Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice (original) (raw)
Introduction
Functional decline and loss of motor neurons are key pathological hallmarks of many human neuromuscular diseases, including motor neuron diseases such as amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy [40, 42, 61]. Degeneration of motor neurons also contributes to age-associated loss of muscle function causing slowing of movement, decline in strength and, eventually, disability [18, 21, 32]. Genotoxic stress resulting from cumulative damage to DNA is regarded as a dominant mechanism underlying aging, and several lines of evidence indicate that it contributes to aging processes in neurons [26, 36, 52]. DNA is subject to spontaneous hydrolytic decay as well as continuous modification from both endogenous reactive chemicals, such as reactive oxygen species and malondialdehyde, and exogenous environmental factors [3, 66]. Several forms of DNA damage, such as strand breaks, base modifications, and cross-links (particularly protein–DNA) have been reported to accumulate with age in a variety of tissues [3, 11, 19, 36, 37]. Being post-mitotic, irreplaceable cells, neurons may accumulate DNA lesions throughout life eventually causing functional deterioration and cell death by interfering with transcription [8, 12, 35, 51, 54].
The vulnerability of neurons to DNA lesions and the importance of DNA repair pathways for neuronal maintenance and survival are highlighted by a number of DNA repair deficiency disorders that are characterized by neurological deficits and neurodegenerative changes, in some instances resembling accelerated aging of the nervous system [7, 8, 37, 52]. DNA repair disorders with neurodegenerative changes include a variety of syndromes associated with single strand break (SSB) and double strand break (DSB) repair deficiency [37, 52], as well as disorders resulting from defects in the nucleotide excision repair (NER) pathway that removes helix-distorting and transcription blocking lesions [30, 50]. Neuropathological data have indicated that different brain regions and neuronal types display differential susceptibility towards specific DNA repair defects, and that the cerebellum is particularly affected in many DNA repair deficiency syndromes [8, 30, 37, 50, 52]. However, for most DNA repair disorders the spatiotemporal distribution of degenerative changes throughout the brain and spinal cord remains to be systematically examined, and the role of DNA damage and repair in maintenance and aging of motor neurons has yet to be investigated.
Here, we have investigated the consequences of a failure to repair DNA damage in motor neurons from DNA repair-deficient Ercc1 _Δ/− mice. These mice lack one allele of the excision repair cross-complementing group 1 (ERCC1) gene, whereas the protein derived from the other allele shows reduced activity, due to a seven amino-acid carboxy-terminal truncation [65]. ERCC1 in complex with XPF acts as a nuclease in the NER pathway [27], but in addition, it is involved in interstrand crosslink and DSB repair [1, 9, 10, 49, 70]. Previous studies with ERCC1- and XPF-deficient patients [2, 29, 49, 55] as well as Ercc1 −/− and XPF −/_− mice [33, 38, 49, 59, 65] have indicated that loss of ERCC1/XPF nuclease function causes a progeroid degenerative phenotype in several organ systems and juvenile death. Ercc1 _Δ/− and Ercc1 Δ/Δ mice show prolonged life span and an attenuated degenerative phenotype compared to Ercc1 −/_− mice, which can be explained by the low level of residual ERCC1/XPF activity [17, 65]. Our data show that Ercc1 _Δ/_− mice develop age-dependent motor abnormalities, disruption of neuromuscular connectivity at the neuromuscular junction, and degeneration of motor neurons. The data support the hypothesis that accumulation of DNA damage and genotoxic stress significantly influences neuronal aging processes and may contribute to motor neuron vulnerability in human neuromuscular disorders.
Methods
Mice
Experiments were performed in accordance with the “Principles of laboratory animal care” (NIH publication no. 86-23) and the guidelines approved by the Erasmus University animal care committee. The generation of Ercc1 − and Ercc1 Δ alleles has been previously described [65]. Ercc1 Δ/− mice were obtained by crossing Ercc1 −/+ with Ercc1 Δ/+ mice of C57Bl6J and FVB backgrounds to yield Ercc1 Δ/− with C57Bl6J/FVB hybrid background [17]. Wild-type, Ercc1 Δ/+, and Ercc1 −/+ littermates were used as controls. Mice were housed in individual ventilated cages with ad libitum access to standard mouse food (CRM pellets, SDS BP Nutrition Ltd; gross energy content 18.36 kJ/g dry mass, digestible energy 13.4 kJ/g) and water. Ercc1 _Δ/_− mice received liquefied food when they were not able to reach the food due to movement disabilities. The mice were weighed and visually inspected weekly, and were scored for gross morphological and motor abnormalities including the onset of kyphosis, abnormalities in hind limb extension when lifted by the tail, and balance and locomotion deficits. Motor performance was further determined using a grip strength test and accelerating Rotarod as described [28, 63].
Antibodies
Primary antibodies (supplier; dilutions) used in this study were as follows: rabbit anti-ATF3 (Santa Cruz; 1:1,000); rabbit anti-calbindin (Swant; 1:10,000); rabbit anti-cleaved caspase 3 (Asp175; Cell Signaling Technology; 1:500); rabbit anti-calreticulin (Affinity BioReagents; 1:5,000); rabbit anti-CGRP (Calbiochem; 1:10,000); goat anti-ChAT (Millipore; 1:500); rat anti-CR3 receptor (clone 5C6; Serotec; 1:500); rabbit anti-FUS (A300-302A RP, Bethyl Laboratories; 1:2,000); rabbit anti-GFAP (DAKO; 1:10,000); mouse anti-GFAP (Sigma; 1:10,000); mouse anti-GM130 (Transduction Laboratories; 1:100); rat anti-Mac2 (Cedarlane; 1:2,000); mouse anti-NeuN (Millipore; 1:1,000); chicken anti-neurofilament-M (Millipore; 1:2,000); rabbit anti-neurofilament160 (Millipore; 1:250); mouse anti-hyperphosphorylated neurofilament (SMI31, Sternberger Monoclonals; 1:20,000); mouse anti-non-phosphorylated neurofilament (SMI32, Sternberger Monoclonals; 1:4,000); rabbit anti-p53 (Leica; 1:1,000); rabbit anti-peripherin (Millipore; 1:1,000); rabbit anti-SCG10 (gift from Dr. A. Sobel; 1:4,000); rabbit anti-TDP43 (Proteintech; 1: 1,000); rabbit anti-ubiquitin (Dako; 1:2,000); rabbit anti-VAPB (#1006-00 [57]; 1:1,000).
For avidin–biotin–peroxidase immunocytochemistry, biotinylated secondary antibodies from Vector Laboratories diluted 1:200 were used. FITC-, Cy3-, and Cy5-conjugated secondary antibodies raised in donkey (Jackson Immunoresearch, USA) diluted at 1:200 were used for immunofluorescence. Cy3-conjugated donkey anti-rabbit Fab fragment (Jackson Immunoresearch) was used in double-labeling experiments with two rabbit primary antibodies.
Histological procedures
Mice were anaesthetized with pentobarbital and perfused transcardially with 4% paraformaldehyde; lumbar and cervical spinal cord were dissected out, post-fixed for 2 h in ice-cold 4% paraformaldehyde, cryoprotected, embedded in 10% gelatine blocks, rapidly frozen, and sectioned at 40 μm using a freezing microtome or stored at −80°C until use [64]. Frozen sections were processed free floating for immunofluorescence or avidin–biotin–immunoperoxidase complex method (ABC, Vector Laboratories, USA) with diaminobenzidine (0.05%) as the chromogen as described previously [64]. In addition, a selected number of frozen gelatin sections was collected in 4% paraformaldehyde, and processed with a modified Gallyas silver impregnation procedure of Nadler and Evenson [43] that selectively labels degenerating neurons and their processes. For double-labeling immunofluorescence with two rabbit antibodies Cy3-conjugated donkey anti-rabbit Fab fragments was used to label and sterically cover the first antibody, as described by the manufacturer (Jackson Immunoresearch). Sections were then incubated with the second antibody that was labeled consecutively with FITC-conjugated donkey anti-rabbit.
Alternatively, brain and spinal cord specimens were paraffin-embedded, sectioned at 4 μm, and mounted on Superfrost slides. For immunohistochemistry, paraffin sections were deparaffinized, rehydrated in decreasing concentrations of ethanol, treated with 10 mM sodium citrate buffer, pH 6, in the microwave for antigen retrieval, and further processed using the ABC method. A series of paraffin sections were employed for TdT-mediated dUTP Nick-End Labeling (TUNEL) assay using a commercial kit (Apoptag Plus Peroxidase in situ apoptosis detection kit, Millipore). Sections were deparaffinized and incubated as described by the manufacturer.
Immunoperoxidase-stained sections were analyzed and photographed using a Leica DM-RB microscope and a Leica DC300 digital camera. Sections stained for immunofluorescence were analyzed with a Zeiss LSM 510 confocal laser scanning microscope. Quantitative analyses of motor neurons were performed on serial lumbar 4 (L4) sections immunoperoxidase stained for ChAT or CGRP as described previously [28]. Parallel series were used for counting ATF3- and p53-labeled cells.
Neuromuscular junction analysis
Neuromuscular junctions were analyzed in deep lumbrical muscles stained for neurofilament 160 and α-bungarotoxin (5 μg/ml for 30 min, Molecular Probes), as described previously [41]. Muscles were whole-mounted in Mowoil® (Calbiochem) on glass slides and cover-slipped for subsequent imaging using a Nikon IX71 epi-fluorescence microscope equipped with a 40× oil immersion objective (NA 0.8) or a BioRad Radiance 2000 Laser Scanning confocal microscope. All analyses were undertaken by an observer blind to the genotype of the specimen. Randomly selected endplates (minimum of 100 per muscle) were categorized as either fully innervated (e.g. ‘normal’: neurofilament staining from a single axon entirely overlies endplate), partially innervated, denervated (no neurofilament overlies endplate), or poly-innervated (more than 1 axon converging on individual endplates).
Statistical analyses
Statistical analyses were performed using GraphPad Prism Software (San Diego, CA, USA). Means from different ages and mouse groups were compared using Student’s t test, Kruskal–Wallis test with Dunn’s post-hoc test or one-way ANOVA with Tukey’s post-hoc test.
Results
Ercc1 _Δ/_− mice show reduced life span and develop progressive motor abnormalities
Ercc1_−/_− and Ercc1 Δ/Δ mice in pure C57Bl/6 or FVB backgrounds are born at a frequency below Mendelian expectation [65]. Therefore, in this study we have used Ercc1 Δ/− mice in a C57Bl6J/FVB hybrid background, generated by crossing Ercc1_−/+ and Ercc1 Δ/+ mice of pure C57Bl6J and FVB backgrounds, respectively, which were born at a predicted Mendelian frequency. Ercc1_−/+ and Ercc1 Δ/+ do not differ from wild-type mice and show normal aging [17, 65], instead Ercc1 _Δ/_− mice were noticeably smaller and showed reduced body weight compared to wild-type littermates (Fig. 1a, c). The mice reached a maximal weight of 10–15 g at 6–8 weeks of age, before gradually losing weight during subsequent aging (Fig. 1c). From 3 to 8 weeks of age Ercc1 Δ/− mice started to show clasping of the hind-limbs when lifted by their tails. Subsequently, Ercc1 Δ/− mice developed fine tremors and kyphosis, and from 16 to 24 weeks they showed severe locomotor deficits and reduced ability to maintain balance (Fig. 1a, b). Grip strength and accelerating rotarod analyses showed reduced motor performance in 16 weeks compared to 8-week-old Ercc1 Δ/− mice (Fig. 1d, e). From 20 to 24 weeks Ercc1 Δ/− mice had problems with feeding themselves and became moribund. From this stage life span could be extended by 2–6 weeks by giving liquefied food, yielding a maximum life span of 24–30 weeks (Fig. 1b).
Fig. 1
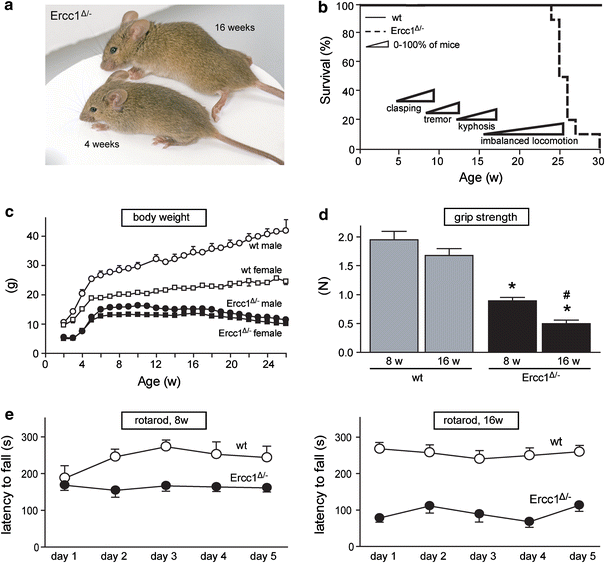
Progressive motor abnormalities and reduced life span in Ercc1 _Δ/_− mice. a Photograph of 4- and 16-week-old Ercc1 _Δ/_− mice, illustrating kyphosis and abnormal limb position in the 16-week-old mouse. b Survival plot of Ercc1 _Δ/_− mice (n = 10) and wild-type (wt) littermates (n = 10) showing reduced life span of Ercc1 _Δ/_− mice. c X_–_Y plot of body weight (mean ± SE, n = 5 per group) of Ercc1 _Δ/_− and wt mice used for survival analysis. d Bar graph (mean ± SE, n = 8 per group) of grip strength of Ercc1 _Δ/_− and wt mice determined by placing mice with four limbs on a grid attached to a force gauge and steadily pulling the mice by their tails. Grip strength is defined as the maximum strength produced by the mouse before releasing the grid. *P < 0.01 compared to wild-types; # P < 0.01 compared to 8-week-old Ercc1 _Δ/_− mice (two-tailed Student’s t test). e Average time spent on an accelerating rotarod of 8- and 16-week-old Ercc1 _Δ/_− mice and wt littermates. Mice were given two trials of maximally 5 min per day with a 45–60-min inter-trial interval for five consecutive days. Values represent mean ± SE (n = 8 per group)
Ercc1 _Δ/_− mice show age-related increase in astrocytosis and microgliosis
Pathologically, Ercc1 _Δ/− mice develop mild histological abnormalities in several tissues, in particular liver and kidney, consistent with previous reported findings in Ercc1 Δ/Δ and Ercc1 −/_− mice [38, 49, 55, 65]. From a neuronal perspective, the brains and spinal cords of Ercc1 Δ/− mice were smaller than those from wild-type mice proportional to their reduced size (Supplementary Fig. 1a, b). The gross histological organization analyzed using thionine-stained sections appeared normal (data not shown). Similarly, the distributions of markers that identify anatomically distinct subpopulations of neurons such as calbindin (Supplementary Fig. 1c, d) or non-phosphorylated neurofilament (Smi32-epitope, data not shown) were indistinguishable in Ercc1 Δ/− and wild-type mice.
To determine whether Ercc1 Δ/− mice develop neurodegenerative changes, we first examined glial markers that are known to be up-regulated in response to neuronal injury and degeneration. In 4-week-old Ercc1 _Δ/_− mice, glial acidic filament protein (GFAP)-immunohistochemistry revealed small areas of increased GFAP staining throughout the brain (Supplementary Fig. 1f) and spinal cord as shown in Fig. 2b. The amount of GFAP staining increased with subsequent aging resulting in high levels of GFAP staining throughout the brain and spinal cord of Ercc1 _Δ/_− of 16 weeks and older (Supplementary Fig. 1h and Fig. 2d). This age-related increase of GFAP staining was particularly evident in the molecular layer of cerebellar cortex where the size and number of areas with intensely GFAP-immunoreactive astrocytes correlated with increased age, and loss of calbindin immunoreactivity which in cerebellum represents a selective marker for Purkinje cells (Supplementary Fig. 2).
Fig. 2
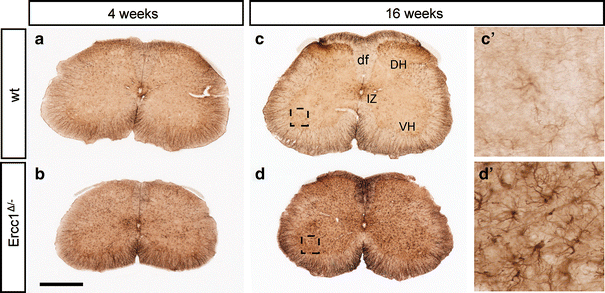
Age-related increase in GFAP immunostaining in spinal cord of Ercc1 _Δ/_− mice. Transverse lumbar L4 sections stained for GFAP illustrating mild increase in GFAP staining in spinal cord of Ercc1 _Δ/_− mice at 4 weeks (b) and robust increase at 16 weeks (d). df dorsal funiculus, DH dorsal horn, IZ intermediate zone, VH ventral horn. Scale bar 500 μm (b)
To examine parallel changes in microglia, we stained for the complement 3 receptor (CR3) present on all microglia cells [5] and Mac2 (also known as galectin-3), a protein that is expressed by activated phagocytosing microglia [53]. Control brain and spinal cord show no or very low levels of Mac2-positive microglial cells (Fig. 3a, c–e), while CR3-positive microglia cells showed a quiescent morphology (Supplementary Fig. 3a). In Ercc1 Δ/− mice, a low level of Mac2-positive microglia cells occurred throughout the brain (not shown) and spinal cord (Fig. 3c, f) at 4 weeks. At older age, Ercc1 Δ/− mice showed an increasing amount of clusters of Mac2-positive microglia cells throughout the nervous system, but in particular in the caudal brain stem and spinal cord (Fig. 3b, g). Within the spinal cord, activated microglia were present both in the gray and white matter, with relatively high levels in the ventral horn, often in close proximity of motor neurons (Supplementary Fig. 3). Taken together, these data indicate that Ercc1 Δ/− mice show age-dependent astrocytosis and microglia cell activation, pointing to an increased occurrence of neuronal degeneration throughout the brain and spinal cord.
Fig. 3
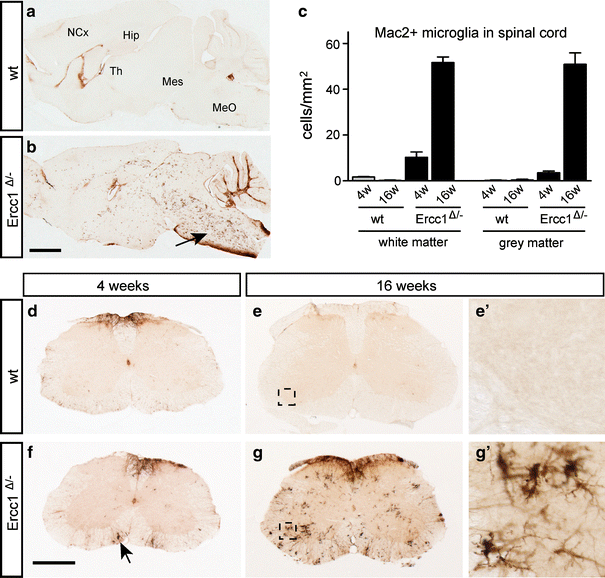
Mac2-positive microgliosis in Ercc1 _Δ/_− brain and spinal cord. Parasagittal brain (a, b) and transverse lumbar L4 sections (d–g) of Ercc1 _Δ/_− and wt mice stained for the phagocytosing microglia marker Mac2 showing Mac2-positive microglia throughout Ercc1Δ/− brain and spinal cord at 16 weeks (b, g). In the brain, the highest levels of Mac2-positive microglia occur in caudal brain stem (arrow, b) and white matter of cerebellar cortex. In spinal cord, highest levels of Mac2-positive microglia occur in white matter and motor columns in the ventral horn (g). Mac2-positive microglia cells are also present in 4-week-old Ercc1 _Δ/_− spinal cord (arrow, f), but at much lower levels than at 16 weeks. c Values in bar graph are mean ± SE of four mice based on countings of three L4 sections per mouse. Hip hippocampus, MeO medulla oblongata, Mes mesencephalon, NCx neocortex, Th thalamus. Scale bars 2 mm (a) and 500 μm (d)
Death of neurons and non-neuronal cells in the spinal cord of Ercc1 _Δ/_− mice
To obtain direct evidence for neuronal degeneration in the spinal cord, we used a silver staining method that selectively labels degenerating neurons and their processes (Fig. 4). Ercc1 Δ/− mice showed an age-related increase in staining of argyrophilic processes and neuronal cell bodies throughout the spinal cord, not observed in wild-type controls (Fig. 4). Argyrophilic processes, mostly representing degenerating axons, were more frequent in the white matter as compared to the gray matter, in particular in the dorsal columns (Fig. 4c–e). In addition, infrequent argyrophilic neuronal cell bodies were observed in the motor columns of spinal cord from 8- and 16-week-old Ercc1 Δ/− mice (Fig. 4b). Analysis of 25 L4 sections of 16-week-old Ercc1 Δ/− mice (n = 4), containing approximately 1,000 motor neurons, identified two argyrophilic motor neurons, suggesting that approximately one in 500 motor neurons was in a moribund stage. The same frequency of dying motor neurons was observed in spinal cord sections stained for active caspase 3, a major proteolytic protein in multiple cell death pathways (Supplementary Fig. 4b). Active caspase 3 staining was also associated with cells with a shrunken apoptotic morphology or with irregular structures, putatively reflecting remnants of dead cells (Supplementary Fig. 4). Apoptotic cells were also identified in the white matter (Supplementary Fig. 4c, d), indicating that glial cell death was likely to be occurring in Ercc1 Δ/− mice. Data from active caspase 3 staining were confirmed by TUNEL staining for detecting DNA fragmentation [23], showing increased levels of TUNEL-positive cells in spinal cord of Ercc1 Δ/− mice (Supplementary Fig. 4e–g). Taken together, these cell death markers provide evidence for a low level of ongoing cell death in Ercc1 Δ/− spinal cord.
Fig. 4
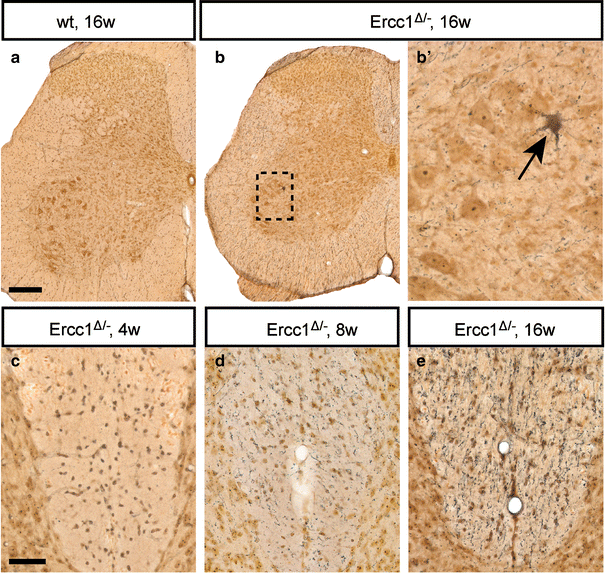
Neuronal degeneration in Ercc1 _Δ/_− spinal cord. Silver degeneration staining visualizing the distribution of neuronal degeneration in Ercc1 _Δ/_− spinal cord. Note an argyrophylic motor neuron (b, arrow) and an age-related increase in the amount of argyrophilic fibers in the dorsal funiculus (c–e). Scale bars 250 μm (a), 50 μm (c)
Loss of motor neurons, axonal pathology, and neuromuscular denervation in Ercc1 _Δ/_− mice
To directly examine the extent of motor neuron loss, we counted motor neurons in serial sections immunostained for choline acetyltransferase (ChAT), or CGRP, a peptide expressed by a subpopulation of large motor neurons [64]. The numbers of both ChAT-labeled and CGRP-labeled motor neurons were significantly reduced in 8- and 16-week-old Ercc1 Δ/− mice compared to 4-week-old Ercc1 Δ/− mice and age-matched wild-type controls (Fig. 5a–e). As motor nerve terminals at the neuromuscular junction are known to be early pathological targets in degenerating lower motor neurons [40, 42], we analyzed neuromuscular junction integrity in deep lumbrical muscles (Fig. 6). There was widespread evidence of denervated muscle fibers where pre-synaptic motor nerve terminals had degenerated, entirely or partially vacating the post-synaptic motor endplate from Ercc1 Δ/− mice at 8 and 16 weeks (Fig. 6). A subset of motor endplates was innervated by more than one incoming axon collateral (Fig. 6b). Quantification of neuromuscular morphology revealed a significant decrease in normal neuromuscular junctions and corresponding increases in the numbers of partially occupied, vacant and poly-innervated neuromuscular junctions in Ercc1 Δ/− mice at 8 and 16 weeks (Fig. 6d–g).
Fig. 5
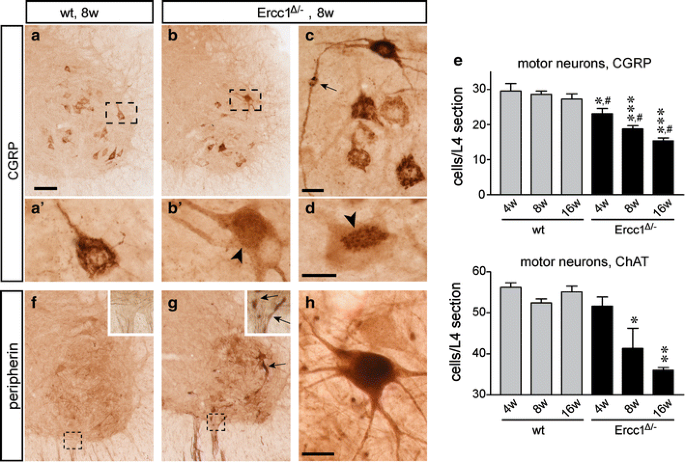
Motor neuron loss in Ercc1 _Δ/_− spinal cord. a–d Low- (a, b) and high-magnification (a′, b′, c, d) photomicrographs of CGRP staining in the ventral horn of wt (a) and Ercc1Δ/− (b–d) spinal cord. In control motor neurons, CGRP is predominantly localized to a network of linear profiles representing the Golgi apparatus. Note a variety of abnormalities of CGRP distribution in a subset of Ercc1 _Δ/_− motor neurons, including diffuse punctate staining in the cell body (b′), axonal accumulation of CGRP (arrow, c), and redistribution of the labeling to part of the cell body (d). e Bar graphs showing the number of CGRP and choline acetyltransferase (ChAT)-labeled motor neurons in L4 spinal cord sections of wt and Ercc1 _Δ/_− mice. Values are mean ± SE of four mice, while value per mouse is the mean of six L4 sections. Note age-related decrease of the number of motor neurons in Ercc1 _Δ/_− mice. *,**,***P < 0.05, 0.01, and 0.001 compared to wt mice of the same age (one-way ANOVA with Tukey’s multiple comparison test); # P = 0.002 (post-test for linear trend). f–h Photomicrographs of peripherin immunostaining in the ventral horn of wt (f) and Ercc1Δ/− (g, h) spinal cord, showing increased peripherin immunostaining in a subset of motor neurons and motor axons (arrows, g). Scale bars 100 μm (a) and 25 μm (c, d, h)
Fig. 6
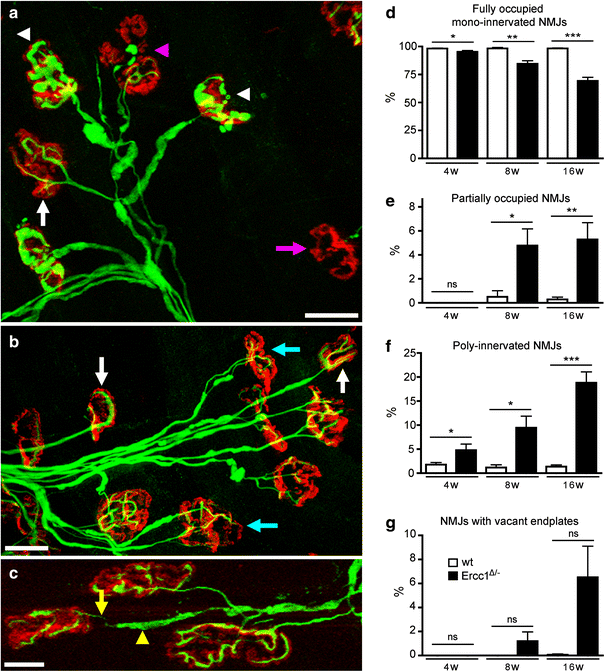
Synaptic pathology at the neuromuscular junction in Ercc1 _Δ/_− mice. a–c Confocal micrographs of neuromuscular junctions from lumbrical muscles of 16-week-old Ercc1 _Δ/_− mice labeled with rhodamine α-bungarotoxin to reveal post-synaptic motor endplates (red) and antibodies against 150 kDa neurofilaments to reveal pre-synaptic axons and motor nerve terminals (green). Examples of ‘normal’ (white arrows), denervated (pink arrow, a), partially denervated (pink arrowhead, a), poly-innervated (cyan arrows, b), and neuromuscular junctions are shown. Examples of motor nerve terminals with conspicuous neurofilament accumulation are indicated by the white arrowheads in a. Also note the presence of axon blebbing in c where the incoming axon is very thin (yellow arrow), but is interspersed by regions of neurofilament accumulations (yellow arrowhead). d–g Quantification of synaptic pathology in 4–16-week-old Ercc1 _Δ/_− mice compared to wild-type controls. *,**,***P < 0.05, 0.01, and 0.001, respectively, compared to wt mice of the same age (Kruskal–Wallis test with Dunn’s post-hoc test). Scale bars 40 μm (a, b) and 20 μm (c)
Analysis of motor nerve terminals revealed signs of neurofilament accumulation (Fig. 6a) as well as axon blebbing in distal motor axons (Fig. 6c). Neurofilament abnormalities also occurred in the proximal motor axons and the cell body (not shown). The same changes were observed with an antibody against peripherin (Fig. 5f–h), a type III intermediate filament protein predominantly expressed in axons of dorsal root ganglion cells and motor neurons that is associated with cytoskeletal pathology in ALS and aging motor neurons [67]. Hence, a subset of motor neurons showed substantial increased peripherin immunoreactivity in the cell body and the proximal axon coursing through the ventral gray and white matter. Axons with increased neurofilament and peripherin immunoreactivity showed an expanded morphology (Fig. 5g, h) reminiscent of axonal swellings such as those observed in ALS and aging spinal cord [67]. The neurofilament and peripherin abnormalities were already present at 4 weeks of age (not shown), and were more frequent at 8 (Fig. 5g, h) and 16 weeks.
p53 and ATF3 activation in partially overlapping populations of Ercc1 _Δ/_− motor neurons
To further examine the link between motor neuron degeneration and DNA damage, we studied the expression of the transcription factor p53, which is known to be activated by multiple types of DNA damage and to mediate neuronal degeneration [34]. P53-immunohistochemistry revealed cells with intense nuclear p53-staining throughout Ercc1 Δ/− spinal cord, while no labeled cells occurred in wild-type or Ercc1 −/+ spinal cords (Fig. 7a–c). Labeled cells were present in the white and the gray matter, and occasionally could be identified as motor neurons on the basis of localization in the ventral horn and somatodendritic morphology and size. Double labeling with the astrocytic marker GFAP, the neuronal marker NeuN or the motor neuron marker ChAT, respectively, confirmed that p53 labeling was present in neurons and astrocytes (Fig. 7g–i), with the latter accounting for most p53-positive cells in white matter. P53-positive astrocytes and neurons were present in spinal cord of 4-week-old Ercc1 Δ/− mice, but were more frequent at 8 and 16 weeks (Fig. 7c).
Fig. 7
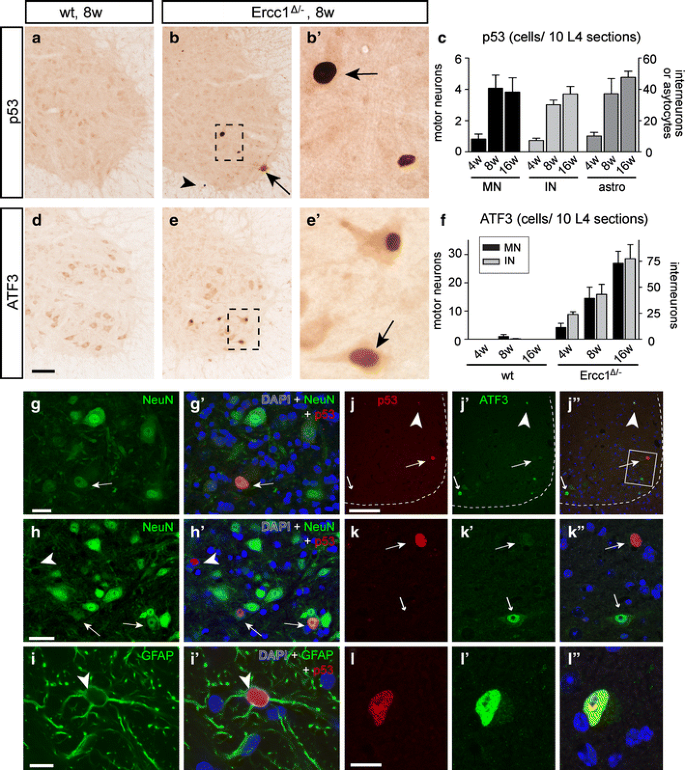
Genotoxic stress-responsive transcription factor p53 and ATF3 expression in Ercc1 _Δ/_− spinal cord. a–f Photomicrographs (a, b, d, e) and bar graphs (c, f) of p53 (a–c) and ATF3 (d–f) immunostaining, illustrating induction of p53 (arrows; b, b′) and ATF3 (arrow, e′) expression in Ercc1 _Δ/_− motor neurons. Prominent p53 labeling also occurred in astrocytes (arrowhead, b), while both p53 and ATF3 were expressed by interneurons in the spinal cord intermediate zone and dorsal horn (c, f). Values in c and f are mean ± SE of four mice, while value per mouse is from ten L4 sections. g–i Double-labeling confocal images showing p53–NeuN (g, h) and p53–GFAP (i) double-labeled cells in Ercc1 _Δ/_− spinal cord. j–l Double-labeling confocal immunofluorescence of p53 and ATF3 showing that p53 and ATF3 in most instances are expressed in distinct cells in Ercc1 _Δ/_− spinal cord sections. ATF3 single-labeled motor neurons (small arrows; j, k) occurred more frequently than p53 single-labeled motor neurons (large arrows; j, k) and double-labeled cells (arrowhead; j, l). Scale bars 100 μm (d, j), 25 μm (g, h) and 10 μm (i, l)
To further explore the DNA damage response in neurons in Ercc1 Δ/− spinal cord, we studied the expression of the stress-inducible transcription factor ATF3, which is induced following genotoxic stress via p53-dependent and -independent pathways [20, 25, 60]. ATF3, while minimally expressed in control spinal cord, was clearly present in an increasing amount of Ercc1 Δ/− motor neurons as well as other spinal cord neurons localized in the intermediate zone and dorsal horn (Fig. 7d–f). Quantitative analysis of L4 lumbar sections indicated that ATF3-positive motor neurons were more frequent than p53-positive motor neurons. Accordingly, double-labeling for p53 and ATF3 indicated that the majority of ATF3-positive motor neurons (33 of 38) were p53-negative, while approximately equal amounts of p53-positive motor neurons were ATF3-negative (6 of 11) or ATF3-positive (5 of 11; Fig. 7j–l).
A variety of Golgi apparatus abnormalities in Ercc1 _Δ/_− motor neurons
Sections stained for CGRP, which in wild-type motor neurons is predominantly localized in the Golgi apparatus (Fig. 5a), revealed subcellular abnormalities suggestive of an abnormal Golgi apparatus in a subset of Ercc1 _Δ/_− motor neurons (Fig. 5d). To systematically characterize Golgi abnormalities, we performed double labeling of p53 or ATF3 with an antibody against the _cis_-Golgi protein GM130 [44]. This analysis showed that ATF3- and p53-labeled motor neurons display a variability of abnormal morphologies of the Golgi apparatus, consisting of fragmentation of Golgi stacks (Fig. 8c, d, f–h, l, m), or the redistribution of unfragmented or fragmented Golgi stacks to part of the cell body (Fig. 8b, i, k, l). In some motor neurons, GM130 labeling was restricted to a very small portion of the cell body, occasionally giving the impression that GM130 labeling had entirely disappeared from the cell (Fig. 8b, i, k). Double labeling of GM130 and CGRP showed that in motor neurons with fragmented Golgi, GM130 and CGRP maintain their adjoining, slightly overlapping localization (Fig. 8e, f) which points to fragmentation into ministacks. Motor neurons with redistribution of GM130 staining to a restricted area of the cell body showed a similar redistribution of CGRP (Fig. 5d), but in some occasions also showed a dramatic accumulation of CGRP in the rest of the cell body (Fig. 5c), putatively representing the accumulation of post-Golgi CGRP vesicles [64]. The same was observed with an antibody against SCG10 (Fig. 8i), a tubulin-binding protein that is associated with the _trans_-Golgi and post-Golgi vesicles [13]. In contrast, no major changes were observed in the intracellular distribution of endoplasmic reticulum proteins calreticulin and VAPB [57], even in Ercc1 _Δ/_− motor neurons with considerably altered GM130 distribution (Fig. 8j, k).
Fig. 8
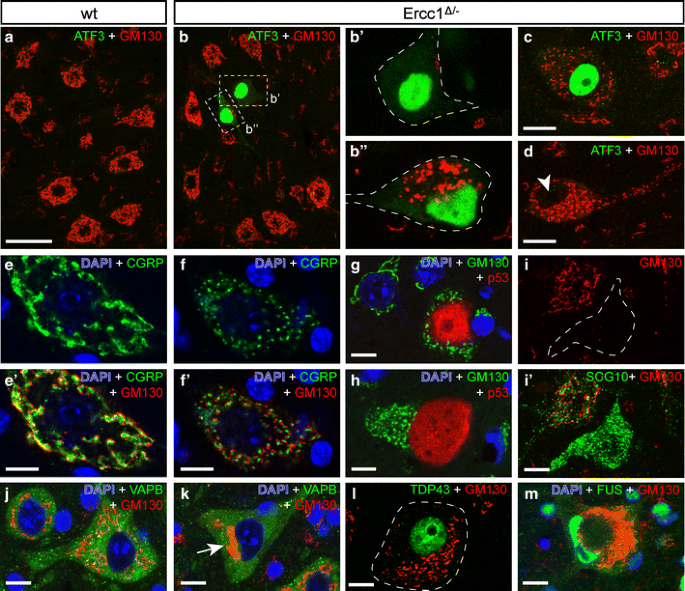
Golgi apparatus abnormalities in Ercc1 _Δ/_− motor neurons. Double-labeling confocal immunofluorescence of the cis-Golgi protein GM130 with ATF3 (a–d), CGRP (e, f), p53 (g, h), SCG10 (i), VAPB (j, k), TDP43 (l), or FUS (m) in motor neurons of wt (a, e, j) and Ercc1 _Δ/_− (b–d, f–i, k–m) spinal cord. A variety of Golgi abnormalities occur in Ercc1 _Δ/_− spinal cord, including a redistribution of GM130 staining to a small part of the cell (b, i, k, l) and fragmentation (c, d, f–h, l, m). Golgi abnormalities may (b, c) or may not (arrowhead, d) be associated with ATF3 expression. Motor neurons that are positive for p53 generally display Golgi fragmentation (g, h). In motor neurons with fragmented Golgi, GM130 and CGRP, which are localized in the _cis_- and _trans_-Golgi, respectively, maintain their adjoining slightly overlapping localization (e, f). Motor neurons with redistributed Golgi apparatus show normal distribution of endoplasmic reticulum-associated protein VAPB (k). Motor neurons with severely abnormal Golgi apparatus morphologies show a normal nuclear distribution of TDP43 and FUS (l, m). Scale bars 50 μm (a), 20 μm (c, d, i) and 10 μm (e–m)
Systematic analysis of 250 lumbar L4 motor neurons in ATF3/GM130-stained sections from 16-week-old Ercc1 Δ/− mice (n = 3) indicated that at this age as many as 25% (63 of 250) of motor neurons had an abnormal Golgi morphology, 22% (14 of 63) of which showed Golgi fragmentation. This analysis also showed that all ATF3-positive Ercc1 Δ/− motor neurons had an abnormal Golgi with various morphologies (Fig. 8b, c). Inversely, only a minority (21 of 63) of motor neurons with an abnormal Golgi was positive for ATF3 (Fig. 8d). Similarly, analysis of p53/GM130-stained sections showed that the majority (36 of 42) of motor neurons with Golgi abnormalities were p53-negative. Inversely, all p53-positive motor neurons (6 of 6) in this analysis had an abnormal Golgi apparatus (Fig. 8g, h). These data indicate that Golgi abnormalities represent a sensitive marker for cellular deficits in motor neurons.
To determine the extent to which p53- and ATF3-positive Ercc1 Δ/− motor neurons and motor neurons with abnormal GM130 labeling were towards the final stages of cell death, we double-labeled for Mac2 outlining phagocytosing microglia (see above). This analysis revealed a small subset (<10%) of afflicted Ercc1 Δ/− motor neurons surrounded by phagocytosing microglia (Supplementary Fig. 5). Taken together, our pathological analyses indicate that Ercc1 _Δ/_− motor neurons may experience a variability of cellular deficits that ultimately lead to cell death.
Motor neurons of Ercc1 _Δ/_− mice do not show TDP-43 or FUS abnormalities
Finally, we examined the extent to which the neurodegenerative phenotype observed in Ercc1 Δ/− motor neurons has features in common with ALS by staining for nuclear TAR-DNA-binding protein-43 (TDP-43), a DNA/RNA-binding protein whose accumulation in the cytoplasm and subsequent aggregation into inclusions represents a key pathological feature in the majority of ALS cases [46], and FUS protein, another DNA/RNA-binding protein that mislocalizes to the cytoplasm and aggregates in a familial form of ALS [31]. Screening of more than 50 spinal cord sections from six 16–20-week-old Ercc1 Δ/− mice indicated that all cells, including motor neurons with abnormal Golgi apparatus and nuclear morphology, showed normal nuclear TDP-43 and FUS staining (Fig. 8l, m). Accordingly, immunostaining with an anti-ubiquitin antibody did not reveal any cells with ubiquitinated pathological protein inclusions, a feature associated with TDP43 pathology in ALS [46].
Discussion
In this study, we provide direct evidence that modifications in DNA repair pathways are a robust modulator of motor neuron vulnerability in vivo. We show that Ercc1 Δ/− mice develop an age-dependent degeneration of motor neurons and progressive loss of neuromuscular connectivity. Motor neuron degeneration occurred in parallel with signs of astrocyte and microglia activation, and was associated with several morphological and molecular changes, including neurofilament and peripherin abnormalities in the cell body and the axon, misplaced and flattened nuclei, morphological changes of the Golgi apparatus, and expression of the transcription factors p53 and ATF3. Neurofilament accumulations in distal axons and motor nerve terminals are a well-documented feature of motor neuron pathology in many different animal models of motor neuron degeneration, thought to occur due to disrupted cytoskeletal organization and impaired axonal transport [40, 69]. P53 and ATF3, which are both known to be activated in response to genotoxic stress [20, 34, 68], were expressed in largely non-overlapping populations of motor neurons. This differential expression of stress genes in different motor neurons may be explained by heterogeneity of cellular damage responses triggered by different cellular deficits. A heterogeneity of cellular deficits was also suggested by the Golgi apparatus abnormalities, occurring in a large proportion of motor neurons on many occasions in the absence of ATF3 or p53 expression, and being variable as compared to the relatively stereotyped Golgi abnormalities known to occur in other neurodegenerative conditions targeting motor neurons, such as ALS [24, 64]. The Golgi apparatus is a highly dynamic organelle whose morphology depends on a large variety of protein components and cellular processes, and alters under a wide variety of conditions, including impaired endoplasmic reticulum function, disruption of the trafficking and sorting machinery, altered lipid metabolism, and activation of cell death pathways [4, 16, 24, 48, 58]. Thus, the large number of factors influencing the morphology and function of the Golgi apparatus may render it relatively vulnerable to stochastic DNA damage afflicting the transcription of randomly distributed genes, and might explain the high frequency and heterogeneity of Golgi abnormalities in Ercc1 _Δ/_− motor neurons. Taken together our pathological data are compatible with a model where motor neurons in Ercc1 Δ/− mice are afflicted by stochastic DNA lesions that block or deregulate gene expression, and indicate that motor neurons require efficient ERCC1-dependent DNA repair pathways for long-term survival.
Our data complement previous observations in ERCC1 and XPF-deficient patients [29, 49] and in Ercc1 −_/− and Xpf −/− mice [33, 38, 49, 59, 65] indicating that loss of ERCC1/XPF nuclease function causes a progeroid degenerative phenotype in several organ systems and juvenile death. Ercc1 −/− mice have also been reported to develop motor problems [38, 49, 65], which were assigned to developmental and degenerative abnormalities of the cerebellar cortex [29]. The short life span of Ercc1 −/_− mice used in these studies precluded the systematic investigation of age-dependent neurodegenerative changes. We did not observe obvious structural abnormalities in the nervous systems of our Ercc1 _Δ/− mice at 4 weeks of age, apart from reduced size and some initial signs of neuronal degeneration. Although at this point subtle neurodevelopmental changes cannot be ruled out, the lack of a neurodevelopmental phenotype such as observed in Ercc1 −/_− mice can be explained by the low level of residual ERCC1/XPF activity in Ercc1 Δ/− mice [50, 65]. Importantly, our data show that the progression of motor abnormalities in Ercc1 Δ/− mice correlated with the time course of neurodegenerative changes, indicating that the motor deficits observed may occur as a result of degenerative changes in the neuromuscular system. As Ercc1 Δ/− mice also develop age-related degenerative changes throughout the rest of the brain, also deficits in other motor areas, including the sensorimotor cortex and the cerebellum may contribute to the progressive motor deficits.
The identity of the DNA lesions triggering neurodegenerative changes in Ercc1 Δ/− mice remains to be determined. Oxidative base damage such as 7,8-dihydro-8-oxoguanine (8-oxoG) is repaired by the base excision repair (BER) pathway and may not accumulate in Ercc1 Δ/− mice. However, other oxidative lesions such as malondialdehyde adducts and 8,5′-cyclopurine-2′-deoxynucleotides are both NER substrates and potential threats to transcription [11, 22, 51]. The involvement of ERCC1/XPF as a nuclease in multiple repair pathways in addition to NER (interstrand crosslink repair and DSB repair) raises the question whether the neurodegenerative changes observed were caused by deficiency in one of these pathways, or by synergistic deleterious actions resulting from defects in multiple pathways [26, 65]. Patients with XPA-mutations that are selectively deficient in NER develop juvenile or adult onset of neuronal degeneration which indicates that NER deficiency alone can cause neuronal degeneration [7, 30]. However, Xpa −_/− mice that are entirely NER deficient have not been reported to develop noticeable neurological or neurodegenerative abnormalities up to 2 years of age [39, 45], and accordingly, extensive analyses of Xpa −/− spinal cords revealed very infrequent p53-positive cells (<1 per mm2; Jaarsma, unpublished observation). Instead, the Xpa −/_− genotype causes a severe progeroid phenotype when crossed into mice that are deficient in transcription coupled repair [6, 62], indicating that NER deficiency may exacerbate the deleterious effects of deficiencies in other repair pathways [26]. Accordingly, the degenerative changes in motor neurons that we observed here may result from combined deficiencies in NER and other repair pathways depending on ERCC1/XPF.
Cumulative DNA damage and abnormalities in DNA repair pathways have been implicated in ALS and other motor neuron diseases, but genetic evidence pointing to DNA repair genes as a risk or causative factor is inconclusive or lacking [15, 36]. A potential exception is provided by mutations in SETX that are linked to a rare dominant slowly progressing early onset motor neuron disease (ALS4) [14]. SETX is a DNA/RNA helicase that has been implicated in the defense against oxidative DNA damage [56]. The ALS4 mutation of SETX may lead to the dysfunction of its helicase activity [14], but how this is connected to motor neuron degeneration and whether it involves DNA damage is not understood. Other evidence involving DNA damage in motor neuron disease comes from pathological studies, suggesting increased levels of DNA lesions and p53 activation in post-mortem ALS motor neurons, but these changes are thought to represent late events rather than causative factors in the degenerative cascade [36]. In the present study, we show that Ercc1 Δ/− motor neurons do not develop TDP43 pathology which is a key pathological feature in the majority of ALS patients [46], a minority of which have mutations in the TDP43 gene, while the majority being caused by mechanisms that remain to be defined [31, 47]. This indicates that DNA damage resulting from ERCC1 deficiency is not sufficient to trigger the events yielding ALS pathology. Rather we propose that cumulative DNA damage may act as one of the age-related risk factors contributing to degenerative disorders such as ALS.
In summary, we propose that DNA repair pathways may have a hitherto unappreciated role in neuromuscular form and function in human neurodegenerative disease, and that the Ercc1 _Δ/_− mouse provides an excellent model to study the effects of accelerated aging on the neuromuscular system in vivo. In addition, the Ercc1 _Δ/_− mice may represent a unique mouse model for testing interventional approaches aimed at reducing genotoxic stress in neurons.
References
- Ahmad A, Robinson AR, Duensing A et al (2008) ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol Cell Biol 28:5082–5092
Article PubMed CAS Google Scholar - Ahmad A, Enzlin JH, Bhagwat NR et al (2010) Mislocalization of XPF-ERCC1 nuclease contributes to reduced DNA repair in XP-F patients. PLoS Genet 6:e1000871
Article PubMed CAS Google Scholar - Akbari M, Krokan HE (2008) Cytotoxicity and mutagenicity of endogenous DNA base lesions as potential cause of human aging. Mech Ageing Dev 129:353–365
Article PubMed CAS Google Scholar - Altan-Bonnet N, Sougrat R, Lippincott-Schwartz J (2004) Molecular basis for Golgi maintenance and biogenesis. Curr Opin Cell Biol 16:364–372
Article PubMed CAS Google Scholar - Andersson PB, Perry VH, Gordon S (1991) The kinetics and morphological characteristics of the macrophage-microglial response to kainic acid-induced neuronal degeneration. Neuroscience 42:201–214
Article PubMed CAS Google Scholar - Andressoo JO, Weeda G, de Wit J et al (2009) An Xpb mouse model for combined xeroderma pigmentosum and cockayne syndrome reveals progeroid features upon further attenuation of DNA repair. Mol Cell Biol 29:1276–1290
Article PubMed CAS Google Scholar - Anttinen A, Koulu L, Nikoskelainen E et al (2008) Neurological symptoms and natural course of xeroderma pigmentosum. Brain 131:1979–1989
Article PubMed Google Scholar - Barzilai A, Biton S, Shiloh Y (2008) The role of the DNA damage response in neuronal development, organization and maintenance. DNA Repair 7:1010–1027
Article PubMed CAS Google Scholar - Bergstralh DT, Sekelsky J (2008) Interstrand crosslink repair: can XPF-ERCC1 be let off the hook? Trends Genet 24:70–76
Article PubMed CAS Google Scholar - Bhagwat N, Olsen AL, Wang AT et al (2009) XPF-ERCC1 participates in the Fanconi anemia pathway of cross-link repair. Mol Cell Biol 29:6427–6437
Article PubMed CAS Google Scholar - Brooks PJ (2008) The 8, 5′-cyclopurine-2′-deoxynucleosides: candidate neurodegenerative DNA lesions in xeroderma pigmentosum, and unique probes of transcription and nucleotide excision repair. DNA Repair 7:1168–1179
Article PubMed CAS Google Scholar - Brooks PJ, Cheng TF, Cooper L (2008) Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage? DNA Repair 7:834–848
Article PubMed CAS Google Scholar - Charbaut E, Chauvin S, Enslen H et al (2005) Two separate motifs cooperate to target stathmin-related proteins to the Golgi complex. J Cell Sci 118:2313–2323
Article PubMed CAS Google Scholar - Chen YZ, Bennett CL, Huynh HM et al (2004) DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 74:1128–1135
Article PubMed CAS Google Scholar - Coppede F, Migheli F, Lo Gerfo A et al (2010) Association study between XRCC1 gene polymorphisms and sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler 11(1–2):122–124
Google Scholar - De Matteis MA, D’Angelo G (2007) The role of the phosphoinositides at the Golgi complex. Biochem Soc Symp (74):107–116
- Dolle ME, Busuttil RA, Garcia AM et al (2006) Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice. Mutat Res 596:22–35
PubMed CAS Google Scholar - Edstrom E, Altun M, Bergman E et al (2007) Factors contributing to neuromuscular impairment and sarcopenia during aging. Physiol Behav 92:129–135
Article PubMed CAS Google Scholar - Evans MD, Dizdaroglu M, Cooke MS (2004) Oxidative DNA damage and disease: induction, repair and significance. Mutat Res 567:1–61
Article PubMed CAS Google Scholar - Fan F, Jin S, Amundson SA et al (2002) ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene 21:7488–7496
Article PubMed CAS Google Scholar - Faulkner JA, Larkin LM, Claflin DR, Brooks SV (2007) Age-related changes in the structure and function of skeletal muscles. Clin Exp Pharmacol Physiol 34:1091–1096
Article PubMed CAS Google Scholar - Fishel ML, Vasko MR, Kelley MR (2007) DNA repair in neurons: so if they don’t divide what’s to repair? Mutat Res 614:24–36
PubMed CAS Google Scholar - Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119:493–501
Article PubMed CAS Google Scholar - Gonatas NK, Stieber A, Gonatas JO (2006) Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J Neurol Sci 246:21–30
Article PubMed CAS Google Scholar - Hai T, Hartman MG (2001) The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene 273:1–11
Article PubMed CAS Google Scholar - Hoeijmakers JH (2009) DNA damage, aging, and cancer. N Engl J Med 361:1475–1485
Article PubMed CAS Google Scholar - Houtsmuller AB, Rademakers S, Nigg AL et al (1999) Action of DNA repair endonuclease ERCC1/XPF in living cells. Science 284:958–961
Article PubMed CAS Google Scholar - Jaarsma D, Teuling E, Haasdijk ED et al (2008) Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J Neurosci 28:2075–2088
Article PubMed CAS Google Scholar - Jaspers NG, Raams A, Silengo MC et al (2007) First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am J Hum Genet 80:457–466
Article PubMed CAS Google Scholar - Kraemer KH, Patronas NJ, Schiffmann R et al (2007) Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience 145:1388–1396
Article PubMed CAS Google Scholar - Lagier-Tourenne C, Cleveland DW (2009) Rethinking ALS: the FUS about TDP-43. Cell 136:1001–1004
Article PubMed CAS Google Scholar - Larsson L, Ansved T (1995) Effects of ageing on the motor unit. Prog Neurobiol 45:397–458
Article PubMed CAS Google Scholar - Lawrence NJ, Sacco JJ, Brownstein DG et al (2008) A neurological phenotype in mice with DNA repair gene Ercc1 deficiency. DNA Repair 7:281–291
Article PubMed CAS Google Scholar - Levine AJ, Hu W, Feng Z (2006) The P53 pathway: what questions remain to be explored? Cell Death Differ 13:1027–1036
Article PubMed CAS Google Scholar - Ljungman M, Lane DP (2004) Transcription—guarding the genome by sensing DNA damage. Nat Rev Cancer 4:727–737
Article PubMed CAS Google Scholar - Martin LJ (2008) DNA damage and repair: relevance to mechanisms of neurodegeneration. J Neuropathol Exp Neurol 67:377–387
Article PubMed CAS Google Scholar - McKinnon PJ (2009) DNA repair deficiency and neurological disease. Nat Rev Neurosci 10:100–112
Article PubMed CAS Google Scholar - McWhir J, Selfridge J, Harrison DJ et al (1993) Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat Genet 5:217–224
Article PubMed CAS Google Scholar - Melis JP, Wijnhoven SW, Beems RB et al (2008) Mouse models for xeroderma pigmentosum group A and group C show divergent cancer phenotypes. Cancer Res 68:1347–1353
Article PubMed CAS Google Scholar - Murray LM, Comley LH, Thomson D et al (2008) Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet 17:949–962
Article PubMed CAS Google Scholar - Murray LM, Lee S, Baumer D et al (2010) Pre-symptomatic development of lower motor neuron connectivity in a mouse model of severe spinal muscular atrophy. Hum Mol Genet 19:420–433
Article PubMed CAS Google Scholar - Murray LM, Talbot K, Gillingwater TH (2010) Neuromuscular synaptic vulnerability in motor neurone disease: amyotrophic lateral sclerosis and spinal muscular atrophy. Neuropathol Appl Neurobiol 36:133–156
PubMed CAS Google Scholar - Nadler JV, Evenson DA (1983) Use of excitatory amino acids to make axon sparing lesions of the hypothalamus. Meth Enzymol 103:393–400
Article PubMed CAS Google Scholar - Nakamura N, Rabouille C, Watson R et al (1995) Characterization of a cis-Golgi matrix protein, GM130. J Cell Biol 131:1715–1726
Article PubMed CAS Google Scholar - Nakane H, Hirota S, Brooks PJ et al (2008) Impaired spermatogenesis and elevated spontaneous tumorigenesis in xeroderma pigmentosum group A gene (Xpa)-deficient mice. DNA Repair 7:1938–1950
Article PubMed CAS Google Scholar - Neumann M, Sampathu DM, Kwong LK et al (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133
Article PubMed CAS Google Scholar - Neumann M (2009) Molecular neuropathology of TDP-43 proteinopathies. Int J Mol Sci 10:232–246
Article PubMed CAS Google Scholar - Ngo M, Ridgway ND (2009) Oxysterol binding protein-related Protein 9 (ORP9) is a cholesterol transfer protein that regulates Golgi structure and function. Mol Biol Cell 20:1388–1399
Article PubMed CAS Google Scholar - Niedernhofer LJ, Garinis GA, Raams A et al (2006) A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444:1038–1043
Article PubMed CAS Google Scholar - Niedernhofer LJ (2008) Nucleotide excision repair deficient mouse models and neurological disease. DNA Repair 7:1180–1189
Article PubMed CAS Google Scholar - Nouspikel T (2007) DNA repair in differentiated cells: some new answers to old questions. Neuroscience 145:1213–1221
Article PubMed CAS Google Scholar - Rass U, Ahel I, West SC (2007) Defective DNA repair and neurodegenerative disease. Cell 130:991–1004
Article PubMed CAS Google Scholar - Rotshenker S, Reichert F, Gitik M et al (2008) Galectin-3/MAC-2, Ras and PI3K activate complement receptor-3 and scavenger receptor-AI/II mediated myelin phagocytosis in microglia. Glia 56:1607–1613
Article PubMed Google Scholar - Saxowsky TT, Doetsch PW (2006) RNA polymerase encounters with DNA damage: transcription-coupled repair or transcriptional mutagenesis? Chem Rev 106:474–488
Article PubMed CAS Google Scholar - Selfridge J, Hsia KT, Redhead NJ, Melton DW (2001) Correction of liver dysfunction in DNA repair-deficient mice with an ERCC1 transgene. Nucleic Acids Res 29:4541–4550
Article PubMed CAS Google Scholar - Suraweera A, Becherel OJ, Chen P et al (2007) Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J Cell Biol 177:969–979
Article PubMed CAS Google Scholar - Teuling E, Ahmed S, Haasdijk E et al (2007) Motor neuron disease-associated mutant vesicle-associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into endoplasmic reticulum-derived tubular aggregates. J Neurosci 27:9801–9815
Article PubMed CAS Google Scholar - Teuling E, van Dis V, Wulf PS et al (2008) A novel mouse model with impaired dynein/dynactin function develops amyotrophic lateral sclerosis (ALS)-like features in motor neurons and improves lifespan in SOD1-ALS mice. Hum Mol Genet 17:2849–2862
Article PubMed CAS Google Scholar - Tian M, Shinkura R, Shinkura N, Alt FW (2004) Growth retardation, early death, and DNA repair defects in mice deficient for the nucleotide excision repair enzyme XPF. Mol Cell Biol 24:1200–1205
Article PubMed CAS Google Scholar - Turchi L, Fareh M, Aberdam E et al (2009) ATF3 and p15PAF are novel gatekeepers of genomic integrity upon UV stress. Cell Death Differ 16:728–737
Article PubMed CAS Google Scholar - Valdmanis PN, Daoud H, Dion PA, Rouleau GA (2009) Recent advances in the genetics of amyotrophic lateral sclerosis. Curr Neurol Neurosci Rep 9:198–205
Article PubMed Google Scholar - van der Pluijm I, Garinis GA, Brandt RM et al (2007) Impaired genome maintenance suppresses the growth hormone—insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol 5:e2
Article PubMed CAS Google Scholar - van Woerden GM, Harris KD, Hojjati MR et al (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci 10:280–282
Article PubMed CAS Google Scholar - Vlug AS, Teuling E, Haasdijk ED et al (2005) ATF3 expression precedes death of spinal motoneurons in amyotrophic lateral sclerosis-SOD1 transgenic mice and correlates with c-Jun phosphorylation, CHOP expression, somato-dendritic ubiquitination and Golgi fragmentation. Eur J Neurosci 22:1881–1894
Article PubMed Google Scholar - Weeda G, Donker I, de Wit J et al (1997) Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol 7:427–439
Article PubMed CAS Google Scholar - Wilson DM 3rd, Bohr VA (2007) The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair 6:544–559
Article PubMed CAS Google Scholar - Xiao S, McLean J, Robertson J (2006) Neuronal intermediate filaments and ALS: a new look at an old question. Biochim Biophys Acta 1762:1001–1012
PubMed CAS Google Scholar - Yan C, Lu D, Hai T, Boyd DD (2005) Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. EMBO J 24:2425–2435
Article PubMed CAS Google Scholar - Zhang B, Tu P, Abtahian F et al (1997) Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol 139:1307–1315
Article PubMed CAS Google Scholar - Zhu XD, Niedernhofer L, Kuster B et al (2003) ERCC1/XPF removes the 3′ overhang from uncapped telomeres and represses formation of telomeric DNA-containing double minute chromosomes. Mol Cell 12:1489–1498
Article PubMed CAS Google Scholar