Mechanisms of sodium channel clustering and its influence on axonal impulse conduction (original) (raw)
Introduction
Electrical axonal propagation of the action potential (AP) leads to chemical neurotransmission through synapses, which drive important nervous system functions such as motor output, control of visceral organs, encoding of sensory stimuli, and higher order cognitive processing. Synaptic input received by the neurons is integrated in the somatodendritic region [1] and the initiation of the AP occurs at a region called the axon initial segment (AIS) [2]. The AIS is enriched in voltage-gated ion channels, particularly voltage-gated sodium channels (Nav) that permit the entry of depolarizing current in the form of Na+ ions [2, 3]. This depolarizing current will then passively spread along the next segment of axonal membrane downstream of the active region, while efflux of K+ from voltage-gated potassium channels (Kv) in the regions trailing the AP will inactivate Nav and slowly bring the patch of axonal membrane back to its resting potential [2, 3].
Once the AP is triggered, it propagates down the axon to reach the synaptic terminals to relay information to the next neuron and carry out proper nervous system functions. Certain neural functions require adjustment of conduction velocity to regulate the synchrony of inputs [4] and fast conduction velocity can be critical for defense and survival. Several biophysical parameters govern the speed by which APs are propagated: axonal diameter, sodium channel density and temperature [3]. In addition, myelination of axons, which is one of the last evolutionary steps in the nervous system of jawed vertebrates [5], ensures rapid propagation of action potentials. Myelin is a lipid-rich, multilamellar membranous structure produced by Schwann cells in the peripheral nervous system (PNS) and oligodendrocytes in the central nervous system (CNS). Myelin provides both insulation of electric current and metabolic support for the axon [6–8]. Importantly, between the segments of myelin are unmyelinated gaps highly enriched with Nav channels, called the nodes of Ranvier, which permit the regeneration of the AP and fast AP propagation through saltatory conduction (Fig. 1a). In addition, studies have shown that clusters of Nav along unmyelinated fibers, but also prior to myelination in fibers destined to be myelinated, can influence axonal impulse conduction [9–12]. In this review, we will outline the molecular architecture of the nodes of Ranvier and mechanisms underlying the clustering of Nav channels in both the PNS and CNS. Further detail will be provided to how APs are propagated along unmyelinated and myelinated fibers, particularly how different biophysical properties of the myelin sheath can affect axonal conduction. Finally, we will describe how the clustering of Nav channels along unmyelinated and myelinated fibers can regulate AP conduction in the physiological and pathological state.
Fig. 1
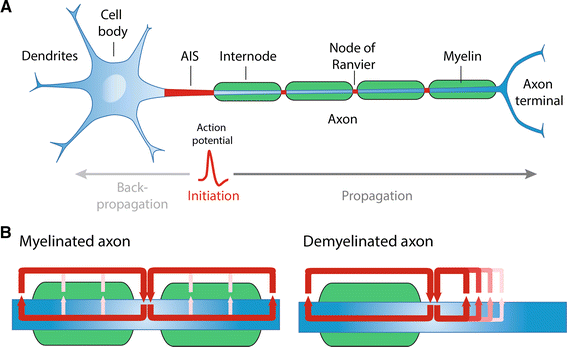
a Structural organization of a myelinated neuron and representation of action potential initiation and propagation. b In myelinated fibers, the action potentials are regenerated at the nodes of Ranvier, where high membrane currents (indicated by the red arrows) are observed. In case of demyelination, the propagation of action potentials is slowed down or blocked
Molecular composition of the nodes of Ranvier
The nodes of Ranvier are macromolecular complexes of ~1 µm length that are highly enriched in voltage-gated sodium channels [6]. These channels consist of a heterotrimeric complex of a pore forming _α_-subunit of approximately 260-kDa, and two differing auxiliary _β_-subunits that can interact with the _α_-subunit through non-covalent interactions (_β_1Nav and _β_3Nav) or covalent disulfide bonds (_β_2Nav and _β_4Nav) [13, 14]. The SCNA family encodes for ten different sodium channel _α_-subunit isoforms [15], yet only Nav1.1, Nav1.2, Nav1.6, Nav1.7, Nav1.8, Nav1.9 have been reported to be clustered at the nodes [16, 17]. Mammalian sodium channel _β_-subunits form a family of five proteins (_β_1Nav, _β_1BNav, _β_2Nav, _β_3Nav, and _β_4Nav) that are encoded by four SCNB genes, but only _β_1Nav, _β_2Nav, and _β_4Nav are expressed at the nodes [14, 18–20]. Nav _β_-subunits are described as “auxiliary” subunits, but they play critical roles in regulating the rate of _α_-subunit activation and inactivation [21, 22], regulating the amount of resurgent sodium current and sodium current density [23–25], and regulating the plasma membrane insertion of _α_-subunits [14, 19].
In addition to Nav channels, Kv channels, cell-adhesion molecules (CAMs), cytoskeletal scaffolding proteins, and extracellular matrix (ECM) components are further clustered at the nodes of Ranvier (Fig. 2a) [6, 16]. The Kv channels Kv7.2, Kv7.3, and Kv3.1b are responsible for the repolarization of the axonodal membrane and regulation of axonal excitability [26–30]. The L1-CAMs neurofascin186 (Nfasc186) and NrCAM are enriched at the nodes of Ranvier both in the PNS and the CNS [31, 32]. In addition, the glycosyl phosphatidylinositol (GPI)-anchored CAM contactin-1 is expressed at nodes only in the CNS [33, 34]. Interaction of Nfasc186, NrCAM or contactin-1 with the Nav β1-subunit through their extracellular immunoglobulin-like domain may increase functional Nav channel expression [18, 34, 35]. On the intracellular side of the nodal axolemma, Nav, Nfasc 186 and NrCAM can bind to the cytoskeletal scaffolding protein ankyrinG, which in turn binds with high affinity to βIV-spectrin, providing a link with the actin cytoskeleton [6, 16, 36]. The glycoprotein-rich ECM forms a negatively charged complex surrounding nodes of Ranvier that is involved in cation buffering and proper stabilization of the nodes [37–41]. Differentiating the PNS and CNS is the presence of Schwann cell microvilli that invade into the PNS nodal extracellular space and secrete several important molecules such as gliomedin and NrCAM, which together play a key role in clustering PNS nodes of Ranvier (see below; [42–44]). Although Schwann cell microvilli are absent from CNS nodes of Ranvier, CNS nodes are occasionally contacted by perinodal astrocytes, but their function at these domains remains to be elucidated [45].
Fig. 2
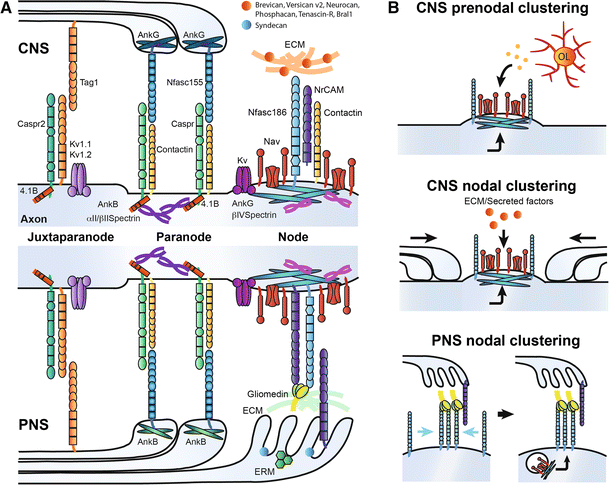
a Molecular organization of the nodes of Ranvier and surrounding domains in the CNS and PNS, respectively. b Mechanisms implicated in Nav channel clustering at node-like clusters and nodes of Ranvier. Node-like clustering depends on both intrinsic and extrinsic cues (oligodendroglial-secreted factors). Nodal clustering differs in the CNS and the PNS. In the CNS, three different components (paranodes, extracellular matrix and scaffold proteins) play a role in Nav channel assembly. In the PNS, early clustering of Nfasc186 through its interaction with glial Gliomedin and NrCAM is followed by targeting of scaffold proteins and voltage-gated channels
The regions flanking the nodes are the paranodes, where axoglial junctions between myelin loops and the axon form a ternary complex via interactions between axonal Caspr/contactin and glial Nfasc155 (Fig. 2a) [31, 46–50]. Importantly, the paranodes act as a molecular sieve to restrict the diffusion of nodal components [51]. Cytoskeletal scaffolding proteins are also enriched at paranodes in both myelinating glia and axons. Glial expression of the 190- and 270-kDa isoforms of ankyrinG is enriched in the CNS paranodes, while the 220-kDa isoform of ankyrinB is clustered at PNS paranodes [52, 53]. Loss of these glial proteins results in a delay of paranodal junction formation [52]. Axonal expression of cytoskeletal scaffolding components in both the CNS and PNS paranodes includes 4.1B, _α_II-spectrin, and ßII-spectrin, which are implicated in the organization and maintenance of the paranodal junction [53–58]. Recently, the Kv channel Slo/BK has been reported to cluster at the paranodes of rodent cerebellar Purkinje neurons, and its clustering at the paranodes is necessary for supporting high-frequency firing that is characteristic of cerebellar Purkinje neurons [59].
Another important function of the paranodes is to also act as a segregation barrier between the nodes and the juxtaparanodal regions that are highly enriched with Kv1 [49, 57, 60, 61]. These Kv channels receive little depolarizing current since they are underneath the multiple layers of compacted myelin. The clustering of Kv1 to the juxtaparanodal regions depends on the cell-adhesion molecules TAG-1 (expressed on both the glial and axonal sides) and axonal Caspr2 [62–64]. Underneath this region is a cytoskeletal complex composed of PSD-93/95, ADAM22, ßII-spectrin, _α_II-spectrin, and 4.1B [16]. The latter protein, 4.1B, plays an essential role in assembling the juxtaparanodal complex [54].
Mechanisms of nodes of Ranvier assembly
Even though the PNS and CNS nodes of Ranvier have only little changes in their molecular composition, the mechanisms underlying their formation are not identical. This dissimilarity between the two nervous systems mainly stems from the differences in cell types producing the myelin sheath that ultimately govern the neuron–glia interactions necessary to form the nodes of Ranvier.
Nav clustering at node of Ranvier in the PNS
During initial myelination of the axon by the Schwann cell, components of the nodal complex cluster to regions adjacent to the myelinating segment called heminodes [65]. The initial steps of heminodal formation, which ultimately gives rise to mature nodes from the fusion of two heminodes at the edges of elongating myelin segments, depend on the interactions between axonal Nfasc186 and gliomedin and NrCAM emanating from Schwann cell microvilli (Fig. 2b) [42–44, 66–69]. This cooperative interaction is highlighted by the fact that Nfasc186 fails to properly localize to heminodes in both _NrCAM_- and _Gliomedin_-deficient mice; however, full nodes of Ranvier ultimately form in these knockouts through paranodal junctions acting as a restriction barrier to assemble the nodal components [44, 70]. For the heminodal assembly, glial NrCAM stabilizes secreted trimerized gliomedin for increased interaction with Nfasc186 [43, 44]. The expression of certain molecules of the basal lamina, microvilli, and ECM also appears to be involved in promoting the heminodal clustering mechanism [71–73]. Then, Nfasc186 plays an important role in nucleating and incorporating the nodal components to the axolemma (Fig. 2b). Nfasc186 interacts with ankyrinG, which in turn is able to recruit Nav channels and its scaffolding partner βIV-spectrin [66, 67, 74, 75]. Axoglial contact mediated by gliomedin, NrCAM and Nfasc186 also contributes to the long-term maintenance of Nav channels at nodes of Ranvier [76].
Nav clustering at node of Ranvier in the CNS
In contrast to the PNS, the molecular mechanisms underlying nodal assembly in the CNS are still partly understood. Three complementary mechanisms have been established to participate in CNS nodal assembly: ECM-induced cell-adhesion molecule clustering, scaffolding molecules anchoring the nodal complex to the actin cytoskeleton, and paranodal barrier formation (Fig. 2b) [41]. However, their relative importance and whether differences exist between neuronal subpopulations is still debated. In contrast to Schwann cells, oligodendrocytes do not contact nodes directly and do not express gliomedin. Other glia-derived ECM proteins (i.e., chondroitin sulfate proteoglycans, tenascin-R, Bral1) form complexes with axonal CAMs such as Nfasc186, NrCAM, contactin-1 and the β-subunits of sodium channels [37–39, 77, 78]. Yet, the clustering of ECM molecules at the nodes of Ranvier in vivo occurs after the clustering of axonodal components in the mouse optic nerve [41], and nodal assembly still occurs in mutant mice lacking these ECM proteins, suggesting that ECM molecules may be involved in the stabilization of the nodes rather than initiating their assembly [37, 39–41, 77].
Axonodal CAMs contribute to the assembly of CNS nodes of Ranvier. Transgenic expression of axonal Nfasc186 or Nfasc140 in _Nfasc_-null mice, which form neither nodes nor paranodes, rescues Nav, CAMs, ECM and cytoskeletal components of the CNS nodes of Ranvier [61, 79]. In addition, loss of the CNS nodal and paranodal GPI-anchored protein Contactin-1 results in reduced numbers of nodes of Ranvier [80].
The paranodal barrier formed through direct axoglial contacts established at the paranodal junctions also participates in the assembly of CNS nodes of Ranvier [41, 81]. In this context, in Nfasc null mice, the reconstitution of paranodes by glial expression of Nfasc155 is sufficient to rescue Nav channel clustering [61]. Double knockout mice for βIV-spectrin and ECM components (thereby leaving the paranodal junctions intact) can still assemble CNS nodes of Ranvier, albeit with reduced Nav clustering compared to wild-type and single knockout mice [41]. However, other studies have shown that the timing or number of developing nodes of Ranvier is unaffected by either suppressing the paranodes through inactivation of genes coding for the paranodal constituents Caspr and Nfasc155 or by disrupting the paranodal junctions through loss of myelin proteins or lipids [49, 61, 82–85]. Overall, these results suggest that, while paranodal junctions have the ability to cluster CNS nodes of Ranvier, they might not be essential for CNS nodal assembly. Conversely, paranodal junctions are particularly important for nodal maintenance, suggesting that mechanisms of nodal stabilization depend on protein–protein interactions that are different from those that dominate initial assembly [51, 83, 86].
CNS nodes of Ranvier can also be assembled through intrinsic neuronal mechanisms directed by axonal scaffolding proteins such as ankyrinG. AnkyrinG is able to bind several membrane-spanning axonodal proteins through its multiple ANK repeats and connects them to the neuronal actin cytoskeleton [87], thereby laying the foundation for a large heterogeneous macromolecular complex at the nodes. The importance of ankyrinG in CNS nodal assembly is highlighted by the fact that loss of the giant 270- and 480-kDa ankyrinG splice variants results in a significant reduction in CNS nodal formation [88]. However, it has also been reported that erythrocyte ankyrin, ankyrinR, can substitute for ankyrinG when ankyrinG is completely lost [89]. AnkyrinG also plays an important role in trafficking Nav to the nodes via its direct interaction with the conventional anterograde microtubule motor kinesin-1 [90]. Taken together, these results point to ankyrinG as an important molecule that directs CNS nodes of Ranvier assembly through linking the nodal components together and through trafficking of Nav channels.
Finally, axonal clustering of Nav channels before myelin deposition and oligodendroglial contact has been shown to occur in retinal ganglion cell cultures, where these clusters were induced by oligodendroglial-secreted factor(s) [91, 92]. More recently, it has been shown that nodal-like clusters (i.e., clusters of Nav channels colocalizing with the scaffold protein ankyrinG and nodal CAMs) are detected before myelin deposition along axons in hippocampal neuron-glia cultures and in the developing hippocampus in vivo. These clusters can be induced by oligodendroglial-secreted factor(s) and depend on ankyrinG for their assembly [11]. Importantly, nodal-like clusters are restricted to hippocampal GABAergic neurons, whereas clustering of nodal proteins along the axons of hippocampal pyramidal neurons occurs concomitantly with myelin ensheathment, suggesting separate mechanisms of assembly among different regional neuronal subpopulations [11].
Clustering of Nav channels will eventually influence AP propagation along axons and the specific role of their clustering, as well as the classical biophysical parameters implicated in conduction velocity will be further highlighted.
Action potential initiation and propagation along unmyelinated fibers
The preferred site of AP initiation is the distal end of the AIS [93, 94], where the density of low-threshold Nav1.6 sodium channels is highest [95]. During AP initiation, the active depolarization backpropagates to the soma (antidromic) and down the axon (orthodromic) [96, 97]. Propagation of the AP along the axon is dependent on how fast the membrane is able to charge, which is determined by the membrane capacitance (how much charge is stored on the axonal membrane per unit area) and the axial resistance (how resistant the interior axonal medium is to electrical current). Factors involved in how quickly the membrane will charge, and consequently increasing conduction velocity, are characterized by either a reduction in the membrane capacitance or a reduction in the internal axial resistance [98]. The reduction in the internal axial resistance may be achieved through increasing the diameter of the axon, which reduces the resistivity Na+ ions must face as they passively spread through the axoplasm. Accordingly, the AP conduction velocity in unmyelinated axons is generally described from theoretical calculations to be proportional to the square root of the axonal diameter [99, 100].
Conduction velocity along unmyelinated vertebrate CNS axons can be measured electrophysiologically using dual soma-axon patch clamp recordings in pyramidal neurons in acute brain slices [94], or by patching axon “blebs” where investigators patch both the soma and the cut end of the axon in pyramidal cells or interneurons [101–106]. Orthodromic conduction velocities along these axons ranged from 0.2 to ~1.45 ms−1 (for review see [107]).
Regulation of action potential propagation along myelinated fibers
While increasing axonal diameter is a viable solution for rapid axonal conduction, it also comes at a price in terms of space constraints and energetics [108]. Maintaining the extracellular Na+ and intracellular K+ ion gradient, mediated through the action of Na+/K+ pumps, is energetically costly [109], even though ion channel kinetics underlying the AP in pyramidal neurons are built to be energy-efficient and to minimize the overlap of Na+ and K+ ion fluxes [110]. Cerebellar Purkinje neurons and fast-spiking interneurons, however, have incomplete inactivation of sodium channels leading to reduced metabolic efficiency [111].
Myelination permits optimization of fast axonal AP propagation over long distances. The insulating properties of the myelin sheath reduce the leakage of Na+ current that flows down the axon (i.e., increase in axial resistance) and reduce the axonal capacitance in conjunction with nodes of Ranvier, thereby allowing for faster charging of the axonal membrane [98]. Myelination is generally beneficial in increasing conduction velocity compared to the unmyelinated nerve when the CNS axonal diameter is above 0.2 µm [112]. Indeed, this fact correlates well with the finding that only axons with a diameter > 0.2 µm can be myelinated in the CNS [112]. The speed of nervous conduction in myelinated axons is linearly proportional to the axonal diameter [99, 113], partly due to increased myelin thickness [113, 114].
Together with axonal diameter, the thickness of the myelin sheath plays an important role in speed of axonal AP propagation. Typically, myelinated axons are classified by their g-ratio, which is a calculation of the ratio between the axonal diameter to the overall diameter of the fiber, and this ratio is optimized (g-ratio between 0.6 and 0.77) to ensure maximal conduction and efficiency [99, 115]. The regulation of myelin thickness is highly important in maintaining this optimum since hypermyelination, such as that observed in the absence of Dlg1-PTEN in peripheral nerves, can lead to unstable myelin sheaths which may ultimately attenuate nerve conduction [116]. The paranodal junctions are key determinants in maintaining rapid axonal conduction through their barrier-like seals that form between the paranodal loops of the myelinating glial cells and the axolemma. These junctions are important for restricting the short-circuiting of the nodes of Ranvier [117] and for metabolic savings for AP regeneration [98].
Conduction timing along myelinated neurons in the CNS ranges from modestly fast to rapidly conducting [107]. Utilizing the dual patch soma-bleb technique or rapid acquisition of voltage changes through voltage-sensitive dyes reports of conduction speeds in pyramidal neurons of the cortex range from ~0.5 to 4.5 ms−1 [96, 97, 118, 119]. Conduction speeds along Purkinje cell axons obtained from bleb recordings and more recently from extracellular antidromic axonal recordings range between 0.52 and 0.77 ms−1 [59, 120]. These values are not much faster compared to those of the unmyelinated axons in the brain, most likely related to the small diameters of these axons. Indeed, in larger diameter axons, such as in motoneurons and along adult mouse sciatic nerve, action potential conduction can reach up to 80 ms−1 [121, 122]. Differences and lower speeds of conduction in the cortex could be related to the fact that these neurons may need to synchronize conduction timing for proper cortical circuit activity, and also that these neurons may be maximizing their metabolic energetic needs in preference to a further increase in axonal conduction.
Nav channel clustering regulates axonal conduction
Concentration of Nav channels and size and structure of the nodes of Ranvier
Not only are axonal diameter and myelin sheath thickness crucial for increasing conduction velocity, but also the density of Nav channels. A recent study has shown that along unmyelinated basket cell hippocampal axons, Nav conductance density increases tenfold in a gradient-wise manner from the soma (2.6 channels/µm2) towards the proximal axon (25 channels/µm2 in the proximal axon), followed by a further increase in Nav density at the distal axon (46.1 channels/µm2) [105]. This increase in distal Nav channels ensures a supercritical density necessary for supporting fast AP propagation along the axons of these fast-spiking GABAergic neurons [105]. In myelinated fibers, however, Nav is highly clustered at the nodes of Ranvier in the order of ~1200 channels/µm2 [123], while internodes contain ~20–25 channels/µm2 [124]. Theoretical and experimental studies show that this asymmetrical concentration of Nav between the nodes of Ranvier and the internodes reduces the axonal capacitance and the concentration of Na+ necessary to regenerate the AP, resulting in improving energy efficiency [9, 125, 126]. Moreover, ECM glycoproteins surrounding the nodes of Ranvier can increase conduction velocity through concentrating Na+ in the vicinity of Nav channels [37, 40]. Another advantage of this restriction of high density of Nav at the nodes of Ranvier is that it permits high-fidelity impulse conduction because the ratio of current available to stimulate the node to the current necessary to depolarize the node, otherwise called the safety factor, is very high (in the order of 5–7). When the safety factor is less than 1, such as in the case of drastic changes in axonal geometry and also in demyelinating pathologies, conduction block occurs and the AP fails to propagate [3].
A second component related to the nodal capacitance is the membrane area occupied by the node of Ranvier. Increasing the size of the nodes of Ranvier can alter conduction velocity, such that an increase in nodal length would result in slowing of the AP being regenerated at the adjacent node due to increased nodal capacitance. Experimental evidence in the electric organ of Sternarchus showed that large nodes of Ranvier have delayed conduction compared to smaller nodes of Ranvier [127]. A recent theoretical study has also demonstrated that conduction velocity decreases when the nodal width is increased [128]. In addition, fenestrated nodes and the wide submyelinic space form the basis for unusually fast impulse conduction in shrimp giant nerve fibers that display remarkable conduction speeds of more than 200 ms−1, making them among the fastest-conducting fibers recorded [129].
Sodium channel isoforms
The biophysical gating properties of the Nav subunits allow for several types of current flow through the pore of the Nav α -subunit, and can therefore support several types of neuronal AP firing properties, which may affect AP conduction. At the AIS of cortical pyramidal neurons, two Nav channel isoforms are asymmetrically distributed: Nav1.2 is enriched in the proximal part of the AIS and it has a high threshold for activation. The low-threshold Nav1.6 accumulates at the distal end, and therefore, favors spike initiation at this region [95]. At the nodes of Ranvier, several different Nav channel isoforms cluster at varying developmental stages [11, 92, 130, 131], and the relative contribution of the various nodal ion channels on axonal conduction is poorly understood. Our understanding of the roles of Nav isoforms in relation to AP conduction is currently limited to electrophysiological studies investigating the biophysical gating and current dynamics of the nodal Nav isoforms. Nav1.2, which is primarily associated to the mammalian immature PNS and CNS nodal-like clusters and nodes of Ranvier [11, 92, 130, 132, 133] needs large depolarizing current to fire and inactivates during high-frequency firing [134]. Nav1.6 and Nav1.1, which are clustered to mature nodes of Ranvier and nodal-like clusters in the PNS and CNS [11, 130, 131, 133, 135], participate in persistent sodium current [134, 136, 137]. This slowly inactivating current may therefore drive faster conduction through the increase of axoplasmic Na+ [134, 137–139]. With their low-threshold voltage dependence, Nav1.6 may favor spike initiation not only in the AIS but also ectopically, at the nodes. Nav1.6 rapidly recovers from inactivation and may also sustain high rates of activity [140, 141]. These data suggest that the sodium channel subunit composition at nodes of Ranvier may contribute to a high safety factor for AP propagation fidelity [107]. Nevertheless, further tools need to be developed to understand the different contributions of each nodal Nav isoform on AP conduction.
Number of nodes and internodal length
Theoretical and experimental evidence suggests that myelinated axons have an optimal internodal length for maximal conduction, but changes in the internodal length may have modest to drastic consequences to conduction velocity based on their shortening or lengthening [113, 114, 142–148]. Court and colleagues (2004) showed that, in _periaxin_-null mice where Schwann cell elongation fails, thereby exhibiting decreased internodal length but normal axon diameter and myelin thickness, conduction velocity along these fibers was significantly decreased, suggesting that conduction velocities are highly sensitive to substantially shorter internodal lengths [143]. Shortened internodal lengths, however, may be physiologically relevant in synchronizing conduction timing of different inputs [149]. In the avian auditory brainstem, nucleus magnocellaris cells in the avian cochlear nucleus have both ipsilateral and contralateral inputs to the nucleus laminaris, which is responsible for the processing of auditory information. Challenges in synchronizing aural input arise when taking the length difference of ~ 1600 µm between the contralateral and ipsilateral axonal branches from nucleus magnocellaris—yet, proper simultaneous timing of input is ensured by the ipsilateral projections having shorter internodes and a smaller axonal diameter than that of the contralateral projections [149]. Thus, in the case of the avian auditory system, shortened internodes act as a physio-anatomical way of slowing down AP conduction and therefore secure correct encoding of auditory information from bilateral inputs [149, 150]. However, recent work from Ford et al., in analogous mammalian auditory brainstem circuits, reports that axons responding to low-frequency sounds had a larger diameter but shorter internodes than high-frequency axons, and higher conduction velocities. Moreover, internode length decreased and nodes of Ranvier diameter increased progressively along the distal axon segments, which simulations predict was important to adjust precisely the conduction velocity of APs within the circuit [148].
Although significant shortening of the internodes gives rise to substantial variations in AP conduction velocity [143, 144], lengthening of the internodal region appears to have only modest effects on conduction velocity at intermediate lengths [98, 144]. Experimental evidence correlates well with these theoretical studies. Mutant mice lacking the N-terminal PDZ domain of periaxin delays Schwann cell elongation and results in shorter internodes and lowered conduction velocities compared to controls at 3 and 6 weeks of age [145]. However, at 16 weeks of age, conduction velocities are indistinguishable between mutant and wild-type mice even though internodal lengths are significantly different, suggesting that conduction velocity speeds reach a “flat maximum” once the distance between nodes have reached a certain threshold. Simpson and colleagues further showed that by increasing internodal length by up to 35 % and keeping axon diameter and myelin sheath thickness constant, there was no significant increase in axonal conduction speeds [146]. Together, these theoretical and experimental data suggest that increases in internodal distance above the optimum result in insignificant changes in axonal impulse conduction.
Finally, a recent work from Tomassy et al. has described a new pattern of myelin distribution along single axons, where myelinated segments are interspaced with long, unmyelinated tracts, in some pyramidal neurons in the neocortex [151]. Although the functional consequences of these heterogeneous profiles of myelination await further identification, the profile of longitudinal distribution of myelin may have evolved as a strategy to modulate long-distance communication in the neocortex [151].
Nav clustering along unmyelinated axons
Several reports have observed nodal-like clusters of Nav along unmyelinated fibers. These have been reported in both the PNS and CNS fibers in a number of different species including the marine invertebrate Aplysia [12], zebrafish mutants lacking Schwann cells in peripheral nerves [152], rodent retinal ganglion cells [85, 86] and hippocampal GABAergic interneurons [11, 91, 92], in dorsal and ventral spinal roots of dystrophic mice lacking merosin [153], in lipid rafts of group-C nerve fibers [154], and unmyelinated axonal segments in non-pathological human dental pulp [155]. In light of these qualitative observations, what could be the possible functional role of these dense aggregates of Nav along unmyelinated axons?
Few studies have attempted to assess the physiological role of these focal Nav clusters. Waxman et al. (1983) using transmission and freeze-fracture electron microscopy proposed that the electron-dense subaxolemmal particles along the axons of the nerve fiber layer of the adult rat retina could correspond to clusters of Nav that act as electrogenic “hot-spots” [156, 157]. Further theoretical work by Johnston and colleagues (1996) modeling their observation of Nav clusters along cultured and ganglion neurons from Aplysia showed that the clustering of Nav along unmyelinated axons required 30–60 % fewer channels to propagate APs compared to an evenly diffuse expression of Nav, suggesting that the physical clustering of these channels acts as a way of optimizing AP conduction [12]. Recent work by Freeman et al. also suggests that these clusters are associated with increased AP conduction. Through simultaneous soma-axon electrophysiological recordings, they observed that nodal-like clusters along GABAergic axons increase conduction velocity by 150 % in comparison to GABAergic axons without clusters [11]. Importantly, this increase is observed independently of axonal caliber, representing a novel means for accelerated axonal propagation of APs before myelin deposition [11]. This augmentation in AP conduction may also underlie what was observed by Foster and colleagues (1982) when they reported that during rat optic nerve development there was an increase in AP conduction prior to myelination that could not be accounted by an increase in axonal caliber [158]. Moreover, experimental and theoretical findings suggest that micro-saltatory conduction may occur in the absence of myelination due to the dense clustering of Nav at nodal-like domains [9, 10, 154]. Lastly, the action of Nav clustering along unmyelinated axons may be important for overcoming axonal branch point failures and maintaining reliable propagation of APs [3]. In this respect, theoretical modeling calculations predict that high-density sodium channel clusters could serve as acceleration points [159] and therefore Nav clustering in the absence of myelination could be a way of maintaining faithful propagation of the AP.
Pathologies leading to alterations of Nav clustering and conduction velocity
Demyelination or alteration of myelin, disruption of paranodal junctions or primary nodal impairment, leading to abnormal ion channel expression and lengthening or disruption of the node of Ranvier may contribute to altering the conduction along myelinated axons in several diseases, including stroke, spinal cord injury, multiple sclerosis (MS), and Guillain–Barré syndrome (GBS) (for review see, [109, 160–162]).
Various dysmyelinating mutant animal models such as Trembler-J mice, characterized by a mutation in the peripheral myelin protein 22 gene, and Shiverer mice, which have a disrupted myelin basic protein gene MBP, have improperly formed and distributed paranodes and abnormal Nav clusters [81, 163]. Similarly, in demyelinated lesions of MS or experimental autoimmune encephalomyelitis (EAE), an animal inflammatory model for MS, a disruption of nodal, paranodal and juxtaparanodal domains has been reported, replaced by diffuse expression of the components of these domains along the denuded portion of the axon [164–167]. More current is required to drive depolarization of the axolemma, and this raises the metabolic cost of maintaining the Na+/K+ gradient. Moreover, the failure of Kv1 to be clustered at the juxtaparanodes or their mislocalization to either the paranodes or nodes has an important impact on axonal conduction [168]. Diffusion of nodal markers is accompanied by a switch from the mature Nav1.6 to the immature Nav1.2 isoform, which could be an adaptive response, since Nav1.6 might favor axonal damage by inducing persistent current that will drive the inversion of the Na+/Ca2+ exchanger and subsequent calcium-mediated axonal damage [165]. Altogether these changes can lead to conduction failures that manifest into substantial neurological deficits in MS patients (Fig. 1b). Axoglial organization is fully restored after remyelination, as observed in shadow plaques in tissue taken from MS patients [164]. Furthermore, clustering of Nav channels has been observed on PLP-negative (i.e., non-remyelinated) fibers within lesions undergoing remyelination [164], suggesting that, as observed for developmental myelination, nodal protein clustering might precede myelin repair. Although the mechanisms of axonal domain reassembly during remyelination are still poorly understood, it can be hypothesized that, similar to early nodal clustering during developmental myelination, these clusters may accelerate conduction velocity before remyelination and therefore participate in functional recovery. The observation of saltatory conduction occurring before remyelination in axons demyelinated with lysophosphatidylcholine may also support this hypothesis [169]. A reduction of the internodal length is observed after remyelination, which may induce changes in conduction velocity but may also have a negative impact due to the increase in the energy needed for AP propagation [109].
Nodal and perinodal proteins are also direct targets of autoimmune reactions, and autoantibodies or specific T cells can be detected notably in patients with MS (reviewed in [160, 161]). These autoantibodies or T cells, administered to the animals with EAE, can induce pathogenic effects such as acute axonal injury [170, 171]. Similarly, in GBS, an acute autoimmune polyradiculoneuropathy, the autoimmune processes specifically target gangliosides (GM1 or GD1a or b) highly enriched at node, which is the site of primary injury. Autoantibodies to gliomedin, Nfasc or contactin have also been found in some patients with GBS. These antibodies can induce lengthening of the nodes of Ranvier and disruption of their molecular organization leading to conduction failure probably due to dysfunction of Nav channels [161].
Axonal degeneration, depending on the specific disorder and its severity, eventually follows conduction block [162]. Indeed, defects in Na+/K+ pump function due to ATP depletion induce axoplasmic Na+ accumulation, which in turn reverses the Na+/Ca2+ exchanger to remove excess Na+. Consequently, Ca2+ accumulation may activate calpain, a protease capable of inducing proteolytic cleavage of neurofilaments, mitochondrial damage and Wallerian degeneration [162].
Conclusion
Beyond axon diameter and the presence of a myelin sheath, Nav channel nodal clustering is also a key process regulating AP propagation along axons. Although the understanding of molecular mechanisms that support nodes of Ranvier assembly and maintenance in physiological conditions is progressing, some questions remain to be clarified regarding the diversity of neuronal responses during development and nodal reassembly after a lesion. An attractive hypothesis is that nodal-like cluster formation on unmyelinated axons might be associated with the need for early establishment of neuronal connections during development on axons with long trajectories. Whether nodal-like cluster formation on unmyelinated fibers will initiate myelination, and how cross-talk between glial cells and specific neuron subpopulations regulates axonal transmission, raise an exciting field of research. Recent findings showing non-uniform myelin distribution along single axons in the neocortex of adult mice, underlie differences in myelination profiles, which is an integral feature of neuronal identity [151]. This suggests further levels of conduction velocity regulation and a possible plasticity in network behavior.
References
- London M, Hausser M (2005) Dendritic computation. Annu Rev Neurosci 28:503–532
Article CAS PubMed Google Scholar - Bean BP (2007) The action potential in mammalian central neurons. Nat Rev Neurosci 8(6):451–465
Article CAS PubMed Google Scholar - Debanne D, Campanac E, Bialowas A, Carlier E, Alcaraz G (2011) Axon physiology. Physiol Rev 91(2):555–602
Article CAS PubMed Google Scholar - Seidl AH (2014) Regulation of conduction time along axons. Neuroscience 276:126–134
Article CAS PubMed Google Scholar - Zalc B, Goujet D, Colman D (2008) The origin of the myelination program in vertebrates. Curr Biol 18(12):R511–R512
Article CAS PubMed Google Scholar - Salzer JL (2003) Polarized domains of myelinated axons. Neuron 40(2):297–318
Article CAS PubMed Google Scholar - Funfschilling U et al (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485(7399):517–521
PubMed Central PubMed Google Scholar - Lee JK, Zheng B (2012) Role of myelin-associated inhibitors in axonal repair after spinal cord injury. Exp Neurol 235(1):33–42
Article PubMed Central CAS PubMed Google Scholar - Waxman SG, Black JA, Kocsis JD, Ritchie JM (1989) Low density of sodium channels supports action potential conduction in axons of neonatal rat optic nerve. Proc Natl Acad Sci USA 86(4):1406–1410
Article PubMed Central CAS PubMed Google Scholar - Neishabouri A, Faisal AA (2014) Saltatory conduction in unmyelinated axons: clustering of Na(+) channels on lipid rafts enables micro-saltatory conduction in C-fibers. Front Neuroanat 8:109
Article PubMed Central PubMed CAS Google Scholar - Freeman SA et al (2015) Acceleration of conduction velocity linked to clustering of nodal components precedes myelination. Proc Natl Acad Sci USA 112(3):E321–E328
Article PubMed Central CAS PubMed Google Scholar - Johnston WL, Dyer JR, Castellucci VF, Dunn RJ (1996) Clustered voltage-gated Na+ channels in Aplysia axons. J Neurosci 16(5):1730–1739
CAS PubMed Google Scholar - Messner DJ, Catterall WA (1985) The sodium channel from rat brain. Separation and characterization of subunits. J Biol Chem 260(19):10597–10604
CAS PubMed Google Scholar - O’Malley HA, Isom LL (2015) Sodium channel beta subunits: emerging targets in channelopathies. Annu Rev Physiol 77:481–504
Article PubMed CAS Google Scholar - Catterall WA (2012) Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol 590(Pt 11):2577–2589
Article PubMed Central CAS PubMed Google Scholar - Chang KJ, Rasband MN (2013) Excitable domains of myelinated nerves: axon initial segments and nodes of Ranvier. Curr Top Membr 72:159–192
Article CAS PubMed Google Scholar - Black JA, Frezel N, Dib-Hajj SD, Waxman SG (2012) Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol Pain 8:82
Article PubMed Central PubMed Google Scholar - Ratcliffe CF, Westenbroek RE, Curtis R, Catterall WA (2001) Sodium channel beta1 and beta3 subunits associate with neurofascin through their extracellular immunoglobulin-like domain. J Cell Biol 154(2):427–434
Article PubMed Central CAS PubMed Google Scholar - Chen C et al (2002) Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel beta 2-subunits. Proc Natl Acad Sci USA 99(26):17072–17077
Article PubMed Central CAS PubMed Google Scholar - Buffington SA, Rasband MN (2013) Na+ channel-dependent recruitment of Navbeta4 to axon initial segments and nodes of Ranvier. J Neurosci 33(14):6191–6202
Article PubMed Central CAS PubMed Google Scholar - Patton DE, Isom LL, Catterall WA, Goldin AL (1994) The adult rat brain beta 1 subunit modifies activation and inactivation gating of multiple sodium channel alpha subunits. J Biol Chem 269(26):17649–17655
CAS PubMed Google Scholar - Kazen-Gillespie KA et al (2000) Cloning, localization, and functional expression of sodium channel beta1A subunits. J Biol Chem 275(2):1079–1088
Article CAS PubMed Google Scholar - Isom LL et al (1995) Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83(3):433–442
Article CAS PubMed Google Scholar - McEwen DP, Meadows LS, Chen C, Thyagarajan V, Isom LL (2004) Sodium channel beta1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin. J Biol Chem 279(16):16044–16049
Article CAS PubMed Google Scholar - Aman TK et al (2009) Regulation of persistent Na current by interactions between beta subunits of voltage-gated Na channels. J Neurosci 29(7):2027–2042
Article PubMed Central CAS PubMed Google Scholar - Schwarz JR et al (2006) KCNQ channels mediate IKs, a slow K+ current regulating excitability in the rat node of Ranvier. J Physiol 573(Pt 1):17–34
Article PubMed Central CAS PubMed Google Scholar - Battefeld A, Tran BT, Gavrilis J, Cooper EC, Kole MH (2014) Heteromeric Kv7.2/7.3 channels differentially regulate action potential initiation and conduction in neocortical myelinated axons. J Neurosci 34(10):3719–3732
Article PubMed Central CAS PubMed Google Scholar - Devaux J et al (2003) Kv3.1b is a novel component of CNS nodes. J Neurosci 23(11):4509–4518
CAS PubMed Google Scholar - Devaux JJ, Kleopa KA, Cooper EC, Scherer SS (2004) KCNQ2 is a nodal K+ channel. J Neurosci 24(5):1236–1244
Article CAS PubMed Google Scholar - Trimmer JS (2015) Subcellular localization of K+ channels in mammalian brain neurons: remarkable precision in the midst of extraordinary complexity. Neuron 85(2):238–256
Article PubMed Central CAS PubMed Google Scholar - Sherman DL et al (2005) Neurofascins are required to establish axonal domains for saltatory conduction. Neuron 48(5):737–742
Article CAS PubMed Google Scholar - Davis JQ, Lambert S, Bennett V (1996) Molecular composition of the node of Ranvier: identification of ankyrin-binding cell adhesion molecules neurofascin (mucin +/third FNIII domain −) and NrCAM at nodal axon segments. J Cell Biol 135(5):1355–1367
Article CAS PubMed Google Scholar - Rios JC et al (2000) Contactin-associated protein (Caspr) and contactin form a complex that is targeted to the paranodal junctions during myelination. J Neurosci 20(22):8354–8364
CAS PubMed Google Scholar - Kazarinova-Noyes K et al (2001) Contactin associates with Na+ channels and increases their functional expression. J Neurosci 21(19):7517–7525
CAS PubMed Google Scholar - McEwen DP, Isom LL (2004) Heterophilic interactions of sodium channel beta1 subunits with axonal and glial cell adhesion molecules. J Biol Chem 279(50):52744–52752
Article CAS PubMed Google Scholar - Sherman DL, Brophy PJ (2005) Mechanisms of axon ensheathment and myelin growth. Nat Rev Neurosci 6(9):683–690
Article CAS PubMed Google Scholar - Weber P et al (1999) Mice deficient for tenascin-R display alterations of the extracellular matrix and decreased axonal conduction velocities in the CNS. J Neurosci 19(11):4245–4262
CAS PubMed Google Scholar - Oohashi T et al (2002) Bral1, a brain-specific link protein, colocalizing with the versican V2 isoform at the nodes of Ranvier in developing and adult mouse central nervous systems. Mol Cell Neurosci 19(1):43–57
Article CAS PubMed Google Scholar - Bekku Y, Rauch U, Ninomiya Y, Oohashi T (2009) Brevican distinctively assembles extracellular components at the large diameter nodes of Ranvier in the CNS. J Neurochem 108(5):1266–1276
Article CAS PubMed Google Scholar - Bekku Y et al (2010) Bral1: its role in diffusion barrier formation and conduction velocity in the CNS. J Neurosci 30(8):3113–3123
Article CAS PubMed Google Scholar - Susuki K et al (2013) Three mechanisms assemble central nervous system nodes of ranvier. Neuron 78(3):469–482
Article PubMed Central CAS PubMed Google Scholar - Eshed Y et al (2005) Gliomedin mediates Schwann cell-axon interaction and the molecular assembly of the nodes of Ranvier. Neuron 47(2):215–229
Article CAS PubMed Google Scholar - Eshed Y, Feinberg K, Carey DJ, Peles E (2007) Secreted gliomedin is a perinodal matrix component of peripheral nerves. J Cell Biol 177(3):551–562
Article PubMed Central CAS PubMed Google Scholar - Feinberg K et al (2010) A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron 65(4):490–502
Article PubMed Central CAS PubMed Google Scholar - Ffrench-Constant C, Miller RH, Kruse J, Schachner M, Raff MC (1986) Molecular specialization of astrocyte processes at nodes of Ranvier in rat optic nerve. J Cell Biol 102(3):844–852
Article CAS PubMed Google Scholar - Peles E et al (1997) Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein–protein interactions. EMBO J 16(5):978–988
Article PubMed Central CAS PubMed Google Scholar - Menegoz M et al (1997) Paranodin, a glycoprotein of neuronal paranodal membranes. Neuron 19(2):319–331
Article CAS PubMed Google Scholar - Tait S et al (2000) An oligodendrocyte cell adhesion molecule at the site of assembly of the paranodal axo-glial junction. J Cell Biol 150(3):657–666
Article PubMed Central CAS PubMed Google Scholar - Bhat MA et al (2001) Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron 30(2):369–383
Article CAS PubMed Google Scholar - Charles P et al (2002) Neurofascin is a glial receptor for the paranodin/Caspr-contactin axonal complex at the axoglial junction. Curr Biol 12(3):217–220
Article CAS PubMed Google Scholar - Rios JC et al (2003) Paranodal interactions regulate expression of sodium channel subtypes and provide a diffusion barrier for the node of Ranvier. J Neurosci 23(18):7001–7011
CAS PubMed Google Scholar - Chang KJ et al (2014) Glial ankyrins facilitate paranodal axoglial junction assembly. Nat Neurosci 17(12):1673–1681
Article PubMed Central CAS PubMed Google Scholar - Ogawa Y et al (2006) Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J Neurosci 26(19):5230–5239
Article CAS PubMed Google Scholar - Horresh I, Bar V, Kissil JL, Peles E (2010) Organization of myelinated axons by Caspr and Caspr2 requires the cytoskeletal adapter protein 4.1B. J Neurosci 30(7):2480–2489
Article PubMed Central CAS PubMed Google Scholar - Buttermore ED et al (2011) The cytoskeletal adaptor protein band 4.1B is required for the maintenance of paranodal axoglial septate junctions in myelinated axons. J Neurosci 31(22):8013–8024
Article PubMed Central CAS PubMed Google Scholar - Cifuentes-Diaz C et al (2011) Protein 4.1B contributes to the organization of peripheral myelinated axons. PLoS ONE 6(9):e25043
Article PubMed Central CAS PubMed Google Scholar - Zhang C, Susuki K, Zollinger DR, Dupree JL, Rasband MN (2013) Membrane domain organization of myelinated axons requires betaII spectrin. J Cell Biol 203(3):437–443
Article PubMed Central CAS PubMed Google Scholar - Einheber S et al (2013) The 4.1B cytoskeletal protein regulates the domain organization and sheath thickness of myelinated axons. Glia 61(2):240–253
Article PubMed Central PubMed Google Scholar - Hirono M et al (2015) BK channels localize to the paranodal junction and regulate action potentials in myelinated axons of cerebellar purkinje cells. J Neurosci 35(18):7082–7094
Article PubMed Central CAS PubMed Google Scholar - Boyle ME et al (2001) Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 30(2):385–397
Article CAS PubMed Google Scholar - Zonta B et al (2008) Glial and neuronal isoforms of Neurofascin have distinct roles in the assembly of nodes of Ranvier in the central nervous system. J Cell Biol 181(7):1169–1177
Article PubMed Central CAS PubMed Google Scholar - Poliak S, Peles E (2003) The local differentiation of myelinated axons at nodes of Ranvier. Nat Rev Neurosci 4(12):968–980
Article CAS PubMed Google Scholar - Traka M et al (2003) Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J Cell Biol 162(6):1161–1172
Article PubMed Central CAS PubMed Google Scholar - Gordon A et al (2014) Caspr and caspr2 are required for both radial and longitudinal organization of myelinated axons. J Neurosci 34(45):14820–14826
Article PubMed Central PubMed CAS Google Scholar - Vabnick I, Novakovic SD, Levinson SR, Schachner M, Shrager P (1996) The clustering of axonal sodium channels during development of the peripheral nervous system. J Neurosci 16(16):4914–4922
CAS PubMed Google Scholar - Dzhashiashvili Y et al (2007) Nodes of Ranvier and axon initial segments are ankyrin G-dependent domains that assemble by distinct mechanisms. J Cell Biol 177(5):857–870
Article PubMed Central CAS PubMed Google Scholar - Zhang Y et al (2012) Assembly and maintenance of nodes of ranvier rely on distinct sources of proteins and targeting mechanisms. Neuron 73(1):92–107
Article PubMed Central CAS PubMed Google Scholar - Labasque M, Devaux JJ, Leveque C, Faivre-Sarrailh C (2011) Fibronectin type III-like domains of neurofascin-186 protein mediate gliomedin binding and its clustering at the developing nodes of Ranvier. J Biol Chem 286(49):42426–42434
Article PubMed Central CAS PubMed Google Scholar - Lustig M et al (2001) Nr-CAM and neurofascin interactions regulate ankyrin G and sodium channel clustering at the node of Ranvier. Curr Biol 11(23):1864–1869
Article CAS PubMed Google Scholar - Custer AW et al (2003) The role of the ankyrin-binding protein NrCAM in node of Ranvier formation. J Neurosci 23(31):10032–10039
CAS PubMed Google Scholar - Saito F et al (2003) Unique role of dystroglycan in peripheral nerve myelination, nodal structure, and sodium channel stabilization. Neuron 38(5):747–758
Article CAS PubMed Google Scholar - Occhi S et al (2005) Both laminin and Schwann cell dystroglycan are necessary for proper clustering of sodium channels at nodes of Ranvier. J Neurosci 25(41):9418–9427
Article PubMed Central CAS PubMed Google Scholar - Colombelli C et al (2015) Perlecan is recruited by dystroglycan to nodes of Ranvier and binds the clustering molecule gliomedin. J Cell Biol 208(3):313–329
Article PubMed Central CAS PubMed Google Scholar - Bennett V, Baines AJ (2001) Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev 81(3):1353–1392
CAS PubMed Google Scholar - Yang Y, Ogawa Y, Hedstrom KL, Rasband MN (2007) betaIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J Cell Biol 176(4):509–519
Article PubMed Central CAS PubMed Google Scholar - Amor V et al (2014) Long-term maintenance of Na+ channels at nodes of Ranvier depends on glial contact mediated by gliomedin and NrCAM. J Neurosci 34(15):5089–5098
Article PubMed Central PubMed CAS Google Scholar - Dours-Zimmermann MT et al (2009) Versican V2 assembles the extracellular matrix surrounding the nodes of ranvier in the CNS. J Neurosci 29(24):7731–7742
Article CAS PubMed Google Scholar - Xiao ZC et al (1999) Tenascin-R is a functional modulator of sodium channel beta subunits. J Biol Chem 274(37):26511–26517
Article CAS PubMed Google Scholar - Zhang A et al (2015) Neurofascin 140 is an embryonic neuronal neurofascin isoform that promotes the assembly of the node of Ranvier. J Neurosci 35(5):2246–2254
Article PubMed Central CAS PubMed Google Scholar - Colakoglu G, Bergstrom-Tyrberg U, Berglund EO, Ranscht B (2014) Contactin-1 regulates myelination and nodal/paranodal domain organization in the central nervous system. Proc Natl Acad Sci USA 111(3):E394–E403
Article PubMed Central CAS PubMed Google Scholar - Rasband MN et al (1999) Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci 19(17):7516–7528
CAS PubMed Google Scholar - Dupree JL, Coetzee T, Blight A, Suzuki K, Popko B (1998) Myelin galactolipids are essential for proper node of Ranvier formation in the CNS. J Neurosci 18(5):1642–1649
CAS PubMed Google Scholar - Ishibashi T et al (2002) A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J Neurosci 22(15):6507–6514
CAS PubMed Google Scholar - Jenkins SM, Bennett V (2002) Developing nodes of Ranvier are defined by ankyrin-G clustering and are independent of paranodal axoglial adhesion. Proc Natl Acad Sci USA 99(4):2303–2308
Article PubMed Central CAS PubMed Google Scholar - Mathis C, Denisenko-Nehrbass N, Girault JA, Borrelli E (2001) Essential role of oligodendrocytes in the formation and maintenance of central nervous system nodal regions. Development 128(23):4881–4890
CAS PubMed Google Scholar - Desmazieres A et al (2014) Differential stability of PNS and CNS nodal complexes when neuronal neurofascin is lost. J Neurosci 34(15):5083–5088
Article PubMed Central PubMed CAS Google Scholar - Bennett V, Lorenzo DN (2013) Spectrin- and ankyrin-based membrane domains and the evolution of vertebrates. Curr Top Membr 72:1–37
Article CAS PubMed Google Scholar - Jenkins PM et al (2015) Giant ankyrin-G: a critical innovation in vertebrate evolution of fast and integrated neuronal signaling. Proc Natl Acad Sci USA 112(4):957–964
Article PubMed Central CAS PubMed Google Scholar - Ho TS et al (2014) A hierarchy of ankyrin-spectrin complexes clusters sodium channels at nodes of Ranvier. Nat Neurosci 17(12):1664–1672
Article PubMed Central CAS PubMed Google Scholar - Barry J et al (2014) Ankyrin-G directly binds to kinesin-1 to transport voltage-gated Na+ channels into axons. Dev Cell 28(2):117–131
Article PubMed Central CAS PubMed Google Scholar - Kaplan MR et al (1997) Induction of sodium channel clustering by oligodendrocytes. Nature 386(6626):724–728
Article CAS PubMed Google Scholar - Kaplan MR et al (2001) Differential control of clustering of the sodium channels Na(v)1.2 and Na(v)1.6 at developing CNS nodes of Ranvier. Neuron 30(1):105–119
Article CAS PubMed Google Scholar - Colbert CM, Johnston D (1996) Axonal action-potential initiation and Na+ channel densities in the soma and axon initial segment of subicular pyramidal neurons. J Neurosci 16(21):6676–6686
CAS PubMed Google Scholar - Stuart G, Schiller J, Sakmann B (1997) Action potential initiation and propagation in rat neocortical pyramidal neurons. J Physiol 505(Pt 3):617–632
Article PubMed Central CAS PubMed Google Scholar - Hu W et al (2009) Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci 12(8):996–1002
Article CAS PubMed Google Scholar - Palmer LM, Stuart GJ (2006) Site of action potential initiation in layer 5 pyramidal neurons. J Neurosci 26(6):1854–1863
Article CAS PubMed Google Scholar - Shu Y, Duque A, Yu Y, Haider B, McCormick DA (2007) Properties of action-potential initiation in neocortical pyramidal cells: evidence from whole cell axon recordings. J Neurophysiol 97(1):746–760
Article PubMed Google Scholar - Castelfranco AM, Hartline DK (2015) The evolution of vertebrate and invertebrate myelin: a theoretical computational study. J Comput Neurosci 38(3):521–538
Article PubMed Google Scholar - Rushton WA (1951) A theory of the effects of fibre size in medullated nerve. J Physiol 115(1):101–122
Article PubMed Central CAS PubMed Google Scholar - Hodgkin AL, Huxley AF (1952) A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117(4):500–544
Article PubMed Central CAS PubMed Google Scholar - Shu Y, Hasenstaub A, Duque A, Yu Y, McCormick DA (2006) Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature 441(7094):761–765
Article CAS PubMed Google Scholar - Kole MH et al (2008) Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci 11(2):178–186
Article CAS PubMed Google Scholar - Schmidt-Hieber C, Jonas P, Bischofberger J (2008) Action potential initiation and propagation in hippocampal mossy fibre axons. J Physiol 586(7):1849–1857
Article PubMed Central CAS PubMed Google Scholar - Sasaki T (2013) The axon as a unique computational unit in neurons. Neurosci Res 75(2):83–88
Article CAS PubMed Google Scholar - Hu H, Jonas P (2014) A supercritical density of Na(+) channels ensures fast signaling in GABAergic interneuron axons. Nat Neurosci 17(5):686–693
Article PubMed Central CAS PubMed Google Scholar - Li T et al (2014) Action potential initiation in neocortical inhibitory interneurons. PLoS Biol 12(9):e1001944
Article PubMed Central PubMed CAS Google Scholar - Kress GJ, Mennerick S (2009) Action potential initiation and propagation: upstream influences on neurotransmission. Neuroscience 158(1):211–222
Article PubMed Central CAS PubMed Google Scholar - Hartline DK, Colman DR (2007) Rapid conduction and the evolution of giant axons and myelinated fibers. Curr Biol 17(1):R29–R35
Article CAS PubMed Google Scholar - Arancibia-Carcamo IL, Attwell D (2014) The node of Ranvier in CNS pathology. Acta Neuropathol 128(2):161–175
Article PubMed Central PubMed Google Scholar - Alle H, Roth A, Geiger JR (2009) Energy-efficient action potentials in hippocampal mossy fibers. Science 325(5946):1405–1408
Article CAS PubMed Google Scholar - Carter BC, Bean BP (2009) Sodium entry during action potentials of mammalian neurons: incomplete inactivation and reduced metabolic efficiency in fast-spiking neurons. Neuron 64(6):898–909
Article PubMed Central CAS PubMed Google Scholar - Waxman SG, Bennett MV (1972) Relative conduction velocities of small myelinated and non-myelinated fibres in the central nervous system. Nat New Biol 238(85):217–219
Article CAS PubMed Google Scholar - Goldman L, Albus JS (1968) Computation of impulse conduction in myelinated fibers; theoretical basis of the velocity-diameter relation. Biophysic J 8(5):596–607
Article CAS Google Scholar - Moore JW, Joyner RW, Brill MH, Waxman SD, Najar-Joa M (1978) Simulations of conduction in uniform myelinated fibers. Relative sensitivity to changes in nodal and internodal parameters. Biophysic J 21(2):147–160
Article CAS Google Scholar - Chomiak T, Hu B (2009) What is the optimal value of the g-ratio for myelinated fibers in the rat CNS? A theoretical approach. PloS one 4(11):e7754
Article PubMed Central PubMed CAS Google Scholar - Cotter L et al (2010) Dlg1-PTEN interaction regulates myelin thickness to prevent damaging peripheral nerve overmyelination. Science 328(5984):1415–1418
Article CAS PubMed Google Scholar - Rosenbluth J (2009) Multiple functions of the paranodal junction of myelinated nerve fibers. J Neurosci Res 87(15):3250–3258
Article CAS PubMed Google Scholar - Kole MH, Letzkus JJ, Stuart GJ (2007) Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron 55(4):633–647
Article CAS PubMed Google Scholar - Popovic MA, Foust AJ, McCormick DA, Zecevic D (2011) The spatio-temporal characteristics of action potential initiation in layer 5 pyramidal neurons: a voltage imaging study. J Physiol 589(Pt 17):4167–4187
Article PubMed Central CAS PubMed Google Scholar - Clark BA, Monsivais P, Branco T, London M, Hausser M (2005) The site of action potential initiation in cerebellar Purkinje neurons. Nat Neurosci 8(2):137–139
Article CAS PubMed Google Scholar - Gogan P, Gueritaud JP, Tyc-Dumont S (1983) Comparison of antidromic and orthodromic action potentials of identified motor axons in the cat’s brain stem. J Physiol 335:205–220
Article PubMed Central CAS PubMed Google Scholar - Xia RH, Yosef N, Ubogu EE (2010) Dorsal caudal tail and sciatic motor nerve conduction studies in adult mice: technical aspects and normative data. Muscle Nerve 41(6):850–856
Article PubMed Google Scholar - Rosenbluth J (1976) Intramembranous particle distribution at the node of Ranvier and adjacent axolemma in myelinated axons of the frog brain. J Neurocytol 5(6):731–745
Article CAS PubMed Google Scholar - Ritchie JM, Rogart RB (1977) Density of sodium channels in mammalian myelinated nerve fibers and nature of the axonal membrane under the myelin sheath. Proc Natl Acad Sci USA 74(1):211–215
Article PubMed Central CAS PubMed Google Scholar - Dimitrov AG (2005) Internodal sodium channels ensure active processes under myelin manifesting in depolarizing afterpotentials. J Theol Biol 235(4):451–462
Article CAS Google Scholar - Zeng S, Jung P (2008) Simulation analysis of intermodal sodium channel function. Phys Rev E 78(6 Pt 1):061916
Article CAS Google Scholar - Waxman SG, Pappas GD, Bennett MV (1972) Morphological correlates of functional differentiation of nodes of Ranvier along single fibers in the neurogenic electric organ of the knife fish Stern archus. J Cell Biol 53(1):210–224
Article PubMed Central CAS PubMed Google Scholar - Babbs CF, Shi R (2013) Subtle paranodal injury slows impulse conduction in a mathematical model of myelinated axons. PLoS ONE 8(7):e67767
Article PubMed Central CAS PubMed Google Scholar - Xu K, Terakawa S (1999) Fenestration nodes and the wide submyelinic space form the basis for the unusually fast impulse conduction of shrimp myelinated axons. J Exp Biol 202(Pt 15):1979–1989
CAS PubMed Google Scholar - Boiko T et al (2001) Compact myelin dictates the differential targeting of two sodium channel isoforms in the same axon. Neuron 30(1):91–104
Article CAS PubMed Google Scholar - Duflocq A, Le Bras B, Bullier E, Couraud F, Davenne M (2008) Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments. Mol Cell Neurosci 39(2):180–192
Article CAS PubMed Google Scholar - Tian C, Wang K, Ke W, Guo H, Shu Y (2014) Molecular identity of axonal sodium channels in human cortical pyramidal cells. Front Cell Neurosci 8:297
Article PubMed Central PubMed CAS Google Scholar - Luo S et al (2014) The sodium channel isoform transition at developing nodes of Ranvier in the peripheral nervous system: dependence on a Genetic program and myelination-induced cluster formation. J Comp Neurol 522(18):4057–4073
Article CAS PubMed Google Scholar - Rush AM, Dib-Hajj SD, Waxman SG (2005) Electrophysiological properties of two axonal sodium channels, Nav1.2 and Nav1.6, expressed in mouse spinal sensory neurones. J Physiol 564(Pt 3):803–815
Article PubMed Central CAS PubMed Google Scholar - Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR (2000) Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci USA 97(10):5616–5620
Article PubMed Central CAS PubMed Google Scholar - Khaliq ZM, Gouwens NW, Raman IM (2003) The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci 23(12):4899–4912
CAS PubMed Google Scholar - Catterall WA, Goldin AL, Waxman SG (2005) International union of pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 57(4):397–409
Article CAS PubMed Google Scholar - Ogiwara I et al (2007) Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci 27(22):5903–5914
Article CAS PubMed Google Scholar - Yu FH et al (2006) Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 9(9):1142–1149
Article CAS PubMed Google Scholar - Herzog RI, Cummins TR, Ghassemi F, Dib-Hajj SD, Waxman SG (2003) Distinct repriming and closed-state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J Physiol 551(Pt 3):741–750
Article PubMed Central CAS PubMed Google Scholar - Zhou W, Goldin AL (2004) Use-dependent potentiation of the Nav1.6 sodium channel. Biophysic J 87(6):3862–3872
Article CAS Google Scholar - Huxley AF, Stampfli R (1949) Evidence for saltatory conduction in peripheral myelinated nerve fibres. J Physiol 108(3):315–339
Article PubMed Central Google Scholar - Court FA et al (2004) Restricted growth of Schwann cells lacking Cajal bands slows conduction in myelinated nerves. Nature 431(7005):191–195
Article CAS PubMed Google Scholar - Brill MH, Waxman SG, Moore JW, Joyner RW (1977) Conduction velocity and spike configuration in myelinated fibres: computed dependence on internode distance. JNNP 40(8):769–774
CAS Google Scholar - Wu LM, Williams A, Delaney A, Sherman DL, Brophy PJ (2012) Increasing internodal distance in myelinated nerves accelerates nerve conduction to a flat maximum. Curr Biol 22(20):1957–1961
Article PubMed Central CAS PubMed Google Scholar - Simpson AH et al (2013) Effect of limb lengthening on internodal length and conduction velocity of peripheral nerve. J Neurosci 33(10):4536–4539
Article PubMed Central CAS PubMed Google Scholar - Young RG, Castelfranco AM, Hartline DK (2013) The “Lillie transition”: models of the onset of saltatory conduction in myelinating axons. J Comp Neurosci 34(3):533–546
Article Google Scholar - Ford MC et al (2015) Tuning of Ranvier node and internode properties in myelinated axons to adjust action potential timing. Nat Com 6:8073
Article CAS Google Scholar - Seidl AH, Rubel EW, Harris DM (2010) Mechanisms for adjusting interaural time differences to achieve binaural coincidence detection. J Neurosci 30(1):70–80
Article PubMed Central CAS PubMed Google Scholar - Seidl AH, Rubel EW, Barria A (2014) Differential conduction velocity regulation in ipsilateral and contralateral collaterals innervating brainstem coincidence detector neurons. J Neurosci 34(14):4914–4919
Article PubMed Central CAS PubMed Google Scholar - Tomassy GS et al (2014) Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science 344(6181):319–324
Article PubMed Central CAS PubMed Google Scholar - Voas MG, Glenn TD, Raphael AR, Talbot WS (2009) Schwann cells inhibit ectopic clustering of axonal sodium channels. J Neurosci 29(46):14408–14414
Article PubMed Central CAS PubMed Google Scholar - Deerinck TJ, Levinson SR, Bennett GV, Ellisman MH (1997) Clustering of voltage-sensitive sodium channels on axons is independent of direct Schwann cell contact in the dystrophic mouse. J Neurosci 17(13):5080–5088
CAS PubMed Google Scholar - Pristera A, Baker MD, Okuse K (2012) Association between tetrodotoxin resistant channels and lipid rafts regulates sensory neuron excitability. PLoS ONE 7(8):e40079
Article PubMed Central CAS PubMed Google Scholar - Henry MA, Luo S, Levinson SR (2012) Unmyelinated nerve fibers in the human dental pulp express markers for myelinated fibers and show sodium channel accumulations. BMC Neurosci 13:29
Article PubMed Central CAS PubMed Google Scholar - Hildebrand C, Waxman SG (1983) Regional node-like membrane specializations in non-myelinated axons of rat retinal nerve fiber layer. Brain Res 258(1):23–32
Article CAS PubMed Google Scholar - Black JA, Foster RE, Waxman SG (1983) Freeze-fracture ultrastructure of developing and adult non-myelinated ganglion cell axolemma in the retinal nerve fibre layer. J Neurocytol 12(2):201–212
Article CAS PubMed Google Scholar - Foster RE, Connors BW, Waxman SG (1982) Rat optic nerve: electrophysiological, pharmacological and anatomical studies during development. Brain Res 255(3):371–386
Article CAS PubMed Google Scholar - Zeng S, Tang Y (2009) Effect of clustered ion channels along an unmyelinated axon. Phys Rev E Stat 80(2 Pt 1):021917
Article CAS Google Scholar - Desmazieres A, Sol-Foulon N, Lubetzki C (2012) Changes at the nodal and perinodal axonal domains: a basis for multiple sclerosis pathology? Mult Scler 18(2):133–137
Article CAS PubMed Google Scholar - Susuki K (2013) Node of Ranvier disruption as a cause of neurological diseases. ASN Neurol 5(3):209–219
Article CAS Google Scholar - Uncini A, Kuwabara S (2015) Nodopathies of the peripheral nerve: an emerging concept. JNNP 86(11):1186–1195
Google Scholar - Devaux JJ, Scherer SS (2005) Altered ion channels in an animal model of Charcot-Marie-Tooth disease type IA. J Neurosci 25(6):1470–1480
Article CAS PubMed Google Scholar - Coman I et al (2006) Nodal, paranodal and juxtaparanodal axonal proteins during demyelination and remyelination in multiple sclerosis. Brain 129(Pt 12):3186–3195
Article CAS PubMed Google Scholar - Craner MJ et al (2004) Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci USA 101(21):8168–8173
Article PubMed Central CAS PubMed Google Scholar - Howell OW et al (2006) Disruption of neurofascin localization reveals early changes preceding demyelination and remyelination in multiple sclerosis. Brain 129(Pt 12):3173–3185
Article CAS PubMed Google Scholar - Wolswijk G, Balesar R (2003) Changes in the expression and localization of the paranodal protein Caspr on axons in chronic multiple sclerosis. Brain 126(Pt 7):1638–1649
Article PubMed Google Scholar - Rasband MN et al (1998) Potassium channel distribution, clustering, and function in remyelinating rat axons. J Neurosci 18(1):36–47
CAS PubMed Google Scholar - Smith KJ, Bostock H, Hall SM (1982) Saltatory conduction precedes remyelination in axons demyelinated with lysophosphatidyl choline. J Neurol Sci 54(1):13–31
Article CAS PubMed Google Scholar - Mathey EK et al (2007) Neurofascin as a novel target for autoantibody-mediated axonal injury. J Exp Med 204(10):2363–2372
Article PubMed Central CAS PubMed Google Scholar - Derfuss T et al (2009) Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc Natl Acad Sci USA 106(20):8302–8307
Article PubMed Central CAS PubMed Google Scholar