Comprehensive resequence analysis of a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers (original) (raw)
Abstract
Recently, genome-wide association studies have identified loci across a segment of chromosome 8q24 (128,100,000–128,700,000) associated with the risk of breast, colon and prostate cancers. At least three regions of 8q24 have been independently associated with prostate cancer risk; the most centromeric of which appears to be population specific. Haplotypes in two contiguous but independent loci, marked by rs6983267 and rs1447295, have been identified in the Cancer Genetic Markers of Susceptibility project (http://cgems.cancer.gov), which genotyped more than 5,000 prostate cancer cases and 5,000 controls of European origin. The rs6983267 locus is also strongly associated with colorectal cancer. To ascertain a comprehensive catalog of common single-nucleotide polymorphisms (SNPs) across the two regions, we conducted a resequence analysis of 136 kb (chr8: 128,473,000–128,609,802) using the Roche/454 next-generation sequencing technology in 39 prostate cancer cases and 40 controls of European origin. We have characterized a comprehensive catalog of common (MAF > 1%) SNPs within this region, including 442 novel SNPs and have determined the pattern of linkage disequilibrium across the region. Our study has generated a detailed map of genetic variation across the region, which should be useful for choosing SNPs for fine mapping of association signals in 8q24 and investigations of the functional consequences of select common variants.
Similar content being viewed by others
Introduction
Recent genome-wide association (GWAS) and linkage studies have identified common genetic variation in at least three independent regions of human chromosome 8q24 (128,100,000–128,700,000) that are associated with risk of prostate cancer (Amundadottir et al. 2006; Freedman et al. 2006; Gudmundsson et al. 2007; Haiman et al. 2007b; Yeager et al. 2007). The first single-nucleotide polymorphism (SNP) marker associated with prostate cancer risk was rs1447295 (Amundadottir et al. 2006; Freedman et al. 2006), and in follow-up GWAS, two studies have reported stronger signals of association in prostate cancer for two markers, rs4242382 (Thomas et al. 2008) and rs4242384 (Eeles et al. 2008), both in strong linkage disequilibrium (LD) with rs1447295. A second independent marker, rs6983267, was identified by GWAS with replication; though the two markers lie in contiguous blocks of LD, they are separated by a recombination hotspot (Yeager et al. 2007). Therefore, the two loci confer independent risk for prostate cancer. A separate study focusing on non-European populations reported that two SNPs (rs10808556 and rs7013278) in LD with rs6983267 are strongly linked with rs6983267 and could capture significant additional risk (Haiman et al. 2007a). Furthermore, rs6983267 has also been identified as a marker for colorectal cancer risk in four independent GWAS (Gruber et al. 2007; Haiman et al. 2007a; Tomlinson et al. 2007; Zanke et al. 2007), and in premalignant colorectal adenoma (Berndt et al. [2008](/article/10.1007/s00439-008-0535-3#ref-CR2 "Berndt SI, Potter JD, Hazra A, Yeager M, Thomas G, Makar KW, Welch R, Cross AJ, Huang WY, Schoen RE, Giovannucci E, Chan AT, Chanock SJ, Peters U, Hunter DJ, Hayes RB (2008) Pooled analysis of genetic variation at chromosome 8q24 and colorectal neoplasia risk. Hum Mol Genet. doi: 10.1093/hmg/ddn166
")). Lastly, a fourth region has been established in breast cancer, marked by rs13281615, which is \~58 kb centromeric to rs6983267 and resides in a distinct block of LD (Easton et al. [2007](/article/10.1007/s00439-008-0535-3#ref-CR8 "Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, Fletcher O, Johnson N, Seal S, Stratton MR, Rahman N, Chenevix-Trench G, Bojesen SE, Nordestgaard BG, Axelsson CK, Garcia-Closas M, Brinton L, Chanock S, Lissowska J, Peplonska B, Nevanlinna H, Fagerholm R, Eerola H, Kang D, Yoo KY, Noh DY, Ahn SH, Hunter DJ, Hankinson SE, Cox DG, Hall P, Wedren S, Liu J, Low YL, Bogdanova N, Schurmann P, Dork T, Tollenaar RA, Jacobi CE, Devilee P, Klijn JG, Sigurdson AJ, Doody MM, Alexander BH, Zhang J, Cox A, Brock IW, MacPherson G, Reed MW, Couch FJ, Goode EL, Olson JE, Meijers-Heijboer H, van den Ouweland A, Uitterlinden A, Rivadeneira F, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Hopper JL, McCredie M, Southey M, Giles GG, Schroen C, Justenhoven C, Brauch H, Hamann U, Ko YD, Spurdle AB, Beesley J, Chen X, Mannermaa A, Kosma VM, Kataja V, Hartikainen J, Day NE et al (2007) Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447:1087–1093")).8q24 is commonly amplified in many different types of cancer, particularly in prostate and colorectal cancer (Cher et al. 1996; Nupponen et al. 1998). Interestingly, in the region of the association signals for breast, colorectal and prostate cancer (128,100,000–128,700,000), there are no known genes with the exception of a processed psuedogene (POU5F1P1). Approximately 300 kb distal to this region is the c-MYC oncogene (MYC) but in multiple large genome-wide scans, so far there is no evidence for LD between genetic variants in the MYC gene and the loci associated with specific cancers.
To identify common alleles with low to moderate effects, GWAS have relied upon replication in independent data sets to confirm true associations (Chanock et al. 2007). Because these studies are designed to discover new genomic regions associated with human disease or traits using SNPs as markers, it is unlikely that the confirmed SNPs are the actual disease-contributory genetic variants. It has also been proposed that there may be significant allelic heterogeneity within regions such as 8q24 and that there may be more than one causal variant present that could regulate a gene or pathway (Camp et al. 2007). Before conducting additional genotyping in large data sets to pinpoint the best markers for further functional study, an important step in following up regions of association identified by GWAS is the characterization of a comprehensive set of common and uncommon genetic polymorphisms within each region. In this regard, the determination that all genetic variation strongly correlated (“tagged”) with the most highly disease-associated SNP markers can focus attention on a subset of variants for laboratory analysis of phenotypic differences.
Recent advances in next-generation sequencing technology promise to accelerate the determination of comprehensive surveys of variation in target regions, because it is possible to resequence regions in multiple individuals (Shaffer 2007). To facilitate the cataloging of common genetic variation in this region of chromosome 8q24, Roche/454 next-generation sequencing technology was utilized to rapidly resequence a ~136 kb region (chr8: 128,473,000–128,609,802) in 39 advanced prostate cancer cases and 40 controls from the Prostate, Lung, Colorectal, and Ovarian Screening Trial (Hayes et al. 2005); these subjects were drawn from cases and controls genotyped in the PLCO GWAS reported in the CGEMS project (http://cgems.cancer.gov) (Yeager et al. 2007). The sample size for analysis was deliberately chosen (158 chromosomes) to identify common SNPs, namely those with a minor allele frequency (MAF) of greater than 1% with a probability of 99% (Kruglyak and Nickerson 2001). Six HapMap CEU samples were also included for quality-control assessment. Furthermore, the sampling strategy using the Roche/454 technology could be compared with the results generated by the Illumina technology in the initial GWAS.
Materials and methods
Samples
Forty advanced prostate cancer cases and forty controls from the National Cancer Institute’s Prostate, Lung, Colorectal, and Ovarian Screening Trial (PLCO) were selected from those DNA samples that were genotyped as part of the Cancer Genetic Markers of Susceptibility (CGEMS) GWAS (http://cgems.cancer.gov). PLCO samples and the subset chosen for the initial GWAS have been described previously (Gohagan et al. 2000; Yeager et al. 2007). One case sample was later determined to be of poor quality and the results were eliminated from the analysis.
Region selection
The region (chr8: 128,473,000–128,609,802) was chosen based on the observed patterns of LD surrounding rs6983267 and rs1447295 in the HapMap CEU samples (n = 60 individuals) such that the disease-linked causative variant(s) is expected to be captured by this analysis; the region was chosen such that the likelihood that the disease-linked variant(s) would not fall within the chosen region is very small (Yeager et al. 2007).
Primer design and PCR optimization process
PCR primers were designed to generate tiled amplicons ranging in size between 2 and 5.5 kb overlapping across 150–600 bp segments. Primers were designed using NetPrimer (http://www.premierbiosoft.com/netprimer/netprlaunch/netprlaunch.html). Primers were synthesized and HPLC-purified (Integrated DNA Technologies, Coralville, IA). Primer testing was performed using 125 ng of a commercial pool of human genomic DNA (Roche Applied Science, Indianapolis, IN) in a PCR core mix containing 200 nM of each forward and reverse primers, 1X AccuPrime™ Buffer II (Invitrogen, Carlsbad, CA) and two units of AccuPrime™ Taq DNA Polymerase High Fidelity (Invitrogen). PCR primers and amplicon lengths are listed in Supplementary Table 1.
Thermal cycling was performed on a DNA Engine Tetrad® (Bio-Rad Laboratories, Hercules, CA) using the following “touch down” cycling profile: one cycle at 95°C for 5 min (initial denaturation), followed by six cycles at 95°C for 30 s (denaturation), at 65°C for 30 s (annealing; −1°C every cycle) at 68°C for 3 min (extension), followed by 24 cycles at 95°C for 30 s (denaturation), at 60°C for 30 s (annealing), at 68°C for 3 min (extension), followed by a final extension step at 68°C for 10 min. Upon visualization of PCR products on 1% agarose gel, amplicons that failed to amplify were attempted with up to three additional modified thermal cycling conditions. The first profile stretched all extension times to 6 min. The second profile maintained the 6-min extension and reduced the annealing temperature for the last 24 cycles to 55°C. The third profile maintained the 6-min extension and dropped the annealing temperature of the last 24 cycles to 50°C. The thermal cycling profile producing the strongest and cleanest amplicon was recorded for each primer set. Any primer pair that failed to amplify with all four PCR profiles was discarded and new primers were designed. In such cases, new primers were designed to maintain a minimum overlap of 150 bp with the upstream amplicon and a minimum amplicon length of 2 kb.
Once all amplicons covering the target region were successfully amplified, PCR products were cleaned up using an equal volume of AMPure Reagent beads (Agencourt, Beverly, MA) following the manufacturer’s protocol. Amplicons were subsequently quantified using the Quant-iT™ PicoGreen® dsDNA reagent (Invitrogen). Amplicons were then pooled at an equimolar ratio. Three micrograms to five micrograms of DNA was used to make a sequencing library.
Sequencing
Library preparation and sequencing was performed on a Genome Sequencer FLX System (Roche Applied Science) as previously described (Gilbert et al. 2007).
Data analysis
The sequence reads that passed QC were aligned to the reference sequence from the target region (chr8: 128,473,000–128,609,802) using a BLAST-based approach. The total numbers of the aligned reads for each position and the proportion of each type of nucleotides were used for calling genotypes for each position using a heuristic approach. Briefly, genotype calls per position were established when total number of aligned reads exceeded a preset threshold (n = 20). For each nucleotide position, genotypes were called based on the proportion of each type of nucleotides. For each sample, homozygote genotype was assigned if the proportion of the nonreference allele was <15% or >85%. Heterozygote genotype was assigned if the proportion is between 30 and 70%. The samples with nonreference allele proportions outside the above ranges were not assigned any genotypes. Insertions and deletions were detected during the alignment, but genotype calls were not made on this class of polymorphisms.
Descriptive statistics
Completion, concordance, MAF estimations, deviations from fitness for Hardy–Weinberg proportion, pair-wise LD, and tag SNP selection were computed using the GLU software package (http://cgfweb.nci.nih.gov/development/tooldev.html).
Enhancer and conservation prediction
SNPs that exhibited the highest correlations (r 2 > 0.9) with the most significant single SNPs in the centromeric and telomeric regions (rs6983267 and rs4242382, respectively) (Thomas et al. 2008) were searched using publicly available bioinformatic tools, particularly searching for highly conserved and/or predicted enhancer regions using the UCSC Genome Browser (http://genome.ucsc.edu/) and Vista Enhancer database (http://enhancer.lbl.gov/). Predicted enhancer elements are highly significantly conserved between humans, mice, and rats. Note that the predicted enhancer that overlaps rs6983267 is conserved in chicken in addition to humans, mice, and rats.
Results
Coverage and depth
Figure 1 shows coverage distribution within the target region. The average coverage depth within the target region is at least fiftyfold (50×), with fluctuations across the region of interest (chr8: 128,473,000–128,609,802). There were three regions that had no coverage for any samples attempted (128,487,643–128,489,703, 128,602,123–128,603,268, 128,609,417–128,609,802, 3,590 bp) and one region of low coverage, less than 20× (128,579,374–128,584,587; cumulatively 5,213 bp).
Fig. 1
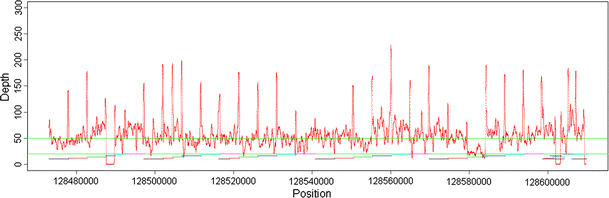
Coverage distribution within the target region (chr8: 128,473,000–128,609,802). The depth of coverage is calculated based on number of reads that mapped to that position. The _X_-axis is the relative position from the start of the target region. The colored horizontal bars at lower portion of the plot indicate the position of the amplicons from long range PCR (Supplementary Table 1)
SNP discovery
Genotype calls were successfully made for 1,004 possible segregating sites. Of these, 562 sites had previously been reported in the dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP), but 213 of the 562 were monomorphic in our study sample set. Four hundred and forty-two novel SNPs were discovered and had not, to our knowledge, been described previously; 174 of these novel SNPs were singletons in our sample set. The mean, median, maximum, and minimum MAF estimates for 791 newly discovered and previously reported SNPs are shown in Table 1, and the distribution of MAFs is shown in Fig. 2; all MAF estimates for the study samples are reported in Supplementary Table 2. Overall, 454 SNPs were observed to occur at a frequency ≥5% across this 136 kb region. It should be noted, however, that because of the relaxation of per-locus completion (as described below), some of these estimates could vary. The position of each SNP with an estimated MAF is depicted per position in Fig. 3. For the SNPs detected in this resequence analysis, the mean, median, maximum, and minimum distances between polymorphic sites were 165, 98, 1,414, and 1 bp, respectively. Chromosomal positions, alleles, MAFs, flanking sequences, completion rates, and concordance rates for all SNPs are included in Supplementary Table 2.
Table 1 Summary of minor allele frequency information for newly-described (“non-dbSNP”) and dbSNP single-nucleotide polymorphisms
Fig. 2
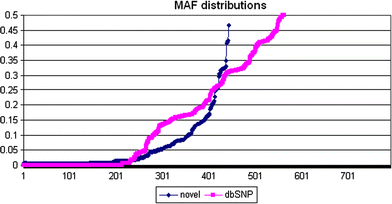
Distribution of minor allele frequencies for newly discovered and previously reported SNPs
Fig. 3
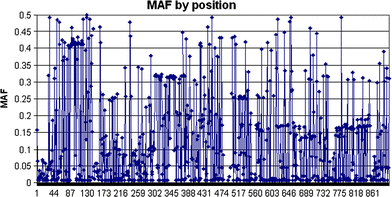
Minor allele frequency estimations for 442 polymorphic novel SNPs and 562 SNPs that had previously been reported in dbSNP across the 8q24 region
Completion rates
For each SNP and sample, percent genotyping completion rates were calculated. One of the 80 study samples, a prostate cancer case, consistently exhibited an extremely low completion rate and, therefore, was dropped from subsequent analyses. After removal, the global completion rate for all remaining samples and polymorphic loci was 93.5% (range 77.1–99.2% for samples and 7.7–100% for loci). Because this study was primarily focused on SNP discovery, no loci were dropped due to low completion rates. Completion rates per locus are included in Fig. 4 and detailed in Supplementary Table 2.
Fig. 4

Completion rates per locus and per sample
Concordance with GWAS
Because 79 DNA study samples in this project were also previously genotyped as a part of the initial CGEMS GWAS, concordance with the genotype data generated using the Illumina Infinium HumanHap550 array was assessed for 39 SNPs across this interval. Overall concordance per locus was 99.45%; genotype concordance for 28 SNPs was 100%, >98% for 10 SNPs, and >95% for the one remaining SNP (rs17467139). For the study samples, the overall concordance rate per sample was 99.45%, the majority (71/79 retained in the analysis) of the samples displayed 100% concordance, six samples were >95% concordant, though one sample was only 77% concordant (described above and dropped from the analysis). These high concordance values are particularly reassuring, since the comparisons are of genotypes derived from multiple technology platforms.
Fitness for Hardy–Weinberg proportions analysis
All polymorphic loci were tested for deviations from fitness for Hardy–Weinberg proportions. Only a small percentage (~2%) of SNPs significantly (P < 0.001) failed to meet fitness for Hardy–Weinberg proportion; the 17 SNPs that failed were removed from subsequent analyses. Of the “SNPs” that failed, nearly all were due to an extreme excess of heterozygotes, which may be due to an early version of the algorithm used to call SNP genotypes and are therefore not reported.
Linkage disequilibrium
Pair-wise LD between all polymorphic sites was estimated using TagZilla (http://tagzilla.nci.nih.gov). The patterns of LD within this region have been refined, particularly in the region surrounding rs6983267, as shown in Fig. 5. The marker that is most highly significantly associated with prostate cancer in the most centromeric portion of this region, rs6983267 (Yeager et al. 2007; (Thomas et al. 2008), is highly correlated (r 2 > 0.9) with one other SNP (rs12682374, r 2 = 0.942), while the most highly significant SNP in the telomeric portion of this region, rs4242382 (Thomas et al. 2008), was highly correlated with 43 SNPs (r 2 = 1.00: rs9297760, rs9297759, rs7824868, rs7814837, rs7812894, rs7812429, rs7017300, rs7005132, rs4641026, rs4582524, rs4515512, rs4498506, rs4314621, rs4297007, rs4242384, rs6470518, rs6470519, rs6470520, rs1447295, rs4871802, rs10109700, rs7826179, rs7832031, rs13255059, rs10090154, rs4242385; r 2 = 0.95: rs11988857, rs9656816, rs9643226, rs1447296, rs12545648, rs10099413, rs10088308, rs7818556, rs8180905, rs1447292, rs4431561, rs7837688, rs4871801, rs4871024, rs3956790; r 2 = 0.91: rs4871798 and rs3999775). Because the polymorphisms within these two groups are so highly correlated with these two associated markers, it is probable that we have determined the SNP sites that will ultimately be established as the actual causal variant(s) in each independent locus. However, because of lower completion rates at select loci, the presented results provide an exploratory analysis of a dense set of SNPs. Pair-wise r 2 and _D_′ values for all SNPs with a MAF >5% between rs6983267 and rs4242382 are included in Supplementary Table 3.
Fig. 5
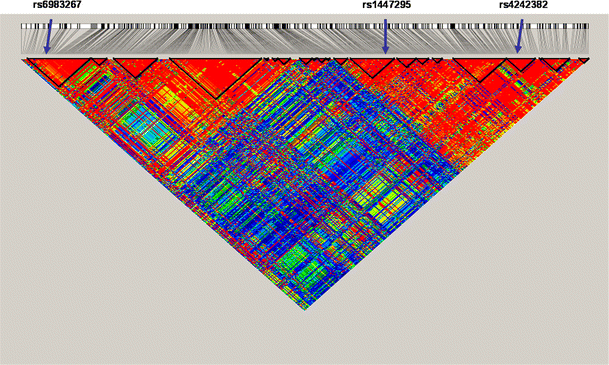
Refinement of linkage disequilibrium among SNPs > 0.05 MAF across the 8q24 region, with approximate positions of SNPs that have been implicated in prostate cancer risk: rs6983267, rs1447295, and rs4242382 are denoted by arrows (from left to right, respectively)
Tag SNP selection
Using the 79 unrelated individuals, we estimated tag SNPs for the entire region (r 2 ≥ 0.9, minimum MAF ≥ 0.05) using the TagZilla software package (http://tagzilla.nci.nih.gov). TagZilla computes tag SNPs based on a modification of an algorithm that has been proposed by Carlson et al. (2004). From the present data, at an r 2 ≥ 0.8, 114 SNPs are necessary to tag 100% of the 454 SNPs that were observed at an estimated MAF ≥ 0.05 (Table 2). Publicly available data is available for 174 SNPs within this region in HapMapII. Using HapMap SNPs alone, 53 bins monitoring 353 SNPs are covered, whereas 62 bins (monitoring 101 SNPs) are not monitored by any known HapMap SNP. Therefore, using HapMap SNPs alone, total coverage of this region is approximately 78%. We observed that an additional 125 SNPs reported in the dbSNP database that were not genotyped as part of the HapMap project had estimated MAFs ≥ 0.05 (Table 2). Genotyping these 125 SNPs + the additional 174 SNPs would provide 90% coverage for this region. However, it should be noted that since complete genotype data are not available for these 125 SNPs, all of them would have to be genotyped (along with the 53 HapMap tags) within a suitable sample to derive detailed tagging information. An additional 34 tags monitoring 44 total SNPs described in this study were not otherwise covered by any entry in dbSNP. All relevant bin information for the 454 SNPs with an estimated MAF ≥ 0.05 is included in Supplementary Table 4.
Table 2 Bin and SNP coverage by data source for chromosome 8: 128,473,000–128,609,802
Enhancer and conservation prediction
Because the expectation is that a causal variant will be in very strong LD with the SNP markers that have been genotyped and found to be associated with prostate cancer, to begin to prioritize SNPs to be followed up with functional and other nonbioinformatic studies, we explored the two regions using the UCSC browser (http://genome.ucsc.edu/) and VISTA Enhancer database (http://enhancer.lbl.gov/) to determine whether the SNPs that are most highly correlated with the top SNPs from the two regions may lie within regions that are highly conserved across mammals and other vertebrate species and/or lie within predicted enhancer elements or other potential regulatory regions. As stated above, for the centromeric and telomeric regions, 2 and 44 SNPs that meet these criteria were nominated, respectively.
For the centromeric region, rs6983267 proved to be the best candidate, falling within a highly conserved (1,083 bp, chr8: 128,481,736–128,482,819, Human Genome Build 36.2) segment that has strong regulatory potential and also contains a putative enhancer chr8: 128,482,390–128,483,622 (UCSC Genome Browser; http://genome.ucsc.edu; May 2004 assembly) (Fig. 6). Under the assumption of a single functional SNP in the LD block, the functional SNP should have a high MAF; therefore, the likelihood that another SNP exists that has been overlooked within this study is extremely small. The enhancer activity of this segment remains to be experimentally assessed. Two other SNPs fall within or very near this segment, rs10808556 and rs7008058. rs10808556 has an r 2 of 0.692 with rs6983267, and rs7008058 is not polymorphic in this sample.
Fig. 6
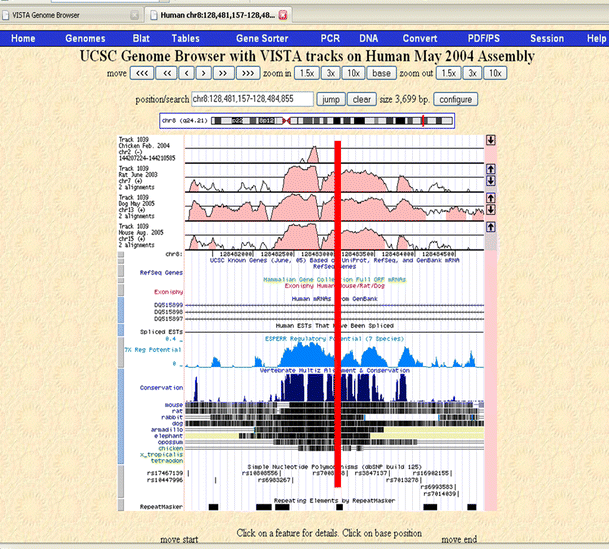
Best SNP from centromeric region (rs6983267), conservation, regulatory potential, and enhancer element probabilities. Vista enhancer tracks are shown on the UCSC browser. The level of conservation is displayed on the vertical axis. Scores for regulatory potential compare frequencies of short alignment patterns between known regulatory elements and neutral DNA. They are computed from human, chimp, macaque, mouse, rat, dog, and cow alignments (King et al. 2005). The location of rs6983267 is indicated with a red line
The 44 SNPs in the telomeric region were similarly examined, though none proved highly compelling using this method. One SNP (rs4431561, r 2 with rs4242382 = 0.949) fell within a highly conserved region, one SNP (rs1447296, r 2 with rs4242382 = 0.952) was located in a putative enhancer region, and one SNP (rs7812894, r 2 with rs4242382 = 1.0) was located in a predicted region of regulatory potential. All other SNPs did not clearly fall within any of these three region types; and although this does not exclude them from being candidates for the causal variant(s), it makes them less likely.
Discussion
Genome-wide association studies with sufficiently large replication components are rapidly identifying regions of the genome that are convincingly associated with risk of cancer and other diseases (Manolio et al. 2008; Rahim et al. 2008). Because the majority of GWAS studies take an approach that is unbiased with respect to function, the genotyped SNPs associated with a disease or trait are not necessarily the functional variants, rather, they are viewed as markers correlated with the true causative variant(s). The identification of a marker association represents the beginning of a process to define the causal variants through functional analyses and molecular phenotyping.
Characterizing all genetic polymorphisms within a region, as we have done here, is a critical next step to GWAS. SNP markers discovered or further characterized as a part of these require subsequent genotyping in sufficiently large sample sets to refine association signals prior to dedicated laboratory analysis. Two advantages of characterizing all common genetic variants prior to undergoing large-scale fine-mapping studies are (1) that all common genetic variants may be represented using a tag SNP approach, and (2) the correlations among all genetic variants will be known, which will allow for rapid nomination of variants for functional studies for those that are most highly correlated with the markers that are most highly associated with disease.
At least four regions of human chromosome 8q24 have recently been implicated in the risk of prostate, breast, and colorectal cancer (Amundadottir et al. 2006; Freedman et al. 2006; Gruber et al. 2007; Gudmundsson et al. 2007; Haiman et al. 2007a, b; Schumacher et al. 2007; Yeager et al. 2007; Zanke et al. 2007) and colorectal adenoma (Berndt et al. [2008](/article/10.1007/s00439-008-0535-3#ref-CR2 "Berndt SI, Potter JD, Hazra A, Yeager M, Thomas G, Makar KW, Welch R, Cross AJ, Huang WY, Schoen RE, Giovannucci E, Chan AT, Chanock SJ, Peters U, Hunter DJ, Hayes RB (2008) Pooled analysis of genetic variation at chromosome 8q24 and colorectal neoplasia risk. Hum Mol Genet. doi: 10.1093/hmg/ddn166
")). Replication of markers in 8q24 has been robust, but candidate variants for functional analyses remain elusive, especially in a region with a dearth of candidate genes. The recent emergence of next-generation sequencing technologies provides an unprecedented avenue to quickly and relatively inexpensively characterize genetic variation in fairly large genomic regions for medium-sized sample sets that are designed to detect with great probability common (>1%) variants. In this report, we have utilized the 454/Roche next-generation sequencing technology to characterize with great certainty and high quality all common variation within two of these regions (chr8: 128,473,000–128,609,802), including what will most likely be the variant(s) associated with prostate and colorectal cancers. We have determined that this region of 8q24 contains 780 common SNPs, 454 of which have a MAF ≥ 0.05.Based on our sequence analysis of 158 chromosomes, we have constructed a map of LD across the region. One hundred and fourteen SNPs are necessary to comprehensively tag this region for further association studies in individuals of European ancestry with an r 2 > 0.8. Genotyping 53 of 174 HapMap tag SNPs alone would cover approximately 78% of SNPs in this region in populations of European ancestry; the addition of 125 non-HapMap SNPs previously reported in dbSNP raises coverage to approximately 90%, though it is worth noting that all 299 SNPs would have to be genotyped to ensure this coverage. The present study not only validates these 299 SNPs, but also provides an additional 10% of information that would have not been monitored.
A PHASE analysis of common genetic variation (MAF > 5%) indicates a complex haplotype structure in which there is recent recombination that generates a large number of rarer haplotypes. Therefore, to interrogate this region efficiently in association studies, it appears that tag SNPs represent a more efficient approach, with respect to the number of required SNPs. Moreover, choosing tagSNPs with a high threshold for r 2 can improve the opportunity to monitor more SNPs and rare haplotypes, but at the cost of an increase in the number of SNPs needed for follow-up genotype analysis.
Preliminary bioinformatic analyses have identified rs6983267 as an excellent SNP for functional assessment. Indeed it lies within a region that is both highly conserved across vertebrates predicted to likely contain regulatory potential and an enhancer-element (see Fig. 6). It has been proposed that variation within evolutionary-conserved regions is likely associated with phenotypic differences that may contribute to human diseases (Dermitzakis et al. 2005). For the telomeric rs1447295 region, three SNPs lie within potentially interesting regions, though strong evidence for nominating one of them as the strongest candidate is still lacking.
In summary, we have extensively characterized the majority of all common SNPs across two high-interest regions, totaling ~136 kb of human chromosome 8q24 that have been reported to be associated with colon and/or prostate cancer as identified by GWAS, replication, and other case–control studies. We have verified that 299 SNPs that have been deposited in dbSNP are polymorphic in our samples (158 chromosomes), and have identified 442 novel polymorphisms, 101 of which have an estimated MAF ≥ 0.05 and are not monitored by HapMap SNPs at an r 2 of 0.8 in our sample population. Our data set provides an important resource that may be used to design fine-mapping projects for this region. Such efforts are critical for providing sufficient information for rapidly following up association findings and for fine mapping project for regions of the genome that are found to be significantly associated with a disease or phenotype. Our results underscore the value of resequence analysis in determining the full catalog of variants necessary to choose for further genotyping and functional analyses. Finally, the determination of the correlations among all genetic variation within this region should expedite the nomination of variants for functional studies post-fine-mapping.
References
- Amundadottir LT, Sulem P, Gudmundsson J, Helgason A, Baker A, Agnarsson BA, Sigurdsson A, Benediktsdottir KR, Cazier JB, Sainz J, Jakobsdottir M, Kostic J, Magnusdottir DN, Ghosh S, Agnarsson K, Birgisdottir B, Le Roux L, Olafsdottir A, Blondal T, Andresdottir M, Gretarsdottir OS, Bergthorsson JT, Gudbjartsson D, Gylfason A, Thorleifsson G, Manolescu A, Kristjansson K, Geirsson G, Isaksson H, Douglas J, Johansson JE, Balter K, Wiklund F, Montie JE, Yu X, Suarez BK, Ober C, Cooney KA, Gronberg H, Catalona WJ, Einarsson GV, Barkardottir RB, Gulcher JR, Kong A, Thorsteinsdottir U, Stefansson K (2006) A common variant associated with prostate cancer in European and African populations. Nat Genet 38:652–658
Article PubMed CAS Google Scholar - Berndt SI, Potter JD, Hazra A, Yeager M, Thomas G, Makar KW, Welch R, Cross AJ, Huang WY, Schoen RE, Giovannucci E, Chan AT, Chanock SJ, Peters U, Hunter DJ, Hayes RB (2008) Pooled analysis of genetic variation at chromosome 8q24 and colorectal neoplasia risk. Hum Mol Genet. doi:10.1093/hmg/ddn166
- Camp NJ, Farnham JM, Allen-Brady K, Cannon-Albright LA (2007) Statistical recombinant mapping in extended high-risk Utah pedigrees narrows the 8q24 prostate cancer locus to 2.0 Mb. Prostate 67:1456–1464
Article PubMed CAS Google Scholar - Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA (2004) Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet 74:106–120
Article PubMed CAS Google Scholar - Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE, Brooks LD, Cardon LR, Daly M, Donnelly P, Fraumeni JF Jr, Freimer NB, Gerhard DS, Gunter C, Guttmacher AE, Guyer MS, Harris EL, Hoh J, Hoover R, Kong CA, Merikangas KR, Morton CC, Palmer LJ, Phimister EG, Rice JP, Roberts J, Rotimi C, Tucker MA, Vogan KJ, Wacholder S, Wijsman EM, Winn DM, Collins FS (2007) Replicating genotype–phenotype associations. Nature 447:655–660
Article PubMed CAS Google Scholar - Cher ML, Bova GS, Moore DH, Small EJ, Carroll PR, Pinn SS, Epstein JI, Isaacs WB, Jensen RH (1996) Genetic alterations in untreated metastases and androgen-independent prostate cancer detected by comparative genomic hybridization and allelotyping. Cancer Res 56:3091–3102
PubMed CAS Google Scholar - Dermitzakis ET, Reymond A, Antonarakis SE (2005) Conserved non-genic sequences—an unexpected feature of mammalian genomes. Nat Rev Genet 6:151–157
Article PubMed CAS Google Scholar - Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, Fletcher O, Johnson N, Seal S, Stratton MR, Rahman N, Chenevix-Trench G, Bojesen SE, Nordestgaard BG, Axelsson CK, Garcia-Closas M, Brinton L, Chanock S, Lissowska J, Peplonska B, Nevanlinna H, Fagerholm R, Eerola H, Kang D, Yoo KY, Noh DY, Ahn SH, Hunter DJ, Hankinson SE, Cox DG, Hall P, Wedren S, Liu J, Low YL, Bogdanova N, Schurmann P, Dork T, Tollenaar RA, Jacobi CE, Devilee P, Klijn JG, Sigurdson AJ, Doody MM, Alexander BH, Zhang J, Cox A, Brock IW, MacPherson G, Reed MW, Couch FJ, Goode EL, Olson JE, Meijers-Heijboer H, van den Ouweland A, Uitterlinden A, Rivadeneira F, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Hopper JL, McCredie M, Southey M, Giles GG, Schroen C, Justenhoven C, Brauch H, Hamann U, Ko YD, Spurdle AB, Beesley J, Chen X, Mannermaa A, Kosma VM, Kataja V, Hartikainen J, Day NE et al (2007) Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447:1087–1093
Article PubMed CAS Google Scholar - Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, Mulholland S, Leongamornlert DA, Edwards SM, Morrison J, Field HI, Southey MC, Severi G, Donovan JL, Hamdy FC, Dearnaley DP, Muir KR, Smith C, Bagnato M, Ardern-Jones AT, Hall AL, O’Brien LT, Gehr-Swain BN, Wilkinson RA, Cox A, Lewis S, Brown PM, Jhavar SG, Tymrakiewicz M, Lophatananon A, Bryant SL, Horwich A, Huddart RA, Khoo VS, Parker CC, Woodhouse CJ, Thompson A, Christmas T, Ogden C, Fisher C, Jamieson C, Cooper CS, English DR, Hopper JL, Neal DE, Easton DF (2008) Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet 40(3):316–321
Article PubMed CAS Google Scholar - Freedman ML, Haiman CA, Patterson N, McDonald GJ, Tandon A, Waliszewska A, Penney K, Steen RG, Ardlie K, John EM, Oakley-Girvan I, Whittemore AS, Cooney KA, Ingles SA, Altshuler D, Henderson BE, Reich D (2006) Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc Natl Acad Sci USA 103:14068–14073
Article PubMed CAS Google Scholar - Gilbert MT, Binladen J, Miller W, Wiuf C, Willerslev E, Poinar H, Carlson JE, Leebens-Mack JH, Schuster SC (2007) Recharacterization of ancient DNA miscoding lesions: insights in the era of sequencing-by-synthesis. Nucleic Acids Res 35:1–10
Article PubMed CAS Google Scholar - Gohagan JK, Prorok PC, Hayes RB, Kramer BS (2000) The Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial of the National Cancer Institute: history, organization, and status. Control Clin Trials 21:251S–272S
Article PubMed CAS Google Scholar - Gruber SB, Moreno V, Rozek LS, Rennert HS, Lejbkowicz F, Bonner JD, Greenson JK, Giordano TJ, Fearon ER, Rennert G (2007) Genetic variation in 8q24 associated with risk of colorectal cancer. Cancer Biol Ther 6(7):1143–1147
Article CAS PubMed Google Scholar - Gudmundsson J, Sulem P, Manolescu A, Amundadottir LT, Gudbjartsson D, Helgason A, Rafnar T, Bergthorsson JT, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, Xu J, Blondal T, Kostic J, Sun J, Ghosh S, Stacey SN, Mouy M, Saemundsdottir J, Backman VM, Kristjansson K, Tres A, Partin AW, Albers-Akkers MT, Godino-Ivan Marcos J, Walsh PC, Swinkels DW, Navarrete S, Isaacs SD, Aben KK, Graif T, Cashy J, Ruiz-Echarri M, Wiley KE, Suarez BK, Witjes JA, Frigge M, Ober C, Jonsson E, Einarsson GV, Mayordomo JI, Kiemeney LA, Isaacs WB, Catalona WJ, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K (2007) Genome-wide association study identifies a second prostate cancer susceptibility variant at 8q24. Nat Genet 39:631–637
Article PubMed CAS Google Scholar - Haiman CA, Le Marchand L, Yamamato J, Stram DO, Sheng X, Kolonel LN, Wu AH, Reich D, Henderson BE (2007a) A common genetic risk factor for colorectal and prostate cancer. Nat Genet 39:954–956
Article PubMed CAS Google Scholar - Haiman CA, Patterson N, Freedman ML, Myers SR, Pike MC, Waliszewska A, Neubauer J, Tandon A, Schirmer C, McDonald GJ, Greenway SC, Stram DO, Le Marchand L, Kolonel LN, Frasco M, Wong D, Pooler LC, Ardlie K, Oakley-Girvan I, Whittemore AS, Cooney KA, John EM, Ingles SA, Altshuler D, Henderson BE, Reich D (2007b) Multiple regions within 8q24 independently affect risk for prostate cancer. Nat Genet 39:638–644
Article PubMed CAS Google Scholar - Hayes RB, Sigurdson A, Moore L, Peters U, Huang WY, Pinsky P, Reding D, Gelmann EP, Rothman N, Pfeiffer RM, Hoover RN, Berg CD (2005) Methods for etiologic and early marker investigations in the PLCO trial. Mutat Res 592:147–154
PubMed CAS Google Scholar - King DC, Taylor J, Elnitski L, Chiaromonte F, Miller W, Hardison RC (2005) Evaluation of regulatory potential and conservation scores for detecting cis-regulatory modules in aligned mammalian genome sequences. Genome Res 15:1051–1060
Article PubMed CAS Google Scholar - Kruglyak L, Nickerson DA (2001) Variation is the spice of life. Nat Genet 27:234–236
Article PubMed CAS Google Scholar - Manolio TA, Brooks LD, Collins FS (2008) A HapMap harvest of insights into the genetics of common disease. J Clin Invest 118:1590–1605
Article PubMed CAS Google Scholar - Nupponen NN, Kakkola L, Koivisto P, Visakorpi T (1998) Genetic alterations in hormone-refractory recurrent prostate carcinomas. Am J Pathol 153:141–148
PubMed CAS Google Scholar - Rahim NG, Harismendy O, Topol EJ, Frazer KA (2008) Genetic determinants of phenotypic diversity in humans. Genome Biol 9:215
Article PubMed CAS Google Scholar - Schumacher FR, Feigelson HS, Cox DG, Haiman CA, Albanes D, Buring J, Calle EE, Chanock SJ, Colditz GA, Diver WR, Dunning AM, Freedman ML, Gaziano JM, Giovannucci E, Hankinson SE, Hayes RB, Henderson BE, Hoover RN, Kaaks R, Key T, Kolonel LN, Kraft P, Le Marchand L, Ma J, Pike MC, Riboli E, Stampfer MJ, Stram DO, Thomas G, Thun MJ, Travis R, Virtamo J, Andriole G, Gelmann E, Willett WC, Hunter DJ (2007) A common 8q24 variant in prostate and breast cancer from a large nested case-control study. Cancer Res 67:2951–2956
Article PubMed CAS Google Scholar - Shaffer C (2007) Next-generation sequencing outpaces expectations. Nat Biotechnol 25:149
Article PubMed CAS Google Scholar - Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, Crenshaw A, Cancel-Tassin G, Staats BJ, Wang Z, Gonzalez-Bosquet J, Fang J, Deng X, Berndt SI, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cussenot O, Valeri A, Andriole GL, Crawford ED, Tucker M, Gerhard DS, Fraumeni JF Jr, Hoover R, Hayes RB, Hunter DJ, Chanock SJ (2008) Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet 40(3):310–315
Article PubMed CAS Google Scholar - Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, Penegar S, Chandler I, Gorman M, Wood W, Barclay E, Lubbe S, Martin L, Sellick G, Jaeger E, Hubner R, Wild R, Rowan A, Fielding S, Howarth K, Silver A, Atkin W, Muir K, Logan R, Kerr D, Johnstone E, Sieber O, Gray R, Thomas H, Peto J, Cazier JB, Houlston R (2007) A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet 39:984–988
Article PubMed CAS Google Scholar - Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF Jr, Hoover R, Hunter DJ, Chanock SJ, Thomas G (2007) Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet 39:645–649
Article PubMed CAS Google Scholar - Zanke BW, Greenwood CM, Rangrej J, Kustra R, Tenesa A, Farrington SM, Prendergast J, Olschwang S, Chiang T, Crowdy E, Ferretti V, Laflamme P, Sundararajan S, Roumy S, Olivier JF, Robidoux F, Sladek R, Montpetit A, Campbell P, Bezieau S, O’Shea AM, Zogopoulos G, Cotterchio M, Newcomb P, McLaughlin J, Younghusband B, Green R, Green J, Porteous ME, Campbell H, Blanche H, Sahbatou M, Tubacher E, Bonaiti-Pellie C, Buecher B, Riboli E, Kury S, Chanock SJ, Potter J, Thomas G, Gallinger S, Hudson TJ, Dunlop MG (2007) Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet 39:989–994
Article PubMed CAS Google Scholar
Acknowledgments
The authors would like to recognize the contribution of the late Robert A. Welch to the conception, execution, and analysis of this project. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
- Core Genotyping Facility, Advanced Technology Program, SAIC-Frederick, Inc., NCI-Frederick, Frederick, MD, 21702, USA
Meredith Yeager, Nianqing Xiao, Laura Burdett, Casey Matthews, Liqun Qi, Andrew Crenshaw & Laufey Amundadottir - Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Department of Health and Human Services, Bethesda, MD, 20892, USA
Meredith Yeager, Nianqing Xiao, Richard B. Hayes, Laura Burdett, Casey Matthews, Liqun Qi, Andrew Crenshaw, Laufey Amundadottir, David J. Hunter, Robert Hoover, Gilles Thomas & Stephen J. Chanock - 454 Life Sciences, Branford, CT, USA
Pascal Bouffard, Brian Desany, Zdenek Markovic & Thomas P. Jarvie - Pediatric Oncology Branch, Center for Cancer Research, NCI, NIH, DHHS, Bethesda, MD, USA
Nick Orr & Stephen J. Chanock - Bioinformed Consulting Services, Gaithersburg, MD, USA
Kevin B. Jacobs - Program in Molecular and Genetic Epidemiology, Department of Epidemiology, Harvard School of Public Health, Boston, MA, USA
David J. Hunter - Roche Applied Science, Indianapolis, IN, USA
Karin M. Fredrikson & Timothy T. Harkins - Advanced Technology Center, NCI, 8717 Grovemont Circle, Gaithersburg, MD, 20877, USA
Meredith Yeager
Authors
- Meredith Yeager
You can also search for this author inPubMed Google Scholar - Nianqing Xiao
You can also search for this author inPubMed Google Scholar - Richard B. Hayes
You can also search for this author inPubMed Google Scholar - Pascal Bouffard
You can also search for this author inPubMed Google Scholar - Brian Desany
You can also search for this author inPubMed Google Scholar - Laura Burdett
You can also search for this author inPubMed Google Scholar - Nick Orr
You can also search for this author inPubMed Google Scholar - Casey Matthews
You can also search for this author inPubMed Google Scholar - Liqun Qi
You can also search for this author inPubMed Google Scholar - Andrew Crenshaw
You can also search for this author inPubMed Google Scholar - Zdenek Markovic
You can also search for this author inPubMed Google Scholar - Karin M. Fredrikson
You can also search for this author inPubMed Google Scholar - Kevin B. Jacobs
You can also search for this author inPubMed Google Scholar - Laufey Amundadottir
You can also search for this author inPubMed Google Scholar - Thomas P. Jarvie
You can also search for this author inPubMed Google Scholar - David J. Hunter
You can also search for this author inPubMed Google Scholar - Robert Hoover
You can also search for this author inPubMed Google Scholar - Gilles Thomas
You can also search for this author inPubMed Google Scholar - Timothy T. Harkins
You can also search for this author inPubMed Google Scholar - Stephen J. Chanock
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toMeredith Yeager.
Electronic supplementary material
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Yeager, M., Xiao, N., Hayes, R.B. et al. Comprehensive resequence analysis of a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers.Hum Genet 124, 161–170 (2008). https://doi.org/10.1007/s00439-008-0535-3
- Received: 23 July 2008
- Accepted: 24 July 2008
- Published: 14 August 2008
- Issue Date: September 2008
- DOI: https://doi.org/10.1007/s00439-008-0535-3