Activated microglia mediate synapse loss and short-term memory deficits in a mouse model of transthyretin-related oculoleptomeningeal amyloidosis (original) (raw)
Main
Oculoleptomeningeal amyloidosis (OA) is a fatal and untreatable form of transthyretin (TTR)-related amyloidosis that is clinically characterized by slow progressive dementia, ataxia, seizures and recurrent subarachnoidal hemorrhaging.1, 2 OA is primarily a hereditary disease, although several reports have described OA symptoms in patients who had received orthotopic liver transplantation for the treatment of familial amyloidotic polyneuropathy (FAP), another TTR-related amyloidosis.3, 4, 5 OA is also considered a form of cerebral amyloid angiopathy (CAA), which is characterized by amyloid deposition in the cerebral vasculature.6 The incidence of CAA is age-dependent, and it is believed that ∼12% of the population over 85 years of age may suffer from CAA-related symptoms.7 Importantly, features of CAA are also found in Alzheimer’s disease (AD) patients and in animal models of AD, and the contribution of CAA to AD progression has been widely studied.8, 9, 10
In the past decade, inflammation has emerged as a key event in the progression of many neurodegenerative diseases, including Alzheimer’s and Parkinson’s disease.11, 12, 13, 14 Microglia, the brain’s resident macrophages, participate in the coordination of events important for the maintenance of neuronal health.15 However, chronically activated microglia may initiate inflammatory processes that lead to neuronal dysfunction and damage.15, 16 Thus, tight regulation of microglial activation is required to maintain the precise balance between normal physiology and pathology in the CNS.
Here, we investigated whether amyloid fibrils composed of A25T TTR, a natural variant of TTR that readily forms fibrils under non-denaturing conditions17 and is associated with OA,1, 2 activate microglia and whether this activation leads to neuronal damage, culminating in cognitive deficits. Although TTR deposits in OA are normally found in subarachnoid spaces and around leptomeningeal vessels, it is conceivable that neuron-produced TTR18 may also aggregate and induce microglia activation in the brain parenchyma. Moreover, subarachnoid bleedings caused by TTR aggregates may contribute to microglia recruitment and activation, thus exacerbating neuroinflammation.19
Our results showed that A25T fibrils were innocuous to primary neurons in culture but caused robust microglial activation in vivo and in vitro. Microglial activation occurred via the Akt signaling pathway, inducing the secretion of TNF-α, IL-6 and nitric oxide (NO). Interestingly, conditioned medium (CM) from fibril-activated microglia was toxic to primary cortical neurons, inducing the loss of synapses and apoptosis via caspase-3 activation. Moreover, mice that received i.c.v. injections of fibrils showed short-term memory deficits that could be prevented by minocycline pretreatment. These results point to a possible mechanism of amyloid-induced toxicity in OA, and possibly other amyloid-related neurodegenerative diseases that are mediated by toxic microglial activation.
Results
A25T amyloid fibrils are internalized by microglia and induce the secretion of pro-inflammatory molecules
We initially investigated the cytotoxicity of amyloid aggregates composed of A25T TTR produced in vitro as previously described.17 A25T aggregates are largely composed of amyloid fibrils, as revealed by TEM, and by their increased capacity to bind amyloid-specific dyes, such as Congo red and Thioflavin-T (Supplementary Figure 1). The toxicity of A25T fibrils was investigated using dissociated mouse cortical neurons exposed to a physiological concentration of TTR: 1 _μ_M A25T fibrils or 1 _μ_M soluble A25T. Neither of these treatments led to neuronal death (Supplementary Figures 2A–E), indicating that A25T (in either soluble or aggregated form) is not inherently toxic to neurons. We next hypothesized that A25T fibrils might exert a neurotoxic effect indirectly via the activation of microglia. To test this hypothesis, we first examined whether A25T fibrils were toxic to microglia in culture. The incubation of microglial cultures with 1 _μ_M A25T fibrils caused no detectable cell death, as probed using three different methods (cell counting and the MTT and Live/Dead assays; Supplementary Figure 3). Soluble A25T, PBS or 100 ng/ml LPS were used as additional controls; none caused microglial death at the concentrations used (Supplementary Figure 3).
Despite the fact that A25T was not toxic to microglia, we found that microglia efficiently internalized acrylodan-labeled A25T fibrils (Figures 1a, b). Fibril internalization was blocked by the application of 2.5 _μ_M cytochalasin D, an inhibitor of actin polymerization (Figures 1c and d). Next, we investigated whether A25T fibrils stimulated microglial secretion of pro-inflammatory molecules. Microglia exposed to A25T fibrils, but not to soluble A25T or PBS, secreted significant amounts of TNF-α, IL-6 and NO (Figures 1e–g), at levels comparable to those secreted when microglia were stimulated with LPS.
Figure 1
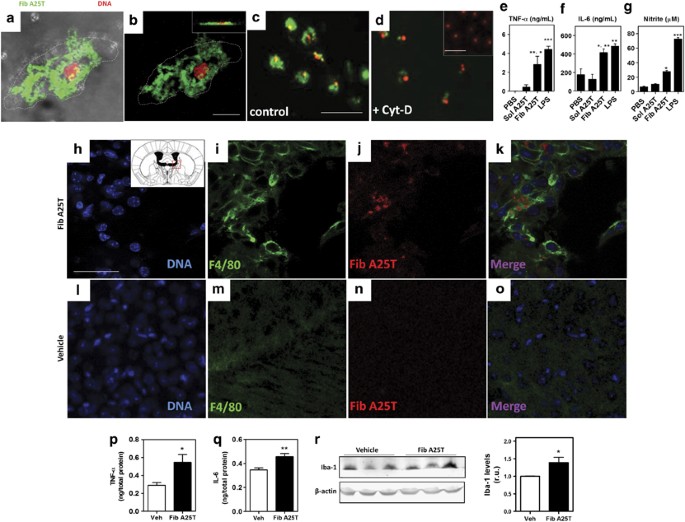
A25T fibrils activate microglia in vitro and in vivo. A25T fibrils (Fib A25T) were fluorescently labeled with acrylodan (pseudocolored in green in panels a–d) and incubated with microglia for 24 h, which nuclei (DNA) were labeled in red with ethidium homodimer-1. (a–d) Two-photon microscopy of microglia shows internalization of fluorescent A25T fibrils (green) together with the cells (DIC; a) and inside the cells (b and inset in panel B). A25T fibrils internalization was inhibited using 2.5 _μ_M cytochalasin D (+Cyt-D) (d) when compared with untreated cells (control; c). Inset in (d) shows microglia incubated with soluble A25T, which is not internalized by microglia in 24 h. Scale bars are 25 _μ_m for panels A-B, 100 _μ_m for panels c and b and 50 _μ_m for inset in panel d. We also measured TNF-α (e), IL-6 (f) and (g) NO (measured as nitrite using Griess reagents) in culture supernatants. As controls, PBS and soluble A25T (Sol A25T) at 1 _μ_M were also incubated with microglia. As positive control, LPS at 100 ng/ml was incubated with microglia. Statistical analysis was performed in three independent experiments using one-way ANOVA with Tukey’s test, and ***P< 0.001, **P<0.01 and *P<0.05. In panel e, ** represents the comparison between PBS and Fib A25T and * represents the comparison between Sol A25T and Fib A25T. In panel f, * represents the comparison between PBS and Fib A25T and ** represents the comparison between Sol A25T and Fib A25T. (h–r) Two-month-old Swiss mice were i.c.v. injected with A25T fibrils or vehicle (PBS). After 24 h, (h–k) A25T fibrils- and (l–o) vehicle-injected mice were killed and brain sections were stained with DAPI (blue; h and l), anti-F4/80 (green, i and m) and anti-human TTR (Fib A25T; red, j and n), and then analyzed using confocal microscopy. Merged images are shown in k and o. Scale bars are 24 _μ_m for all images. After 4 h, mice were euthanized and brain homogenates were analyzed for TNF-α (p) and IL-6 (q). Statistical analysis of panel p and q was performed using Student’s _t_-test were *P<0.05, **P<0.01. (r) Iba-1 levels were analyzed in lysates from A25T fibrils- and vehicle-injected mice by western blotting. Statistical analysis was performed using Student’s _t_-test were *P<0.05
Mice injected with A25T fibrils exhibit microglial activation and increased brain levels of TNF-α and IL-6
Next, we sought to determine whether the inflammatory mechanisms observed in vitro were also active in vivo. To tackle this question, we performed i.c.v. injections of A25T fibrils (Fib TTR) or vehicle (PBS) in 2-month-old male Swiss mice. Microglial activation in the brain was assessed 24 h post-injection using an anti-F4/80 antibody. There was an increase in F4/80 immunoreactivity when animals were injected with A25T fibrils (Figures 1h–k), which was corroborated by western blotting using another microglial marker, Iba-1 (Figure 1r). In addition, 4 h after the i.c.v. injection of A25T, a group of mice was killed and their brain extracts analyzed for pro-inflammatory cytokines (TNF-α and IL-6). Compared with vehicle-injected mice, A25T fibril-injected mice showed increased brain levels of TNF-α and IL-6 4 h after treatment with A25T (Figures 1p and q).
Interestingly, we noted that A25T fibrils did not migrate deeply into the brain parenchyma and remained in the area surrounding the lateral ventricles until 7 days after the injection (Supplementary Figure S4B). After 7 days, it was also possible to detect activated microglia around the injected A25T fibrils (Supplementary Figure S4A–D), suggesting that microglial activation and, possibly, inflammation was sustained for several days post injection. We also observed that the amount of fibrils had diminished considerably 7 days after the injection compared with 24 h after the injection (not shown).
Notably, we could also detect the presence of cells that were positively stained for anti-F4/80 and contained A25T fibrils within their cell bodies (Supplementary Figure S5). Both at 24 h and 7 days post-injection, microglial activation and anti-TTR immunoreactivity were absent in vehicle-injected mice (Figures 1l–o and Supplementary Figure S4E–H).
Microglial activation by A25T fibrils is mediated by the phosphorylation of Akt and inactivation of GSK-3_β_ and results in the translocation of NFkB to the nucleus
To investigate the signaling pathway involved in the activation of microglia by A25T fibrils, we analyzed the cellular lysates of microglia incubated for 30 min with soluble A25T, PBS or A25T fibrils. It has been shown that phosphatidylinositol-3-kinase (PI3K)/Akt signaling modulates the release of cytokines by activated cells by inflammatory molecules such as LPS.20 To determine whether Akt mediates the activation of microglia by A25T fibrils, we examined the activation of Akt through phosphorylation at Ser473.21 Activated Akt in turn inactivates GSK-3_β_ through phosphorylation at Ser9,22 enhancing TNF-α secretion.23 A25T fibrils significantly activated microglial Akt (Figures 2a and b), and this led to the inactivation of GSK-3_β_ (Figures 2a and c).
Figure 2
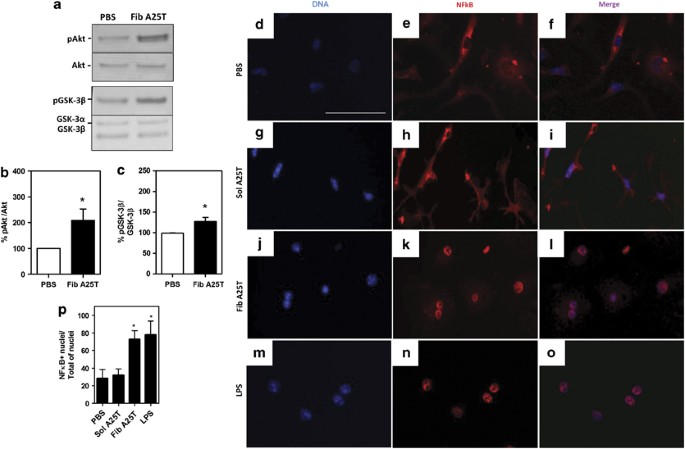
A25T fibrils induce microglia activation via Akt phosphorilation at Ser473, GSK-3_β_ phosphorilation at Ser9 and NFκB translocation to cell nucleus. Primary microglia cells were incubated with 1 μ_M of fibrillar A25T (Fib A25T) or PBS for 30 min. Cells were then lysed and protein lysates analyzed by (a) western blotting using anti-phosphorilated Akt (pAkt) at Ser473 and anti-phosphorilated GSK-3_β (pGSK-3_β_) at Ser9. Levels of phosphorilated protein (b; % of pAkt/Akt) or (c; % of pGSK-3_β_/GSK-3_β_) were analyzed relative to PBS (control; 100%). Statistical analysis was performed in three independent experiments using Student’s _t_-test; *P<0.05. Panels d–p depict NF_κ_B translocation to the nucleus: d–f shows microglia incubated with PBS, soluble A25T (Sol A25T; g–i), fibrillar A25T (Fib A25T; j–l) and LPS (m–o). NF_κ_B+ nuclei/total of nuclei cells were quantified in p. Statistical analysis was performed in three independent experiments using one-way ANOVA, with *P<0.05. Scale bars are 25 _μ_m for panels d–o
Akt signaling also modulates the activation and translocation of NFκB to the cell nucleus, where it activates the transcription of a variety of genes, including inflammation-related genes.24 We thus investigated whether A25T fibrils would induce the translocation of NFκB to the nucleus in microglia. Interestingly, the translocation of NFκB to the nucleus was observed when microglia were incubated with A25T fibrils (Figures 2j–l and p), but not with soluble A25T (Figures 2g–i and p) or PBS (Figures 2d–f and p). As expected, when incubated with 100 ng/ml LPS, ∼80% of cells exhibited nuclear NF_κ_B immunoreactivity (Figures 2m–o and p). These data indicate that A25T fibrils induce the activation of microglia by modulating Akt and NF_κ_B signaling.
CM from A25T fibril-activated microglia induces synapse loss in neuronal cultures
Pro-inflammatory cytokines have been associated with a variety of CNS disorders, including trauma, ischemia, multiple sclerosis and Alzheimer’s disease.14 Some cytokines, such as TNF-α and IL-6, can adversely modulate synaptic plasticity and induce synapse dysfunction.25 The results described above demonstrated that A25T fibrils induce the secretion of pro-inflammatory molecules by microglia (Figure 1). Therefore, we investigated whether CM from A25T fibril-activated microglia would affect synapses in 2-week-old cortical neuron cultures. To address this question, we incubated neurons for 3 h with either CM from A25T fibril- or LPS-activated microglia, or control CM (from microglial cultures treated with PBS or soluble A25T) and analyzed synaptic density using both presynaptic (synaptophysin) and postsynaptic (PSD-95) markers. After 3 h of incubation, CM from A25T fibril-activated microglia induced severe synapse loss in the neuronal cultures, similar to the effects of CM from LPS-activated microglia (Figures 3c, d). In contrast, CM from control (non-activated) microglia had no effect on synaptophysin or PSD-95 levels (Figures 3a and b), suggesting that soluble factors released by activated microglia modulate neuronal synapses, inducing synapse loss well before neuronal death.
Figure 3
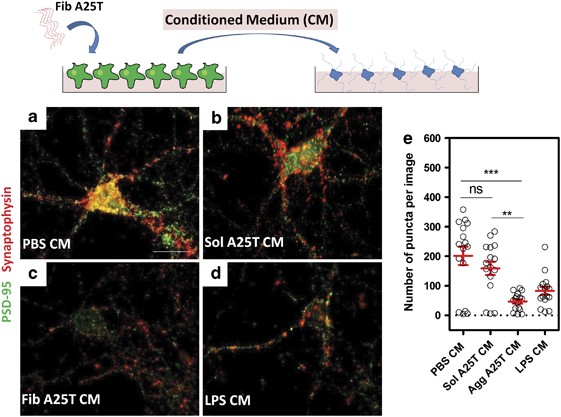
CM from A25T fibrils-activated microglia induce synapse loss in primary cortical neurons. A25T fibrils or soluble A25T at 1 _μ_M were incubated for 48 h with microglia cells and then CM (Fib A25T CM and Sol A25T CM) was transferred to primary neurons for 3 h. As controls, we used CM from PBS- or LPS (100 ng/ml)-activated microglia. Cells were fixed and immunostained with antibodies anti-PSD-95 (red) and anti-synaptophysin (green), and the colocalization (puncta; yellow) of both proteins suggest the presence of a functional synapse. Images show a representative neuron incubated with (a) PBS CM, (b) Sol A25T CM, (c) Fib A25T CM, (d) LPS CM. (e) Quantification of colocalization of PSD-95 and synaptophysin (number of puncta per image). Each experiment was performed in duplicates and 12–20 images were captured, quantified for colocalization and the mean of all images was plotted for each experiment. Statistical analysis was performed in three independent experiments using Kruskal–Wallis test and Dunn’s post hoc test, and ***P<0.01 when PBS CM and Agg A25T CM were compared and **P<0.01 when Sol A25T CM and Agg A25T CM were compared. Scale bars are 25 _μ_m for all panels
CM from A25T fibril-activated microglia is neurotoxic
To test whether longer exposure to CM from A25T fibril-activated microglia caused additional damage to neurons, mature cortical neuronal cultures were exposed for 48 h to CM from microglia previously incubated for 48 h with 1 _μ_M A25T fibrils. After 48 h, we observed extensive neuronal death under these conditions (Figures 4c and e). Similar neurotoxicity was observed when neurons were treated with CM from microglia incubated with LPS (Figures 4d and e), but not with CM from microglial cultures treated with PBS or soluble A25T (Figures 4a and b). Similar results were also obtained in experiments using immature cortical cultures (data not shown). These results suggest that LPS and A25T fibrils induce microglia to secrete sufficient quantities of a variety of neurotoxic molecules to have detrimental effects on cortical neurons in vitro.
Figure 4
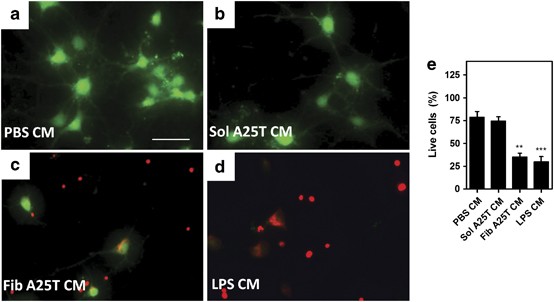
CM from A25T fibril-activated microglia induce neuron death. Fibrillar A25T (Fib A25T) or soluble A25T (Sol A25T) were incubated with primary microglia cells for 48 h to generate fibrillar A25T-conditioned media (Fib A25T CM) or soluble A25T-CM (Sol A25T CM) and then, CM were incubated with primary cortical neuron cultures for 48 h. Cell viability was assessed using Live/Dead assay, where live cells are stained in green and dead cells in red (a–e). Also as controls, PBS and 100 ng/ml LPS were incubated first with microglia to generate CMs (PBS CM; A and LPS CM; d) and then incubated with neurons. To test microglia-mediated cell death, (b) Sol A25T CM or (c) Fib A25T CM was incubated with neurons for 48 h. Panel (e) show the percentage (%) of live cells compared with medium alone (considered as 100% viability; not shown) and statistical analysis was performed in three independent experiments using one-way ANOVA, with Tukey’s test, and **P<0.01, ***P<0.001. Scale bars are 50 _μ_m
CM from A25T fibril-activated microglia induces neuronal cell death via apoptosis
Next, we investigated whether neuronal death induced by treatment with CM derived from A25T fibril-treated microglia took place via apoptosis. To address this question, apoptotic markers, including caspase-3 activation and DNA fragmentation, were evaluated.
Caspase-3 is an important executer of apoptosis and its activation requires proteolytic processing at aspartic acid 175 (Asp175).26 Interestingly, CM from LPS- or A25T fibril-activated microglia induced caspase-3 activation in >50% of the cells, while in neuronal cultures treated with control CM (CM from PBS- or soluble A25T-treated microglia) <20% of cells were positive for activated caspase-3 (Figures 5a–d, quantified in e). To assess DNA fragmentation, we used the TUNEL assay. As expected, no TUNEL-positive cells were observed when neurons were treated with CM derived from microglia incubated with PBS or soluble A25T (Figures 5f and g). However, when neurons were incubated with CM from LPS- or A25T fibril-activated microglia, >50% of the cells were TUNEL-positive (Figures 5h and i, quantified in j), suggesting DNA fragmentation and apoptosis.
Figure 5
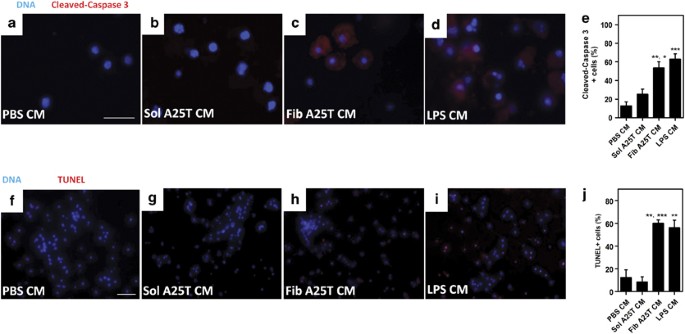
CM from A25T fibril-activated microglia induce caspase-3 activation and fragmentation of DNA in neurons. A25T fibrils (Fib A25T CM) or soluble A25T (Sol A25T CM) at 1 _μ_M were incubated with primary microglia for 48 h and CM was transferred to cortical neurons for 24 h. As controls, we used conditioned media from PBS- or LPS (100 ng/ml)-activated microglia. Caspase-3 activation was analyzed using an antibody anti-cleaved caspase-3 (red, a–e) and DNA fragmentation was monitored by TUNEL assay (f–j). Cells were also stained with Hoescht (blue; a–d and f–i). In (e) cleaved caspase-3-positive cells were quantified and compared with total cells (Hoescht-positive cells) and % of cleaved caspase-3-positive cells were plotted relative to neurons incubated with LPS CM. In (j) TUNEL-positive cells were quantified and compared to total cells (Hoescht-positive cells) and % of TUNEL-positive cells was plotted relative to positive control, DNAse I treated cells (not shown; considered 100%). Statistical analysis was performed in three independent experiments using one-way ANOVA, with Tukey’s test, and ***P<0.001 and **P<0.01. In panel e, ** represents the comparison between PBS and Fib A25T and * represents the comparison between Sol A25T and Fib A25T. In panel F, ** represents the comparison between PBS and Fib A25T and *** represents the comparison between Sol A25T and Fib A25T. Scale bars are 100 _μ_m for panels a–d and 25 _μ_m for panels (e–h)
Mice injected with A25T fibrils exhibit short-term memory deficits that could be prevented by minocycline treatment
Finally, we sought to determine whether the activation of microglia in vivo led to changes in behavior. To answer that question, we performed i.c.v. injections of either A25T fibrils or vehicle (PBS) in 2-month-old male Swiss mice and evaluated short-term memory 24 h later using the novel object recognition test. Interestingly, A25T fibril injection impaired short-term memory (Figure 6), but not locomotor or exploratory activity (Supplementary Figure S6). Next, animals were pretreated for 3 consecutive days with 50 mg/kg minocycline to determine whether the short-term memory impairment was a consequence of microglia-mediated inflammation. Minocycline is a broad-spectrum tetracycline antibiotic that has anti-inflammatory properties and is an effective inhibitor of microglial activation.27 We first tested the efficacy of minocycline on reducing inflammation by pretreating microglial cultures with 10 _μ_M minocycline for 1 h (Supplementary Figure S7). We confirmed that minocycline prevented the secretion of NO by microglia cells exposed to A25T fibrils (Supplementary Figure S7). Surprisingly, minocycline pretreatment prevented the short-term memory deficits caused by A25T fibril injection in mice (Figure 6), suggesting the participation of microglial-derived inflammation in A25T fibril-induced memory loss.
Figure 6
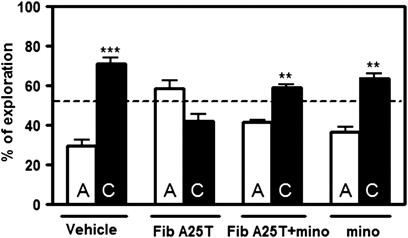
Minocycline prevented A25T fibrils-induced short-term memory loss in the novel object recognition test. Two-month-old Swiss mice were pretreated with 50 mg/kg/day minocycline for 3 consecutive days and then i.c.v. injected with A25T fibrils or vehicle (PBS). After 24 h, animals were subjected to the novel object recognition test in which a familiar object (A) was substituted by a novel object (C). Exploratory activity was measured and % of exploration was analyzed in all animals. Statistical analysis was performed using one-sample _t_-test applying a hypothetical value of 50.0; ***P<0.001 and **P<0.05
Discussion
OA is a form of CAA caused by the accumulation of TTR amyloid fibrils in the leptomeninges, vitreous humor and subarachnoid space.1 CAA can be caused not only by TTR but also by other amyloidogenic proteins/peptides such as amyloid-β, cystatin C, aDan and aBri.28 Twelve different naturally occurring mutant forms of TTR have been found to be associated with OA,29, 30, 31, 32, 33 and common variants such as V30M TTR, which is mainly associated with sympathetic and autonomic neuropathy, have been shown to cause OA in patients who received liver transplants.3, 4 Liver transplantation is a clinically established procedure for removing mutant protein from the blood,34 but not from the CNS, as TTR is also produced by the choroid plexus epithelia, hippocampal and cortical neurons.18 Thus, accumulation of the mutant protein in the CSF leads to the development of OA in transplanted patients. For that reason, there is an urgent need to understand the cellular and molecular mechanisms involved in the pathogenesis of OA, which could operate in other neurodegenerative diseases as well.
Here, we show that fibrils formed from a natural variant of TTR (A25T), but not from soluble WT TTR protein, induce microglial activation in vitro and in vivo. We also found that A25T fibrils are internalized by microglia through a yet unknown mechanism, inducing the activation of Akt and the translocation of NFκB to the nucleus, followed by the secretion of pro-inflammatory molecules including TNF-α, IL-6 and NO. Release of these pro-inflammatory cytokines by microglia fuels a cycle of neuroinflammation that can cause bystander damage to neurons.15 Indeed, we found that when immature (1 DIV) or mature (14 DIV) neurons were exposed to CM from A25T fibril-activated microglia, <50% of the neurons remained viable after 48 h. We also searched for apoptotic markers, such as DNA fragmentation and caspase-3 activation, to determine whether CM from fibril-activated microglia induced apoptosis in neuronal cells. After 24 h, we observed both DNA fragmentation and activated caspase-3 (cleavage at Asp175) in neurons exposed to CM from A25T-activated microglia. Interestingly, significantly before neuronal death took place, CM from A25T fibrils-activated microglia-induced synapse loss in neurons, which might lead to cognitive deficits. Notably, patients with OA exhibit diverse neurological symptoms, including hearing loss, ataxia, dementia and seizures,33 which could be explained, at least in part, by the neurotoxic activation of microglia in the presence of TTR fibrils. Although histopathological alterations in OA mainly comprise TTR deposits in subarachnoid spaces and around leptomeningeal vessels, it is conceivable that TTR produced by neurons18 might undergo oligomerization/aggregation within the brain parenchyma, where they might induce resident microglia activation. Furthermore, subarachnoid bleedings caused by aggregates may contribute to microglia recruitment and activation, thus exacerbating neuroinflammation.19 The synapse loss observed when neurons were exposed to CM from A25T fibril-activated microglia may occur through the remodeling of both presynaptic and postsynaptic compartments. Pro-inflammatory cytokines, such IL-6, can cause reduced synaptic release of glutamate and decreased neuronal activity, which could lead to more fragile synapses and, ultimately, to synapse loss.35
Our current results showed that A25T fibrils alone were not directly toxic to primary cortical neurons; their toxicity required microglial activation. In line with our findings, amyloid fibrils have been shown to exert their toxicity by modulating the immune system.11, 14 Amyloid fibrils formed from different pathological or non-pathological proteins or peptides activate macrophages and microglia, leading to the secretion of pro-inflammatory molecules, such as TNF-α, IL-6, IL-1_β_ and NO.15, 36, 37, 38 In the case of neurodegenerative diseases, such as AD, amyloid-β fibril-activated microglia initiate a self-perpetuating neurotoxicity cycle, in which toxic molecules secreted by microglia induce neuronal damage and, in turn, the damaged neurons release danger signals that maintain the reactive state of the microglia.14, 39
Finally, in the current study, we demonstrated that i.c.v. injection of A25T fibrils-induced an increase in TNF-α and IL-6 levels in mouse brains 4 h after the injection. At 24 h post-injection, we found that the injected fibrils retained a periventricular distribution, suggesting that these fibrils might be too large to diffuse into the brain parenchyma. Although we did observe that the amount of fibrils near the injection site diminished over 7 days compared with results obtained 4 h post-injection, the mechanism by which this clearance takes place remains to be elucidated. It seems possible, based on our findings, that fibrils are internalized by microglia (or other cell types) and are then degraded or digested into smaller fragments that cannot be detected by our antibody. Remarkably, A25T fibril-injected mice exhibited short-term memory deficits, while no change in behavior was observed in vehicle-injected mice. Memory deficits could be prevented in A25T-injected mice by a 3-day pre-treatment with minocycline, suggesting that microglia-mediated inflammation had an important role in the cognitive deficit.
In conclusion, our results show that TTR fibrils induce the neurotoxic activation of microglia in vitro and in vivo, revealing a novel cellular mechanism that could be involved in the pathogenesis of OA, and possibly of other vascular amyloid dementias, because amyloid fibrils formed from different proteins have been shown to share common structural features and toxic mechanisms. More generally, our findings indicate that amyloid fibrils modulate the immune system, especially microglial cells, thereby contributing to a vicious cycle of neurodegeneration.
Materials and Methods
Ethics statement and animal facilities
All animal procedures complied with Brazilian legislation and were reviewed and approved by the Ethics Committee for Animal Experimentation (CEUA) of the Federal University of Rio de Janeiro.
Mice
Two-month-old male Swiss mice were stereotaxically injected i.c.v. with either 400 pmol of A25T amyloid fibrils or vehicle (sterile phosphate-buffered saline, pH 7.2 (PBS)) using coordinates from the mouse brain Atlas.40 Briefly, mice were treated with diazepam (5 mg/kg) 30 min before the injection to minimize stress and anesthetized with ketamine (100 mg/kg) and xylazine (25 mg/kg). The hair overlying the skull was shaved and the cranial bone exposed. The bone was then perforated to expose the meninges and 4 _μ_l of a suspension of A25T aggregates was injected at a flow rate of 1 _μ_l/10 min using the following stereotaxic coordinates: AP=−0.22 mm; L=−1.0 mm left; and DV=−2.0 mm. The mice were then returned to their cages to rest and recover from anesthesia. Either 24 h or 7 days after the injection, the mice were euthanized and transcardially perfused with 4% paraformaldehyde. The fixed brains were removed, and 40-_μ_m sections were obtained using a vibratome (Leica VT1000S, Leica, Wetzlar, Germany). The sections were transferred to 24-well plates containing PBS. For the novel object recognition test, the mice were treated (i.p) for 3 consecutive days with either minocycline (50 mg/kg/day) or saline and then analyzed as described.
Expression and purification of recombinant TTR
The plasmid containing the A25T TTR mutant was amplified in E. coli DH_α_5 cells and further purified using the Wizard Plus SV Miniprep DNA Purification System kit (Promega, Fitchburg, WI, USA). The expression and purification of recombinant A25T were performed as described previously.41 Protein concentration was measured using an extinction coefficient of 7.76 × 104 M−1 cm−1 at 280 nm. A25T was further purified by elution through a polymixin B-conjugated resin (Detoxi-gel endotoxin removing column, Thermo Scientific, Waltham, MA, USA) to remove possible endotoxin contaminants.42 The samples were stored in liquid N2 to prevent protein aggregation. To induce protein aggregation, samples were aged for 15 days at 37 °C. Samples were characterized as fibrillar or soluble using transmission electron microscopy (TEM), Congo red and thioflavin-T binding as described.42 For internalization assays, A25T was labeled with the fluorescent probe acrylodan as described previously.43 In all assays, fibrillar and soluble A25T were used at 1 _μ_M.
Primary microglial culture
Microglia were obtained as described previously.44 Briefly, cortices from neonatal Swiss mice were dissociated, and the resulting cells were plated on poly-L-lysine-coated 75-cm2 flasks. The cells were maintained in Dulbecco’s Modified Eagle Medium supplemented with F12 medium (1 : 1 ratio), 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin for 2 weeks at 37 °C in a humidified chamber with 5% CO2. The medium was changed after 1 week and before cell harvesting. After 14 days, microglia were harvested using an orbital shaker. The cells were counted and plated in 6-, 24- or 96-well plates for assays. To produce CM, aggregated TTR samples were incubated with microglia for 48 h in Neurobasal medium (Gibco, Carlsbad, CA, USA). The medium was then centrifuged, and the supernatant was incubated with neurons for 3, 24 or 48 h.
Primary neuronal culture
Two different types of primary neuronal cultures were used: neonatal immature cortical neurons (days in vitro; DIV 1) and E14 mature cortical neurons (DIV 14). In both cases, cortices were gently dissected, and the resulting neurons were counted and maintained in 24-well plates (previously coated with poly-L-lysine) in Neurobasal medium supplemented with B27 (Gibco) for 24 h (neonatal neurons) or 14 days (E14 neurons). The cells were kept at 37 °C in a humidified chamber with 5% CO2 until use.
Phagocytosis assay
A25T samples were fluorescently labeled with acrylodan and incubated with primary microglia for 24 h in 5% CO2 at 37 °C. The cells were then fixed with 4% paraformaldehyde and stained with ethidium homodimer-1 to visualize cell nuclei. As phagocytosis depends on actin polymerization, 2.5 _μ_M of cytochalasin D (Cyt-D; Sigma, St Louis, MO, USA) was added to the wells. Internalization was assessed in both the treated and non-treated cells.
Quantification of TNF-α, IL-6 and NO
Cells were plated in 96-well plates at a density of 5 × 104 cells per well and incubated with A25T fibrils for 24 h (for cytokine measurements) or 48 h (for NO measurements). Aliquots of the culture medium were then withdrawn for assays. For the measurement of cytokines in brain extracts, brains were removed from euthanized animals and mechanically homogenized in 1 ml of 50 mM Tris-HCl (pH 8.0) containing a protease inhibitor cocktail (Roche, Basel, Switzerland). Homogenates were vortexed and centrifuged for 10 min at 10 000 rpm at 4 °C. The soluble fraction was immediately analyzed or frozen until use. NO (detected as nitrite) was measured using the Griess reagent, and cytokines were measured using sandwich ELISA kits (Peprotech, Rocky Hill, NJ, USA). When minocycline was used, the cells were incubated for 1 h with 10 _μ_M minocycline and subsequently incubated with the aggregated TTR samples.
Viability assays
For the MTT reduction assay (Promega), microglia were plated in 96-well plates at a density of 3 × 104 cells per well. After 48 h of incubation with A25T fibrils, 5 _μ_g/ml MTT was added and the cells incubated for 2 h at 37 °C in a humidified chamber with 5% CO2. Purple formazan was solubilized using DMSO and absorbance measured at 570 nm. For the Live/Dead assay (Invitrogen, Carlsbad, CA, USA), cells were plated on coverslips in 24-well plates at a density of 7 × 104 cells/well for neurons and 3 × 104 cells/well for microglia, and the assay was performed according to the manufacturer’s instructions. For Live/Dead assay, 9–10 images were acquired in each condition, with 35–61 cells/image for microglia and 12–20 cells/image for neurons. The percentage of live cells was calculated relative to the number of control (PBS-treated) cells.
TUNEL assay
Primary neurons were plated in 24-well plates, exposed for 48 h to CM from microglia incubated for 48 h with either protein samples or a control solution (PBS or lipopolyssaccharide (LPS) from E. coli at 100 ng/ml; Sigma) and fixed in 4% paraformaldehyde. The TUNEL assay (Invitrogen) was performed according to the manufacturer’s instructions. In each independent experiment, 10 images were acquired per experimental condition, with 67–84 cells/image.
Cleaved caspase-3 (Asp175) immunocytochemistry
Primary neurons were plated in 24-well plates and exposed for 24 h to CM from microglia incubated for 48 h with either protein samples or a control solution (PBS or LPS). The cells were then fixed in 4% paraformaldehyde and incubated in blocking/permeabilizing solution (PBS containing 5% normal goat serum and 0.3% Triton X-100) for 1 h at 25 °C. The cells were incubated with rabbit anti-cleaved caspase-3 (Asp175; 1 : 400 dilution; Cell Signaling, Danvers, MA, USA) overnight at 4 °C. The cells were then washed three times in PBS containing 0.3% Triton X-100. Texas Red-conjugated goat anti-rabbit IgG (1 : 200; Santa Cruz Biotechnology, Dallas, TX, USA) was then added for 2 h at 25 °C. The coverslips were mounted in Prolong Gold antifade reagent (Invitrogen). For the cleaved-caspase-3 assay, 10 images were acquired in each experimental condition, with 4–18 cells/image.
Western blotting
Primary microglial cells (106) were plated in six-well plates and supplemented with DMEM/F12 with 10% FBS. After cell adhesion, 1 μ_M fibrillar A25T or soluble A25T was added for 30 min. The cells were lysed with cold RIPA buffer, vortexed and then centrifuged for 10 min at 10 000 r.p.m. at 4 °C. The protein content of the soluble fraction was assessed using Bradford’s reagent, and 15 μ_g protein was resolved in 15% SDS-PAGE. The gels were blotted onto low-fluorescence PVDF membranes (Millipore, Billerica, MA, USA), blocked for 1 h in blocking buffer (Li-cor Biosciences, Lincoln, NE, USA) and incubated overnight at 4 °C with anti-Iba-1 (1 : 1000; Wako, Chuo-Ku, Japan), anti-pAkt (Ser473) (1 : 1000), anti-Akt pan (1 : 1000), anti-pGSK-3_β (Ser9) (1 : 1000) and anti-GSK-3_β pan (1 : 1000) antibodies (all from Cell Signaling). The membranes were thoroughly washed with PBS containing 0.1% Tween-20 and incubated for 45 min with anti-rabbit IgG-Dylight700 (1 : 20 000; Li-cor Biosciences). The membranes were then washed in PBS and scanned on an Odyssey Infrared Imaging System (Li-cor Biosciences).
NFκB p65 translocation assay
Primary microglia were plated in 24-well plates and exposed for 2 h to protein samples or controls (PBS or LPS). The cells were then fixed in 4% paraformaldehyde and incubated in blocking/permeabilizing solution (PBS containing 5% normal goat serum and 0.3% Triton X-100) for 1 h at 25 °C. The cells were incubated with rabbit anti-NF_κ_B p65 (1 : 50 dilution; Cell Signaling) overnight at 4 °C. The cells were then washed and incubated with Alexa 546-conjugated goat anti-rabbit IgG (1 : 500; Cell Signaling) for 2 h at 25 °C. For the NF_κ_B p65 translocation assay, 12–15 images were acquired in each experimental condition, with 4–8 cells/image.
Synapse quantification
After being fixed with 4% paraformaldehyde for 15 min, cultures were permeabilized with 0.2% Triton X-100 for 5 min at room temperature. Nonspecific binding sites were blocked with 10% bovine serum albumin (Sigma) for 1 h before immunoreacting the cultures with mouse anti-_α_-synaptophysin (1 : 300; Chemicon International, Billerica, MA, USA) and rabbit anti-PSD-95 (1:300; Abcam, Cambridge, UK). After the primary antibody incubation, the cells were washed with PBS and incubated with secondary antibodies, Alexa Fluor 546-conjugated goat anti-rabbit IgG (Molecular Probes, Carlsbad, CA, USA; 1 : 1000) or Alexa Fluor 488 goat anti-mouse IgG (Molecular Probes; 1 : 300), for 2 h at room temperature. The nuclei were counterstained with DAPI (4’, 6-diamidino-2-phenyindole, dilactate; Sigma). After the immunostaining, the cells were imaged and counted on a TE2000 Nikon microscope (Nikon, Tokyo, Japan). For the analysis of synapses, the green and red channels from 12–20 neurons (one neuron per image) per experimental condition were aligned and the colocalization of the signals quantified using the Puncta Analyzer plugin in ImageJ (NIH, Bethesda, MD, USA), as previously described.45 In each experiment, at least 20 images were analyzed per experimental condition. The results reported are the mean values from three independent experiments.
Immunohistochemistry
Mouse brain sections were permeabilized in Triton X-100 (1% in PBS) for 30 min at room temperature and pre-incubated with 5% bovine serum albumin (BSA) and normal goat serum (NGS) for 3 h. They were then incubated with rabbit polyclonal anti-TTR (1 : 400, DAKO, Dako, Glostrup, Denmark) or mouse anti-F4/80 (1 : 200; ABD Serotec, Oxford, UK) in blocking buffer. After being washed with PBS, the sections were incubated with secondary antibodies conjugated with Alexa Fluor 488 (goat anti-mouse 1 : 500; Molecular Probes) or Alexa Fluor 546 (goat anti-rabbit 1 : 500; Molecular Probes) for 2 h at room temperature. The sections were then washed with PBS, stained with DAPI, washed three times with PBS and mounted. Negative controls were performed using rabbit or mouse IgG. The slices were imaged using either an epifluorescence microscope (Nikon TE300, Nikon) or a confocal microscope (Leica TCS-SP5, Leica) equipped with a × 63 1.40 NA oil-immersion objective. The images were processed using Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA, USA).
Novel object recognition test
The novel object recognition test was performed according to Figueiredo et al.46 Using a 30 × 30 × 45 cm box that had lines on the floor delimiting a total of nine squares the task was divided into three stages: habituation, training and test. Habituation consisted of the acclimation of the animal to the arena for 5 min, during which the number of lines each animal crossed on the floor of the arena (number of crossings) and the number of rearings (elevation on rear paws, denoting exploratory behavior) were recorded using manual stopwatches to verify locomotor/exploratory activities. In the training session, the animals were exposed to two identical objects (A and B; placed side-by-side), and the time spent exploring each object was counted over a 5 min period. Two hours after the training session, the test session was performed by replacing one of the familiar objects (B) with a novel object (C) and the exploration times for object A (familiar) and C (novel) were counted. Time recording in the training and test stages was performed using stopwatches and the experiment was blind to the researchers who performed it. Results were expressed as percentage of time exploring each object and were analyzed using a one-sample Student’s _t-_test comparing the mean exploration time for each object with the fixed value of 50%. The tests were repeated twice, using four to five animals per group.
Statistical analysis
Experiments were performed at least in triplicate using different batches of protein samples. Statistical analysis was performed using a one-way ANOVA followed by Tukey’s post test, or with the Kruskal–Wallis test followed by Dunn’s post hoc test as appropriate. For the novel object recognition test, statistical analysis was performed using a one-sample _t_-test comparing the percentage of time spent exploring the novel object to the theoretical value of 50% (which corresponds to random exploration of the two objects).
Abbreviations
OA:
oculoleptomeningeal amyloidosis
CNS:
central nervous system
CSF:
cerebral spinal fluid
TTR:
transthyretin
LPS:
lipopolysaccharide
NO:
nitric oxide
TNF-α:
tumor necrosis factor alpha
IL-6:
interleukin-6
GSK-3_β_:
glycogen synthase kinase -3beta
MTT:
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
TUNEL:
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
References
- Shimizu Y, Takeuchi M, Matsumura M, Tokuda T, Iwata M . A case of biopsy-proven leptomeningeal amyloidosis and intravenous Ig-responsive polyneuropathy associated with the Ala25Thr transthyretin gene mutation. Amyloid 2006; 13: 37–41.
Article CAS Google Scholar - Hagiwara K, Ochi H, Suzuki S, Shimizu Y, Tokuda T, Murai H et al. Highly selective leptomeningeal amyloidosis with transthyretin variant Ala25Thr. Neurology 2009; 72: 1358–1360.
Article CAS Google Scholar - De Carolis P, Galeotti M, Ficarra G, Masetti M, Grimaldi D, Cortelli P . Fatal cerebral haemorrhage after liver transplantation in a patient with transthyretin variant (gly53glu) amyloidosis. Neurol Sci 2006; 27: 352–354.
Article CAS Google Scholar - Rinaldi L, Busani S, Pasetto A, Girardis M . Posttransplant cerebral hemorrhage in a patient with familial amyloidotic polyneuropathy. Transplantation 2006; 81: 134–135.
Article Google Scholar - Owa M, Takei Y, Hashikura Y, Kawasaki S, Koyama M, Ikeda S . Recurrent cerebral embolism in a familial amyloid polyneuropathy patient who received partial liver transplantation from a living donor. Intern Med 2001; 40: 259–264.
Article CAS Google Scholar - Revesz T, Holton JL, Lashley T, Plant G, Frangione B, Rostagno A et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 2009; 118: 115–130.
Article CAS Google Scholar - Greenberg SM, Vonsattel JP . Diagnosis of cerebral amyloid angiopathy. Sensitivity and specificity of cortical biopsy. Stroke 1997; 28: 1418–1422.
Article CAS Google Scholar - Bell RD, Zlokovic BV . Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol 2009; 118: 103–113.
Article CAS Google Scholar - Yates PA, Sirisriro R, Villemagne VL, Farquharson S, Masters CL, Rowe CC . Cerebral microhemorrhage and brain _β_-amyloid in aging and Alzheimer disease. Neurology 2011; 77: 48–54.
Article CAS Google Scholar - Peuralinna T, Tanskanen M, Mäkelä M, Polvikoski T, Paetau A, Kalimo H et al. APOE and A_β_PP gene variation in cortical and cerebrovascular amyloid-β pathology and Alzheimer's disease: a population-based analysis. J Alzheimers Dis 2011; 2: 377–385.
Article Google Scholar - Sondag CM, Dhawan G, Combs CK . Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation 2009; 5: 6–1.
Google Scholar - Mitrasinovic OM, Murphy GM Jr . Accelerated phagocytosis of amyloid-beta by mouse and human microglia overexpressing the macrophage colony-stimulating factor receptor. J Biol Chem 2002; 277: 29889–29896.
Article CAS Google Scholar - Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M et al. expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40. J Neurosci 2006; 26: 3345–3356.
Article CAS Google Scholar - Surace MJ, Block ML. . Targeting microglia-mediated neurotoxicity: the potential of NOX2 inhibitors. Cell Mol Life Sci 2012; 69: 2409–2427.
Article CAS Google Scholar - Saijo K, Glass CK . Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol 2011; 11: 775–787.
Article CAS Google Scholar - Luo XG, Ding JQ, Chen SD . Microglia in the aging brain: relevance to neurodegeneration. Mol Neurodegener 2010; 24: 5–12.
CAS Google Scholar - Azevedo EP, Pereira HM, Garratt RC, Kelly JW, Foguel D, Palhano FL . Dissecting the structure, thermodynamic stability, and aggregation properties of the A25T transthyretin (A25T-TTR) variant involved in leptomeningeal amyloidosis: identifying protein partners that co-aggregate during A25T-TTR fibrillogenesis in cerebrospinal fluid. Biochemistry 2011; 50: 11070–11083.
Article CAS Google Scholar - Li X, Masliah E, Reixach N, Buxbaum JN . Neuronal production of transthyretin in human and murine Alzheimer′s disease: is it protective? J Neurosci 2011; 35: 12483–12490.
Article Google Scholar - Murakami K, Koide M, Dumont TM, Russell SR, Tranmer BI, Wellman GC. . Subarachnoid hemorrhage induces gliosis and increased expression of the pro-inflammatory cytokine high mobility Group Box 1 Protein. Transl Stroke Res 2011; 1: 72–79.
Article Google Scholar - He Z, Gao Y, Deng Y, Li W, Chen Y, Xing S et al. Lipopolysaccharide induces lung fibroblast proliferation through Toll-like receptor 4 signaling and the phosphoinositide3-kinase-Akt pathway. PLoS One 2012; 7: e35926.
Article CAS Google Scholar - Sarbassov DD, Guertin DA, Ali SM, Sabatini DM . Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 5712: 1098–1101.
Article Google Scholar - Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. . Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995; 6559: 785–789.
Article Google Scholar - Shen E, Fan J, Peng T . Glycogen synthase kinase-3beta suppresses tumor necrosis factor-alpha expression in cardiomyocytes during lipopolysaccharide stimulation. J Cell Biochem 2008; 104: 329–338.
Article CAS Google Scholar - Manning BD, Cantley LC . AKT/PKB signaling: navigating downstream. Cell 2007; 129: 1261–1274.
Article CAS Google Scholar - Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M et al. Control of synaptic strength by glial TNFα. Science 2002; 5563: 2282–2285.
Article Google Scholar - Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995; 376: 37–43.
Article CAS Google Scholar - Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis 2013; 4: e525.
Article CAS Google Scholar - Chiti F, Dobson CM . Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 2006; 75: 333–366.
Article CAS Google Scholar - Liepnieks JJ, Dickson DW, Benson MD . A new transthyretin mutation associated with leptomeningeal amyloidosis. Amyloid 2011; Suppl 1: 160–162.
Article Google Scholar - Hagiwara K, Ochi H, Suzuki S, Shimizu Y, Tokuda T, Murai H _et al_2009 Highly selective leptomeningeal amyloidosis with transthyretin variant Ala25Thr. Neurology 72: 1358–1360.
Article CAS Google Scholar - Nakagawa K, Sheikh SI, Snuderl M, Frosch MP, Greenberg SM . A new Thr49Pro transthyretin gene mutation associated with leptomeningeal amyloidosis. J Neurol Sci 2008; 272: 186–190.
Article CAS Google Scholar - Roe RH, Fisher Y, Eagle RC Jr., Fine HF, Cunningham ET Jr . Oculoleptomeningeal amyloidosis in a patient with a TTR Val30Gly mutation in the transthyretin gene. Ophthalmology 2007; 114: e33–e37.
Article Google Scholar - Mitsuhashi S, Yazaki M, Tokuda T, Sekijima Y, Washimi Y, Shimizu Y et al. Biochemical characteristics of variant transthyretins causing hereditary leptomeningeal. Amyloid 2005; 12: 216–225.
Article CAS Google Scholar - Ando Y, Tanaka Y, Nakazato M, Ericzon BG, Yamashita T, Tashima K et al. Change in variant transthyretin levels in patients with familial amyloidotic polyneuropathy type I following liver transplantation. Biochem Biophys Res Commun 1995; 211: 354–358.
Article CAS Google Scholar - Nelson TE, Olde Engberink A, Hernandez R, Puro A, Huitron-Resendiz S, Hao C et al. Altered synaptic transmission in the hippocampus of transgenic mice with enhanced central nervous systems expression of interleukin-6. Brain Behav Immun 2012; 26: 959–971.
Article CAS Google Scholar - Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG . Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 2005; 280: 17294–17300.
Article CAS Google Scholar - Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B et al. TLR2 is a primary receptor for Alzheimer's amyloid β peptide to trigger neuroinflammatory activation. J Immunol 2012; 188: 1098–1107.
Article CAS Google Scholar - Nishimori JH, Newman TN, Oppong GO, Rapsinski GJ, Yen JH, Biesecker SG et al. Microbial amyloids induce interleukin 17A (IL-17A) and IL-22 responses via toll-like receptor 2 activation in the intestinal mucosa. Infect Immun 2012; 80: 4398–4408.
Article CAS Google Scholar - Block ML, Zecca L, Hong JS . Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 2007; 8: 57–69.
Article CAS Google Scholar - Paxinos G, KBJ Franklin In The Mouse Brain in Stereotaxic Coordinates. San Diego, USA. : Academic Press, 2001.
Google Scholar - Lashuel HA, Lai Z, Kelly JW . Characterization of the transthyretin acid denaturation pathways by analytical ultracentrifugation: implications for wild-type, V30M, and L55P amyloid fibril formation. Biochemistry 1998; 37: 17851–17864.
Article CAS Google Scholar - Azevedo EP, Guimarães-Costa AB, Torezani GS, Braga CA, Palhano FL, Kelly JW et al. Amyloid fibrils trigger the release of neutrophil extracellular traps (NETs), causing fibril fragmentation by NET-associated elastase. J Biol Chem 2012; 287: 37206–37218.
Article CAS Google Scholar - Palhano FL, Leme LP, Busnardo RG, Foguel D. . Trapping the monomer of a non-amyloidogenic variant of transthyretin: exploring its possible use as a therapeutic strategy against transthyretin amyloidogenic diseases. J Biol Chem 2009; 284: 1443–1453.
Article CAS Google Scholar - Lima FR, Gervais A, Colin C, Izembart M, Neto VM, Mallat M . Regulation of microglial development: a novel role for thyroid hormone. J Neurosci 2001; 21: 2028–2038.
Article CAS Google Scholar - Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 2005; 120: 421–433.
Article CAS Google Scholar - Figueiredo CP, Clarke JR, Ledo JH, Ribeiro FC, Costa CV, Melo HM et al. Memantine rescues transient cognitive impairment caused by high-molecular-weight a_β_ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J Neurosci 2013; 23: 9626–9634.
Article Google Scholar
Acknowledgements
This research was supported by grants from Conselho Nacional de Desenvolvimento Científico e Tecnologico (CNPq), Fundação Carlos Chagas Filho de Amparo a Pesquisa do Estado do Rio de Janeiro (FAPERJ), and Coordena a Aperfeiçoamento de Pessoal de Nível Superior (CAPES). We are also very grateful to Dr. Andre Gomes (Federal University of Rio de Janeiro) for two-photon assistance, Dr. Evan Powers (The Scripps Research Institute) for guidance on statistical analysis, and to ER Gonçalves and S Alonso for competent technical assistance.
Author information
Authors and Affiliations
- Instituto de Bioquímica Médica, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ 21941-590, Brazil
E P Azevedo, J H Ledo, F L Palhano, S T Ferreira & D Foguel - Instituto de Ciências Biomédicas, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ 21941-590, Brazil
G Barbosa, M Sobrinho, L Diniz, A C C Fonseca, F Gomes, L Romão & F R S Lima - Pólo de Macaé, Universidade Federal do Rio de Janeiro, Macaé, RJ 27930-560, Brazil
L Romão
Authors
- E P Azevedo
You can also search for this author inPubMed Google Scholar - J H Ledo
You can also search for this author inPubMed Google Scholar - G Barbosa
You can also search for this author inPubMed Google Scholar - M Sobrinho
You can also search for this author inPubMed Google Scholar - L Diniz
You can also search for this author inPubMed Google Scholar - A C C Fonseca
You can also search for this author inPubMed Google Scholar - F Gomes
You can also search for this author inPubMed Google Scholar - L Romão
You can also search for this author inPubMed Google Scholar - F R S Lima
You can also search for this author inPubMed Google Scholar - F L Palhano
You can also search for this author inPubMed Google Scholar - S T Ferreira
You can also search for this author inPubMed Google Scholar - D Foguel
You can also search for this author inPubMed Google Scholar
Corresponding authors
Correspondence toF L Palhano or D Foguel.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by A Verkhratsky
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Azevedo, E., Ledo, J., Barbosa, G. et al. Activated microglia mediate synapse loss and short-term memory deficits in a mouse model of transthyretin-related oculoleptomeningeal amyloidosis.Cell Death Dis 4, e789 (2013). https://doi.org/10.1038/cddis.2013.325
- Received: 21 May 2013
- Revised: 24 July 2013
- Accepted: 29 July 2013
- Published: 05 September 2013
- Issue Date: September 2013
- DOI: https://doi.org/10.1038/cddis.2013.325