Lytic cell death induced by melittin bypasses pyroptosis but induces NLRP3 inflammasome activation and IL-1β release (original) (raw)
Main
Inflammasomes are multiprotein complexes activated in response to multiple pathogen and noninfectious stimuli, including pore-forming toxins, venoms, changes in extracellular osmolarity or extracellular accumulation of metabolites such as ATP or crystalline uric acid.1, 2, 3, 4, 5 Inflammasomes are formed by a sensor protein that usually belongs to the nucleotide-binding domain and leucine-rich repeat-containing receptor (NLR) family, among them, the sensor NLR with a pyrin domain 3 (NLRP3), the inflammasome most thoroughly investigated and the only one able to respond to sterile stimulation.1, 2 The canonical NLRP3 inflammasome is formed by the sensor protein NLRP3, the adaptor protein apoptosis-associated speck-like protein with a caspase recruitment domain (ASC) and the effector protease caspase-1.1, 2, 6 NLRP3 is activated by a wide plethora of stimuli that induce a common intracellular signaling, including a decrease in intracellular K+, production of reactive oxygen species, plasma membrane permeabilization, lysosomal destabilization and metabolic disruption.6, 7, 8 NLRP3 inflammasome induces ASC assembly and oligomerization by a prion-like process into large aggregates that amplify caspase-1 activation.9, 10, 11 Caspase-1 induces maturation and release of the pro-inflammatory cytokines IL-1_β_ and IL-18 through a noncanonical (endoplasmic reticulum- and Golgi-independent) release pathway.12 The release of these cytokines is mainly due to plasma membrane permeabilization,13 induced by the processing of gasdermin D by caspase-1 and by insertion of the lytic amino-terminal fragment in the plasma membrane.14, 15, 16 This process also results in a specific type of cell death termed pyroptosis, characterized by the release of intracellular content due to plasma membrane permeabilization.14, 15, 16
Antimicrobial peptides form a chemical barrier against invading pathogens. These peptides must be massively inserted into the targeted membrane to attain their lethal mechanism, a process facilitated by their amphipathic nature. In many cases, the final step is severe membrane distortion or pore formation in the target cell, with lethal leakage of intracellular metabolites and ions.17
To avoid indiscriminate cellular lysis, the targeted microorganism must be recognized. Most antimicrobial peptides are strongly cationic and, therefore, preferentially accumulate in bacterial and lower eukaryote plasma membranes with exposure of anionic phospholipids to the external medium. In higher eukaryotes, acidic phospholipids are confined on the cytoplasmic side of the membrane, exposing a zwitterionic surface to the external medium. Accordingly, antimicrobial peptides specificity relies on a subtle structural blend of amphipaticity and cationic character. In fact, these characteristics are shared with other eukaryotic peptide toxins, such as melittin, endowed with antimicrobial activity aside from their toxic effect on higher eukaryotic cells.18 Similarly, at higher concentrations antimicrobial peptides can also permeabilize the plasma membrane of mammalian cells, and it is known that the human antimicrobial peptide LL-37 (a 37-amino acid peptide) and the insect peptide toxin melittin (a 26-amino acid cationic peptide) may be able to release IL-1_β_.5, 19 It is also known that LL-37 modulates NLRP3 inflammasome by amplification of purinergic P2X7 receptor response to extracellular ATP;19, 20 however, little is known about the mechanism of NLRP3 activation induced by melittin. Melittin is the main component of bee venom, and venoms cause an allergic reaction characterized by local inflammation consisting of redness, pain, heat and swelling.21 The NLRP3 inflammasome and IL-1_β_ are important for this inflammatory response in vivo.5 Melittin has a hydrophobic amino-terminal region (1–20 amino acids) and a hydrophilic cationic carboxyl-terminal region (21–26 amino acids); this amphipathic nature allow melittin to be a potent hemolytic molecule by interacting and permeabilizing eukaryotic membranes forming pores.22, 23 In this study, we aimed to investigate the mechanism of melittin-induced NLRP3 inflammasome activation. We found that melittin induces K+ efflux from macrophages and NLRP3 inflammasome formation. This NLRP3 inflammasome requires the adaptor protein ASC to activate caspase-1 and induce IL-1_β_ release; however, melittin was unable to induce large ASC aggregates. Melittin-induced plasma membrane permeabilization and subsequent cell death was independent of caspase-1, showing that rapid cell lysis driven by melittin excludes pyroptosis execution by caspase-1.
Results
Melittin, but no other antimicrobial peptides, induces robust release of IL-1_β_
Cationic amphipathic antimicrobial peptides may permeabilize plasma membrane and are candidates for NLRP3 inflammasome activation. Different antimicrobial peptides were tested at 10 μ_M on macrophages for IL-1_β release (Table 1), with melittin being the only one able to induce robust release of IL-1_β_ in both human THP-1 and mouse BMDM macrophages (Figures 1a and b). In THP-1, melittin-induced IL-1_β_ release was similar to treatment with nigericin (Figure 1a), a classical NLRP3 inflammasome activator. In contrast, at this concentration, LL-37 – a human peptide reported to induce IL-1_β_ release19 – induced only scant IL-1_β_ release from THP-1 but none from mouse BMDMs (Figures 1a and b). Temporin A and indolicidin failed to trigger significant secretion of IL-1_β_ from macrophages (Figures 1a and b), although indolicidin did have strong effects on the membrane of mammalian cells.24 To gain further insight into the melittin sequence responsible for IL-1_β_ release, we tested chimeric peptides comprising different stretches of the hydrophobic melittin sequence fused to the N-terminus of the antimicrobial peptide cecropin A (Table 1).25 These chimeric peptides improve the microbicidal activity of cecropin A, which is otherwise unable to kill Gram-positive bacteria, while decreasing melittin toxicity by excluding the hemolytic part of melittin.25 All chimeric peptides (peptides 1 to 3) resulted in IL-1_β_ release (Figure 1c), suggesting that the smallest melittin motif necessary to trigger IL-1_β_ release is VLKVL (residues 5–9) (Table 1). IL-1_β_ release was not attained when disturbing the alpha helix conformation of melittin by replacing the native residues in the VLKVL motif by d-amino acids (Figure 1c and Table 1, peptide 4). However, similar IL-1_β_ induction to that of all l-amino acid control peptides did occur when d-amino acids were introduced in the cecropin A moiety of chimeric peptide 5 (Figure 1c and Table 1). This suggests that in chimeric peptides the melittin sequence, rather than the cecropin A region, is responsible for inducing IL-1_β_ release.
Table 1 Peptide sequences used in this study
Figure 1
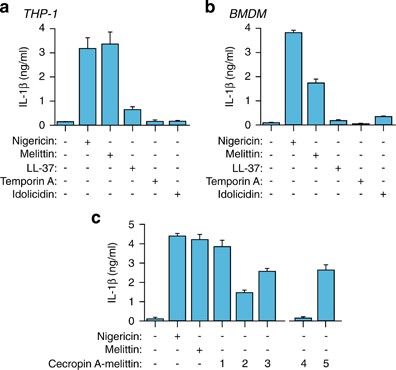
Melittin induces the release of IL-1_β_. (a and b) ELISA for IL-1_β_ release from THP-1 (a) or BMDMs (b) primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with nigericin (25 _μ_M), melittin, LL-37, temporin A or indolicidin (10 μ_M each). (c) ELISA for IL-1_β release from THP-1 treated as in panel (a) but with the chimeric peptides shown (10 _μ_M each); chimeric peptides are described in Table 1
Bee venom induces IL-1_β_ release
Melittin is the main active component of apitoxin (bee venom), and the application of bee venom to lipopolysaccharide (LPS)-primed THP-1 macrophages at concentrations >10 μ_g/ml induced a robust release of IL-1_β, comparable to 20 μ_M of melittin treatment (Figure 2a). Bee venom-induced release of IL-1_β occurs during the first 30 min of treatment and then reaches a plateau (Figures 2b and c). These kinetics were similar to the kinetics for IL-1_β_ melittin-induced release in THP-1 (Figure 2b); however, in mouse BMDMs, bee venom was more potent in inducing IL-1_β_ than melittin (Figure 2c), suggesting that other bee venom components could activate different pathways besides melittin in mouse macrophages. Moreover, many antimicrobial peptides have antitumoral activity due to the exposure of phosphatidylserine residues at the surface of these cells, whereas this characteristic is absent in nontransformed cells.26 Consequently, susceptibility of THP-1 to other components of the bee venom may differ from BMDMs.
Figure 2
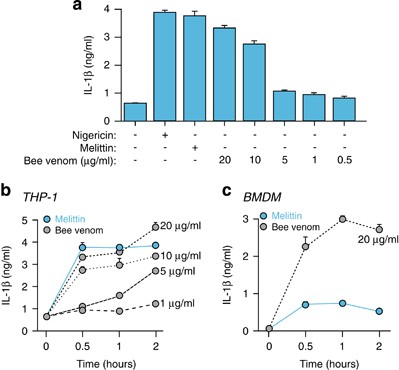
Bee venom induces the release of IL-1_β_. (a) ELISA for IL-1_β_ release from THP-1 primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with nigericin (25 _μ_M), melittin (20 μ_M) or bee venom at the doses shown. (b and c) Time-course of IL-1_β release from THP-1 (b) or BMDMs (c) primed as in panel (a), and then stimulated with melittin (20 _μ_M) or bee venom at the doses and for the times shown
Melittin activates the NLRP3 inflammasome by potassium efflux
Low intracellular K+ concentration is a key step in NLRP3 activation,27, 28 and it has previously reported that bee venom-induced release of IL-1_β_ was blocked using a high extracellular K+ solution.5 In our study, we found that THP-1 macrophage incubation with melittin induced a significant decrease in intracellular K+ concentration (Figure 3a). Furthermore, macrophage incubation with melittin in a buffer with a high K+ concentration reduced IL-1_β_ release from THP-1 macrophages (Figure 3b). An analysis of the NLRP3 bioluminescence resonance energy transfer (BRET) signal during inflammasome activation showed that melittin treatment is able to change the conformation of NLRP3, with a similar but smaller profile than nigericin treatment (Figure 3c). BMDMs from NLRP3-deficient mice failed to release IL-1_β_ after melittin treatment (Figure 3d). Melittin also induced an increase in caspase-1 activity in macrophage supernatants that was dependent on NLRP3 (Figure 4a), suggesting that NLRP3 inflammasome was activating caspase-1. The p10 subunit of active caspase-1 was also found in macrophage supernatants after melittin treatment; however, the detection of caspase-1 p10 subunit was weak when compared to nigericin-treated macrophages (Figure 4b). The melittin-induced IL-1_β_ that was released was the mature p17 form of the cytokine (Figure 4c) and was dependent on caspase-1 activity (Figure 4d). These data suggest that the decrease in melittin-induced intracellular K+ in LPS-primed macrophages triggers the NLRP3 inflammasome to activate caspase-1.
Figure 3
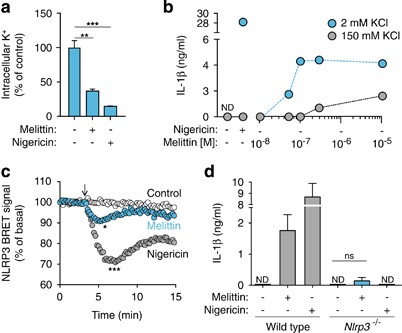
Melittin induces K+ depletion and NLRP3 activation. (a) Relative intracellular K+ concentration from THP-1 primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with nigericin (25 _μ_M) or melittin (5 μ_M). (b) ELISA for IL-1_β release from THP-1 primed as in panel (a), and then stimulated or not for 30 min with nigericin (25 _μ_M) or melittin in low K+ buffer (2 mM KCl, blue circles) or high K+ buffer (150 mM KCl, gray circles) at the doses shown. (c) Kinetics of net BRET signal for the NLRP3 protein in unstimulated control cells (white circles) or cells treated with nigericin (10 _μ_M, gray circles) or melittin (5 μ_M, blue circles); nigericin or melittin was added as indicated by the arrow. (d) ELISA for IL-1_β release from wild-type or _Nlrp3_−/− BMDMs primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with nigericin (25 _μ_M) or melittin (0.5 _μ_M); ND, not detected
Figure 4
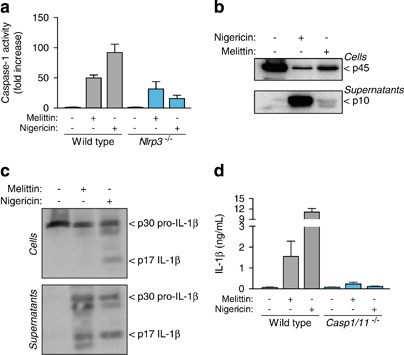
Melittin activates caspase-1 in macrophages. (a) Caspase-1 activity in supernatants from wild-type or _Nlrp3_−/− BMDMs primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with nigericin (25 _μ_M) or melittin (0.5 μ_M); data are shown as fold increase from unstimulated macrophages for each genotype. (b and c) Western blot analysis of caspase-1 (b) or IL-1_β (c) in cell extracts or supernatants from BMDMs primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with nigericin (25 _μ_M) or melittin (5 μ_M). (d) ELISA for IL-1_β release from wild type or _Casp1/11_−/− BMDMs primed and activated as in (a)
Melittin does not induce IL-18 release
Despite the induction of IL-1_β_ release via caspase-1 activation, melittin failed to induce IL-18 release (Figure 5a). The differential secretion of both caspase-1 dependent cytokines was not due to differences in Il18 gene expression (not shown) and might be explained by specific degradation of IL-18 protein after melittin treatment. It is known that melittin can induce caspase-3 activation,29 and caspase-3 can degrade IL-18, resulting in the inactivation of this cytokine.30 In macrophages, melittin induced the activation of caspase-3 and to a lesser extent, also of caspase-8 (Figure 5b). However, nigericin also induced the activation of these caspases (Figure 5c) and the release of IL-18 (Figure 5a); hence, it does not seem feasible that caspase-3 can degrade IL-18 in melittin-treated macrophages. To rule out possible degradation of IL-18 by caspase-3, we used caspase-3-deficient macrophages. The deficiency of caspase-3 was not able to rescue IL-18 release from melittin-treated macrophages (Figure 5d). Similarly, caspase-8 inhibition was not able to increase IL-18 release after melittin treatment (Figure 5e). Neither caspase-3 or caspase-8 inhibition affected IL-1_β_ or IL-18 release induced by nigericin (Figures 5d and e), denoting a minimal contribution of these caspases to cytokine release after NLRP3 inflammasome activation.
Figure 5
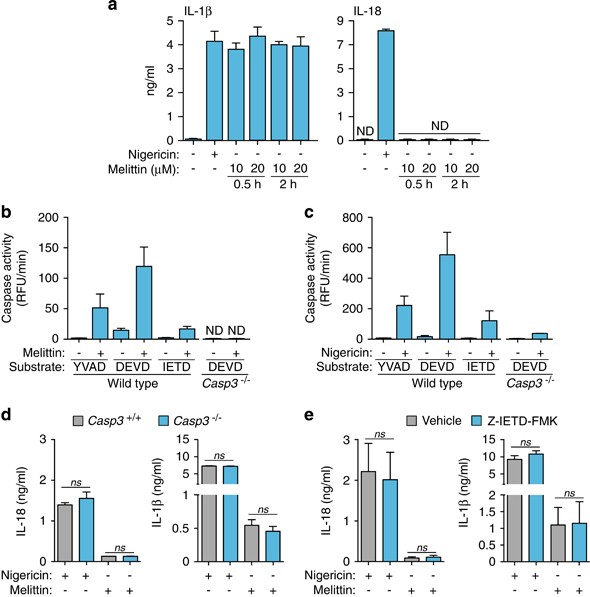
Melittin does not induce IL-18 release. (a) ELISA for IL-1_β_ or IL-18 release from BMDMs primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with nigericin (25 _μ_M) or the melittin concentrations and times shown; ND, not detected. (b and c) Caspase activity measured by the fluorescent peptide substrates indicated in the supernatants from wild type or _Casp3_−/− BMDMs primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with melittin (0.5 _μ_M) (b) or nigericin (25 μ_M) (c). (d and e) ELISA for IL-1_β or IL-18 release from wild type (d,e) or _Casp3_−/− (d) BMDMs, treated or not with the caspase-8 inhibitory peptide Z-IETD-FMK (e) treated as in (b and c)
Melittin does not induce pyroptotic cell death
The NLRP3 inflammasome requires the adaptor protein ASC to activate caspase-1;1, 2, 6 accordingly, melittin-induced IL-1_β_ release was reduced in ASC-deficient macrophages (Figure 6a). Canonical NLRP3 inflammasome activation induces ASC oligomerization in large aggregates, which are responsible for amplifying caspase-1 activation and inducing subsequent pyroptotic cell death.9, 10, 11 Despite ASC dependence in melittin-induced NLRP3 inflammasome, we only detected less than 10% of melittin-treated macrophages that form ASC specks, compared with the robust and significant induction of ASC specks by nigericin (Figures 6b and c). This was also confirmed by crosslinking experiments, where melittin failed to induce ASC oligomerization (Figure 6d). This suggests that, although melittin may activate caspase-1 via NLRP3 inflammasome using the ASC adaptor protein, ASC failed to form large oligomeric signaling complexes.
Figure 6
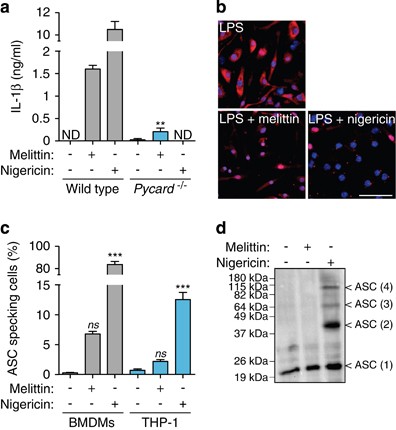
Melittin does not induce ASC oligomerization. (a) ELISA for IL-1_β_ release from wild type or _Pycard_−/− BMDMs primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with nigericin (25 _μ_M) or melittin (0.5 _μ_M); ND, not detected. (b) Deconvolved representative images of BMDMs treated as in panel (a) and stained for ASC (red) and nuclei (blue; DAPI); scale bar, 50 _μ_M. (c) Quantification of intracellular ASC speck from BMDMs or THP-1 from pictures as shown in (b). (d) Western blot from crosslinked ASC from BMDMs stimulated as in (a)
The lack of robust ASC oligomerization suggests that melittin might not induce pyroptosis, which is a cell death process dependent on caspase-1. However, melittin induced higher levels of cell death when compared with nigericin treatment (Figures 7a and b). Cell death induced by melittin was dose-dependent reaching a plateau (Figures 7c and d), with a melittin LD50 of ∼5 μ_M in both THP-1 and BMDMs. Similarly, the release of IL-1_β was dose-dependent for small concentrations of melittin, but cytokine release decreased at high concentrations of melittin (≥ 10 μ_M) (Figures 7e and f), suggesting that an excessive rate of cell death by rapid cellular lysis leads to reduced NLRP3 inflammasome activation. While nigericin-induced cell death was dependent on NLRP3 and ASC (Figure 8a), melittin-induced cell death was independent on the NLRP3 inflammasome (Figure 8a). In line with this result, caspase-1 deficiency did not affect melittin cell death (Figure 8a). Furthermore, high extracellular K+ reduced nigericin but not melittin-induced cell death (Figure 8b). This result confirms that nigericin was inducing pyroptosis dependent on inflammasome and caspase-1 activation, whereas melittin-induced cell death was independent of the inflammasome. Finally, addition of melittin to macrophages immediately induced rapid (≤1 min) plasma membrane permeabilization that was dose-dependent (Figures 8c and d). However, nigericin induced a delayed plasma membrane permeabilization (after 5 min) (Figure 8e), consistent with the time required to activate caspase-1 and induce pyroptosis. In fact, caspase-1 deficiency impaired plasma membrane permeabilization induced by nigericin (Figure 8f), meanwhile melittin-induced plasma membrane permeabilization was independent on caspase-1 (Figure 8g). Altogether, our data show that melittin directly permeabilizes the plasma membrane of macrophages, inducing K+ leakage and rapid activation of the NLRP3 inflammasome in order to activate caspase-1 via ASC and induce IL-1_β maturation and release. Because macrophage viability and plasma membrane integrity were rapidly and directly compromised by melittin, this impairs the subsequent formation of large ASC aggregates and execution of pyroptosis by caspase-1.
Figure 7
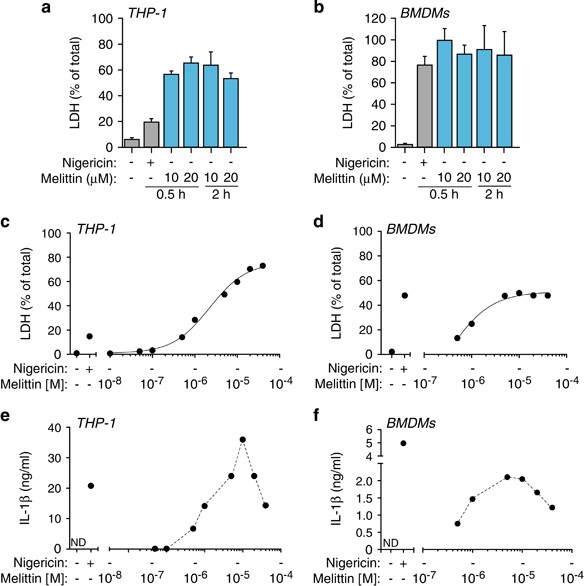
Melittin induces macrophage cell death. (a and b) Cell death measured as percentage of extracellular LDH in THP-1 (a) or BMDMs (b) primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with nigericin (25 μ_M) or melittin at the concentrations and for the times shown. (c and d) Dose–response curves for cell death in THP-1 (c) or BMDMs (d) treated as in (a and b) for 30 min with the indicated concentrations of melittin. (e and f) Dose–response for IL-1_β release from THP-1 (e) or BMDMs (f) treated as in (c and d)
Figure 8
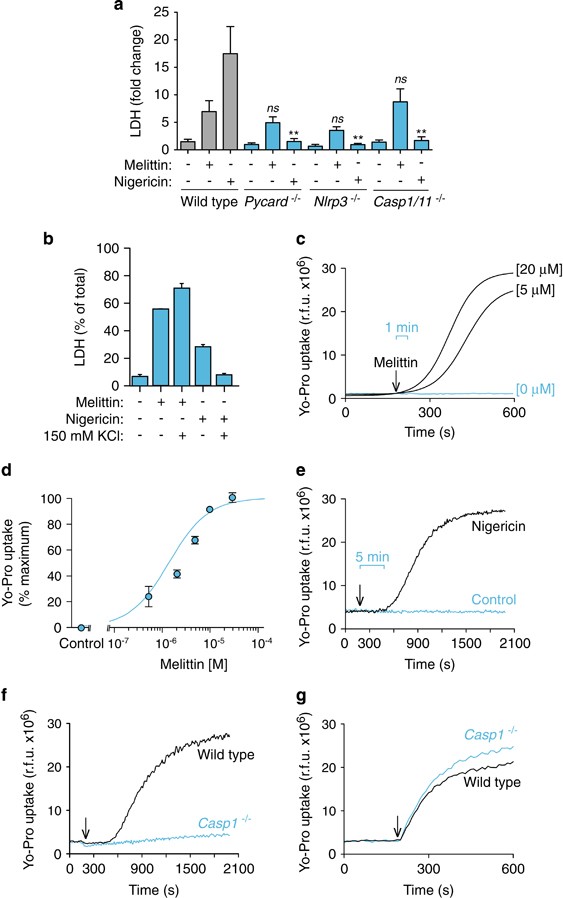
Melittin-induced cell death is independent of the inflammasome. (a) Cell death measured as fold increase of extracellular LDH in wild type, _Pycard_−/−, _Nlrp3_−/− or _Casp1/11_−/− BMDMs primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with nigericin (25 _μ_M) or melittin (0.5 _μ_M). (b) Cell death measured as fold increase of extracellular LDH in THP-1 primed with LPS (1 _μ_g/ml, 4 h), and then stimulated or not for 30 min with nigericin (25 _μ_M) or melittin (5 _μ_M) in low K+ buffer (2 mM KCl) or high K+ buffer (150 mM KCl). (c) Kinetics of Yo-Pro uptake in BMDMs primed as in (a) and stimulated with the melittin concentrations shown; melittin was added as shown by the arrow. (d) Dose–response curve for Yo-Pro uptake in BMDMs treated for 20 min as in (c) with the melittin concentrations shown. (e) Kinetics of Yo-Pro uptake in BMDMs primed as in (a) and stimulated with nigericin (5 _μ_M) as shown by the arrow. (f and g) Kinetics of Yo-Pro uptake in wild type or _Casp1/11_−/− BMDMs primed as in (a) and stimulated with nigericin (5 _μ_M) (f) or melittin (20 _μ_M) (g) as shown by the arrow
Discussion
We have described that the cytotoxic antimicrobial peptide known as melittin produces plasma membrane disruption with a concomitant decrease in intracellular K+ concentration, driving the canonical NLRP3 inflammasome activation. However, melittin-induced plasma membrane permeabilization also leads to rapid cytotoxicity that impairs the formation of large ASC aggregates and the execution of pyroptosis, denoting that necrotic cell death induced by this antimicrobial peptide toxin, similar to other plasma membrane disrupting agents, could preclude pyroptosis.
Antimicrobial peptides are produced by different cells from distinct organisms to aid in the clearance of pathogen infections.17 In this study, we examined the function of four cationic natural amphipathic antimicrobial peptides in the activation of the NLRP3 inflammasome: (a) LL-37, the only human cathelicidin peptide produced by a variety of cells, including monocytes; (b) temporin A, an antimicrobial peptide produced by the skin of the European frog Rana temporaria; (c) Indolicidin, a peptide present in the cytoplasmic granules of bovine neutrophils; and (d) melittin, a peptide toxin produced by bees and with high microbicidal activity.17, 23, 31 The lytic mechanism of these peptides is driven by their small, cationic and amphipathic structures that insert and destabilize membrane structures by different mechanisms.17 Although this mechanism of action is shared by these four antimicrobial peptides, we identified melittin as the most active, inducing IL-1_β_ release from LPS-primed macrophages, confirming previous work.5 LL-37 is also known to induce IL-1_β_ release via modulation of the P2X7 receptor in human monocytes;19 however, while we also confirm that LL-37 was able to induce IL-1_β_ release from human THP-1, it has no activity in mouse macrophages. This may be due to different activation pathways to induce the NLRP3 inflammasome in both cell types; in fact, strong differences were reported in NLRP3 activation after cell swelling when THP-1 and BMDMs were compared.3 Furthermore, THP-1 is a derived monocytic cell line, and although treatment with phorbol esters differentiates THP-1 into a macrophage-like type, it is known that monocytes regulate the NLRP3 inflammasome in a different manner.32 While in macrophages, NLRP3 requires two-step signaling for its activation, monocytes activate NLRP3 through an alternative pathway that involves the TRIF–RIPK1–FADD–CASP8 pathway upon TLR4 engagement.33 Therefore, the differences in activating NLRP3 among monocytes and macrophages could help explain why LL-37 is only able to induce IL-1_β_ release in the THP-1 monocytic line. Furthermore, THP-1 is a tumor cell line and, as such, more susceptible to the action of membrane-active antimicrobial peptides than primary cells such as BMDMs.
Melittin is the main component of bee venom and is responsible for inducing an allergic reaction characterized by local inflammation.21 NLRP3 inflammasome is known to play a role in this inflammatory response in vivo,5 and in our study we found that melittin activates the canonical NLRP3 inflammasome pathway via intracellular K+ depletion, a common key step for activation of this inflammasome.27, 28 NLRP3 activates caspase-1 by recruitment of the adaptor protein ASC via homotypic domain interactions,6, 7 and although melittin-induced caspase-1 activation and IL-1_β_ secretion required the adaptor ASC protein,5 we also found that melittin fails to induce ASC specks, and all cell death driven by melittin was independent of caspase-1. ASC aggregation into large speck-like structures is the result of a prion-like oligomerization process that amplifies inflammasome signaling and executes a specific type of cell death called pyroptosis, driven by gasdermin D cleavage by caspase-1.9, 10, 11 The lack of caspase-1-induced pyroptosis after NLRP3 activation by melittin could likely be associated to its rapid effect on plasma membrane destabilization that occurs in parallel to NLRP3 activation. Melittin is a peptide able to induce cell death by the formation of pores in the plasma membrane eukaryotic cells, leading to osmotic cell lysis.22, 23, 34, 35, 36 We found plasma membrane destabilization by melittin to occur more rapidly than caspase-1 plasma membrane disruption by the production of the lytic amino-terminal fragment of gasdermin D. However, melittin-induced plasma membrane pore formation is important for inducing caspase-1 activation and IL-1_β_ secretion by K+ efflux. Similarly, mixed-lineage kinase domain-like protein induces necroptosis and activates the NLRP3 inflammasome, and in this process, IL-1_β_ release is independent of gasdermin D plasma membrane permeabilization.37 Melittin-induced cell lysis is independent of pyroptosis and excludes pyroptosis because when the plasma membrane is already permeabilized by melittin caspase-1 activates and processes gasdermin D and, therefore, pyroptosis execution by caspase-1 is bypassed. However, melittin is still the initial trigger for NLRP3 activation and IL-1_β_ release. It also has been reported that LL-37 inhibits pyroptosis after NLRP3 activation through caspase-1 blocking,38 denoting different mechanisms of action for antimicrobial peptides, preventing pyroptosis. Hydroxyapatite crystals also induce the activation of both NLRP3 inflammasome and IL-1_β_ release independently of cell lysis, and caspase-1 is not required for hydroxyapatite induced cytotoxicity.39
Following caspase-1 activation, together with IL-1_β_ there is also a processing and release of the pro-inflammatory cytokine IL-18.1, 2 These two inflammasome-derived cytokines, IL-1_β_ and IL-18, are differentially induced in vivo and may even drive different disease symptoms.40 IL-18 has been specifically implicated in the development of age-related macular degeneration, ischemic acute renal failure and NLRC4-associated hyperinflammation.41, 42, 43 Furthermore, in mouse models of autoinflammatory cryopyrin-associated periodic syndromes, IL-1_β_ and IL-18 drive pathological conditions at different stages of the disease process.40 However, in vitro both cytokines are usually detected in macrophage supernatants after NLRP3 inflammasome activation, that is, after ATP, nigericin treatment or hypotonicity stimulation, but levels of IL-18 are around 10 times lower than IL-1_β_,3, 42, 44 consistent with our findings. Melittin failed to induce IL-18 release, an effect not due to IL-18 degradation by caspase-3, because caspase-3 is a negative regulator mechanism for IL-18, inducing its degradation.30 The lack of IL-18 secretion by melittin-treated macrophages could be attributed to low caspase-1 activation induced by melittin treatment, which may be enough to induce IL-1_β_, but not IL-18 release. We surmise that lack of ASC-speck formation, important to amplify caspase-1 activation, may explain this observation.9 Overall, melittin induced a differential release of IL-1_β_ over IL-18, and this could be important for the resulting response to bee venom. In fact, inflammatory response associated with bee venom has been found to be dependent on IL-1 receptor signaling.5
In summary, our work found that the antimicrobial peptide melittin induces the canonical NLRP3 inflammasome activation by permeabilizing the plasma membrane and decreasing intracellular K+, but melittin-induced lytic cell death was incompatible with the formation of ASC aggregates, amplification of caspase-1 activation, IL-18 release and execution of pyroptosis. Melittin reveals a pathway for NLRP3 function, where cell death is not driven by caspase-1 and inflammasome signaling is attenuated.
Materials and methods
Reagents
The following reagents were used in this study: Escherichia coli LPS O55:B5, nigericin and bee venom (Sigma-Aldrich, Madrid, Spain); caspase-8 inhibitor II X-IETD-FMK (Merck-Millipore, Darmstadt, Germany); fluorogenic substrates for caspase-1 YVAD-AFC, caspase-3 DEVD-AFC and caspase-8 IETD-AFC (PromoKine, Heidelberg, Germany); crosslinking reagent SDA (Thermo Scientific, Waltham, MA, USA); rabbit polyclonal antibody against IL-1_β_, caspase-1 p10 (M-20) and anti-ASC (N-15)-R (Santa Cruz Biotechnology, Dallas, TX, USA); ECL horseradish peroxidase-conjugated secondary antibody for immunoblot analysis (GE Healthcare, Uppsala, Sweden); and Alexa Fluor 647-conjugated donkey anti rabbit IgG secondary antibody and ProLong Diamond Antifade Mountant with 4′,6-diamidino-2-phenylindole (DAPI) (Life Technologies, Paisley, UK).
Cell culture
Bone marrow was obtained from wild-type C57BL/6, _Nlrp3_−/−, _Pycard_−/− or _Casp1_−/−_Casp11_−/− mice4, 45, 46 and differentiated to bone marrow derived-macrophages (BMDMs) using standard protocols.47 All animals were kept under controlled pathogen-free conditions (20±2 °C and 12-h light–dark cycle), with free access to sterile food and water. THP-1 cells were stored in Roswell Park Memorial Institute (RPMI) 1640 media and human embryonic kidney 293 (HEK293) cells in Dulbecco’s modified Eagle’s medium media, both supplemented with 10% fetal calf serum). Cell lines were routinely checked to ensure they were mycoplasma-free.
Peptides
The antimicrobial peptides tested (Table 1) were produced by solid-phase synthesis in ABI433 (Applied Biosystems, Foster City, CA, USA) or Prelude (Protein Technologies, Manchester, UK) synthesizers running optimized Fmoc protocols. After assembly, the peptide resins were treated with TFA/H2O/triisopropylsilane (95:2.5:2.5, by vol.) for 90 min for side-chain deprotection and cleavage from the resin. The crude peptides were isolated by precipitation with cold ether and centrifugation (3000 × g, 20 min, 4 °C), dissolved in 0.1 M acetic acid, lyophilized and purified by reverse-phase HPLC to >95% homogeneity. The identity of these peptides was confirmed by matrix-assisted laser desorption/ionization coupled time-of-flight mass spectrometry (MALDI–TOF MS) using _α_-hydroxycinnamic acid as the matrix.18
Enzyme-linked immunosorbent assay
IL-1_β_ and IL-18 release were measured using the respective kits for human or mouse (R&D Systems, Minneapolis, MN, USA) following the manufacturer’s instructions.
Lactate dehydrogenase-release measurements
Pyroptotic cell death was determined by measuring the amount of extracellular lactate dehydrogenase (LDH) using the Cytotoxicity Detection Kit (Roche, Mannheim, Germany) following the manufacturer’s instructions. Extracellular LDH activity was expressed as a percentage of the total amount of intracellular LDH under nonstimulated conditions.
Caspase activity
Supernatants from stimulated cells were monitored every 30 min for 6 h for the cleavage of fluorescent substrates for caspase-1 YVAD-AFC, caspase-3 DEVD-AFC or caspase-8 IETD-AFC, at 400 nm excitation and 505 nm emission using a Synergy Mx plate reader (BioTek, Winooski, VT, USA).
Yo-Pro uptake
Cells were incubated with 2.5 _μ_M Yo-Pro-1 (Life Technologies) for 5 min, and fluorescence was recorded every 10 s for 35 min at 37 ºC, before and after the addition of melittin or nigericin at different concentrations as annotated in the figure legends. Fluorescence of DNA-bound Yo-Pro-1 was measured in a Synergy Mx plate reader (BioTek) at 485±9 nm excitation and 515±9 nm emission.
Intracellular K+ determination
THP-1 cells were lysed in ultrapure water by five freezing–thaw cycles. The lysates were centrifuged, and K+ was quantified by indirect potentiometry using a Cobas 6000 with ISE module (Roche).
ASC crosslinking and western blot
ASC crosslinking was performed following the standard procedure.48 Briefly, cells were lysed with (3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate) (CHAPS) buffer (20 mM (4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid potassium salt (HEPES-KOH) pH 7.5, 5 mM MgCl2, 0.5 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′_′,N′-tetraacetic acid, 0.1% CHAPS and incubated 45 min with 2 mM succinimidyl 4,4′-azipentanoate (SDA), then quenched with 50 mM Tris-HCl pH 8.8 for 15 min and crosslinked for 15 min under 365 nm UV light. Crosslinked samples, or cell lysates and concentrated cell-free supernatants were all resolved in 4–12% polyacrylamide gels and electrotransferred. Membranes were probed with different antibodies for ASC, IL-1_β or caspase-1.
BRET assay
HEK293 cells were transfected with a vector encoding for human NLRP3 tagged with luciferase (N-terminus) and YFP (C-terminus)49 using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. After 24 h, transfected cells were seeded on a poly-l-lysine-coated white 96-well plate the day before the assay. The BRET signal was read after 5 min of coelenterazine-h (5 _μ_M; Invitrogen) addition. Luminescence was detected at 37 °C in a Synergy Mx plate reader (Biotek) using two filters for 485±20 and 528±20 nm emission. The BRET ratio was calculated as the difference between the 528 and 485 nm emission ratio of R-Luc and YFP-NLRP3 fusion protein and the 530 and 485 nm emission ratio of the R-Luc protein alone. Results are expressed in milliBRET (mBRET) units normalized to the basal signal, as we have previously described.50
Immunofluorescence
Macrophages were seeded on poly-l-lysine coverslips 24 h before use. After stimulation, cells were fixed with 4% formaldehyde, blocked using 1% bovine serum albumin (Sigma-Aldrich) and permeabilized with 0.2% saponin (Sigma-Aldrich). Cells were stained using the rabbit polyclonal antibody anti-ASC (1:500 dilution) and the Alexa Fluor 467 conjugated secondary antibody (1:200 dilution). The coverslips were mounted on slides with ProLong Diamond Antifade Mountant with DAPI. Images were acquired with an Eclipse Ti microscope (Nikon, Tokyo, Japan) equipped with a × 20 S Plan Fluor objective (numerical aperture, 0.45), a × 40 S Plan Fluor objective (numerical aperture, 0.6) and a × 60 S Plan Apo Vc objective (numerical aperture, 1.40) and a digital Sight DS-QiMc camera (Nikon) with a Z optical spacing of 0.2 _μ_M and 387 /447, 482/536, 543/593 and 650 nm/668 nm filter sets (Semrock, Lake Forest, IL, USA). Images were processed using ImageJ software (NIH-National Institutes of Health (Bethesda, MD, USA)).
Statistical analysis
Data are shown as the mean±S.E.M. from at least three independent experiments. The data were analyzed using Prism (GraphPad, La Jolla, CA, USA) software, by one-way ANOVA with Bonferroni multiple-comparison post-test to determine the differences among more than two groups (***P<0.001; **_P_<0.01; *_P_<0.05; NS, not significant (_P_>0.05) difference).
References
- Schroder K, Tschopp J . The inflammasomes. Cell 2010; 140: 821–832.
Article CAS Google Scholar - Strowig T, Henao-Mejia J, Elinav E, Flavell R . Inflammasomes in health and disease. Nature 2012; 481: 278–286.
Article CAS Google Scholar - Compan V, Baroja-Mazo A, López-Castejón G, Gomez AI, Martínez CM, Angosto D et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 2012; 37: 487–500.
Article CAS Google Scholar - Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J . Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440: 237–241.
Article CAS Google Scholar - Palm NW, Medzhitov R . Role of the inflammasome in defense against venoms. Proc Natl Acad Sci USA 2013; 110: 1809–1814.
Article CAS Google Scholar - De Torre C, Mesa del Castillo P, Pelegrin P . The NLRP3 and Pyrin inflammasomes: implications in the pathophysiology of autoinflammatory diseases. Front Immunol 2017; 8: 43.
Google Scholar - Próchnicki T, Mangan MS, Latz E . Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Res 2016; 5: 1–15.
Article Google Scholar - Pelegrin P . Many ways to dilate the P2X7 receptor pore. Br J Pharmacol 2011; 163: 908–911.
Article CAS Google Scholar - Dick MS, Sborgi L, Rühl S, Hiller S, Broz P . ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun 2016; 7: 11929.
Article CAS Google Scholar - Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R et al. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014; 156: 1207–1222.
Article CAS Google Scholar - Lu A, Magupalli VG, Magupalli VG, Ruan J, Ruan J, Yin Q et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014; 156: 1193–1206.
Article CAS Google Scholar - López-Castejón G, Brough D . Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev 2011; 22: 189–195.
Article Google Scholar - Martín-Sánchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ 2016; 23: 1219–1231.
Article Google Scholar - Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016; 535: 153–158.
Article CAS Google Scholar - Ding J, Wang K, Liu W, She Y, Sun Q, Shi J et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016; 535: 111–116.
Article CAS Google Scholar - He W, Wan H, Hu L, Chen P, Wang X, Huang Z et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res 2015; 25: 1285–1298.
Article CAS Google Scholar - Zasloff M . Antimicrobial peptides of multicellular organisms. Nature 2002; 415: 389–395.
Article CAS Google Scholar - Boman H, Wade D, Boman I, Wåhlin B, Merrifield R . Antibacterial and antimalarial properties of peptides that are cecropin-melittin hybrids. FEBS Lett 1989; 259: 103–106.
Article CAS Google Scholar - Elssner A, Duncan M, Gavrilin M, Wewers MD . A novel P2X7 receptor activator, the human cathelicidin-derived peptide LL37, induces IL-1 beta processing and release. J Immunol (Baltimore, Md 1950) 2004; 172: 4987–4994.
Article CAS Google Scholar - Tomasinsig L, Pizzirani C, Skerlavaj B, Pellegatti P, Gulinelli S, Tossi A et al. The human Cathelicidin LL-37 modulates the activities of the P2X7 receptor in a structure-dependent manner. J Biol Chem 2008; 283: 30471–30481.
Article CAS Google Scholar - Müller UR . Insect venoms. Chem Immunol Allergy 2010; 95: 141–156.
Article Google Scholar - Van Den Bogaart G, Guzmán JV, Mika JT, Poolman B . On the mechanism of pore formation by melittin. J Biol Chem 2008; 283: 33854–33857.
Article CAS Google Scholar - Raghuraman H, Chattopadhyay A . Melittin: a membrane-active peptide with diverse functions. Biosci Rep 2007; 27: 189–223.
Article CAS Google Scholar - Subbalakshmi C, Krishnakumari V, Nagaraj R, Sitaram N . Requirements for antibacterial and hemolytic activities in the bovine neutrophil derived 13-residue peptide indolicidin. FEBS Lett 1996; 395: 48–52.
Article CAS Google Scholar - Andreu D, Ubach J, Boman A, Wåhlin B, Wade D, Merrifield R et al. Shortened cecropin A-melittin hybrids. Significant size reduction retains potent antibiotic activity. FEBS Lett 1992; 296: 190–194.
Article CAS Google Scholar - Papo N, Shai Y . Host defense peptides as new weapons in cancer treatment. Cell Mol Life Sci 2005; 62: 784–790.
Article CAS Google Scholar - Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G . K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013; 38: 1142–1153.
Article CAS Google Scholar - Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J . Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 2007; 14: 1583–1589.
Article CAS Google Scholar - Moon DO, Park SY, Choi YH, Kim ND, Lee C, Kim GY . Melittin induces Bcl-2 and caspase-3-dependent apoptosis through downregulation of Akt phosphorylation in human leukemic U937 cells. Toxicon 2008; 51: 112–120.
Article CAS Google Scholar - Akita K, Ohtsuki T, Nukada Y, Tanimoto T, Namba M, Okura T et al. Involvement of caspase-1 and caspase-3 in the production and processing of mature human interleukin 18 in monocytic THP-1 cells. J Biol Chem 1997; 272: 26595–26603.
Article CAS Google Scholar - Rinaldi AC, Mangoni ML, Rufo A, Luzi C, Barra D, Zhao H et al. Temporin L: antimicrobial, haemolytic and cytotoxic activities, and effects on membrane permeabilization in lipid vesicles. Biochem J 2002; 368: 91.
Article CAS Google Scholar - Netea MG, Nold-Petry CA, Nold MF, Joosten LAB, Opitz B, Opitz B et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009; 113: 2324–2335.
Article CAS Google Scholar - Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F et al. Human monocytes engage an alternative inflammasome pathway. Immunity 2016; 44: 833–846.
Article CAS Google Scholar - Oren Z, Hong J, Shai Y . A repertoire of novel antibacterial diastereomeric peptides with selective cytolytic activity. J Biol Chem 1997; 272: 14643–14649.
Article CAS Google Scholar - Yokum TS, Hammer RP, McLaughlin ML, Elzer PH . Peptides with indirect in vivo activity against an intracellular pathogen: selective lysis of infected macrophages. J Pept Res 2002; 59: 9–17.
Article CAS Google Scholar - Gilbert RJC, Serra MD, Froelich CJ, Wallace MI, Anderluh G . Membrane pore formation at protein-lipid interfaces. Trends Biochem Sci 2014; 39: 510–516.
Article CAS Google Scholar - Gutierrez KD, Davis MA, Daniels BP, Olsen TM, Ralli-Jain P, Tait SWG et al. MLKL activation triggers NLRP3-mediated processing and release of IL-1β independently of Gasdermin-D. J Immunol 2017; 198: 2156–2164.
Article CAS Google Scholar - Hu Z, Murakami T, Suzuki K, Tamura H, Kuwahara-Arai K, Iba T et al. Antimicrobial cathelicidin peptide LL-37 inhibits the LPS/ATP-induced pyroptosis of macrophages by dual mechanism. PLoS ONE 2014; 9: e85765.
Article Google Scholar - Jin C, Frayssinet P, Pelker R, Cwirka D, Hu B, Vignery A et al. NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proc Natl Acad Sci USA 2011; 108: 14867–14872.
Article CAS Google Scholar - Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM . Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Invest 2013; 123: 4695–4705.
Article CAS Google Scholar - Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with Interleukin-18 inhibition. J Allergy Clin Immunol 2016; 139: 1698–1701.
Article Google Scholar - Doyle SL, Campbell M, Ozaki E, Salomon RG, Salomon RG, Mori A et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med 2012; 18: 791–798.
Article CAS Google Scholar - Melnikov VY, Ecder T, Fantuzzi G, Siegmund B, Lucia MS, Dinarello CA et al. Impaired IL-18 processing protects caspase-1–deficient mice from ischemic acute renal failure. J Clin Invest 2001; 107: 1145–1152.
Article CAS Google Scholar - Perregaux DG, McNiff P, Laliberte R, Conklyn M, Gabel CA . ATP acts as an agonist to promote stimulus-induced secretion of IL-1 beta and IL-18 in human blood. J Immunol 2000; 165: 4615–4623.
Article CAS Google Scholar - Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 1995; 267: 2000–2003.
Article CAS Google Scholar - Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004; 430: 213–218.
Article CAS Google Scholar - de Torre-Minguela C, Barberà-Cremades M, Gomez AI, Martin-Sanchez F, Pelegrin P . Macrophage activation and polarization modify P2X7 receptor secretome influencing the inflammatory process. Sci Rep 2016; 6: 22586.
Article CAS Google Scholar - Compan V, Martin-Sanchez F, Baroja-Mazo A, López-Castejón G, Gomez AI, Verkhratsky A et al. Apoptosis-associated Speck-like protein containing a CARD forms specks but does not activate caspase-1 in the absence of NLRP3 during macrophage swelling. J Immunol 2015; 194: 1261–1273.
Article CAS Google Scholar - Martín-Sánchez F, Diamond C, Zeitler M, Gomez-Sánchez A, Peñalver M, Paszek P et al. Inflammasome-dependent IL-1 β release depends upon membrane permeabilisation. Cell Death Differ 2016; 23: 1219–1231.
Article Google Scholar - Martin-Sanchez F, Compan V, Pelegrin P . Measuring NLR oligomerization III: detection of NLRP3 complex by bioluminescence resonance energy transfer. Methods Mol Biol 2016; 1417: 159–168.
Article CAS Google Scholar
Acknowledgements
We sincerely thank María Carmen Banños and Ana I Gómez (IMIB-Arrixaca, Murcia, Spain) for technical assistance with the cell cultures. We also wish to thank Dr. Isabelle Couillin (Experimental and Molecular Immunology and Neurogenetics, University of Orleans, Orleans, France) for supplying NLRP3, ASC and caspase-1-deficient mice and Prof. Richard Flavell (Yale School of Medicine, New Haven, CT, USA) and Dr. Anthony Rongvaux (Program in Immunology, Fred Hutchinson Cancer Research Center, Seattle, WA, USA) for supplying caspase-3-deficient bone marrow. LR was supported by SAF2015-65740-R and _Subdirección General de Redes y Centros de Investigación Cooperativa_-FEDER, RICET RD12/0018/0007 and RD16/0027/0010. This work was supported by grants from the European Research Council (ERC-2013-CoG 614578 to PP) and the Instituto Salud Carlos III-Fondo Europeo de Desarrollo Regional (PI13/00174 to PP). FM-S was supported by the Sara Borrell postdoctoral grant from the Instituto Salud Carlos III (CD12/00523).
Author contributions
FM-S, JJM-G and MM-G performed the experiments; MM-V and JAN-V measured ions on macrophage lysates; DA and LR produced the peptides; FM-S, JJM-G and PP analyzed the data and prepared the figures. FM-S and PP wrote the paper. PP conceived the experiments and provided funding, design and overall supervision for this study.
Author information
Author notes
- María Muñoz-García
Present address: 6Current address: Unidad de Ensayos Clínicos e Investigación Clínica, Biomedical Research Institute of Murcia IMIB-Arrixaca, University Clinical Hospital Virgen de la Arrixaca, Murcia 30120, Spain., - Fátima Martín-Sánchez and Juan José Martínez-García: These authors contributed equally to this work.
Authors and Affiliations
- Inflammation and Experimental Surgery Unit, Biomedical Research Institute of Murcia IMIB-Arrixaca, University Clinical Hospital Virgen de la Arrixaca, Murcia, 30120, Spain
Fátima Martín-Sánchez, Juan José Martínez-García, María Muñoz-García & Pablo Pelegrín - Clinical Laboratory, University Clinical Hospital Virgen de la Arrixaca, Murcia, 30120, Spain
Miriam Martínez-Villanueva & José A Noguera-Velasco - Department of Experimental and Health Sciences, Pompeu Fabra University, Barcelona Biomedical Research Park, Barcelona, 08003, Spain
David Andreu - Department of Physico-Chemical Biology, Centro de Investigaciones Biológicas (C.S.I.C.), Madrid, 28040, Spain
Luís Rivas
Authors
- Fátima Martín-Sánchez
You can also search for this author inPubMed Google Scholar - Juan José Martínez-García
You can also search for this author inPubMed Google Scholar - María Muñoz-García
You can also search for this author inPubMed Google Scholar - Miriam Martínez-Villanueva
You can also search for this author inPubMed Google Scholar - José A Noguera-Velasco
You can also search for this author inPubMed Google Scholar - David Andreu
You can also search for this author inPubMed Google Scholar - Luís Rivas
You can also search for this author inPubMed Google Scholar - Pablo Pelegrín
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toPablo Pelegrín.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by H-U Simon
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article
Cite this article
Martín-Sánchez, F., Martínez-García, J., Muñoz-García, M. et al. Lytic cell death induced by melittin bypasses pyroptosis but induces NLRP3 inflammasome activation and IL-1_β_ release.Cell Death Dis 8, e2984 (2017). https://doi.org/10.1038/cddis.2017.390
- Received: 25 April 2017
- Revised: 05 July 2017
- Accepted: 06 July 2017
- Published: 10 August 2017
- Issue Date: August 2017
- DOI: https://doi.org/10.1038/cddis.2017.390