect may reflect chromatin architecture. If Abp1p plays a role at matrix attachment regions, lack of this protein may perturb gene expression by causing aberrant compartmentalization of genome segments. Alternatively, Abp1p and Cbh1p may directly affect gene expression, as they share significant blocks of homology with CENP-B- and _pogo_-related transcription factors Pdc2p and Rag3p in Saccharomyces cerevisiae and Kluyveromyces lactis, respectively (24, 66).
It is not unprecedented for a DNA-binding protein to play roles at both centromeric and noncentromeric locations. The centromere-binding factor Cbf1p is not an essential protein, but it modulates centromere function in budding yeast and also acts as a transcription factor at noncentromeric locations. Depletion of S. cerevisiae Cbf1p has a pleiotropic effect: strains exhibit an elevated rate of mitotic chromosome loss, increased generation time, and methionine auxotrophy (5). Deletion of the Cbf1p binding site from the S. cerevisiae point centromere has allowed confirmation of its role in centromere function. An analogous assessment of the role of fission yeast Cbh1p at the S. pombe regional centromere, which is several orders of magnitude larger and carries repeated protein binding sites, has not yet been possible.
A regular array of protein binding sites may be crucial for efficient heterochromatin formation, a universal feature of regional centromeres. Artificial human chromosomes that are dependent on the presence of CENP-B box DNA have recently been described (40). This CENP-B box dependency is somewhat analogous to the centromere enhancer function in fission yeast, which results in the efficient conversion of an inactive centromere to an active conformation by an epigenetic mechanism (50). Although a less favored occurrence, alternate sequences are able to give rise to active centromeres in fission yeast, and this process may also account for neocentromere formation in other systems (17, 50, 68, 70). So far, the only common feature among centromeric DNAs from budding yeast to humans is A+T richness. Thus, centromere formation may be specified by an epigenetic mechanism that acts on A+T-rich DNA, perhaps preferring DNA that can accommodate natural curvature (49).
Centromeric DNAs from fission yeast (S. pombe), filamentous fungi (Neurospora crassa), Drosophila, and mammals are also characterized by transposon-derived or noncoding transposon-like DNA elements. Structurally similar to retrotransposons that contain terminal direct repeats, the functionally important S. pombe 6.4-kb K repeat contains a 150-bp sequence that is duplicated at each terminus, although computer and Northern blot analyses indicate the K repeat does not encode any proteins (6, 20, 50). Analysis of N. crassa centromeric DNA indicates a complex arrangement of retrotransposon-like elements and LINE-like retrotransposon segments clustered at this region, including the first example of a novel centromere-specific transposon (10). Given the prevalence of transposable-element-like sequences at centromeres, it is perhaps not surprising that divergent transposase-like proteins bind centromeric DNA in vivo.
In addition to the redundancy within the central core and K repeat sequences supporting centromere formation in S. pombe (6, 50), apparent redundancy is also observed at the protein level between Abp1p and Cbh1p. A Δ_abp1 Δcbh1_ double mutation has a synergistic effect on chromosome missegregation, but is not lethal. This suggests that an additional CENP-B-related family member may partially compensate for the absence of Abp1p and Cbh1p. A BLAST (2) search of the translated database nucleotide sequences in fact identifies a third fission yeast CENP-B-like homolog (EMBL accession no. AL023780; nucleotides 32131 to 30590), which we will refer to as Cbh2p (514 aa), that is 41% identical and 62% similar to Cbh1p (514 aa) and 47% identical and 68% similar to Abp1p (522 aa) over 513 aa and 510 aa, respectively (D. Halverson and L. Clarke, unpublished observations). Although Abp1p, Cbh1p, and Cbh2p from fission yeast, as well as CENP-B from humans, mice, and hamsters, contain altered residues or spacing at an invariant 3-aa motif associated with transposase strand cleavage and strand transfer activities (24, 35), the proposed initial step of transposition, which brings together two noncontiguous terminal repeats in a manner similar to the folding envisioned for centromeric higher-order structures, may be uncompromised in these proteins.
The redundancy observed in fission yeast at both the DNA and protein levels may have important parallels in mammalian systems. Paradoxically, α-satellite DNA is the only family of satellite DNA that is present at each human centromere, but CENP-B binding sites are not found on Y chromosomes or some neocentromeres (37). In African green monkey cells, CENP-B is barely detectable by immunolocalization at centromeres, in agreement with the low number of CENP-B binding sites that have been detected in monkey cell satellite DNA, even though CENP-B levels are similar to those in humans (21, 71). Recent studies have shown that, analogous to Δ_abp1_ or Δ_cbh1_ mutants of fission yeast, CENP-B null mice are viable, leaving open the possibility that other functionally redundant proteins may compensate in the absence of CENP-B function (29, 33, 53). In addition to the _jerky_-like proteins described in the introduction, another probable candidate for functional redundancy with CENP-B is the recently identified autoantigen CENP-G, which binds to the same α-I subfamily of α-satellite DNA as CENP-B but to an as-yet-unidentified site. Unlike CENP-B, CENP-G immunolocalizes to the Y chromosome in humans and other mammals, indicating that CENP-G and CENP-B recognize distinct sites in the α-I subfamily of alphoid repeats (27). Thus, multiple CENP-B-like proteins may play conserved structural roles in chromatin organization at the centromere in organisms as diverse as fission yeast and mammals.
While a family of centromeric chromatin proteins may share overlapping functions, accounting for the redundancy observed between Abp1p and Cbh1p, not all of these proteins are necessarily centromere specific. Proteins with dual roles, such as that presumed for Abp1p and Cbh1p, could perhaps lead to neocentromere formation in rare cases. Numerous instances of neocentromere formation at seemingly noncentric regions have already been well documented in Drosophila and humans, supporting the concept that structural motifs, such as A+T richness or repeated elements capable of forming heterochromatin, rather than primary DNA sequence, are the overriding determinants of centromere specification. Once epigenetically marked as nucleation sites, such regions apparently remain imprinted on the DNA. Fission yeast, which bears regional centromeres exhibiting functional redundancy at both the DNA and protein levels and subject to epigenetic regulation, provide a useful centromere model with clear parallels to mammalian systems.
ACKNOWLEDGMENTS
We thank John Carbon, Susan Myers, and Dana Halverson for critical reading of the manuscript and members of the Clarke and Carbon laboratories and Norbert Reich for helpful discussions. Janet Stryker performed the electrophoretic mobility shift assays presented here. Nazanin Firouztaleh, Linda Jacobson, and Gary Gutkin contributed expert technical assistance, and Dottie McLaren prepared the figures.
This work was supported by Public Health Service grant GM-33783 from the National Institutes of Health.
REFERENCES
- 1.Allshire R C. Centromeres, checkpoints and chromatid cohesion. Curr Opin Genet Dev. 1997;7:264–273. doi: 10.1016/s0959-437x(97)80137-2. [DOI] [PubMed] [Google Scholar]
- 2.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baker R E, Gabrielsen O, Hall B D. Effects of tRNATyr point mutations on the binding of yeast RNA polymerase III transcription factor C. J Biol Chem. 1986;261:5275–5282. [PubMed] [Google Scholar]
- 4.Baker R E, Fitzgerald-Hayes M, O'Brien T C. Purification of the yeast centromere binding protein CP1 and a mutational analysis of its binding site. J Biol Chem. 1989;264:10843–10850. [PubMed] [Google Scholar]
- 5.Baker R E, Masison D C. Isolation of the gene encoding the Saccharomyces cerevisiae centromere-binding protein CP1. Mol Cell Biol. 1990;10:2458–2467. doi: 10.1128/mcb.10.6.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baum M, Ngan V K, Clarke L. The centromeric K-type repeat and the central core are together sufficient to establish a functional Schizosaccharomyces pombe centromere. Mol Biol Cell. 1994;5:747–761. doi: 10.1091/mbc.5.7.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boulanger P A, Yoshinaga S K, Berk A J. DNA-binding properties and characterization of human transcription factor TFIIIC2. J Biol Chem. 1987;262:15098–15105. [PubMed] [Google Scholar]
- 8.Boulikas T. Nature of DNA-sequences at the attachment regions of genes to the nuclear matrix. J Cell Biochem. 1993;52:14–22. doi: 10.1002/jcb.240520104. [DOI] [PubMed] [Google Scholar]
- 9.Cahill D P, Lengauer D, Yu J, Riggins G J, Willson J K V, Markowitz S D, Kinzler K W, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 10.Cambareri E B, Aisner R, Carbon J. Structure of the chromosome VII centromere region in Neurospora crassa: degenerate transposons and simple repeats. Mol Cell Biol. 1998;18:5465–5477. doi: 10.1128/mcb.18.9.5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chalfie M, Tu Y, Euskirchen G, Ward W W, Prasher D C. Green fluorescent protein as a marker for gene expression. Science. 1994;263:802–805. doi: 10.1126/science.8303295. [DOI] [PubMed] [Google Scholar]
- 12.Chodosh L A, Carthew R W, Sharp P A. A single polypeptide possesses the binding and transcription activities of the Adenovirus major late transcription factor. Mol Cell Biol. 1986;6:4723–4733. doi: 10.1128/mcb.6.12.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clarke L. Centromeres: proteins, protein complexes, and repeated domains at centromeres of simple eukaryotes. Curr Opin Genet Dev. 1998;8:212–218. doi: 10.1016/s0959-437x(98)80143-3. [DOI] [PubMed] [Google Scholar]
- 14.Clarke L, Baum M, Marschall L G, Ngan V K, Steiner N C. Structure and function of Schizosaccharomyces pombe centromeres. Cold Spring Harbor Symp Quant Biol. 1993;58:687–695. doi: 10.1101/sqb.1993.058.01.076. [DOI] [PubMed] [Google Scholar]
- 15.Coleman R A, Pugh B F. Evidence for functional binding and stable sliding of the TATA binding protein on nonspecific DNA. J Biol Chem. 1995;270:13850–13859. doi: 10.1074/jbc.270.23.13850. [DOI] [PubMed] [Google Scholar]
- 16.Craig J M, Earnshaw W C, Vagnarelli P. Mammalian centromeres: DNA sequence, protein composition, and role in cell cycle progression. Exp Cell Res. 1999;246:249–262. doi: 10.1006/excr.1998.4278. [DOI] [PubMed] [Google Scholar]
- 17.du Sart D, Cancilla M R, Earle E, Mao J, Saffery R, Tainton D M, Kalitsis P, Martyn J, Barry A E, Choo K H A. A functional neo-centromere formed through activation of a latent human centromere and consisting of non-alpha-satellite DNA. Nat Genet. 1997;16:144–153. doi: 10.1038/ng0697-144. [DOI] [PubMed] [Google Scholar]
- 18.Earnshaw W C, Ratrie III H, Stetten G. Visualization of centromere proteins CENP-B and CENP-C on a stable dicentric chromosome in cytological spreads. Chromosoma. 1989;98:1–12. doi: 10.1007/BF00293329. [DOI] [PubMed] [Google Scholar]
- 19.Fantes P, Nurse P. Control of cell size at division in fission yeast by a growth-modulated size control over nuclear division. Exp Cell Res. 1977;107:377–386. doi: 10.1016/0014-4827(77)90359-7. [DOI] [PubMed] [Google Scholar]
- 20.Fishel B, Amstutz H, Baum M, Carbon J, Clarke L. Structural organization and functional analysis of centromeric DNA in the fission yeast Schizosaccharomyces pombe. Mol Cell Biol. 1988;8:754–763. doi: 10.1128/mcb.8.2.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldberg I G, Sawhney H, Pluta A F, Warburton P E, Earnshaw W C. Surprising deficiency of CENP-B binding sites in African green monkey α-satellite DNA: implications for CENP-B function at centromeres. Mol Cell Biol. 1996;16:5156–5168. doi: 10.1128/mcb.16.9.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gutz H, Heslot H, Leupold U, Loprieno N. Schizosaccharomyces pombe. In: King R D, editor. Handbook of genetics. New York, N.Y: Plenum Publishing; 1974. pp. 394–446. [Google Scholar]
- 23.Hahnenberger K M, Carbon J, Clarke L. Identification of DNA regions required for mitotic and meiotic functions within the centromere of Schizosaccharomyces pombe chromosome I. Mol Cell Biol. 1991;11:2206–2215. doi: 10.1128/mcb.11.4.2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Halverson D, Baum M, Stryker J, Carbon J, Clarke L. A centromere DNA-binding protein from fission yeast affects chromosome segregation and has homology to human CENP-B. J Cell Biol. 1997;136:487–500. doi: 10.1083/jcb.136.3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanas J S, Bogenhagen D F, Wu C-W. Cooperative model for the binding of Xenopus transcription factor A to the 5S RNA gene. Proc Natl Acad Sci USA. 1983;80:2142–2145. doi: 10.1073/pnas.80.8.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartwell L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell. 1992;71:543–546. doi: 10.1016/0092-8674(92)90586-2. [DOI] [PubMed] [Google Scholar]
- 27.He D, Zeng C, Woods K, Zhong L, Turner D, Busch R K, Brinkley B R, Busch H. CENP-G: a new centromeric protein that is associated with the α-1 satellite DNA subfamily. Chromosoma. 1998;107:189–197. doi: 10.1007/s004120050296. [DOI] [PubMed] [Google Scholar]
- 28.Hecht A, Strahl-Bolsinger S, Grunstein M. Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature. 1996;383:92–96. doi: 10.1038/383092a0. [DOI] [PubMed] [Google Scholar]
- 29.Hudson D F, Fowler K J, Earle E, Saffrey R, Kalitsis P, Trowell H, Hill J, Wreford N G, de Kretser D M, Cancilla M R, Howman E, Hii L, Cutts S M, Irvine D V, Choo K H A. Centromere protein B null mice are mitotically and meiotically normal but have lower body and testis weights. J Cell Biol. 1998;141:309–319. doi: 10.1083/jcb.141.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ikeno M, Grimes B, Okazaki T, Nakano M, Saitoh K, Hoshino H, McGill N I, Cooke H, Masumoto H. Construction of YAC-based mammalian artificial chromosomes. Nat Biotech. 1998;16:431–439. doi: 10.1038/nbt0598-431. [DOI] [PubMed] [Google Scholar]
- 31.Ikeno M, Masumoto H, Okazaki T. Distribution of CENP-B boxes reflected in CREST centromere antigenic sites on long-range α-satellite DNA arrays of human chromosome 21. Hum Mol Genet. 1994;3:1245–1257. doi: 10.1093/hmg/3.8.1245. [DOI] [PubMed] [Google Scholar]
- 32.Iwahara J, Kigawa T, Kitagawa K, Masumoto H, Okazaki T, Yokoyama S. A helix-turn-helix structure unit in human centromere protein B (CENP-B) EMBO J. 1998;17:827–837. doi: 10.1093/emboj/17.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kapoor M, Montes de Oca Luna R, Liu G, Lozano G, Cummings C, Mancini M, Ouspenski I, Brinkley B R, May G S. The cenpB gene is not essential in mice. Chromosoma. 1998;107:570–576. doi: 10.1007/s004120050343. [DOI] [PubMed] [Google Scholar]
- 34.Karpen G H, Allshire R C. The case for epigenetic effects on centromere identity and function. Trends Genet. 1997;13:489–496. doi: 10.1016/s0168-9525(97)01298-5. [DOI] [PubMed] [Google Scholar]
- 35.Kipling D, Warburton P E. Centromeres, CENP-B and tigger too. Trends Genet. 1997;13:141–145. doi: 10.1016/s0168-9525(97)01098-6. [DOI] [PubMed] [Google Scholar]
- 36.Koehler K E, Hawley R S, Sherman S, Hassold T. Recombination and nondisjunction in humans and flies. Hum Mol Genet. 1996;5:1495–1504. doi: 10.1093/hmg/5.supplement_1.1495. [DOI] [PubMed] [Google Scholar]
- 37.Lee C, Wevrick R, Fisher R B, Ferguson-Smith M A, Lin C C. Human centromeric DNAs. Hum Genet. 1997;100:291–304. doi: 10.1007/s004390050508. [DOI] [PubMed] [Google Scholar]
- 38.Lee J-K, Huberman J A, Hurwitz J. Purification and characterization of a CENP-B homologue protein that binds to the centromeric K-type repeat DNA of Schizosaccharomyces pombe. Proc Natl Acad Sci USA. 1997;94:8427–8432. doi: 10.1073/pnas.94.16.8427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marschall L G, Clarke L. A novel cis-acting centromeric DNA element affects S. pombe centromeric chromatin structure at a distance. J Cell Biol. 1995;128:445–454. doi: 10.1083/jcb.128.4.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Masumoto H, Ikeno M, Nakano M, Okazaki T, Grimes B, Cooke H, Suzuki N. Assay of centromere function using a human artificial chromosome. Chromosoma. 1998;107:406–416. doi: 10.1007/s004120050324. [DOI] [PubMed] [Google Scholar]
- 41.Masumoto H, Masukata H, Muro Y, Nozaki N, Okazaki T. A human centromere antigen (CENP-B) interacts with a short specific sequence in alphoid DNA, a human centromeric satellite. J Cell Biol. 1989;109:1963–1973. doi: 10.1083/jcb.109.5.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McDonald I J, Tsai C S. Continuous culture and intermediary carbon metabolism. In: Nasim A, Young P, Johnson B F, editors. Molecular biology of the fission yeast. San Diego, Calif: Academic Press; 1989. pp. 367–396. [Google Scholar]
- 43.McNabb D S, Pak S M, Guarente L. Cassette for the generation of sequential gene disruptions in the yeast Schizosaccharomyces pombe. BioTechniques. 1997;22:1134–1139. doi: 10.2144/97226rr01. [DOI] [PubMed] [Google Scholar]
- 44.Mitchison J M. Physiological and cytological methods for Schizosaccharomyces pombe. In: Prescott D M, editor. Methods in cell physiology. Vol. 4. New York, N.Y: Academic Press; 1970. pp. 131–165. [Google Scholar]
- 45.Mitchison J M. Cell cycle growth and periodicities. In: Nasim A, Young P, Johnson B F, editors. Molecular biology of the fission yeast. San Diego, Calif: Academic Press; 1989. pp. 205–242. [Google Scholar]
- 46.Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- 47.Murakami Y, Huberman J A, Hurwitz J. Identification, purification, and molecular cloning of autonomously replicating sequence-binding protein 1 from fission yeast Schizosaccharomyces pombe. Proc Natl Acad Sci USA. 1996;93:502–507. doi: 10.1073/pnas.93.1.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muro Y, Masumoto H, Yoda K, Nozaki N, Ohashi M, Okazaki T. Centromere protein B assembles human centromeric α-satellite DNA at the 17-bp sequence, CENP-B box. J Cell Biol. 1992;116:585–596. doi: 10.1083/jcb.116.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy T D, Karpen G H. Centromeres take flight: alpha satellite and the quest for the human centromere. Cell. 1998;93:317–320. doi: 10.1016/s0092-8674(00)81158-7. [DOI] [PubMed] [Google Scholar]
- 50.Ngan V K, Clarke L. The centromere enhancer mediates centromere activation in Schizosaccharomyces pombe. Mol Cell Biol. 1997;17:3305–3314. doi: 10.1128/mcb.17.6.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Orlando V, Paro R. Mapping polycomb-repressed domains in the bithorax complex using in vivo formaldehyde cross-linked chromatin. Cell. 1993;75:1187–1198. doi: 10.1016/0092-8674(93)90328-n. [DOI] [PubMed] [Google Scholar]
- 52.Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997;11:205–214. doi: 10.1006/meth.1996.0407. [DOI] [PubMed] [Google Scholar]
- 53.Perez-Castro A V, Shamanski F L, Meneses J J, Lovato T L, Vogel K G, Moyzis R K, Pedersen R. Centromeric protein B null mice are viable with no apparent abnormalities. Dev Biol. 1998;201:135–143. doi: 10.1006/dbio.1998.9005. [DOI] [PubMed] [Google Scholar]
- 54.Pluta A F, Mackay A M, Ainsztein A M, Goldberg I G, Earnshaw W C. The centromere: hub of chromosomal activities. Science. 1995;270:1591–1594. doi: 10.1126/science.270.5242.1591. [DOI] [PubMed] [Google Scholar]
- 55.Polizzi C, Clarke L. The chromatin structure of centromeres from fission yeast: differentiation of the central core that correlates with function. J Cell Biol. 1991;112:191–201. doi: 10.1083/jcb.112.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rhind N, Russell P. The Schizosaccharomyces pombe S-phase checkpoint differentiates between different types of DNA damage. Genetics. 1998;149:1729–1737. doi: 10.1093/genetics/149.4.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rothstein R. Targetting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 1991;194:281–301. doi: 10.1016/0076-6879(91)94022-5. [DOI] [PubMed] [Google Scholar]
- 58.Saitoh S, Takahashi K, Yanagida M. Mis6, a fission yeast inner centromere protein, acts during G1/S and forms specialized chromatin required for equal segregation. Cell. 1997;90:131–143. doi: 10.1016/s0092-8674(00)80320-7. [DOI] [PubMed] [Google Scholar]
- 59.Smit A F, Riggs A D. Tiggers and other transposon fossils in the human genome. Proc Natl Acad Sci USA. 1996;93:1443–1448. doi: 10.1073/pnas.93.4.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith J G, Caddle M S, Bulboaca G H, Wohlgemuth J G, Baum M, Clarke L, Calos M P. Replication of centromere II of Schizosaccharomyces pombe. Mol Cell Biol. 1995;15:5165–5172. doi: 10.1128/mcb.15.9.5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Steiner N C, Clarke L. A novel epigenetic effect can alter centromere function in fission yeast. Cell. 1994;79:865–874. doi: 10.1016/0092-8674(94)90075-2. [DOI] [PubMed] [Google Scholar]
- 62.Strausbaugh L D, Williams S M. High density of an SAR-associated motif differentiates heterochromatin from euchromatin. J Theor Biol. 1996;183:159–167. doi: 10.1006/jtbi.1996.0209. [DOI] [PubMed] [Google Scholar]
- 63.Strissel P L, Espinosa R, Rowley J D, Swift H. Scaffold attachment regions in centromere-associated DNA. Chromosoma. 1996;105:122–133. doi: 10.1007/BF02509522. [DOI] [PubMed] [Google Scholar]
- 64.Swedlow J R, Hirano T. Chromosome dynamics: fuzzy sequences, specific attachments? Curr Biol. 1996;6:544–547. doi: 10.1016/s0960-9822(02)00538-9. [DOI] [PubMed] [Google Scholar]
- 65.Takahashi K, Murakami S, Chikashige Y, Funabiki H, Niwa O, Yanagida M. A low copy number central sequence with strict symmetry and unusual chromatin structure in fission yeast centromere. Mol Biol Cell. 1992;3:819–835. doi: 10.1091/mbc.3.7.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Toth M, Grimsby J, Buzsaki G, Donovan G P. Epileptic seizures caused by inactivation of a novel gene, jerky, related to centromere binding protein-B in transgenic mice. Nat Genet. 1995;11:71–75. doi: 10.1038/ng0995-71. [DOI] [PubMed] [Google Scholar]
- 67.van Holde K E. Chromatin. New York, N.Y: Springer-Verlag; 1989. pp. 70–73. [Google Scholar]
- 68.Wandall A, Tranebjaerg L, Tommerup N. A neocentromere on human chromosome 3 without detectable α-satellite DNA forms morphologically normal kinetochores. Nat Genet. 1998;107:359–365. doi: 10.1007/s004120050319. [DOI] [PubMed] [Google Scholar]
- 69.Wevrick R, Willard H F. Long-range organization of tandem arrays of α satellite DNA at the centromeres of human chromosomes: high-frequency array-length polymorphism and meiotic stability. Proc Natl Acad Sci USA. 1989;86:9394–9398. doi: 10.1073/pnas.86.23.9394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Williams B C, Murphy T D, Goldberg M L, Karpen G H. Neocentromere activity of structurally acentric mini-chromosomes in Drosophila. Nat Genet. 1998;18:30–37. doi: 10.1038/ng0198-30. [DOI] [PubMed] [Google Scholar]
- 71.Yoda K, Nakamura R, Masumoto H, Suzuki N, Kitagawa K, Nakano M, Shinjo A, Okazaki T. Centromere protein B of African green monkey cells: gene structure, cellular expression, and centromeric localization. Mol Cell Biol. 1996;16:5169–5177. doi: 10.1128/mcb.16.9.5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoda K, Ando S, Okudo A, Kikuchi A, Okazaki T. In vitro assembly of the CENP-B/α-satellite DNA/core histone complex: CENP-B causes nucleosome positioning. Genes Cells. 1998;3:553–548. doi: 10.1046/j.1365-2443.1998.00210.x. [DOI] [PubMed] [Google Scholar]
- 73.Zeng X, Kahana J A, Silver P A, Morphew M K, McIntosh J R, Fitch I T, Carbon J, Saunders W S. Slk19p is a centromere protein that functions to stabilize mitotic spindles. J Cell Biol. 1999;26:415–425. doi: 10.1083/jcb.146.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]