Prostate cancer and metastasis initiating stem cells (original) (raw)
Main
Prostate cancer is the most commonly diagnosed malignancy in men in the United States and is projected to be the third most frequent cause of male cancer-related deaths in 2007 (http://www.cancer.org/docroot/stt/stt_0.asp). For organ-confined disease, initial treatment is prostatectomy or radiation, which usually is curative. However, approximately 20% of patients are not cured by such treatments and their cancer recurs, sometimes with long latencies, and some patients are diagnosed only after the cancer has spread. Progressive prostate cancer is almost always treated with androgen deprivation therapy, which causes an initial regression, due to the androgen-dependent nature of the vast majority of prostate cancer cells. Very frequently, however, androgen-independent cancers emerge and, subsequently, wide-spread metastasis occurs.
Metastases are found most commonly in the bone, lymph nodes, liver, lungs, and dura 1, 2, 3. Bone metastases occur in approximately 90% of patients with advanced disease and are a leading cause of morbidity. Replacement of hematopoietic tissues with tumor cells leads to anemia and increased susceptibility to infection. Bone remodeling and osteoblast-driven new bone overgrowth lead to pain, fractures, and spinal cord compression 4. Data collected from rapid autopsy programs have revealed a remarkable degree of phenotypic heterogeneity among tumor cells within bone metastatic sites when comparing different patients as well as at multiple sites within individual patients 2, 3. This heterogeneity includes differences in morphology as well as immunophenotypes for differentiation markers.
Understanding the mechanistic basis that underlies the development of prostate cancer metastasis is fundamental to the development of treatment and preventive therapies. The identification of the cell of origin for metastatic prostate cancer must be a major goal of research in this area, in order to properly target therapy. Obtaining such a goal is still mostly in the conceptual stages. Properties of the metastatic prostate cancer-initiating cell that may provide insight to its origin are an apparent plasticity with respect to the expression of differentiation markers and a propensity to develop androgen independence. Here we discuss current data and theories concerning a stem or progenitor cell of origin for metastatic prostate cancer. Another important property of metastatic prostate cancer cells is their selectivity for growth in the bone microenvironment, a topic which has recently been reviewed and will be only briefly addressed here 5, 6.
The property of secondary tumor initiation
Metastatic colonization requires that the initiating cell have self-renewal properties following its migration to a secondary site. It is apparent from both clinical and experimental data that only a small proportion of the cells from a primary tumor are able to initiate secondary growth 7, 8. For example, there is evidence that tumor cell dissemination occurs early in disease progression for a variety of cancers, including prostate cancer, but the vast majority of these cells do not establish metastases 9, 10. In addition, minor populations of cells in various types of tumor samples have the capacity to form replicative colonies in vitro or to form tumors upon transplantation into a secondary host 11, 12, 13. Thus, the combination of properties required for growth outside the primary tumor occurs in rare cells within a tumor population.
Mechanistic insight into these observations with respect to tumor samples originally came from studies in human leukemia. It was demonstrated by John Dick and colleagues that the long-term tumorigenic potential of human AML resides in a rare subpopulation, which is characterized by surface markers overlapping with normal hematopoietic stem cells but distinct from the bulk of the tumor 14. Because normal stem cells have substantial self-renewing capability and give rise to phenotypically distinct differentiated progeny, these findings suggested the concept of a cancer stem cell (CSC) in which the tumor-initiating properties reside. Similarly, more recent studies have demonstrated that a minor population of tumor-initiating cells resides within solid tumors, including brain, breast, and colon tumors 15, 16, 17. These tumor-initiating cells were identified by their unique and rare constellation of surface markers that are different from those expressed on the majority of cells in the tumor. The defining surface markers for the tumor-initiating population are thought to overlap with stem cell markers in the respective normal tissues. Investigations in a mouse model system of leukemia demonstrated a difference between the drug sensitivities of normal and leukemic stem cells 18. This finding suggests that targeted therapies can be developed for CSC's without damaging normal stem cells.
A major question with respect to CSC's is their origin, i.e. in which cell type do mutations occur that lead to transformation and tumor-initiating properties. Reasonable possibilities include the stem cell compartment itself, as well as more mature progenitor cells that acquire at least some stem cell characteristics following mutation. In fact, experimental leukemia models suggest that both mechanisms occur, depending upon the initiating mutation 19, 20, 21. Because markers that delineate stem cells and their intermediate progeny in epithelial organ systems are not nearly as finely defined as those for hematopoietic cell differentiation, addressing such mechanistic questions in solid tumors awaits the discovery of additional markers and the construction of tissue-specific lineage maps.
Stem cells in the normal prostate
The prostate is a glandular organ comprised of three distinct cell types (basal, luminal, and neuroendocrine cells) embedded in a fibro-muscular stroma 22. The lineage relationships of the three nonmesenchymal cell types are relevant for understanding the origins of prostate cancer. The epithelium is composed of two histologically distinct layers that perform different functions. Prostate basal cells form a layer along the basement membrane of each duct, and luminal cells form a layer above the basal cells. Neuroendocrine cells are a minor population scattered throughout the basal layer and are identified by the expression of neuroendocrine markers such as chromogranin A and synaptophysin (Figure 1). As will be discussed, direct evidence based upon prospective isolation suggests a common origin for basal and luminal cells. In addition, indirect evidence supports the notion that prostate neuroendocrine and epithelial cells share a common progenitor.
Figure 1
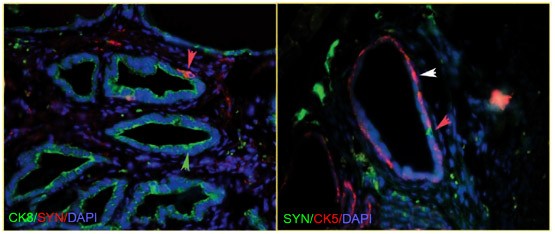
Immunofluorescent double-labeling of basal, luminal, and neuroendocrine populations in the mouse dorsal prostate gland. Left and right panels: Luminal and neuroendocrine cells or basal and neuroendocrine cells, respectively, are labeled within a background of all cells stained with DAPI, a general nuclear stain. Cytokeratin 8 (CK8): luminal, Cytokeratin 5 (CK5): basal, Synaptophysin (syn): neuroendocrine, DAPI: general nuclear stain. Colored arrowheads indicate the following: green – a luminal cell, white – a basal cell, red – a neuroendocrine cell. Images were kindly provided by Drs Zongxiang Zhou and Alexander Nikitin, Cornell University, Ithaca, New York.
Basal cells express cytokeratins 5 and 14, CD44, and integrin α6β1. Basal cells also express the anti-apoptotic protein BCL-2 (human and some mouse basal cells), and a subpopulation of basal cells may express androgen receptor (AR) 23, 24, 25. Luminal cells are dependent upon androgen for viability and produce prostatic secretory proteins such as PSA and PAP. They are characterized by expression of cytokeratins 8 and 18 and AR. Immunohistological analyses of human prostate tissue also have identified two intermediate phenotypes based upon cytokeratin expression patterns that are hypothesized to represent cells intermediate in differentiation between basal and luminal cells and possibly signify a progenitor or transit population 26, 27. These rare cells are positive for combinations of CK5/14/18 and CK5/18. Likewise, another marker, prostate stem cell antigen (PSCA), may also first appear on intermediate cells 28. Another rare intermediate cell type has been identified in the normal mouse prostate that co-expresses luminal CK8 and AR markers in addition to neuroendocrine markers, but not CK5 29.
The strongest evidence for the existence of prostate stem cells comes from the remarkable regenerative capacity of both human and mouse prostates 30. John Isaacs and colleagues investigating the effects of androgen cycling on regression and regeneration of rat prostate first demonstrated the presence of prostate epithelial stem cells 31, 32. Androgen withdrawal leads to apoptosis of the majority of luminal epithelial cells and the survival of basal cells. Androgen replacement in previously androgen-depleted animals results in regeneration of the luminal cell layer. This cyclical response can be repeated multiple times. The fact that the basal layer remains intact and that luminal cells can be regenerated has led to the suggestion that the epithelial regenerative, i.e. stem cell, activity resides within the basal layer.
Given the central role played by androgen in the developing and regenerating prostate, important questions are which cell types express AR's and what potential mechanisms mediate androgen responsiveness. Different tissue types, mesenchymal and epithelial, play a role in androgen-driven growth and differentiation of the prostate. Cunha and colleagues developed tissue recombination studies, which utilized microdissected and enzymatically digested embryonic urogenital sinus (UGS) mesenchyme and epithelium, transplanted under the kidney capsule of immunocompromised mice. These studies demonstrated that AR function in the mesenchymal component, but not the epithelial component, of the developing prostate is sufficient for growth of the prostate gland 33, 34. Androgen stimulation of stromal cells is thought to stimulate growth factor and cytokine expression, which subsequently act upon epithelial progenitor cells. With respect to differentiating function, it was shown that androgen acting through AR expressed in the luminal cells mediates their secretory function 35, 36. Similarly, AR-positive mesenchymal cells are required to obtain regeneration of adult epithelium, suggesting mechanistic similarities between prostate development and regeneration 37. Therefore, it appears that androgen indirectly affects the growth and early differentiation, through the nonsecretory stage, of prostate epithelium. Although AR expression in basal and intermediate cells may not be required for regenerative activity, AR expression in these cell populations has been described 23, 25. An adaptation of the tissue recombination procedure in which UGS mesenchyme is mixed with manipulated mouse epithelial populations has been used to assay growth and differentiative potential of candidate progenitor populations 38.
Progress has been made in establishing markers that distinguish mouse prostate cells with progenitor activity. The majority of the regenerative capacity in the mouse prostate has been localized anatomically to the proximal region adjacent to the urethra 24, 39, 40. The regenerative activity of the proximal region is maintained during conditions of androgen depletion. The proximal region contains a high proportion of castration-resistant CK5, BCL-2, SCA1, α6 integrin-positive cells 39, 41, 42. SCA1 and α6 integrin expression are characteristic of stem cell populations from some other tissues 43, 44, 45, 46. BCL-2 expression may help cells resist apoptotic stimuli such as high TGFβ production resulting from androgen depletion. As few as 400 isolated proximal prostate epithelial cells recombined with rat mesenchyme were able to regenerate prostate tubular structures with a basal layer and a secretory AR+ luminal layer 39.
Mouse prostate epithelial cells have been prospectively isolated using SCA1 and α6 integrin as markers 41, 42. These cells express CK5 but not AR, and they clonally regenerate tubules containing both basal and luminal cells, some of which are weakly positive for AR expression. NE cells are not observed. These studies directly demonstrate that basal and luminal cells share a common precursor cell. It should be noted that the SCA1+ α6+ progenitors appear to have less regenerative capacity than unseparated proximal epithelial cells 39. An explanation for such a loss of regenerative potential is that the separation procedure damages or prematurely differentiates stem cells.
Using a separation and reconstitution approach with human prostate tissue similar to that described for mouse cells, it has been found that epithelial cells expressing CD44, CD133, and high levels of α2β1 integrin give rise to prostatic-like acini containing cells expressing differentiation markers such as AR, PAP, and luminal keratins 47. Similarly to the mouse progenitors, the majority of prospectively isolated human progenitor cells expressed basal keratins and no AR. The relative heterogeneity of prospectively isolated mouse and human progenitor populations to date suggests that care be taken in assigning correlative non-selected expression characteristics (e.g. AR or p63) to these populations. As yet, assays that allow the quantification of prostate stem cell/regenerative activity have not been developed, and the proportion of stem cells within the prospectively isolated populations is unknown. The identification of a relatively unique stem cell-specific marker in the prostate, as has been done for Lgr5 that marks small intestine and colonic epithelial stem cells, would greatly facilitate the molecular and biological characterization of a prostate stem cell 48.
The overlap between anatomically defined CK5+ basal cells and stem progenitor cells in the prostate has been addressed by investigations that make use of mice that have lost p63 gene function 49, 50. However, the interpretation of these experiments is controversial. p63 is a marker for mouse basal cells, and the prostate is not formed in embryonic p63−/− mice 51, 52. Rescue of p63−/− blastocysts with beta-galactosidase marked p63+ cells demonstrated that basal and luminal cells have a common ancestor that develops from the p63+ cells 50. That is, there was no evidence for luminal cell clones that developed independently of basal cells. In apparent contradiction to this conclusion, transplantation of p63−/− UGS under the kidney capsule of immunocompromised rats gave rise to abnormal mucin-producing cells and a proportion of cells expressing either luminal or NE markers 49. These results suggest that luminal cell progenitors do not require p63 for their development, and therefore luminal cells do not develop in a linear fashion from p63+ basal cells. Taken together, it appears that the efficient physiological development of normal luminal epithelium is clonally dependent upon p63 expression. However, it is possible that p63+ daughter cells influence the differentiation of p63− progenitors. Such a scenario does not eliminate the possibility that a p63− progenitor for both luminal and basal cells may well exist.
Prostate cancer stem cells
The simplest definition of a CSC is that of a cell able to regenerate a tumor, i.e. a tumor-initiating cell. As discussed earlier, the expectation is that CSC's will express overlapping markers with normal stem cells from the same tissue and that CSC's will display high clonogenic activity in vitro. However, the most definitive property with respect to defining CSC's is that of tumor initiation, and this is also the property that directly links CSC's to metastatic colonizing cells. Therefore, although data have been generated using in vitro clonogenicity to characterize human prostate CSC's, we will concentrate the following discussion upon tumor-initiating cells (for a discussion of in vitro properties, see 26, 53, 54, 55). Human prostate adenocarcinomas have a mature luminal phenotype characterized by CK8/18, AR, PAP, and PSA expression. Like normal luminal cells, prostate cancer cells initially are androgen-dependent for survival. With tumor progression and metastasis, tumors become more heterogeneous with admixtures of luminal cells and one or multiple other cell types that can include scattered basal cells, neuroendocrine cells, and/or intermediate cells 2, 3, 56. In addition, following androgen ablation therapy, androgen-independent tumors emerge 30.
The direct demonstration of purified tumor-initiating cells from primary human prostate tumors has not been reported. Limited access to biopsy material, potential contamination with normal stem cells, and the expected rarity of putative CSC's have led to an alternative approach in which primary prostate tumor tissue is cultured in epithelial-selective media, immortalized with human telomerase-expressing lentiviruses, and subsequently clonally derived 57, 58. Continuously proliferating lines of the phenotype CD44+, CK8/18±, p63−, AR− gave rise upon recombination with rat UGS mesenchyme to tumors that were mostly AR+ and displayed regions representing luminal (CK8/18+), basal (CK14+), and NE (synaptophysin+) cells 57. This directly demonstrates the existence of a cloned cell with pluripotent differentiative capability. It will be important to analyze further the tumorigenic and metastatic properties of such cells in xenograft models. Prior studies demonstrated that a CD44+, α2β1hi, CD133+, AR− basal-like cell is rare in primary tumors, and such a cell may be overlapping with the immortalized tumor-initiating cell described above 59.
Concerns exist for the immortalization model described above. One issue is the contribution of in vitro selection to the transformed phenotype. The existence of abnormal karyotypes in telomerase-immortalized lines from non-cancerous derived epithelium underscores this problem 58. Another concern is the potential genetic heterogeneity of human prostate cancer and the need to determine the underlying carcinogenic events in individual cell lines in order to generalize and place results in context. Thus, the advent of relatively affordable whole genome sequencing should contribute to the utility of human cell lines. Even though the precise markers expressed by the immortalized progenitor cell may be modulated by in vitro culture and/or hTERT introduction, this system may be an entry for establishing cell lineage relationships in addition to allowing mechanistic questions about prostate cancer development.
Tumor-initiating cells have not been prospectively isolated from mouse models of prostate cancer. However, the ability to analyze the development of disease over time has led to some insights into the potential cell of origin for specific prostate cancer models. PTEN deletion is common in human prostate cancer, and the deletion of PTEN in the adolescent mouse prostate leads over a period of months to the development of localized and then invasive luminal type adenocarcinoma, which is accompanied by occasional microscopic metastases to the lymph nodes, lung and liver 60. Disease progression over time is characterized by the expansion of cell populations, which were analyzed in the proximal region of the prostate 25. These expanded populations included CK5+/p63+ basal cells as well as an intermediate population of CK5/8+ cells. The latter also contains cells expressing BCL-2, SCA1, and AR, consistent with a potential luminal progenitor phenotype. Because PTEN deletion appears to occur in both basal and luminal cells as well as various progenitor populations, it is not clear whether the CK5 and CK5/8 cell populations expand independently or whether CK5/8 expansion follows from CK5 expansion.
Another model that demonstrates the expansion of a potential progenitor population develops in mice following prostate-specific deletion of Rb and p53 29, 61. Interestingly, early invasive adenocarcinomas are observed in the proximal region of the prostate and are characterized by the co-expression of CK8, the neuroendocrine marker synaptophysin, AR, and SCA1. This model leads to the development of large metastases in the majority of animals. Metastases are found in the lung, liver, lymph nodes, and adrenal gland, but not the bone. Because rare cells co-expressing CK8, AR, and NE markers are found in normal mouse prostate, this model may represent the dysregulated expansion of a pre-existing intermediate progenitor population of cells. Based upon the PTEN and the Rb/p53 deletion models discussed above, a speculative lineage model is shown in Figure 2. The correctness of the model is partly dependent upon the assumption that mutations leading to cancer expand an existing intermediate population and do not endow new differentiative properties to the transformed cells.
Figure 2
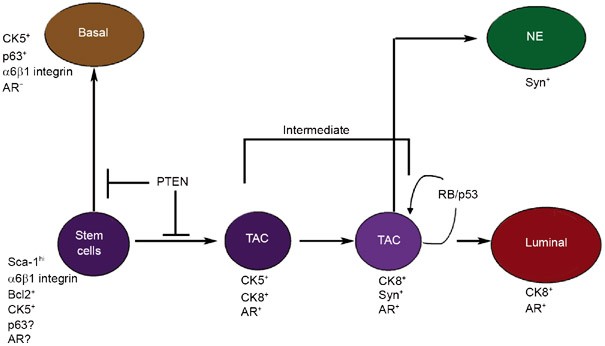
A proposed lineage map for epithelial/neuroendocrine prostate cell differentiation based upon the amplification of intermediate cell populations in human and mouse models of prostate cancer. Regulatory mutations (loss of PTEN or Rb/p53) that result in increased intermediate populations are indicated. TAC: transit amplifying cells, NE: neuroendocrine, AR: androgen receptor, Syn: synaptophysin.
Castrate-resistant prostate cancer
The transient response of human prostate cancers to androgen ablation therapy has led widely to the speculation that prostate tumors arise from a small population of androgen-independent cells, often presumed to be androgen-independent prostate stem cells with predominantly basal cell characteristics. In support of the concept that androgen-independent cells exist as an inherent component of prostate adenocarcinoma, mouse models have suggested that a pre-existing population of androgen-independent prostate cancer cells exists and emerges uncoupled from disease progression 62, 63. However, it is important to consider the principles that have been established with respect to the emergence of androgen-refractory human prostate cancer 30. AR signaling is maintained, and through a variety of potential mechanisms, some of which directly involve mutation of the AR, AR-dependent signaling evolves to androgen-independence in castrate-resistant prostate cancer. There are few AR mutations in primary cancer in comparison to castrate-resistant, metastatic prostate cancer. One study found that 50% of bone metastatic lesions from different patients demonstrated AR mutations, implying that AR mutations pre-exist in metastatic, tumor-initiating cells 64. If AR mutations are selected, such selection must occur in a self-renewing population. Therefore, the cell of origin for prostate cancer metastases appears to express AR and presumably is sensitive to AR-mediated signaling, although AR-dependence is not necessarily required. An alternative possibility is that the prostate cancer progenitor is influenced in its selection in a nonautonomous manner by progeny that express AR 65.
The most straightforward model suggests that an AR-expressing intermediate cell acquires mutations that endow it with the unlimited self-renewal property of stem cells 66, 67. The progenitor nature of intermediate cells is consistent with the observed plasticity of morphology and immunophenotype in prostate cancer metastases. One finding that supports the notion that the cell of origin for human prostate cancer is AR sensitive is the existence of an androgen-regulated gene fusion in a high proportion of prostate cancers 68. Fusion of the androgen-regulated TMPRSS2 promoter with the coding region of various ETS family transcription factors (TF) is thought to result in over-expression of the translocated ETS TF. Such translocations occur in about 50% of prostate cancers and have been found in 20% of PIN lesions, early localized neoplastic prostate cells. Therefore, androgen-regulated ETS TF expression appears to be an early genetic event. As a fuller understanding of the developmental lineage of prostate epithelial cells emerges, the important questions of which cells express androgen responsiveness and how androgen independence is selected will hopefully become clearer.
Growth factor signaling systems implicated in prostate cancer stem cell function and metastasis
A stem or progenitor cell origin for prostate cancer metastases predicts that drugs which target pathways relevant for stem cell proliferation or maintenance may inhibit the expansion of metastases. Two signaling pathways that have been implicated in prostate stem cell regulation are the Hedgehog (Hh) and TGFβ pathways. The predicted role or mechanism of action in prostate cancer metastasis is significantly different for the two pathways. Hh is predicted to be a positive regulator of prostate stem cell growth. On the other hand, TGFβ is thought to maintain prostate stem cells in a quiescent state, subject to regulatory feedback upon the need to expand the epithelial population. In prostate cancer progression, loss of sensitivity to inhibitory autocrine TGFβ is hypothesized to lead to paracrine activity of TGFβ resulting in positive conditioning of the metastatic microenvironment.
The Hh signaling pathway has been implicated in prostate development, adult prostate homeostasis, prostate cancer growth, and prostate cancer metastasis formation 69. Hh signaling has been connected to stem cell proliferation in tissues that include the central nervous system, skin, mammary gland, gut, and pancreas 70. Therefore, the reasonable possibility exists that Hh signaling plays a role in the regulation of growth and/or differentiation of prostate CSC's.
The Hh family of secreted proteins includes Sonic (Shh), Indian (Ihh), and Desert (Dhh). Shh is the most widely expressed during development, but it appears that Ihh provides some functional redundancy for Shh during mouse prostate development 71. The Hh proteins exert their activity by binding to a 12-pass transmembrane protein Patched (PTCH), which constitutively represses Hh pathway activity through its interaction with a second 7-transmembrane protein Smoothened (SMO). Binding of Hh ligand to PTCH derepresses SMO, which activates the Gli family of TF, leading to specific target gene induction or repression. There are a number of cytoplasmic regulatory proteins and feedback loops that modulate Hh signaling 72. Because Hh signaling results in increased Gli1 and PTCH expression, the presence of these gene products is used as an assay of Hh pathway activation. In addition, the drug cyclopamine binds to SMO and blocks ligand-dependent SMO activation 73. Cyclopamine has been widely used to probe Hh pathway function.
Shh is a secreted factor produced by prostate epithelial cells. Histological studies that localize Shh production and pathway activation suggest that Shh mediates its functions through both autocrine stimulation of prostate epithelial cells and paracrine stimulation of stromal elements 74, 75. Developmental studies in the mouse have shown that Hh/Gli signaling is required for normal ductal bud proliferation and for glandular morphogenesis 76, 77, 78, 79. In addition, the human adult prostate demonstrates high levels of expression of Shh, PTCH, Gli1, and SMO, but the exact role of Hh signaling in normal adult prostate homeostasis has not been established 80. A role for Hh signaling in prostate stem/progenitor cell maintenance or proliferation is suggested by the observation that testosterone-induced prostate regeneration in rodents is blocked by cyclopamine 81.
Various lines of evidence suggest that the Hh signaling pathway functions in prostate cancer tumorigenesis and metastasis. Shh expression and signaling are often increased in localized prostate cancer as compared to normal or nontumorigenic hyperproliferative tissue, although there is a wide range of expression in all conditions 74, 75, 81. Based upon histological analyses using PTCH and Gli expression as markers of pathway signaling, data exist for both Shh-mediated prostate adenocarcinoma autocrine and paracrine signaling. RT-PCR analyses of metastatic prostate tumors demonstrated increased PTCH expression, taken as an indicator of Hh pathway activation, which was correlated with increased SMO expression, suggested to be a determinant of pathway activation 81. Due to the heterogeneous cell types in tumor samples, it is possible that either autocrine and/or paracrine signaling is upregulated in metastasis. Given the predilection of prostate cancer to metastasize to bone, it is of interest that bone marrow stromal cells are responsive to Hh ligands and that Shh and Ihh stimulate bone remodeling 82, 83.
It is difficult to interpret experiments addressing the functional consequences of autocrine ligand-dependent Hh signaling in prostate cancer tumorigenesis or metastasis due to the fact that appropriately regulated syngeneic or xenograft models do not exist. That is, the available human prostate xenograft models are at best marginally responsive to either the addition or inhibition of Hh ligands 75, 80. On the other hand, Hh-dependent paracrine stimulation has been shown to increase xenograft growth of LnCaP tumors 74. Although cyclopamine is growth inhibitory for prostate cell lines, the lack of ligand-dependent signaling in these cell lines suggests at least one mechanism of action for the drug that does not directly follow from SMO inhibition 74, 75, 80, 81. Although Hh ligand-dependent signaling is nonfunctional in prostate cancer cell lines, the manipulation of Gli1 expression levels suggests a positive correlation between tumor cell growth and levels of Gli expression 74, 75, 81. Constitutive Gli1 expression may represent Hh-pathway-dependent or independent genetic or epigenetic effects upon Gli1 regulation. The potential role of Hh signaling in prostate stem cell growth regulation combined with the existing suggestion that Hh signaling is modified in prostate tumorigenesis and metastasis promises that Hh signaling will continue to be of great interest in questions surrounding the cell of origin for prostate cancer metastasis. The development of genetically defined mouse models with metastatic properties will likely be one approach to unraveling the role of Hh pathway components in prostate cancer transformation and progression.
Another secreted factor that has been shown by the work of Lynette Wilson and colleagues to be important in regulating androgen-mediated prostate involution and regeneration is TGFβ 24, 40. A concept that has developed for stem cell homeostasis in a variety of systems, including epidermis and prostate, is that stem cell proliferation is regulated by a balance between the inhibitory effects of TGFβ and the stimulatory effects of growth factors and cytokines 84, 85. Mouse prostate stem cells are protected from a high local concentration of TGFβ by high endogenous BCL2 expression. An inverse relationship exists between TGFβ signaling and TGFβ receptor expression, which has led to the suggestion that cell autonomous regulatory loops exist in prostate epithelial stem cells to modulate TGFβ signaling pathways.
TGFβ signals are mediated by heterodimeric type I and type II serine/threonine kinase receptors. There is strong selective pressure for loss of the TGFβ growth inhibitory response when prostate cancer progresses. Clinical studies have shown that TGFβ overexpression and loss of expression of TGFβ-RI or TGFβ RII are associated with greater pathological Gleason scores, higher vascular counts, and progression to metastasis. In the absence of inhibitory autocrine TGFβ signaling, high levels of secreted TGFβ may promote prostate epithelial tumorigenesis and metastasis through paracrine stimulation of cells in the microenvironment 86, 87. TGFβ signaling is central to the pathogenesis of osteolytic breast cancer metastasis 88, 89. Although the role of TGFβ in prostate cancer bone metastasis is unclear, some evidence exists for the promotion of an osteolytic bone response by TGFβ in a xenograft prostate cancer model system 90. It will be of interest to establish whether TGFβ blockade may be a useful therapeutic tool in prostate cancer bone metastasis.
Summary
Metastasis is the main reason for prostate-cancer-related deaths, and current therapies for metastatic prostate cancer have short-term benefits at best. Developing appropriate therapies requires a mechanistic understanding of how growth is regulated in the metastasis-initiating cell. The discovery that a small subset of tumor cells are responsible for initiating and sustaining the growth of other epithelial tumors, such as mammary and colon cancers, suggests the same may be true for prostate cancer. Metastasis-initiating cells are rare within the tumor population, and, at a minimum, such cells must have tumor-initiating properties. The challenge is to decipher the lineage relationships of normal prostate stem and progenitor cells and the potential cell lineages that upon transformation give rise to metastatic prostate cancer. Although the existence of a prostate CSC in either mouse or human models has not been definitively demonstrated, indirect evidence supports this concept. Kinetic analyses of mouse models of prostate cancer have shown the early abnormal expansion of cells with progenitor phenotypes. Similarly, hTERT immortalized human cell clones derived from prostate cancer specimens are able to form xenograft tumors containing multiple prostate epithelial cell types. The discovery of specific markers for prostate stem and progenitor populations is needed. The ability to identify and manipulate specific populations will allow a more refined analysis of clinical samples and the development of mouse models that directly address the consequences of stem/progenitor cell transformation.
References
- Bubendorf L, Schopfer A, Wagner U, et al. Metastatic patterns of prostate cancer: an autopsy study of 1 589 patients. Hum Pathol 2000; 31:578–583.
Article CAS PubMed Google Scholar - Roudier MP, True LD, Higano CS, et al. Phenotypic heterogeneity of end-stage prostate carcinoma metastatic to bone. Hum Pathol 2003; 34:646–653.
Article PubMed Google Scholar - Shah RB, Mehra R, Chinnaiyan AM, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res 2004; 64:9209–9216.
Article CAS PubMed Google Scholar - Logothetis CJ, Lin SH . Osteoblasts in prostate cancer metastasis to bone. Nat Rev Cancer 2005; 5:21–28.
Article CAS PubMed Google Scholar - Morrissey C, Vessella RL . The role of tumor microenvironment in prostate cancer bone metastasis. J Cell Biochem 2007; 101:873–886.
Article CAS PubMed Google Scholar - Storey JA, Torti FM . Bone metastases in prostate cancer: a targeted approach. Curr Opin Oncol 2007; 19:254–258.
Article PubMed Google Scholar - Chambers AF, Groom AC, MacDonald IC . Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002; 2:563–572.
Article CAS PubMed Google Scholar - Gupta GP, Massague J . Cancer metastasis: building a framework. Cell 2006; 127:679–695.
Article CAS PubMed Google Scholar - Aguirre-Ghiso JA . Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 2007; 7:834–846.
Article CAS PubMed PubMed Central Google Scholar - Ellis WJ, Pfitzenmaier J, Colli J, Arfman E, Lange PH, Vessella RL . Detection and isolation of prostate cancer cells from peripheral blood and bone marrow. Urology 2003; 61:277–281.
Article PubMed Google Scholar - Al-Hajj M, Clarke MF . Self-renewal and solid tumor stem cells. Oncogene 2004; 23:7274–7282.
Article CAS PubMed Google Scholar - Huntly BJ, Gilliland DG . Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer 2005; 5:311–321.
Article CAS PubMed Google Scholar - Reya T, Morrison SJ, Clarke MF, Weissman IL . Stem cells, cancer, and cancer stem cells. Nature 2001; 414:105–111.
Article CAS PubMed Google Scholar - Bonnet D, Dick JE . Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3:730–737.
Article CAS PubMed Google Scholar - Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF . Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100:3983–3988.
Article CAS PubMed PubMed Central Google Scholar - O'Brien CA, Pollett A, Gallinger S, Dick JE . A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007; 445:106–110.
Article CAS PubMed Google Scholar - Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature 2004; 432:396–401.
Article CAS PubMed Google Scholar - Yilmaz OH, Valdez R, Theisen BK, et al. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006; 441:475–482.
Article CAS PubMed Google Scholar - Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL . Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev 2003; 17:3029–3035.
Article CAS PubMed PubMed Central Google Scholar - Huntly BJ, Shigematsu H, Deguchi K, et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004; 6:587–596.
Article CAS PubMed Google Scholar - Krivtsov AV, Twomey D, Feng Z, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006; 442:818–822.
Article CAS PubMed Google Scholar - Abate-Shen C, Shen MM . Molecular genetics of prostate cancer. Genes Dev 2000; 14:2410–2434.
Article CAS PubMed Google Scholar - Nakada SY, di Sant'Agnese PA, Moynes RA, et al. The androgen receptor status of neuroendocrine cells in human benign and malignant prostatic tissue. Cancer Res 1993; 53:1967–1970.
CAS PubMed Google Scholar - Salm SN, Burger PE, Coetzee S, Goto K, Moscatelli D, Wilson EL . TGF-{beta} maintains dormancy of prostatic stem cells in the proximal region of ducts. J Cell Biol 2005; 170:81–90.
Article CAS PubMed PubMed Central Google Scholar - Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON, Wu H . Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci USA 2006; 103:1480–1485.
Article CAS PubMed PubMed Central Google Scholar - Long RM, Morrissey C, Fitzpatrick JM, Watson RW . Prostate epithelial cell differentiation and its relevance to the understanding of prostate cancer therapies. Clin Sci (Lond) 2005; 108:1–11.
Article CAS Google Scholar - Verhagen AP, Ramaekers FC, Aalders TW, Schaafsma HE, Debruyne FM, Schalken JA . Colocalization of basal and luminal cell-type cytokeratins in human prostate cancer. Cancer Res 1992; 52:6182–6187.
CAS PubMed Google Scholar - Tran CP, Lin C, Yamashiro J, Reiter RE . Prostate stem cell antigen is a marker of late intermediate prostate epithelial cells. Mol Cancer Res 2002; 1:113–121.
CAS PubMed Google Scholar - Zhou Z, Flesken-Nikitin A, Corney DC, et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res 2006; 66:7889–7898.
Article CAS PubMed Google Scholar - Feldman BJ, Feldman D . The development of androgen-independent prostate cancer. Nat Rev Cancer 2001; 1:34–45.
Article CAS PubMed Google Scholar - English HF, Santen RJ, Isaacs JT . Response of glandular versus basal rat ventral prostatic epithelial cells to androgen withdrawal and replacement. Prostate 1987; 11:229–242.
Article CAS PubMed Google Scholar - Kyprianou N, Isaacs JT . Activation of programmed cell death in the rat ventral prostate after castration. Endocrinology 1988; 122:552–562.
Article CAS PubMed Google Scholar - Cunha GR, Lung B . The possible influence of temporal factors in androgenic responsiveness of urogenital tissue recombinants from wild-type and androgen-insensitive (Tfm) mice. J Exp Zool 1978; 205:181–193.
Article CAS PubMed Google Scholar - Kurita T, Wang YZ, Donjacour AA, et al. Paracrine regulation of apoptosis by steroid hormones in the male and female reproductive system. Cell Death Differ 2001; 8:192–200.
Article CAS PubMed Google Scholar - Cunha GR, Donjacour AA, Cooke PS, et al. The endocrinology and developmental biology of the prostate. Endocr Rev 1987; 8:338–362.
Article CAS PubMed Google Scholar - Donjacour AA, Rosales A, Higgins SJ, Cunha GR . Characterization of antibodies to androgen-dependent secretory proteins of the mouse dorsolateral prostate. Endocrinology 1990; 126:1343–1354.
Article CAS PubMed Google Scholar - Gao J, Arnold JT, Isaacs JT . Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells. Cancer Res 2001; 61:5038–5044.
CAS PubMed Google Scholar - Xin L, Ide H, Kim Y, Dubey P, Witte ON . In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proc Natl Acad Sci USA 2003; 100(Suppl 1):11896–11903.
Article CAS PubMed PubMed Central Google Scholar - Goto K, Salm SN, Coetzee S, et al. Proximal prostatic stem cells are programmed to regenerate a proximal-distal ductal axis. Stem Cells 2006; 24:1859–1868.
Article CAS PubMed Google Scholar - Tsujimura A, Koikawa Y, Salm S, et al. Proximal location of mouse prostate epithelial stem cells: a model of prostatic homeostasis. J Cell Biol 2002; 157:1257–1265.
Article CAS PubMed PubMed Central Google Scholar - Burger PE, Xiong X, Coetzee S, et al. Sca-1 expression identifies stem cells in the proximal region of prostatic ducts with high capacity to reconstitute prostatic tissue. Proc Natl Acad Sci USA 2005; 102:7180–7185.
Article CAS PubMed PubMed Central Google Scholar - Lawson DA, Xin L, Lukacs RU, Cheng D, Witte ON . Isolation and functional characterization of murine prostate stem cells. Proc Natl Acad Sci USA 2007; 104:181–186.
Article CAS PubMed Google Scholar - Fortunel NO, Otu HH, Ng HH, et al. Comment on “'Stemness': transcriptional profiling of embryonic and adult stem cells” and “a stem cell molecular signature”. Science 2003; 302:393.
Article CAS PubMed Google Scholar - Jackson KA, Majka SM, Wulf GG, Goodell MA . Stem cells: a minireview. J Cell Biochem Suppl 2002; 38:1–6.
Article CAS PubMed Google Scholar - Suzuki A, Zheng Y, Kondo R, et al. Flow-cytometric separation and enrichment of hepatic progenitor cells in the developing mouse liver. Hepatology 2000; 32:1230–1239.
Article CAS PubMed Google Scholar - Tumbar T, Guasch G, Greco V, et al. Defining the epithelial stem cell niche in skin. Science 2004; 303:359–363.
Article CAS PubMed Google Scholar - Richardson GD, Robson CN, Lang SH, Neal DE, Maitland NJ, Collins AT . CD133, a novel marker for human prostatic epithelial stem cells. J Cell Sci 2004; 117(Part 16):3539–3545.
Article CAS PubMed Google Scholar - Barker N, van Es JH, Kuipers J, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007; 449:1003–1007.
Article CAS PubMed Google Scholar - Kurita T, Medina RT, Mills AA, Cunha GR . Role of p63 and basal cells in the prostate. Development 2004; 131:4955–4964.
Article CAS PubMed Google Scholar - Signoretti S, Pires MM, Lindauer M, et al. p63 regulates commitment to the prostate cell lineage. Proc Natl Acad Sci USA 2005; 102:11355–11360.
Article CAS PubMed PubMed Central Google Scholar - Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A . p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999; 398:708–713.
Article CAS PubMed Google Scholar - Yang A, Schweitzer R, Sun D, et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999; 398:714–718.
Article CAS PubMed Google Scholar - Collins AT, Maitland NJ . Prostate cancer stem cells. Eur J Cancer 2006; 42:1213–1218.
Article CAS PubMed Google Scholar - Maitland NJ, Bryce SD, Stower MJ, Collins AT . Prostate cancer stem cells: a target for new therapies. Ernst Schering Found Symp Proc 2006; 5:155–179.
Google Scholar - Tang DG, Patrawala L, Calhoun T, et al. Prostate cancer stem/progenitor cells: identification, characterization, and implications. Mol Carcinog 2007; 46:1–14.
Article CAS PubMed Google Scholar - van Leenders GJ, Aalders TW, Hulsbergen-van de Kaa CA, Ruiter DJ, Schalken JA . Expression of basal cell keratins in human prostate cancer metastases and cell lines. J Pathol 2001; 195:563–570.
Article CAS PubMed Google Scholar - Gu G, Yuan J, Wills M, Kasper S . Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res 2007; 67:4807–4815.
Article CAS PubMed Google Scholar - Gu Y, Li H, Miki J, et al. Phenotypic characterization of telomerase-immortalized primary non-malignant and malignant tumor-derived human prostate epithelial cell lines. Exp Cell Res 2006; 312:831–843.
Article CAS PubMed Google Scholar - Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ . Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005; 65:10946–10951.
Article CAS PubMed Google Scholar - Wang S, Gao J, Lei Q, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003; 4:209–221.
Article CAS PubMed Google Scholar - Zhou Z, Flesken-Nikitin A, Nikitin AY . Prostate cancer associated with p53 and Rb deficiency arises from the stem/progenitor cell-enriched proximal region of prostatic ducts. Cancer Res 2007; 67:5683–5690.
Article CAS PubMed Google Scholar - Banach-Petrosky W, Jessen WJ, Ouyang X, et al. Prolonged exposure to reduced levels of androgen accelerates prostate cancer progression in Nkx3.1; Pten mutant mice. Cancer Res 2007; 67:9089–9096.
Article CAS PubMed Google Scholar - Shen MM, Abate-Shen C . Pten inactivation and the emergence of androgen-independent prostate cancer. Cancer Res 2007; 67:6535–6538.
Article CAS PubMed Google Scholar - Taplin ME, Bubley GJ, Shuster TD, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med 1995; 332:1393–1398.
Article CAS PubMed Google Scholar - Sharifi N, Kawasaki BT, Hurt EM, Farrar WL . Stem cells in prostate cancer: resolving the castrate-resistant conundrum and implications for hormonal therapy. Cancer Biol Ther 2006; 5:901–906.
Article CAS PubMed Google Scholar - De Marzo AM, Nelson WG, Meeker AK, Coffey DS . Stem cell features of benign and malignant prostate epithelial cells. J Urol 1998; 160(Part 2):2381–2392.
Article CAS PubMed Google Scholar - Isaacs JT . The biology of hormone refractory prostate cancer. Why does it develop? Urol Clin North Am 1999; 26:263–273.
Article CAS PubMed Google Scholar - Demichelis F, Rubin MA . TMPRSS2-ETS fusion prostate cancer: biological and clinical implications. J Clin Pathol 2007; 60:1185–1186.
Article PubMed PubMed Central Google Scholar - Shaw A, Bushman W . Hedgehog signaling in the prostate. J Urol 2007; 177:832–838.
Article CAS PubMed Google Scholar - Beachy PA, Karhadkar SS, Berman DM . Tissue repair and stem cell renewal in carcinogenesis. Nature 2004; 432:324–331.
Article CAS PubMed Google Scholar - Doles J, Cook C, Shi X, Valosky J, Lipinski R, Bushman W . Functional compensation in Hedgehog signaling during mouse prostate development. Dev Biol 2006; 295:13–25.
Article CAS PubMed Google Scholar - Hooper JE, Scott MP . Communicating with Hedgehogs. Nat Rev Mol Cell Biol 2005; 6:306–317.
Article CAS PubMed Google Scholar - Taipale J, Chen JK, Cooper MK, et al. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000; 406:1005–1009.
Article CAS PubMed Google Scholar - Fan L, Pepicelli CV, Dibble CC, et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004; 145:3961–3970.
Article CAS PubMed Google Scholar - Sanchez P, Hernandez AM, Stecca B, et al. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc Natl Acad Sci USA 2004; 101:12561–12566.
Article CAS PubMed PubMed Central Google Scholar - Berman DM, Desai N, Wang X, et al. Roles for Hedgehog signaling in androgen production and prostate ductal morphogenesis. Dev Biol 2004; 267:387–398.
Article CAS PubMed Google Scholar - Freestone SH, Marker P, Grace OC, et al. Sonic hedgehog regulates prostatic growth and epithelial differentiation. Dev Biol 2003; 264:352–362.
Article CAS PubMed Google Scholar - Lamm ML, Catbagan WS, Laciak RJ, et al. Sonic hedgehog activates mesenchymal Gli1 expression during prostate ductal bud formation. Dev Biol 2002; 249:349–366.
Article CAS PubMed Google Scholar - Podlasek CA, Barnett DH, Clemens JQ, Bak PM, Bushman W . Prostate development requires Sonic hedgehog expressed by the urogenital sinus epithelium. Dev Biol 1999; 209:28–39.
Article CAS PubMed Google Scholar - Zhang J, Lipinski R, Shaw A, Gipp J, Bushman W . Lack of demonstrable autocrine hedgehog signaling in human prostate cancer cell lines. J Urol 2007; 177:1179–1185.
Article CAS PubMed Google Scholar - Karhadkar SS, Bova GS, Abdallah N, et al. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004; 431:707–712.
Article CAS PubMed Google Scholar - Edwards PC, Ruggiero S, Fantasia J, et al. Sonic hedgehog gene-enhanced tissue engineering for bone regeneration. Gene Ther 2005; 12:75–86.
Article CAS PubMed Google Scholar - St-Jacques B, Hammerschmidt M, McMahon AP . Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev 1999; 13:2072–2086.
Article CAS PubMed PubMed Central Google Scholar - Fortunel NO, Hatzfeld A, Hatzfeld JA . Transforming growth factor-beta: pleiotropic role in the regulation of hematopoiesis. Blood 2000; 96:2022–2036.
CAS PubMed Google Scholar - Fuchs E, Tumbar T, Guasch G . Socializing with the neighbors: stem cells and their niche. Cell 2004; 116:769–778.
Article CAS PubMed Google Scholar - Shariat SF, Menesses-Diaz A, Kim IY, Muramoto M, Wheeler TM, Slawin KM . Tissue expression of transforming growth factor-beta1 and its receptors: correlation with pathologic features and biochemical progression in patients undergoing radical prostatectomy. Urology 2004; 63:1191–1197.
Article PubMed Google Scholar - Wikstrom P, Stattin P, Franck-Lissbrant I, Damber JE, Bergh A . Transforming growth factor beta1 is associated with angiogenesis, metastasis, and poor clinical outcome in prostate cancer. Prostate 1998; 37:19–29.
Article CAS PubMed Google Scholar - Kang Y, He W, Tulley S, et al. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci USA 2005; 102:13909–13914.
Article CAS PubMed PubMed Central Google Scholar - Yin JJ, Selander K, Chirgwin JM, et al. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest 1999; 103:197–206.
Article CAS PubMed PubMed Central Google Scholar - Zhang J, Lu Y, Dai J, et al. In vivo real-time imaging of TGF-beta-induced transcriptional activation of the RANK ligand gene promoter in intraosseous prostate cancer. Prostate 2004; 59:360–369.
Article CAS PubMed Google Scholar
Acknowledgements
The authors acknowledge the support of the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, USA.
Author information
Authors and Affiliations
- Cell and Cancer Biology Branch, Center for Cancer Research, National Cancer Institute, Building 37, Room 1068, Bethesda, 20892, MD, USA
Kathleen Kelly & Juan Juan Yin
Authors
- Kathleen Kelly
You can also search for this author inPubMed Google Scholar - Juan Juan Yin
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toKathleen Kelly.
Rights and permissions
About this article
Cite this article
Kelly, K., Yin, J. Prostate cancer and metastasis initiating stem cells.Cell Res 18, 528–537 (2008). https://doi.org/10.1038/cr.2008.50
- Published: 15 April 2008
- Issue Date: May 2008
- DOI: https://doi.org/10.1038/cr.2008.50