Mice Lacking Major Brain Gangliosides Develop Parkinsonism (original) (raw)
Abstract
Parkinson’s disease (PD) is the second most prevalent late-onset neurodegenerative disorder that affects nearly 1% of the global population aged 65 and older. Whereas palliative treatments are in use, the goal of blocking progression of motor and cognitive disability remains unfulfilled. A better understanding of the basic pathophysiological mechanisms underlying PD would help to advance that goal. The present study provides evidence that brain ganglioside abnormality, in particular GM1, may be involved. This is based on use of the genetically altered mice with disrupted gene Galgt1 for GM2/GD2 synthase which depletes GM2/GD2 and all the gangliotetraose gangliosides that constitute the major molecular species of brain. These knockout mice show overt motor disability on aging and clear indications of motor impairment with appropriate testing at an earlier age. This disability was rectified by L-dopa administration. These mice show other characteristic symptoms of PD, including depletion of striatal dopamine (DA), loss of DA neurons of the substantia nigra pars compacta, and aggregation of alpha synuclein. These manifestations of parkinsonism were largely attenuated by administration of LIGA-20, a membrane permeable analog of GM1 that penetrates the blood brain barrier and enters living neurons. These results suggest that perturbation of intracellular mechanisms mediated by intracellular GM1 may be a contributing factor to PD.
Similar content being viewed by others
Introduction
Gangliosides are well recognized mediators of certain essential signaling pathways in the nervous system and other organs [1], and a growing number of proteins involved in such pathways are seen to function in association with one or another ganglioside. Considerable insight into ganglioside-protein interactions has been gained from a study of animals with mutated genes required for ganglioside biosynthesis. One of the most studied is a mouse with disrupted gene Galgt1 which encodes the enzyme UDP-GalNAc:lactosylceramide/GM3/GD3 β-1,4-N-acetylgalactosaminyltransferase (GM2/GD2 synthase; GalNAcT; EC 2.4.1.92). This mutation results in elimination of GM2 and GM1 along with other members of the gangliotetraose family which compromise >90% of CNS gangliosides. Such mice suffer a slight reduction in neural conduction velocity [2] in addition to decreased central myelination and axonal degeneration [3]. They also show progressive behavioral neuropathies including deficits in reflexes, strength coordination and balance [4]. At the cellular level, cultured neurons from such mice were found deficient in Ca2+ regulation as reflected in vitro by degeneration in the presence of depolarizing levels of K+ [5] and in vivo by enhanced susceptibility to kainate-induced seizures [6].
The present study has probed further into the movement disorder and pathophysiological manifestations of such knockout (KO) mice and found a constellation of symptoms strongly representative of Parkinson’s disease (PD). A primary neuropathological feature of the sporadic form of PD is the substantial depletion of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc), although loss of other neuronal types can also occur prior or subsequent to that in the SNpc [7]. In addition to accompanying decrease of dopamine (DA) in SNpc and striatum, a prominent neuropathological feature is accumulation of alpha-synuclein (a-syn) within cytoplasmic inclusions termed Lewy bodies. In contrast to the much rarer familial cases, which show generally similar neuropathology, the etiology of sporadic PD is not well understood as reflected in the several theories that have been proposed. Most of these are based on a variety of interactions between environmental factors and genetic predisposition.
Rodents subjected to neurotoxins have been widely used to model some of the key features of PD, the most prominent being 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), 6-hydroxy dopamine, rotenone, and paraquat [8]. The above mentioned GalNAcT−/− KO mouse has the advantage as a model in its spontaneous acquisition of parkinsonian symptoms, thus avoiding the potentially confounding influence of extraneous toxins. The present study chronicles these parallel symptoms and provides evidence suggesting GM1 as the crucial missing ganglioside. This idea correlates with the many previous studies demonstrating GM1 amelioration of parkinsonism in mouse and primate models of PD [9–13] as well as in PD patients [14, 15]. In that light such treatment might eventually be viewed as a form of replacement therapy pending verification of the hypothesis that susceptible DA cells in PD suffer deficiency of GM1.
Methods
Animals and Behavioral tests
A breeding pair of heterozygotes with disrupted gene for GM2/GD2 synthase (C57BL/6 background), created by Dr. Richard Proia and coworkers [16] was provided as a gift by Dr. Ronald Schnaar (Johns Hopkins University School of Medicine). Animals were maintained in the University of Medicine and Dentistry of New Jersey Research Animal Facility with 12 h light/dark cycles. They were genotyped by PCR analysis as described [5]. All animal procedures were in accord with the guidelines of the UMDNJ Animal Care and Use Committee (IACUC).
Wild type (WT) and KO mice of both genders at 35 and 200 days of age (DOA) were used. To determine physical impairment, a grip-hanging test was employed in which the mouse clings with forepaws to a narrow horizontal rod positioned 50 cm above a pillow and hang time to fall is measured [17]; the mouse’s tail is gently held by experimenter to prevent climbing with hind legs. Hang times in three consecutive trials, with rest intervals of 20 min, were averaged. The second test, adhesive removal, was used to determine motor response to sensory stimulus [18]. A small piece of adhesive is applied to the snout and the time to removal by forepaw is recorded. Durations longer than 120 s were counted as 120 s. Each animal was measured five times and the shortest three times were averaged. In rescue experiments, animals were injected intraperitoneally (IP) with GM1 (30 mg/kg), LIGA-20 (2.5 mg/kg) or balanced salt solution (BSS; Alcon, Fort Worth, TX) 3×/week over 5 weeks and then subjected to behavioral tests. GM1 and LIGA-20 were gifts from the Fidia Research Laboratories (Abano Terme, Italy). To test the rescue effect of L-dopa on impairment, KO mice were IP injected with L-dopa (20 mg/kg) plus carbidopa (3 mg/kg) in BSS and subjected to behavioral tests 3 h after injection; this was compared to test results before L-dopa administration. Statistical analysis was via two-tailed Student’s t test. After behavior tests, mice were perfused (see below) and the frozen brains stored prior to histochemical and biochemical assays.
Histochemical Analyses
Animals were subjected to cardiac perfusion with phosphate buffered saline (PBS) followed by 4% parafomaldehyde in PBS. The brains were fixed overnight or longer in 4% paraformaldehyde followed by transfer to 30% sucrose (cryoprotection) in PBS and storage at 4°C. Coronal frozen sections (25 μm) were cut from the midbrain and stained with CtxB-FITC (1 μg/ml; Sigma; St. Louis, MO) and anti-tyrosine hydroxylase (TH) antibody (Ab) (Millipore; Billerica, MA; 1:1000) plus 2nd Ab linked to Texas red. Other sections were stained with above anti-TH Ab plus Texas red 2nd Ab and anti-a-syn Ab (BD Science, 1:2000) plus 2nd Ab linked to FITC. Immuno- fluorescence in the SNpc region was photographed with 40X objective lens. Stereological analysis was carried out on the SNpc and ventral tegmental area (VTA) of brains from three KO treated with BSS, three KO mice treated with LIGA-20 (2.5 mg/kg), and three WT brains; this was done at the Rutgers University Molecular Histology Center (Dr. Eric Richfield; http://eohsi.rutgers.edu/mhc/). In preparation the brains were perfused, fixed and cryoprotected as above. Using the above anti-TH Ab with Texas red 2nd Ab, the total number of TH+ neurons in the SNpc and VTA were counted using the optical fractionator and previously described counting criteria [19, 20]. The right and left sides were counted in each brain. Regions of interest (ROI) were outlined at low magnification (4X objective) and sampled at high magnification (100X oil immersion objective) using StereoInvestigator (MicroBrightField, VT). ROI were selected based on literature descriptions and their consistent visual identification at 4X. For the area sampling fraction (asf) a sampling frame of size of 55 × 55 μm (3,025 μm2) and a grid area of 123 × 123 μm (15,129 μm2) yielding an asf of 20% were employed. All choices were made to obtain sufficient precision for TH+ neuron counts in the SNpc and VTA.
Immunoblot Assay for Alpha-synuclein
Freshly collected brains from PBS perfused mice were employed and the midbrain cut into 250 μm coronal sections with a vibratome sectioning system in cold PBS. The entire SN region, including compact and reticulum, was isolated with a dissecting microscope. The pooled sections were extracted with Cell Lysis Buffer (Cell Signaling, Danvers, MA) and aliquots containing 20 μg protein were resolved with SDS–PAGE on a 4–15% gradient gel (Bio-Rad, Hercules, CA). After transfer to PVDF membrane, proteins were blotted with anti-a-syn antibody (1:1000; Santa Cruz Biotec; Sanda Cruz, CA) followed by 2nd Ab linked to horseradish peroxidase (HRP). Protein bands were revealed by ECL reagent (Amersham; Piscataway, NJ) on film. Anti-actin Ab (Sigma) was run in parallel for loading control. The optical density of actin and a-syn polymer (ranging from 34 to 170 Kd) blots was scanned and quantified by AlphaEase FC imaging system (Alpha Innotech, San Leandro, CA). Three brains in each group were assayed, the data normalized with respect to actin and statistically analyzed with the Student’s t-test.
Neurochemistry
For DA assay, striatal samples prepared as above were each homogenized and sonicated in 0.7 ml cold 0.4 N perchloric acid containing 70 ng dihydroxybenzylamine as internal standard for 20 min. This was centrifuged for 20 min at 16,000 g, the supernatant filtered through a Millex-GV syringe filter (0.22 μm; Millipore) and the filtrate stored at −20°C prior to analysis. One hundred μl was applied to a DIONEX ICS-3000 HPLC system with a 5-μm C8 Acclaim 120 column and electrochemical detector (0.7 V). Concentrations of DA and dihydroxyphenylacetic acid (DOPAC) were determined from peak areas adjusted to aliquot size, internal standard, and protein content. Also assayed by HPLC was serotonin (5-HT) and its metabolite, 5-hydroxyindole acetic acid (5-HIAA). Protein content was determined by the Lowry method following digestion of the HClO4 pellet several hrs in NaOH/SDS [21].
Results
The impaired movement of KO mice that became more evident with age was quantified by the two methods described above. Such mice at 200 DOA retained their forepaw grasp on a horizontal bar for 20 s or less in contrast to WT mice which maintained their grasp for 150 s or more (Fig. 1A_b_). Surprisingly, the same was true of relatively young KO mice, 35 DOA (Fig. 1A_a_). Grip duration was fully restored to the latter mice by serial IP injections (3×/week) of LIGA-20 over the 5 week period, while grip duration was significantly improved by LIGA-20, though less dramatically, for the older mice. GM1 similarly administered to the younger KO mice had relatively little effect. The second impairment test, determining motor response to sensory stimulus by measuring time for removal of adhesive irritant from the snout, gave similar results: older KO mice required 60 s in contrast to WT time of ~5 s (Fig. 1B_b_). The younger KO mice effected removal in ~30 s, significantly more than the ~2 s required by the younger WT mice. As before, GM1 administered to the younger KO mice had virtually no effect. In both tests the ameliorative effect of LIGA-20 rose gradually over the 5 weeks, reaching a plateau at ~3 weeks (not shown).
Fig. 1
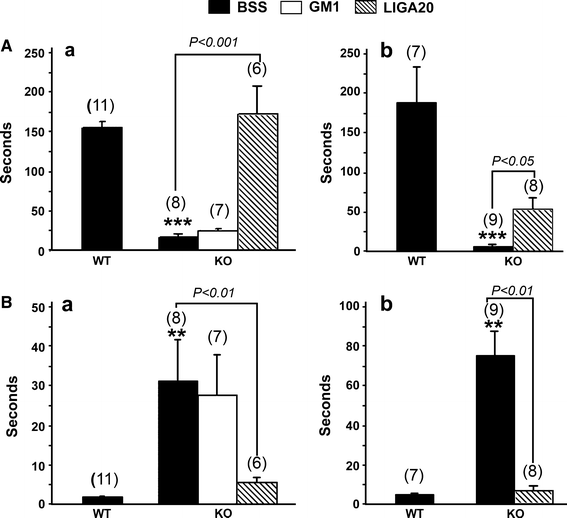
Movement impairment in KO mice. Mice at 35 (A a, B a) and 200 (A b, B b) days of age were IP injected with GM1, LIGA20 or BSS for 5 weeks. They were subjected to the grip-hanging (A) and adhesive removal (B) tests. () = n. Data are average ± SEM. ** and *** represent P < 0.01 and 0.001 compared to BSS-treated WT with Student’s two-tailed t test
Comparison of TH-expressing neurons in SNpc of KO versus WT brains of ~200 day old mice suggested significant deficiency in the mutant (Fig. 2). Tyrosine hydroxylase (TH), an enzyme that catalyzes the first step in conversion of tyrosine to DA, is a marker for DA neurons. This was quantified by unbiased stereology employing anti-TH Ab to count DA neurons in the SNpc and VTA. Decrease in TH-expressing neurons was significant in the SNpc, and although the VTA showed a decrease this did not reach significance (Fig. 3). These changes accord with the significant loss of DA neurons in the SNpc and relative sparing of DA neurons in the VTA reported for murine models of PD [22]. Treatment of the KO mice with LIGA-20 over the 5 week period elevated TH+ neurons in the SNpc to the point where the TH+ count in SNpc of LIGA-20-treated mice was not significantly different from WT (P = 0.059), even though the difference between KO and KO + LIGA-20 did not reach significance.
Fig. 2
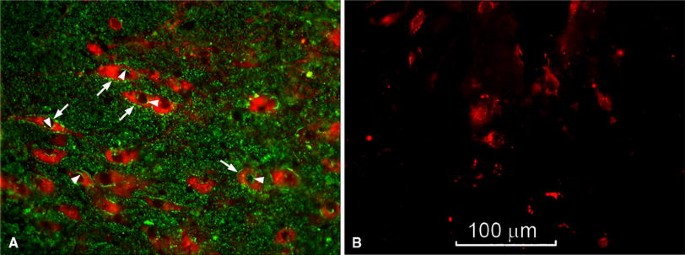
Degeneration of TH+ neurons in SNpc of KO mice. Brain sections containing SNpc from 200 day old WT (A) and KO (B) mice were stained with anti-TH antibody (2nd Ab with Texas red) and CtxB-FITC for GM1 (green), showing reduced number of TH+ neurons in KO mice. Expression of GM1 in WT TH+ neurons is indicated by arrows (plasma membrane) and arrowheads (nuclear membrane). Photographed with 40X objective lens
Fig. 3
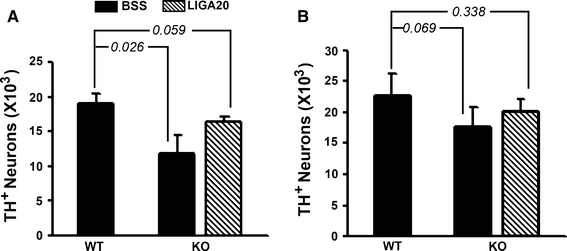
Stereological analysis. WT and KO mice (200 days old) were IP injected with LIGA-20 or BSS for 5 weeks. Brain sections were immunostained with anti-TH as shown in Fig. 2 and subjected to stereological analysis. Two regions, SNpc (A) and VTA (B), containing TH+ neurons were counted. The data are average (±SEM) of 3 animals in each group (n = 3). Significant decrease in DA neurons of SNpc but not VTA was indicated by Student’s two-tailed t test. These changes accord with the significant loss of DA neurons in the SNpc and relative sparing of DA neurons in the VTA reported for PD and murine models of PD [22]. Although the difference between KO and KO + LIGA-20 did not reach significance, the treatment rendered the difference between WT and KO + LIGA-20 insignificant
Alpha synuclein expression was greatly elevated in SNpc of KO brain, as revealed by immunocytochemistry; 5 weeks of LIGA-20 treatment attenuated a-syn levels while restoring much of the depleted TH expression (Fig. 4). Quantitative verification of a-syn elevation was obtained by Western blot analysis, which also indicated aggregation by marked increase of higher molecular weight bands at 34- and 170 kDa as well as less dense bands in between; this applied to mice 200 DOA (Fig. 5A_b_) as well as younger mice at 35 DOA (Fig. 5A_a_). Densitometric quantification revealed significant reduction of aggregated forms of a-syn following 5 weeks of LIGA-20 treatment of both age groups, in contrast to GM1 which produced no significant reduction (Fig. 5B).
Fig. 4
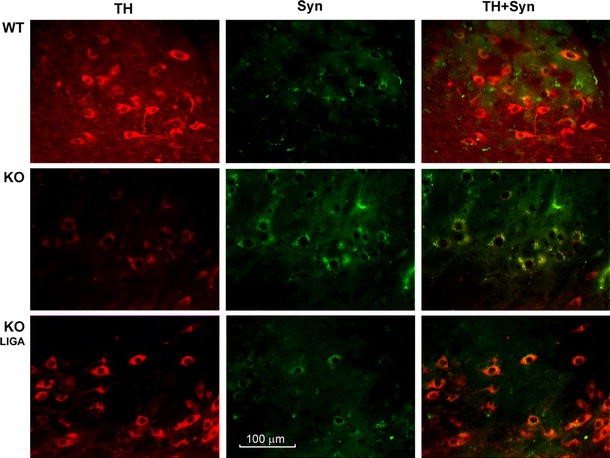
Accumulation of a-syn in TH+ neurons. Brain sections were co-stained with anti-TH (2nd Ab with Texas red) and anti-a-syn (2nd Ab with FITC), showing reduced TH+ neurons and aggregated a-syn in remaining TH+ neurons in KO mouse. IP injection of KO mouse with LIGA-20 for 5 weeks attenuated a-syn accumulation while rescuing TH+ neurons
Fig. 5
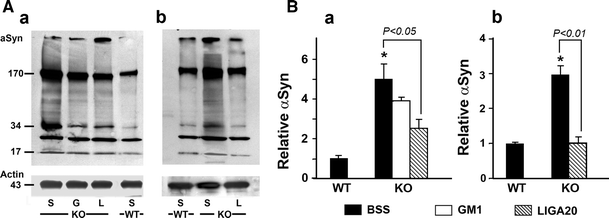
Immunoblot analysis for a-syn accumulation. WT and KO mice at 35 (A a, B a) and 200 (A b, B b) days of age were treated with BSS, GM1 (G), or LIGA-20 (L). Brain tissue of SN regions was micro-dissected, and subjected to immuno-blotting for a-syn. House-keeping protein actin was run in parallel for loading control. A Blot images showing polymerization of a-syn ranging form 34- to 170 kD. B Densitometry quantification of a-syn polymers. Data are mean ± SEM from 3 mice in each group (n = 3). Student’s two-tailed t test was employed. *P < 0.01 versus BSS-treated WT. The results show enhanced aggregation of a-syn in KO, which is significantly reduce by LIGA-20 but not GM1
Dopamine levels in the striatum, measured as described by HPLC, were significantly reduced in KO mice compared to WT, as was also observed for DOPAC, a principal DA metabolite (Fig. 6a). Serotonin, another neurotransmitter in the striatum, and 5-HIAA, its metabolite, were moderately reduced by an amount that did not reach significance (Fig. 6b). To test whether the movement disorders were due to depleted DA, KO mice were administered L-dopa as described. The animals thus treated showed highly significant recuperation from physical impairment as determined by both tests (Fig. 7). Mice in both age groups showed approximately equal rescue from physical impairment in both tests.
Fig. 6
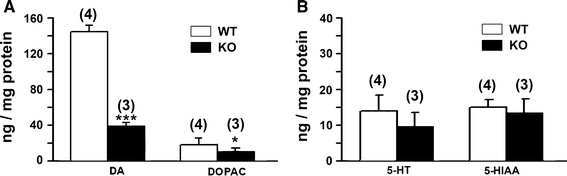
Biochemical analyses. Dissected striata were extracted with 0.4 N HClO4, spiked with dihydroxybenzylamine as internal standard and subjected to HPLC. (A) Striatal DA and DOPAC, that were significantly reduced in the KO; (B) 5-HT and 5-HIAA, that were not significantly reduced. Data are averages ± SEM; # of animals indicated in (). Statistical significance was determined by Student’s two-tailed t test: * and *** represent P < 0.05 and 0.001, respectively
Fig. 7
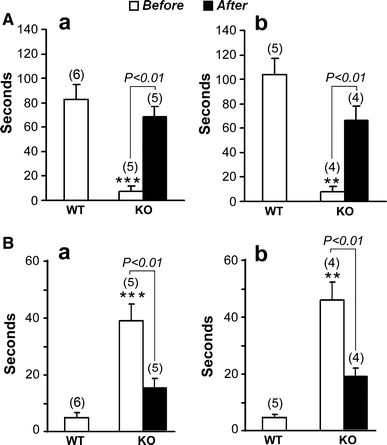
Effect of L-dopa on physical impairment. Mice at 35 (A a, B a) and 200 (A b, B b) days of age were IP injected with L-dopa (20 mg/kg) and carbidopa (3 mg/kg). Behavior tests, grip-hanging (A) and adhesive removal (B), were performed before and 3 h after injection. Data are average ± SEM; ( ) = n. Two-tailed Student’s t test was used for statistical analysis. ** and *** represent P < 0.01 and 0.001 respectively compared to WT before L-dopa treatment
Discussion
Parkinson’s disease is the second most prevalent late-onset neurodegenerative disorder that affects nearly 1% of the global population aged 65 and older. It is a progressive neurodegenerative disease characterized by resting tremor, postural rigidity, bradykinesia, autonomic instability and in its later stages by cognitive and emotional disorders [23, 24]. A principal feature is loss of pigmented DA neurons in the SNpc that project to the striatum, resulting in DA deficit and aberrations in the activity of the neural circuits within the basal ganglia that regulate movement. Cytosolic accumulation of Lewy bodies that contain aggregated a-syn is an additional hallmark of the disease [25]. The large majority of PD cases have been described as sporadic, the remainder as familial based on a variety of genetic mutations. Symptomatic treatments of the motor features are in clinical use but no neuroprotective treatment to prevent progressive loss of DA neurons has yet become available.
This study has provided evidence for the presence of key features of parkinsonism in genetically modified mice with disrupted gene for Galgt1 (GalNAcT; GM2/GD2 synthase). This included behavioral, histopathological, and biochemical parameters that are the defining symptoms of PD. These findings thus amplify and extend previous observations of behavioral and cellular pathologies of these mutants, which included progressive behavioral neuropathies involving deficits in reflexes, strength, coordination, balance, and gait [4]. A morphological study revealed axonal degeneration and decreased central myelination [3]. These earlier reports thus indicated a variety of neuropathies and neuropathologies attendant to these mutant mice while the present study has elucidated PD-like symptoms not previously reported. Disruption of this gene for a key enzyme in synthesis of GM2 results in ablation of that molelcule and the entire gangliotetraose series [3, 16] which comprise >90% of the complex gangliosides of brain; corresponding elevations of GM3 and GD3 have been observed [5, 26].
We hypothesize that much of the PD-specific symptomatology observed here derives from the GM1 deficit, based in part on the rescue effects of LIGA-20. This GM1 analog, developed by Costa and coworkers [27], contains the same oligosaccharide chain as GM1 but a modified hydrophobic moiety (replacement of ceramide stearoyl by dichloroacetyl) that induces membrane permeability. The latter property has been affirmed both in vitro with primary neurons [28] and in vivo with CNS neurons of ganglio-ganglioside-deficient mice suffering enhanced kainate-induced seizures [6]. LIGA-20 proved more potent than GM1 in partially restoring depleted DA levels in MPTP-treated mice [11] and, significantly, was able to accomplish this via oral administration [29]. These manifestations of LIGA-20 efficacy, believed to derive from its membrane permeability, result in enhanced ability to cross the blood brain barrier and the neuronal plasma membrane. When applied to cells or mice deficient in GM1 it thus serves as replacement for the missing ganglioside in both the plasma membrane and intracellular sites where GM1 has functional roles. Intracellulaar loci include the nuclear Na+/Ca2+ exchanger which is potentiated by GM1 [30, 31], the autophagy pathway including promotion of lysosomal integrity [32], and a-syn which requires GM1 association to prevent fibrillation and maintenance of helical conformation [33]. Long term Ca2+ dysregulation in the DA neurons of the SNpc, associated with their autonomous pacemaker activity and resultant mitochondrial impairment, has been suggested as a major contributor to the selective vulnerability of those neurons over time [34]. An additional reason for ascribing primacy to GM1 is the finding that knockout mice with disrupted GD3 synthase, resulting in deletion of all b- and c-series and retention of a-series gangliosides, showed virtually intact nervous system morphology with no apparent abnormal behavior despite some aberrations in pain perception and nerve regeneration [35, 36]. GD1a, the other prominent ganglioside of the a-series, often functions as metabolic precursor to GM1 through the action of membrane-bound sialidase [37, 38]. The complexity of ganglioside changes that occur in the Galgt1 mutant and the uncertainty as to which GM1 functions are efficiently restored by LIGA-20 require caution at this stage in ascribing a primary role to GM1 deficiency in relation to parkinsonism. However, the results of this and the other mentioned studies point to that conclusion.
Additional evidence of a key role for Ca2+ dysregulation in PD is the finding that elevated Ca2+ promoted a-syn aggregation in 1321N1 cells expressing an a-syn-GFP construct [39]. Alpha synuclein in aggregated form, together with ubiquitin, is a prominent component of Lewy bodies which are pathological hallmarks of PD. Lewy bodies as such are not usually found in rodent models of PD, but inclusion bodies staining for a-syn and ubiquitin have been observed [40]. The present study obtained Western blot evidence for a-syn aggregation in the prominence of higher molecular weight bands between 34- and 170- kDa in KO striatum compared to WT (Fig. 5). As shown, the densities of such bands were noticeably diminished by LIGA-20 treatment of the KO mice. Significantly, a-syn aggregation was inhibited specifically by GM1 which promoted its alpha-helical structure [33].
A randomized double blind placebo controlled study reported that GM1-treated PD patients showed significant improvements on Unified Parkinson’s Disease Rating Scale (UPDRS) motor scores and performance of timed motor tasks, compared to baseline performance [14]. Subsequently a 5 year open study revealed that patients using GM1 had lower UPDRS motor scores than at baseline [15]. These are interesting findings in view of preliminary results in our lab showing a marked deficiency of GM1 in DA neurons of the SNpc of PD patients, compared to age-matched controls. This suggests that applied GM1 may be functioning as a form of replacement therapy to the limited extent it is able to enter the brain of PD patients. This leads to the further suggestion that membrane permeable derivatives of GM1 may prove more efficacious in preserving DA neuronal viability and alleviating PD pathology.
Abbreviations
Ab:
antibody
a-syn:
alpha-synuclein
BSS:
balanced salt solution
CtxB:
B subunit of cholera toxin
DA:
dopamine or dopaminergic
DOA:
days of age
DOPAC:
dihydroxyphenyl acetic acid
ER:
endoplasmic reticulum
HRP:
horseradish peroxidase
5-HT:
5-hydroxytryptamine (serotonin)
5-HIAA:
5-hydroxyindole acetic acid
IP:
intraperitoneally
KO:
knockout
MPTP:
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
PBS:
phosphate buffered saline
PD:
Parkinson’s disease
SNpc:
substantia nigra pars compacta
TH:
tyrosine hydroxylase
VTA:
ventral tegmental area
WT:
wild type
References
- Allende ML, Proia RL (2002) Lubricating cell signaling pathways with gangliosides. Current Opinion in Struct Biol 12:587–592
Article CAS Google Scholar - Takamiya K, Yamamoto A, Furukawa K et al (1996) Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc Nat Acad Sc USA 93:10662–10667
Article CAS Google Scholar - Sheikh KA, Sun J, Liu Y et al (1999) Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Nat Acad Sci USA 96:7532–7537
Article PubMed CAS Google Scholar - Chiavegatto S, Sun J, Nelson RJ et al (2000) A functional role for complex gangliosides: motor deficits in GM2/GD2 synthase knockout mice. Exp Neurol 166:227–234
Article PubMed CAS Google Scholar - Wu G, Xie X, Lu Z-H et al (2001) Cerebellar neurons lacking complex gangliosides degenerate in the presence of depolarizing levels of potassium. Proc Natl Acad Sci USA 98:307–312
Article PubMed CAS Google Scholar - Wu G, Lu Z-H, Wang J et al (2005) Enhanced susceptibility to kainate-induced seizures, neuronal apoptosis, and death in mice lacking gangliotetraose gangliosides: protection with LIGA 20, a membrane-permeant analog of GM1. J Neurosci 25:11014–11022
Article PubMed CAS Google Scholar - Braak H, Del Tredici K, Rub U et al (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Article PubMed Google Scholar - Meredith GE, Sonsalla PK, Chesselet M-F (2008) Animal models of Parkinson’s disease progression. Acta Neuropathol 115:385–398
Article PubMed Google Scholar - Hadjiconstantinou M, Rossetti ZL, Paxton RC et al (1986) Administration of GM1 ganglioside restores the dopamine content in striatum after chronic treatment with MPTP. Neuropharm 25:1075–1077
Article CAS Google Scholar - Schneider JS, Kean A, DiStefano L (1995) GM1 ganglioside rescues substantia nigra pars compacta neurons and increases DA synthesis in residual nigrostriatal dopaminergic neurons in MPTP-treated mice. J Neurosci Res 42:117–123
Article PubMed CAS Google Scholar - Schneider JS, DiStefano L (1995) Response of the damaged dopamine system to GM1 and semisynthetic gangliosides: effects of dose and extent of lesion. Neuropharm 34:489–493
Article CAS Google Scholar - Hadjiconstantinou M, Mariani AP, Neff NH (1989) GM1 ganglioside-induced recovery of nigrostriatal dopaminergic neurons after MPTP: an immunohistochemical study. Brain Res 484:297–303
Article PubMed CAS Google Scholar - Pope-Coleman A, Schneider JS (1998) Effects of chronic GM1 ganglioside treatment on cognitive and motor deficits in a slowly progressing model of Parkinsonism in non-human primates. Restorative Neurol Neurosci 12:255–266
CAS Google Scholar - Schneider JS, Roeltgen DP, Mancall EL (1998) Parkinson’s disease: improved function with GM1 ganglioside treatment in a randomized placebo-controlled study. Neurology 50:1630–1636
PubMed CAS Google Scholar - Schneider JS, Sendek S, Daskalakis C et al (2010) GM1 ganglioside in Parkinson’s disease: results of a five year open study. J Neurol Sciences 292:45–51
Article CAS Google Scholar - Liu Y, Wada R, Kawai H et al (1999) A genetic model of substrate deprivation therapy for a glycosphingolipid storage disorder. J Clin Invest 103:497–505
Article PubMed CAS Google Scholar - Sedelis M, Hofele K, Auburger GW et al (2000) MPTP susceptibility in the mouse: behavioral, neurochemical, and histological analysis of gender and strain differences. Behav Genet 30:171–182
Article PubMed CAS Google Scholar - Lu XH, Fleming SM, Meurers B et al (2009) Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant alpha-synuclein. J Neurosci 29:1962–1976
Article PubMed Google Scholar - Prasad K, Richfield EK (2008) Sporadic midbrain dopamine neuron abnormalities in laboratory mice. Neurobiol Disease 32:262–272
Article CAS Google Scholar - Prasad K, Richfield EK (2010) Number and nuclear morphology of TH+ and TH− neurons in the mouse ventral midbrain using epifluorescence stereology. Exp Neurol 225:328–340
Article PubMed CAS Google Scholar - Lees M, Paxman S (1972) Lipid composition of the normal human brain: gray matter, white matter, and myelin. J Lipid Res 6:537–544
Google Scholar - German DC, Manaye KF, Sonsalla PK, Brooks BA (1992) Midbrain dopaminergic cell loss in Parkinson’s disease and MPTP-induced Parkinsonism: sparing of calbindin-D28k-containing cells. Ann NY Acad Sci 648:42–62
Article PubMed CAS Google Scholar - Fahn S (2006) Description of Parkinson’s disease as a clinical syndrome. Ann NY Acad Sci 991:1–14
Article Google Scholar - Jankovic J (2008) Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psych 79:368–376
Article CAS Google Scholar - Eriksen JL, Dawson TM, Dickson DW et al (2003) Caught in the act: alpha-synuclein is the culprit in Parkinson’s disease. Neuron 40:453–456
Article PubMed CAS Google Scholar - Matsuda J, Vanier MT, Popa J et al (2006) GD3- and O-acetylated GD3-gangliosides in the GM2 synthase-deficient mouse brain and their immunohistochemical localization. Proc Jpn Acad Ser B 82:189–196
Article CAS Google Scholar - Manev H, Favaron M, Vicini S et al (1990) Glutamate-induced neuronal death in primary cultures of cerebellar granule cells: protection by synthetic derivatives of endogenous sphingolipids. J Pharmacol Exp Ther 252:419–427
PubMed CAS Google Scholar - Wu G, Lu Z-H, Xie X et al (2004) Susceptibility of cerebellar granule neurons from GM2/GD2 synthase-null mice to apoptosis induced by glutamate excitotoxicity and elevated KCl: rescue by GM1 and LIGA20. Glycoconj J 21:305–313
Article PubMed CAS Google Scholar - Schneider JS, DiStefano L (1994) Oral administration of semisynthetic sphingolipids promotes recovery of striatal dopamine concentrations in a murine model of parkinsonism. Neurology 44:748–750
PubMed CAS Google Scholar - Xie X, Wu G, Lu Z-H et al (2002) Potentiation of a sodium-calcium exchanger in the nuclear envelope by nuclear GM1 ganglioside. J Neurochem 81:1185–1195
Article PubMed CAS Google Scholar - Wu G, Xie X, Lu Z-H et al (2009) Sodium-calcium exchanger complexed with GM1 ganglioside in nuclear membrane transfers calcium from nucleoplasm to endoplasmic reticulum. Proc Natl Acad Sci USA 106:10829–10834
Article PubMed CAS Google Scholar - Wei J, Fujita M, Nakai M et al (2009) Protective role of endogenous gangliosides for lysosomal pathology in a cellular model of synucleinopathies. Amer J Pathol 174:1891–1909
Article CAS Google Scholar - Martinez Z, Zhu M, Han S et al (2007) GM1 specifically interacts with alpha-synuclein and inhibits fibrillation. Biochemistry 46:1868–1877
Article PubMed CAS Google Scholar - Chan CS, Gertler TS, Surmeier DJ (2009) Calcium homeostasis, selective vulnerability and Parkinson’s disease. Trends Neurosci 32:249–256
Article PubMed CAS Google Scholar - Okada M, Itoh M, Haraguchi M et al (2002) b-series Ganglioside deficiency exhibits no definite changes in the neurogenesis and the sensitivity to Fas-mediated apoptosis but impairs regeneration of the lesioned hypoglossal nerve. J Biol Chem 277:1633–1636
Article PubMed CAS Google Scholar - Handa Y, Ozaki N, Honda T et al (2005) GD3 synthase gene knockout mice exhibit thermal hyperalgesia and mechanical allodynia but decreased response to formalin-induced prolonged noxious stimulation. Pain 117:271–279
Article PubMed CAS Google Scholar - Miyagi T, Wada T, Iwamatsu A et al (1999) Molecular cloning and characterization of plasma membrane-associated sialidase specific for gangliosides. J Biol Chem 274:5004–5011
Article PubMed CAS Google Scholar - Wang J, Wu G, Miyagi T, Lu Z-H, Ledeen RW (2009) Sialidase occurs in both membranes of the nuclear envelope and hydrolyzes endogenous GD1a. J Neurochem 111:547–554
Article PubMed CAS Google Scholar - Goodwin NS, Engelborghs Y, Pountney DL (2010) Raised calcium promotes α-synuclein aggregate formation. Mol Cell Neurosci (2010 Dec 8) [Epub ahead of print]
- Yazdani U, German DC, Liang C-L et al (2006) Rat model of Parkinson’s disease: chronic central delivery of 1-methyl-4-phenylpyridinium (MPP+). Exp Neurol 200:172–183
Article PubMed CAS Google Scholar
Acknowledgments
This work was supported by NIH grant 2 RO1 NS33912.
It is a pleasure to contribute to this special issue of Neurochemical Research honoring Dr. Robert Yu for his many seminal contributions to the field of Neurochemistry. His studies on the biochemistry, cell biology, and pathophysiology of glycosphingolipids have been well recognized and honored over the years. Bob was my first postdoc (and his first such experience) more than a few years ago, coming from the lab of Herbert Carter, an iconic sphingolipid pioneer at the University of Illinois. Herbert responded to my inquiry with the affirmation that Bob was indeed a promising young neuroscientist who would likely contribute to my program. That proved quite a good prophecy; everyone should be so lucky as to have someone like Bob as their first posdoc. Those years were memorable for the quality and quantity of the work he produced as well the beginning of a life long friendship. I’m convinced there are many more miles to go in terms of Bob’s contributions to neuroscience.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
- Department of Neurology and Neurosciences, New Jersey Medical School, UMDNJ, 185 So. Orange Ave., MSB-H506, Newark, NJ, 07103, USA
Gusheng Wu, Zi-Hua Lu, Neil Kulkarni, Ruchi Amin & Robert W. Ledeen
Authors
- Gusheng Wu
You can also search for this author inPubMed Google Scholar - Zi-Hua Lu
You can also search for this author inPubMed Google Scholar - Neil Kulkarni
You can also search for this author inPubMed Google Scholar - Ruchi Amin
You can also search for this author inPubMed Google Scholar - Robert W. Ledeen
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toGusheng Wu.
Additional information
Special Issue: In Honor Dr. Robert Yu.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wu, G., Lu, ZH., Kulkarni, N. et al. Mice Lacking Major Brain Gangliosides Develop Parkinsonism.Neurochem Res 36, 1706–1714 (2011). https://doi.org/10.1007/s11064-011-0437-y
- Accepted: 24 February 2011
- Published: 12 March 2011
- Issue Date: September 2011
- DOI: https://doi.org/10.1007/s11064-011-0437-y