TGF-β-induced epithelial to mesenchymal transition (original) (raw)
EMT: loss of epithelial and acquisition of mesenchymal characteristics
Epithelial cells typically form sheets, tubes or vesicles, in which the cells establish an apical-basal polarity. Their apical and basolateral cell surfaces possess a distinct appearance and serve different functions. In tube- or vesicle-like structures, the apical side is exposed to the lumen, whereas the basolateral surface rests on a basement membrane. Epithelial cells intimately associate with each other through laterally located, specialized cell-cell contact structures, i.e. tight junctions, adherens junctions and desmosomes 1.
Tight junctions are membrane fusions at the lateral side close to the apical surface that provide intercellular sealing and protect against paracellular diffusion, and separate the distinct functions of the apical and basolateral surfaces 2. Occludin and claudins are important components of the intercellular tight junction strands, while the cytoplasmic components Zonula Occludens (ZO)-1, -2, -3 and p120 are integral to an undercoat structure. The cytoplasmic tails of ZO proteins attach to actin filaments, thus contributing to the strength and integrity of tight junctions 2. Par3 and Par6, key components of the polarity protein complex that maintains the apical compartment, are recruited to tight junctions through their PDZ domains 3, 4. One of the early events in EMT is the disassembly of tight junctions, which results in the redistribution of ZO proteins, claudins and occludin, concomitant with disruption of the polarity complex and initiation of cytoskeleton reorganization 1.
Adherens junctions are located adjacent to the tight junctions in the basolateral surface compartments of epithelial cells, and connect to cytoskeletal microfilaments. Similarly to the tight junctions, adherens junctions form a belt-like structure at the lateral interface of epithelial cells 5. The transmembrane adhesion receptor E-cadherin, which characterizes this type of epithelial junction, engages in homotypic interactions through its ectodomain, thus allowing E-cadherin proteins to span the intercellular space between neighboring cells at opposing adherens junctions. The cytoplasmic domains of E-cadherin bind tightly to β-catenin, a cytoplasmic protein that interacts with α-catenin, which in turn anchors to the actin cytoskeleton, either directly or indirectly via actin-binding proteins α-actinin and vinculin 5, 6. During EMT, the adherens junction complexes disassemble and the actin cytoskeleton reorganizes from an epithelial cortical alignment associated with cell-cell junctions into actin stress fibers that are anchored to focal adhesion complexes 1. Loss of E-cadherin is considered as a hallmark event of EMT that initiates a series of signaling events and major cytoskeletal reorganization. However, loss of E-cadherin should not be considered as the sole pivotal event in EMT, since blocking E-cadherin expression by transfection of antisense RNA does not induce a full EMT 7. Furthermore, forced E-cadherin expression is insufficient to restore the epithelial phenotype in spindle carcinoma cells and does not reverse induced EMT 8, 9, 10, 11, 12.
Also located at the lateral side are the desmosomes, which provide additional strength for intercellular adhesion. Desmosomes are structurally similar to adherens junctions, containing transmembrane cadherins and linker proteins that connect the cadherins to the intermediate filament cytoskeleton. However, unlike the adherens junctions, they are organized as individual patches and not as a belt 13, 14. The desmosome cadherins are paired transmembrane proteins composed of a desmoglein and a desmocollin. Their cytoplasmic tails interact with the Armadillo family proteins, plakophilin and plakoglobin, which also associate with desmoplakins, thus providing a link to keratin intermediate filaments. Decrease in desmoplakin expression has been reported in various EMT settings and downregulation of the expression of desmosome components is apparent in microarray analyses that evaluate changes in gene expression associated with EMT 11, 15, 16, 17. Hence, dissolution of the desmosomes accompanies the EMT process, but how this change fits into the sequential events resulting in EMT and whether it has a causative effect in EMT remain unknown.
Concomitant with the loss of epithelial cell-cell contact structures and actin reorganization, cells undergoing EMT acquire a mesenchymal identity (Figure 1). The mesenchymal phenotype is apparent from the expression of mesenchymal cytoskeletal proteins, such as vimentin, and the increased deposition of extracellular matrix proteins, including collagens and fibronectin. These extracellular matrix components stimulate integrin signaling and induce the formation of focal adhesion complexes, which facilitate cell migration 18, [19](/articles/cr20095#ref-CR19 "Zhao Y, Min C, Vora S, et al. The lysyl oxidase pro-peptide attenuates fibronectin-mediated activation of FAK and p130CAS in breast cancer cells. J Biol Chem 2008 Nov 21. doi: 10.1074/jbc.M802612200
."). The generally observed downregulation of E-cadherin also promotes the assembly of focal adhesion through activation of focal adhesion kinase [20](/articles/cr20095#ref-CR20 "Frame MC, Inman GJ . NCAM is at the heart of reciprocal regulation of E-cadherinand integrin-mediated adhesions via signaling modulation. Dev Cell 2008; 15:494–496."). Furthermore, decreased expression of E-cadherin during EMT is accompanied by increased expression of N-cadherin, which renders the cell more motile and invasive [21](/articles/cr20095#ref-CR21 "Shirakihara T, Saitoh M, Miyazono K . Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-β. Mol Biol Cell 2007; 18:3533–3544."), [22](/articles/cr20095#ref-CR22 "Deckers M, van Dinther M, Buijs J, et al. The tumor suppressor Smad4 is required for transforming growth factor β-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res 2006; 66:2202–2209."), [23](/articles/cr20095#ref-CR23 "Cavallaro U, Christofori G . Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer 2004; 4:118–132."). These different events result in a loss of apical-basal polarity that is critical to the maintenance of epithelial morphology as well as the function of the epithelial sheet. Furthermore, the loss of specialized cell-cell contact structures facilitates the increased migration, a defining behavioral property of cells that have undergone EMT [24](/articles/cr20095#ref-CR24 "Grunert S, Jechlinger M, Beug H . Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol 2003; 4:657–665."). With the loss of apical-basal polarity, cells acquire a front-back polarity that allows them to migrate in a directional fashion. The increased expression and activity of extracellular proteases, such as matrix metalloproteinases, allow the cells to degrade extracellular matrix proteins, thus allowing the migration to translate into an invasive behavior, whereby the cells delaminate and escape from their epithelial structures [25](/articles/cr20095#ref-CR25 "Moustakas A, Heldin CH . Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci 2007; 98:1512–1520.").Figure 1
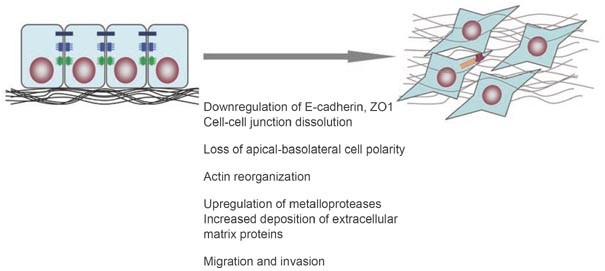
Epithelial-mesenchymal transition (EMT) occurs when epithelial cells lose their epithelial cell characteristics, including dissolution of cell-cell junctions, i.e. tight junctions (black), adherens junctions (blue) and desmosomes (green), and loss of apical-basolateral polarity, and acquire a mesenchymal phenotype, characterized by actin reorganization and stress fiber formation (red), migration and invasion.
This complex and multifaceted process that defines EMT results from a plexus of changes in transcriptional regulation, in which epithelial gene expression is repressed and expression of mesenchymal genes is activated. In fact, several families of transcription factors, including the Snail family, ZEB family and basic helix-loop-helix (bHLH) family, act in concert to control the EMT process 26. These changes in the transcription program are complemented by non-transcriptional changes that help define the changes in cytoskeletal organization and cell shape and the interaction of the cell with its environment.
EMT can be induced or regulated by various growth and differentiation factors, including TGF-β, growth factors that act through receptor tyrosine kinases, such as fibroblast growth factor, hepatic growth factor and platelet derived growth factor, and Wnt and Notch proteins 25. Among these, TGF-β has received much attention as a major inducer of EMT during embryogenesis, cancer progression and fibrosis. How TGF-β induces this orchestrated program of transcriptional changes and integrates these changes with the non-transcriptional regulation will be discussed below.
EMT in development and pathology
The induction of EMT by TGF-β was first recognized in cell culture. Upon TGF-β treatment, epithelial cells changed from cuboidal to an elongated spindle shape, and showed decreased expression of epithelial markers and enhanced expression of mesenchymal markers fibronectin and vimentin 17. These changes were accompanied by increased motility. Consistent with their binding to the same receptor complexes, TGF-β1, TGF-β2 and TGF-β3 share the capacity to induce EMT in epithelial cells 17, 27, 28. Subsequent studies have demonstrated an involvement of TGF-β and TGF-β-related proteins in EMT in normal development and in pathological processes.
In triploblastic metazoans, EMT first occurs at gastrulation and allows the formation of a third germ layer, called the mesoderm, between the ectoderm and the endoderm 29. While lower vertebrates acquire those internal layers through movement of epithelial cell sheets, EMT is an integral process in gastrulation in higher vertebrates 29. BMPs are believed to participate in the EMT associated with gastrulation initiation and mesoderm patterning 30. At a later stage in embryogenesis, neural crest cells, arising from the dorsal part of the neural tube, undergo EMT and migrate to contribute as different cell types to the formation of various tissues, including bones and smooth muscle 31. In cardiogenesis, TGF-β has been shown to play a key role in the EMT that occurs in the atrioventricular canal and the outflow tract region. Accordingly, the expression of TGF-β1 and TGF-β2 increases at the onset of EMT in the atrioventricular canal endothelium and myocardium, respectively 32. In mice, TGF-β1 null embryos present severe cardiac abnormalities including defective atrioventricular junction. Moreover, TGF-β2-deficient mice present atrioventricular and outflow tract defects. Finally, blocking TGF-β activity using antibodies or antisense oligonucleotides inhibits EMT in chicken atrioventricular explants and TGF-β2-induced EMT in mouse explant cultures 33, 34. TGF-β signaling has also been implicated in the EMT that is associated with palatal morphogenesis. EMT occurs to induce mesenchyme continuity during the fusion of the two palatal shelves with the nasal septum, and high TGF-β3 expression is apparent at these sites 35. TGF-β3 null mice present a cleft palate, resulting from the lack of fusion of the two palatal shelves 36. At a later developmental stage, EMT occurs in the coelomic epithelium during the second phase of Müllerian duct regression upon signaling by anti-Müllerian hormone, a member of the TGF-β family 37.
While EMT is spatially and temporally highly regulated in normal development, EMT also occurs in pathological contexts in the adult organism, specifically in cancer progression and fibrosis. In cancer, the epithelial tumor cells become more invasive after undergoing EMT and access the circulatory system through intravasation, resulting in dissemination of cancer cells to distal loci from the primary tumor. Consequent metastatic colonization of secondary sites by cancer cells then involves the reverse MET process 38. Although it has been difficult to observe EMT during cancer development, presumably a result of its transient and reversible nature, recent progress in in vivo imaging techniques may facilitate the study of this process 25. Several lines of evidence implicate increased TGF-β signaling as a key effector of EMT in cancer progression and metastasis. Chemical carcinogenesis studies in vivo showed that transgenic expression of activated TGF-β1 correlates with the conversion of squamous into more invasive spindle cell carcinomas 39. Cancer cells often increase their production of active TGF-β, which not only triggers EMT and allows the cells to become invasive, but also enhances angiogenesis in close proximity to the tumor microenvironment, providing an exit route for migratory mesenchymal cells 40.
EMT also occurs following tissue injury and contributes to organ fibrosis. In renal fibrosis, renal interstitial fibroblasts derive not only from mesenchymal stem cells in the bone marrow, but also from proximal tubular kidney epithelial cells that undergo EMT 41. Several observations indicate that TGF-β is causally associated with this process. Abnormally high levels of TGF-β are expressed in renal fibrotic sites of patients with kidney diseases. Moreover, transgenic mice with increased expression of TGF-β1 develop renal fibrosis 42.
TGF-β has also been shown to play a key role in pulmonary and hepatic fibrosis, not only through its ability to attract fibroblasts and to stimulate their proliferation, but also through induction of EMT in alveolar epithelial cells and transdifferentiation of quiescent hepatic stellate cells into myofibroblasts respectively 43, 44. Finally, the recently described endothelial to mesenchymal transition may contribute to TGF-β-induced cardiac fibrosis 45.
TGF-β-activated Smad signaling in EMT
The dissection of the signaling mechanisms that are activated in response to TGF-β and lead to EMT has been largely conducted in cells in culture, predominantly using the NMuMG, MDCK and HaCaT cell lines as model systems. Parallel studies have allowed an evaluation of the role of TGF-β signaling components in EMT in vivo. TGF-β signals through a heteromeric complex of two type I and two type II transmembrane serine-threonine kinase receptors. In response to TGF-β, the type II receptor kinases phosphorylate the type I receptors, which then leads to activation of the cellular responses to TGF-β 46. Dominant negative interference with the TGF-β type II receptor function reverses EMT in colon cancer cells in culture and inhibits EMT in skin and mammary cancer models in vivo 47, 48. Furthermore, loss of the TGF-β type II receptor has been associated with decreased EMT at cancer loci in a mouse skin carcinoma model 49. The key role of the type I receptor in TGF-β-induced EMT is also revealed in cell culture and in vivo studies. Indeed, expression of an activated version of the TβRI receptor ALK-5, the major TGF-β type I receptor, or of ActRIB/ALK-4, the major type I receptor for activin and nodal, recapitulates TGF-β-induced EMT in NMuMG cells 27, 28, while dominant negative forms of either type I receptor block TGF-β-induced EMT 28. Inhibition of TβRI function using a chemical inhibitor also blocks EMT and promotes an enhanced epithelial phenotype in cell culture 50, 51, while interference with TβRI function in vivo suppresses EMT and decreases mesenchymal differentiation during cardiac valve formation 52.
TGF-β-induced activation of the receptor complex leads to activation of Smad2 and Smad3 through direct C-terminal phosphorylation by TβRI. Phosphorylated Smad2 and Smad3 then form trimers with Smad4, and translocate into the nucleus, where they associate and cooperate with DNA binding transcription factors to activate or repress target gene transcription (Figure 2). Consequently, Smad2 and Smad3 function in cooperation with Smad4 as TGF-β-induced transcription regulators. In contrast, the inhibitory Smad6 and Smad7 inhibit activation of the receptor-regulated Smads 46.
Figure 2
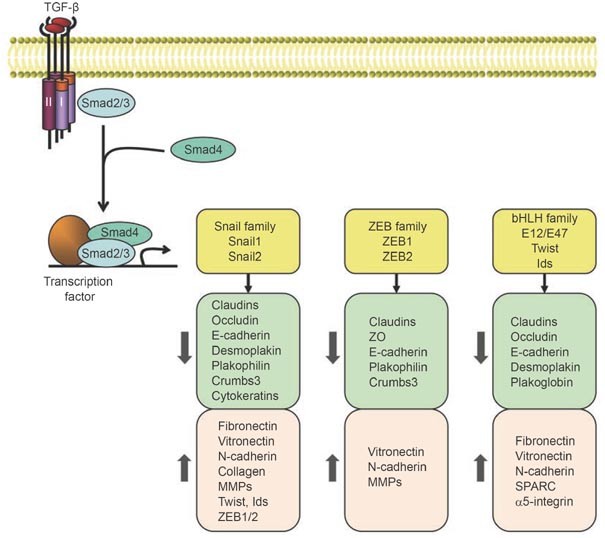
Transcriptional regulation of EMT induced by TGF-β. In response to TGF-β, Smad2 and 3 are activated, and form complexes with Smad4, which then regulate transcription of target genes through interactions with other DNA binding transcription factors. In the induction of EMT, the activated Smads mediate transcriptional regulation through three families of transcription factors, resulting in repression of epithelial marker gene expression and activation of mesenchymal gene expression.
Various studies have explored the roles of TGF-β-activated Smads in EMT. Increased expression of Smad2 or Smad3 with Smad4 induces EMT, or enhances the induction of EMT by the activated form of TβRI, in NMuMG cells 27, 28, whereas expression of dominant negative versions of Smad2 or Smad3 blocks TGF-β-induced EMT in this cell system 28. Consistent with the pivotal role of Smad3 in EMT, renal tubular epithelial cells deficient in Smad3 fail to undergo EMT in response to TGF-β or mechanical stress 53, and keratinocytes derived from Smad3−/− mice show reduced migration in response to TGF-β 54. Compared with Smad3, Smad2 may play an antagonistic role in the EMT process in vivo. Loss of Smad2 is frequently noted in human skin cancer patients, and Smad2 deficiency in keratinocytes promotes EMT and accelerates skin tumor formation. This has been explained by increased binding of the Smad3/4 complex to the promoter of the Snail gene and by increased Snail expression in the absence of Smad2, thus enhancing the progression of EMT 55. Similarly, Smad2−/− hepatocytes appear mesenchymal and migrate faster than wild-type cells, while Smad3−/− hepatocytes retain their epithelial characteristics 56. However, expression of activated Smad2 promotes spindle tumor cell invasion, and a dominant negative form of Smad2 inhibits it, suggesting that Smad2 may promote EMT in vivo 57. Similarly to Smad3, Smad4 is indispensable for EMT. RNA interference-mediated knockdown of Smad4 expression or expression of a dominant negative mutant of Smad4 results in preserved E-cadherin expression 22, 28, 58, 59, suppression of fibrotic type I collagen synthesis in vitro 59, and decreased bone metastasis in vivo 22. Furthermore, genetic ablation of Smad4 leads to preservation of epithelial markers and a lower degree of EMT in adenocarcinoma 60. Conversely, the inhibitory Smads function as negative regulators of and thus repress TGF-β-induced EMT. Smad6 controls the timing and extent of EMT during cardiac valve formation 61, 62, while increased expression of Smad7 blocks TGF-β-induced EMT in multiple tissues 28, 63, 64, 65, 66.
Transcriptional regulation of EMT
Consistent with the important roles of Smads, the loss of epithelial markers and acquisition of mesenchymal features are achieved through a well-orchestrated transcription program that involves three families of transcription factors, the Snail, ZEB and bHLH families (Figure 2). Their expression is induced in response to TGF-β, either through a Smad-dependent mechanism (in the case of Snail proteins) or indirectly through activation of other transcription factors or relief of repression. Upon activation, these transcription factors in turn repress epithelial marker gene expression and concomitantly activate mesenchymal gene expression. Additionally, these factors elaborate tissue-specific or developmental stage-specific functions associated with their distinct expression profiles 26.
Snail family transcription factors
The Snail transcription factors share extensive structural similarity, containing a characteristic C-terminal domain with four to six zinc fingers that mediates sequence-specific DNA binding to E-box elements C/A(CAGGTG) 67. Three Snail family proteins have been identified in vertebrates: Snail1 (first described as Snail), Snail2 (also known as Slug) and a more recently characterized Snail3. Snail proteins function as transcription repressors and their activities depend on the zinc finger domain and an N-terminal SNAG (Snail/Gfi) domain.
Induction of Snail1 expression has been noted in all EMT processes that have been studied 67, 68, and increased Snail1 levels have been correlated with more invasive tumor types 15, 69. Snail2 is more broadly expressed than Snail1, and its expression appears in some cases unrelated to EMT 68, 70. Both genes are induced in response to TGF-β in cells that undergo TGF-β-induced EMT. Furthermore, TGF-β induces Snail1 expression in skin 71 and palate development 72, in mesothelial cells during pathological fibrosis 73, in cultured hepatocytes 59, and in multiple epithelial cell lines 74, as well as during heart development 75. The induction of Snail1 expression in response to TGF-β is mediated by Smad3; Smad3 binds to the Snail1 promoter and activates its transcription 55, 76. Renal tubular epithelial cells deficient in Smad3 expression fail to activate Snail1 expression upon TGF-β treatment 53, and interference with Smad4 expression attenuates TGF-β1-induced Snail1 expression in epithelial cell lines 22, 58, 59. Smad3 also mediates TGF-β-induced expression of Snail2 in MDCK cells, whereby Smad3 forms a complex with myocardin-related transcription factors and binds to the Snail2 promoter to activate transcription 77.
Induction of EMT by hepatocyte growth factor (HGF) 78, fibroblast growth factor (FGF), or epidermal growth factor (EGF), which all act through the Ras-MAPK or PI3K-Akt pathway, also results in the induction of Snail expression 68. Furthermore, the TGF-β-Smad pathway also cooperates with Ras, Notch and Wnt signaling in inducing Snail expression in development and in tumor metastasis 68. Additionally, post-translational modification defines Snail subcellular localization, stability and transcription activity 26, 68.
Several lines of evidence highlight the key roles of Snail transcription factors in the execution of the EMT program 15, 69. Ectopic expression of Snail1 or Snail2 suppresses E-cadherin and plakoglobin expression and enhances vimentin and fibronectin expression, leading to a full EMT phenotype, whereas silencing of Snail expression reverses this process 15, 69, 79, 80, 81. Snail1-deficient mouse embryos are unable to complete the EMT process and form a defective mesodermal layer that maintains E-cadherin expression, causing the mutant embryos to die at gastrulation 82. Silencing of Snail2 expression in chick embryos results in mesodermal malformation and neural crest emigration failure 83.
Substantial efforts have gone into defining the targets of the Snail transcription factors. Snail1 and 2 both repress the expression of the epithelial marker gene CDH1 that encodes E-cadherin. At the E-cadherin promoter, Snail1 binds to E-box elements, recruits a complex consisting of HDAC1, HDAC2 and mSin3A, and thus represses gene transcription 15, 69, 81. Snail2 similarly represses E-cadherin through binding to the same E-box elements, but recruits a different combination of co-repressors, i.e. HDAC1/3 and CTBP 70, 80, 81. The expression of Snail proteins appears to inversely correlate with E-cadherin expression, and silencing of the Snail1 gene can restore E-cadherin levels 69, 81. However, forced expression of E-cadherin is unable to counteract Snail1-induced EMT 11, 12, suggesting an involvement of additional Snail target genes in the elaboration of EMT. In fact, Snail proteins repress a spectrum of genes involved in maintaining epithelial structure and function, including genes encoding claudins and occludin, major transmembrane components of tight junctions. Snail1 represses the expression of claudin-3, -4 and -7 at their promoters 11, 84, whereas Snail1 and Snail2 both repress the expression of claudin-1 and occludin 84, 85, 86. In contrast, Snail1 expression does not dramatically decrease the levels of cytoplasmic components of tight junctions, such as ZO-1 and p120, but their distribution is altered from peripheral localization to a diffused cytoplasmic pattern 84. Snail proteins regulate the expression of desmosome proteins as well. Ectopic expression of Snail1 results in decreased desmoplakin and plakophilin levels, and causes disorganized plakoglobin distribution 11, 15, 16, 87. Furthermore, Snail1 disrupts epithelial polarity by repressing the expression of Crumbs3, which in complex with two other proteins, PALS1 and PATJ, is required for the maintenance of apical-basal polarity. Snail1 binds to the E-box element in the Crumbs3 promoter and inhibits Crumbs3 expression, leading to disassembly of the Crumbs complex 88. However, Snail1 expression does not affect the Par complex, another important polarity complex 88. Snail1 expression also results in decreased expression of a subset of cytokeratins, i.e. cytokeratin 17, 18, 19 and 20, thus affecting the epithelial cytoskeletal organization. Cytokeratins 17 and 18 have been identified as direct targets of Snail1 by chromatin immunoprecipitation 11, 84, 89. Recent microarray analyses have unveiled additional genes encoding epithelial or basal lamina proteins that are downregulated by Snail1, suggesting an even broader range of regulation of the epithelial program by Snail proteins 90.
Other Snail target genes show a more tissue-restricted distribution and are hence associated with tissue-specific EMT processes. One such target of Snail1 is HNF-1β, a transcription factor that is expressed in kidney epithelial cells and activates the expression of the kidney-specific epithelial marker cadherin-16. Through direct repression of HNF-1β transcription, Snail1 suppresses cadherin-16 expression and induces EMT in cell culture, reminiscent of the development of kidney fibrosis in vivo 91, 92. In hepatocytes, Snail1 represses another HNF transcription factor, HNF-4α, resulting in loss of epithelial markers and expression of mesenchymal proteins 93. Snail proteins also repress the expression of mucin-1, a transmembrane protein on the apical surface of pulmonary airway epithelium that serves to hydrate, lubricate and protect the epithelium 94.
While repressing epithelial gene expression, Snail proteins activate the expression of the mesenchymal proteins fibronectin 15, 79, vitronectin 15, 79 and N-cadherin 90, the extracellular matrix proteins collagen type III and V 90, and proteins involved in migration and invasion, such as RhoB, plasminogen activator inhibitor-1 and matrix metalloproteinases 68, 79. These effects of Snail may be indirect and involve other transcription factors, such as Ets-1 which has been proposed to mediate the induction of MMP-2 expression 95 or Sp-1 and Ets-1 which are thought to be partially responsible for the upregulation of MMP-9 expression 96. Snail1 also regulates the expression of multiple actin-modulating proteins to facilitate the rearrangement of actin filaments from cortical distribution to stress fibers anchored to focal adhesions 11. Consistent with the indirect induction of gene expression by Snails, Snail transcription factors induce the expression of other EMT-related transcription factors, such as Twist and Ids in MDCK cells 90, and ZEB1 and ZEB2 in squamous carcinoma cell lines 89, 95.
In addition to regulating the expression of epithelial or mesenchymal genes, Snail also regulates genes required for cell survival 68, which are frequently intertwined with EMT during embryonic development or in pathological conditions. Finally, in some cases Snail1 and Snail2 cooperate in the control of the transcription network that regulates EMT. For example, in breast tumors, Snail1 expression correlates with mesenchymal transition and metastasis, and Snail2 expression associates with the repression of tumor suppressor gene BRCA2 68.
ZEB family transcription factors
Two ZEB family transcription factors are known in vertebrates: ZEB1, also known as δEF1 or AREB6, and ZEB2, also known as Smad-interacting protein 1 (SIP1). They have two zinc-finger clusters at each end, whose simultaneous binding to bipartite E-boxes mediates the interaction with regulatory DNA sequences. The central region contains a Smad-interaction domain, a homeodomain and a CTBP binding domain. Repression of gene transcription by ZEB1 or ZEB2 is mediated by repressor motifs in the central homeodomain and through recruitment of CTBP as a co-repressor. However, interaction of ZEB1 with co-activators PCAF and p300 switches ZEB1 function from repression to activation 26, 97. ZEB proteins are expressed during development in various tissues, including the central nervous system, the heart, skeletal muscle and haematopoietic cells. They partially compensate for each other in these tissues, although in other cases, such as during neural crest emigration, and in lymphocytes, ZEB proteins exhibit distinct expression patterns and do not functionally compensate 26.
TGF-β signaling induces the expression of ZEB proteins during EMT through an indirect mechanism mediated in part by Ets-1 21. ZEB proteins then interact with Smad3 and directly repress the expression of epithelial marker genes, possibly by recruiting the co-repressor CTBP 97, 98. ZEB1 is also induced by TGF-β during smooth muscle differentiation, in which it synergistically interacts with Smad3 and SRF to transactivate the genes encoding smooth muscle α-actin and smooth muscle myosin heavy chain 99. As is in the case of the Snails, the expression of ZEB proteins is not only activated by TGF-β but also by other growth factors that activate Ras-MAPK signaling and by Wnt/β-catenin signaling 26. The expression of ZEB factors is also post-transcriptionally repressed by microRNAs, miR-200 family and miR-205 100, 101, 102. This group of microRNAs is dramatically downregulated in cells undergoing EMT, allowing expression of ZEB transcription factors. Thus, forced expression of miR-200 family is sufficient to block TGF-β-induced EMT 100, 101, 102. In addition, ZEB2 is subject to post-translational regulation, whereby Pc2-mediated sumoylation impairs its repressor activity 103.
The induction of ZEB proteins is necessary for the downregulation of E-cadherin expression and promotion of cell migration 21, 104, 105, 106. ZEB proteins directly repress E-cadherin expression independently of the Snail transcription factors in mouse mammary epithelial cells 21, 106. Concomitantly, they confer a delocalization of β-catenin in a cell context-dependent manner and promote cell migration 104, 107. Besides its effects on adherens junctions, ZEB2 directly represses the expression of the tight junction proteins claudin-4 and ZO-3 107. ZEB2 also suppresses the expression of the desmosome protein plakophilin-2 107 and induces the expression of the mesenchymal proteins vimentin 108, N-cadherin 107 and matrix metalloproteinase-2 95 through as yet unknown mechanism(s). Both ZEB proteins promote cell migration and induce invasion 104, 107, 109. ZEB1 has also been implicated in the downregulation of epithelial polarity through the direct repression of Crumbs3 expression; depletion of ZEB1 restores the expression of Crumbs3 and PATJ, major components of the Crumbs3 polarity complex 109, 110. ZEB1 also directly represses the expression of mucin-1 89.
Helix-loop-helix family factors
The HLH family is a large family of transcription factors controlling a wide array of developmental and pathological processes. The basic structure of HLH family members includes two parallel α-helices linked by a loop required for dimerization. HLH factors are divided into seven categories based on their tissue distribution, dimerization capability and DNA-binding specificity 111. Among these, the class I proteins E12 and E47, class II proteins Twists and class V proteins Ids are involved in the elaboration of EMT. The class I proteins E12 and E47 form homodimers or heterodimers with class II proteins, and the class II proteins Twists (Twist1 and Twist2) always heterodimerize with class I proteins. In contrast to these, the class V Id proteins (Id1, Id2, Id3 and Id4) lack the basic domain and thus are incapable of DNA binding. They function as dominant negative inhibitors through high affinity binding to class I proteins 26, 111.
E12 and E47 are encoded by alternative splicing products of the E2A gene 26, 111. They directly repress E-cadherin expression through DNA binding at the E-box element in the proximal promoter. Ectopic expression of E12 or E47 represses E-cadherin and plakoglobin expression, induces vimentin and fibronectin expression, and promotes migration and invasion 80, 112, 113. It remains unclear whether E12 and E47 form homodimers or heterodimerize with other HLH factors for E-cadherin repression. However, their repression of E-cadherin transcription can be antagonized by Id factors, which interact with E2A proteins 113. The expression of Id1, Id2 and Id3 is repressed in response to TGF-β, which in the case of Id1, occurs through the rapid activation of expression of the transcription repressor ATF3 by TGF-β and the subsequent binding of an ATF3/Smad3/Smad4 complex to the Id1 promoter 114, 115. Consequently, loss of Id protein expression correlates with a decrease in E-cadherin expression, and ectopic expression of Id2 or Id3 dose-dependently blocks TGF-β-induced repression of E-cadherin expression, inhibits TGF-β-induced ZO-1 delocalization and represses TGF-β-induced smooth muscle actin expression 113, 114. Besides E-cadherin, E47 also represses desmoplakin expression and induces the expression of N-cadherin, SPARC and α5-integrin 90.
The HLH protein Twist1 is a major regulator of mesoderm formation in Drosophila and neural tube closure in mice, suggesting its involvement in developmental EMT 116. Mutation of the gene encoding Twist1 in human has been associated with Saethre-Chotzen syndrome, which is characterized by cleft palate and possibly caused by an inhibition of EMT 117, 118. Conversely, both Twist1 and Twist2 expression are upregulated in a large fraction of human tumors 119. Ectopic expression of Twist1 or Twist2 decreases E-cadherin, occludin and claudin-7 expression, increases vimentin and N-cadherin expression, and enhances migration and invasion 9, 10. In addition, Twist1 and Twist2 both synergize with H-Ras to induce a full EMT 10.
HMGA2
HMGA2 (high mobility group A2) is another downstream effector of TGF-β during EMT. HMGA2 belongs to a group of transcription factors that bind AT-rich DNA sequences to form nucleoprotein complexes. HMGA2 is expressed at high levels during embryogenesis and at low levels in adulthood, and is expressed aberrantly in some transformed cells or cancer tissues. TGF-β induces a drastic increase in HMGA2 expression through a Smad3/Smad4-dependent mechanism, and ectopic HMGA2 expression is able to induce the expression of Snail1/2 and Twist1 120.
Non-Smad signaling in TGF-β-induced EMT
TGF-β also elicits signaling responses through pathways that are generally considered as important effector pathways for tyrosine kinase receptors in response to ligands that do not belong to the TGF-β family. The rapid activation of these non-Smad signaling pathways by TGF-β often follows similar kinetics as Smad signaling, and attenuation of Smad signaling does not generally affect the activation of these pathways (Figure 3). In addition, non-Smad signaling responses to TGF-β can also occur with delayed kinetics and are then often indirect, presumably as a result of Smad-mediated changes in gene expression 121. Direct activation of non-Smad signaling pathways by TGF-β occurs through interactions of signaling mediators either directly with the TβRII and/or TβRI receptors or through adaptor proteins. Among the non-Smad signaling responses, activation of Erk MAP kinases, Rho GTPases and the PI3 kinase/Akt pathway in response to TGF-β has been linked to TGF-β-induced EMT through their regulation of distinct processes, such as cytoskeleton organization, cell growth, survival, migration or invasion 28, 121. Treatment of cells with chemical inhibitors that selectively or specifically block one or several of these pathways dramatically affects the induction of the EMT phenotype and downstream transcriptional responses by TGF-β, strongly suggesting that activation of non-Smad signaling complements Smad signaling in the elaboration of the EMT response.
Figure 3
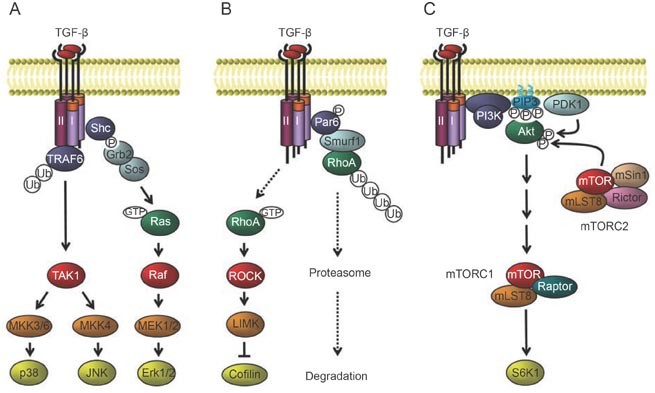
Non-Smad signaling in response to TGF-β. (A) TGF-β activates p38 MAP kinase and JNK MAP kinase signaling through the activation of TAK1 by receptor-associated TRAF6, and Erk MAP kinase signaling through recruitment and phosphorylation of Shc by the TβRI receptor. (B) Activation of RhoA in response to TGF-β and induction of ubiquitin-mediated RhoA degradation at tight junctions. (C) TGF-β induces PI3-kinase signaling, leading to the activation of Akt-mTOR signaling, and consequently to increased translation.
Ras and MAP kinase activation in TGF-β-induced EMT
Growth factor signaling through tyrosine kinase receptors leads to MAP kinase signaling, and MAP kinase pathways have been shown to regulate TGF-β-induced Smad signaling. Erk MAP kinases phosphorylate R-Smads in their linker region thus inhibiting their nuclear translocation and decreasing their activities 122. Other reports, however, support a cooperation between Erk MAP kinase signaling and TGF-β/Smad signaling in TGF-β-induced gene expression 123, 124, 125. TGF-β, acting through its distinct class of receptors, is also capable of directly activating MAP kinase signaling pathways, although the activation levels are generally much lower than that by tyrosine kinase receptors. The activation of Erk1 and Erk2 MAP kinases in response to TGF-β is initiated by Ras, leading to activation of Raf and MEK1/2 kinases, following a similar activation cascade as in response to growth factors 121, 126. This signaling pathway is linked to the receptor complex by ShcA. In response to TGF-β, ShcA associates with and is phosphorylated on tyrosine by TβRI, thus offering a docking site for the recruitment of Grb2 and Sos, and allowing the Shc/Grb2/Sos complex to initiate Ras activation upstream from the kinase cascade 126. In addition to this role of Shc, the interaction of the integrin αvβ3 with TβRII was shown to result in tyrosine phosphorylation of TβRII by Src, leading in turn to association of Shc and Grb2 with the TβRII receptor 127.
Various observations support a cooperation of the Ras-Erk MAP kinase pathway with TGF-β signaling in the induction of EMT. Increased Ras-Erk MAP kinase signaling, e.g. in response to growth factor stimulation or due to expression of mutant Ras, enhances TGF-β-induced EMT, as apparent by the morphological changes and downregulation of E-cadherin expression 124, 128, 129, 130. Consistent with this cooperation, blocking the kinase function of MEK1/2 using a chemical inhibitor, thus resulting in inactivation of the Erk1/2 MAP kinases, inhibits TGF-β-induced EMT 131. Since Erk1/2 MAP kinases target various transcription factors, this cooperation with TGF-β signaling may occur at the level of gene expression, which would be consistent with the key role of Smads in activating the EMT process. Accordingly, activation of MEK/Erk MAP kinase signaling enhances TGF-β-induced transcription responses, leading to downregulation of E-cadherin, upregulation of N-cadherin and upregulation of matrix metalloproteinase expression 128, 130. In addition, this cooperation may result in part from the increased expression of Snail2 leading to downregulation of E-cadherin expression 132, and oncogenic Ras hyperactivity was shown to result in enhanced TGF-β signaling concomitant with increased autocrine TGF-β secretion, phosphorylation and nuclear accumulation of Smads, and TGF-β-specific transcriptional responses 124. In vivo, ectopic expression of an activated version of Smad2 enhances the metastatic potential of cells expressing activated Ras 57. Additionally, activation of Erk5 MAP kinase, another member of Erk family, which can also occur in response to TGF-β, stabilizes Snail1 expression 133. Taken together, these results suggest that activation of Erk MAP kinase signaling contributes to and may even be required for TGF-β-induced EMT. Therefore, the induction of this signaling pathway by TGF-β itself may be important in the EMT response.
TGF-β treatment also induces activation of p38 MAP kinase. In fact, TGF-β was shown to induce phosphorylation of p38 MAP kinase 134, 135. The observation that a chemical inhibitor of p38 MAP kinase prevents TGF-β-induced EMT in mammary epithelial cells without affecting Smad phosphorylation suggests a role of p38 MAP kinase in TGF-β-induced EMT 134, 135. This may be explained by the activation of the transcription factor ATF-2 by p38 MAP kinase, since ATF-2 mediates TGF-β-induced transcription responses 136.
Activation of JNK MAP kinase, which occurs in response to TGF-β in several cell systems, has also been shown to be required for TGF-β-induced EMT. Indeed, epithelial cells deficient in JNK1 expression, and keratinocytes transfected with antisense oligonucleotides targeting JNK, were unable to undergo EMT upon TGF-β treatment 137, 138. Moreover, a chemical inhibitor of JNK blocks the TGF-β-induced increase in fibronectin, vimentin and α-smooth muscle actin expression, and decrease in E-cadherin expression observed during EMT 137, 139. This requirement may reflect the role of JNK in cytoskeleton organization, cell migration and invasion through regulation of matrix metalloproteinase expression 140. JNK also activates c-Jun, a component of the transcription complex AP-1 that cooperates with TGF-β-induced Smads in mediating TGF-β-induced transcription responses, such as plasminogen activator like-urokinase (uPA) expression, and plays an important role in EMT 137.
Consistent with the activation of p38 MAP kinase in response to TGF-β, TGF-β has been shown to induce phosphorylation of p38 MAP kinase, as well as upstream kinases that mediate p38 activation, i.e. MKK3 and MKK6, and TAK1, which can activate MKK3 and MKK6 137, 138. In addition, TAK1 has been shown to be required for JNK MAPK activation in response to TGF-β signaling through activation of MKK4 141, 142. The activation of TAK1 was reported to require the interaction of the ubiquitin ligase TRAF6 with the TGF-β receptor complex followed by a consequent direct activation of TAK1 by TRAF6 143, 144.
Activation of Rho-like GTPases in TGF-β-induced EMT
Rho-like GTPases, comprised of the Rho, Rac and Cdc42 subfamilies of proteins, are key regulators of cytoskeleton organization, cell migration and gene regulation. TGF-β has been found to induce activation of Rho, Rac and Cdc42 in different cell systems, however, most studies have focused on the role of RhoA and its effector kinase ROCK in TGF-β-induced EMT. During embryonic chick heart development, RhoA is necessary for normal EMT in the atrioventricular canal and antibodies against TGF-β lead to a dramatic reduction of RhoA mRNA 145. TGF-β induces increased levels of active GTP-RhoA, which in lens epithelial cells occurs in a biphasic manner within the first 30 minutes and then after 1 to 2 days 146. The activation of RhoA in response to TGF-β in turn results in activation of ROCK, which induces the formation of actin stress fibers 147. Moreover, TGF-β can activate LIM kinase, a downstream target of ROCK, which inactivates the actin-depolymerizing factor cofilin. Downregulation of LIM kinase expression using siRNA inhibits TGF-β-induced actin reorganization in fibroblasts 148. Chemical inhibition of ROCK activity or downregulating Rho mRNA levels using antisense oligonucleotides results in inhibition of TGF-β-induced actin reorganization and α-smooth muscle actin expression that accompany EMT, without affecting Smad activation 145, 146, 149. Complementary to these observations, TGF-β also directly regulates RhoA activity at the tight junctions of epithelial cells. At these junctions, Par6 was found to interact with TβRI and TGF-β induces the association of TβRII with the TβRI/Par6 complex, permitting TβRII to phosphorylate Par6 at a defined serine 150. This event in turn recruits the E3 ubiquitin ligase Smurf1, resulting in enhanced RhoA ubiquitination by Smurf1 and degradation at tight junctions. This Smad-independent, TGF-β-induced mechanism thus participates in the disassembly of the junctions during TGF-β-mediated EMT, yet contrasts with the overall activation of RhoA by TGF-β, which may be explained by the spatio-temporal regulation of Rho during EMT.
Activation of PI3 kinase/Akt signaling in TGF-β-induced EMT
Similarly to various growth factors that act through tyrosine kinase receptors, TGF-β has been shown to rapidly activate PI3 kinase, leading to activation of the Akt kinase, in diverse cell systems 151, 152, 153, 154, 155, 156. Consistent with this observation, the regulatory subunit of PI3 kinase was found to interact with the TβRII and TβRI receptors, and the PI3 kinase activity increases upon TGF-β stimulation 157. Akt is a central regulator of several pathways involved in cell survival, cell size control and cell migration. Downstream from Akt, mTOR is activated resulting in activation of S6 kinase 1, which in turn regulates the translational machinery and confers increased cell size 158.
Activation of the PI3 kinase/Akt pathway by TGF-β plays a major role in EMT. Inhibitors of PI3 kinase and Akt, or a dominant negative form of Akt, were found to inhibit TGF-β-induced morphological transition, α-smooth muscle actin expression and E-cadherin downregulation 151, 159. We found that during EMT, TGF-β induces activation of the mTOR/S6 kinase 1 pathway through PI3 kinase and Akt, resulting in increased protein synthesis and cell size. Furthermore, rapamycin, an inhibitor of mTOR, inhibits the increased cell migration and invasion that are associated with TGF-β-induced EMT, but does not block the phenotypic changes characteristic of EMT 50.
During EMT, activation of S6 kinase 1 is required for the induction of Snail1 expression whereas Snail2 expression is not affected 160. The observation that S6 kinase 1 regulates gene transcription in addition to its effects on protein translation requires further mechanistic characterization. In a lens epithelial cell EMT model, it has been shown that the Smad-activated expression of Snail1 is required for Akt activation via PI3 kinase 76. Furthermore, blocking the PI3 kinase/Akt pathway inhibits TGF-β-induced Smad2 but not Smad3 phosphorylation in a mammary gland epithelial cell line, demonstrating crosstalk and feedback loops between Smad and PI3 kinase/Akt signaling that are not yet well understood 50, 151.
Other mechanisms of signaling crosstalk in TGF-β-induced EMT
As illustrated above, TGF-β signaling activates non-Smad pathways that are also activated by tyrosine kinase receptors or other receptor types in response to their respective ligands. These pathways cooperate with TGF-β/Smad signaling in the execution of the plethora of responses that constitute TGF-β-induced EMT. Thus, activation of the Ras-Erk MAP kinase pathway, p38 MAP kinase and JNK signaling, as well as Rho GTPase signaling and the PI3 kinase/Akt pathway all enhances and contributes to TGF-β-induced EMT. The activation of these pathways by TGF-β, albeit at a lower level than in response to ligands such as growth factors, strongly suggests that TGF-β signaling provides an inherent complementarity that supports the initiation of the EMT process. Other families of ligands activate signaling pathways that are specific to these classes of ligands and receptors, and are not activated by TGF-β family ligands. Some of these, specifically Wnt signaling and Notch signaling, also cooperate with TGF-β signaling in the elaboration of the EMT response (Figure 4).
Figure 4
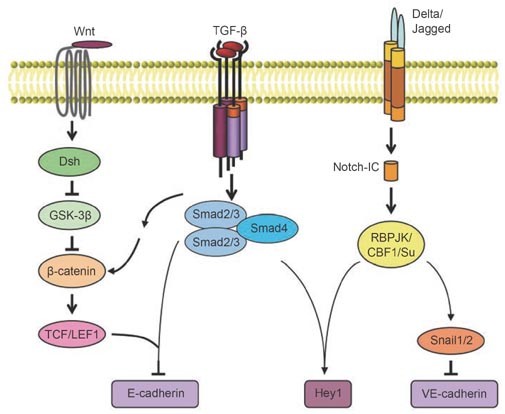
Signaling crosstalk between the TGF-β-activated Smad pathway and the Wnt- and Notch-activated signaling pathways during EMT. TGF-β signaling interacts with the Wnt pathway at multiple levels, by regulating the activity of β-catenin, or through interactions of Smad complexes with β-catenin and/or TCF/LEF1. TGF-β signaling crosstalks with the Notch pathway, through interactions of Smad complexes with RBPJK/CBF1/Su to regulate EMT-related gene transcription.
Wnt signaling
Secreted Wnt proteins do not induce EMT per se, but their “canonical” signaling pathway which controls target gene transcription cooperates with TGF-β signaling to control EMT in some tissues. The canonical Wnt signaling pathway is mediated by β-catenin, which also functions as a component of adherens junctions and links E-cadherin to the cytoskeleton. In the absence of Wnt signaling, GSK-3β phosphorylates β-catenin and targets it for ubiquitination and degradation, thus maintaining cytoplasmic β-catenin at a low level. Activation of Wnt signaling inhibits GSK-3β, leading to accumulation of cytoplasmic β-catenin. This allows the formation of complexes of β-catenin with TCF or LEF1, two closely related transcription factors, which then translocate into the nucleus and regulate target gene transcription in response to Wnt ligands.
Crosstalk between TGF-β/Smad signaling and Wnt signaling have been documented in developmental and pathological events. The mechanistic basis for this crosstalk has been attributed to interactions of activated Smad complexes with the Wnt-activated transcription factor complexes, either with TCF or LEF1 or with β-catenin 161, 162, 163. In the context of EMT, it has been reported that Smad2 and Smad4 form a complex with LEF1 at the E-cadherin promoter, resulting in transcriptional repression in palate medial-edge epithelial cells. Smad4 and LEF1 are also required for the upregulation of mesenchymal markers vimentin and fibronectin, as well as for the acquisition of migratory behavior that characterizes cells that have undergone EMT 163.
During cardiac morphogenesis, which involves EMT induced by TGF-β, β-catenin is an indispensable component downstream of TGF-β. TGF-β2 induces endocardial cells to undergo EMT and invade the cardiac jelly to form the cardiac cushion, which will give rise to atrio-ventricular valves and part of the septum. Mouse embryos that lack β-catenin expression showed defective septum formation, suggesting an impaired EMT process. In fact, β-catenin deficient endocardial cells showed dramatically reduced actin stress fiber formation and failed to invade ex vivo 162. The molecular basis for the role of β-catenin in TGF-β signaling remains to be elucidated.
Notch signaling
Notch signaling occurs in a highly localized manner, when receptors and ligands, both membrane-bound, interact through cellular contacts between neighboring cells. Four receptors (Notch1-4) and two families of ligands (Delta or Delta-like and Serrate or Jagged) have been identified. Notch signaling has been shown to contribute to EMT in both tumor progression and cardiac development. When a ligand binds to the Notch receptor, Notch undergoes two proteolytic cleavage events, first an extracellular cleavage by TACE, a protease from the ADAM family, and then an intracellular cleavage by the γ-secretase complex, to liberate the Notch intracellular domain (NIC) from its membrane anchoring. Upon activation, the NIC enters the nucleus, where it binds to the transcription factor RBPJK/CBF1/Su(H) and converts it from a repressor to an activator for target genes, including the Hes and Hey family genes.
Signaling crosstalk between Notch and TGF-β pathways has been demonstrated, and may be associated with tumor progression. In epithelial cells, Hey1, a well-known target gene of Notch signaling, was found to be required for TGF-β-induced EMT and migration. TGF-β induces a biphasic expression of Hey1: the immediate-early induction is Smad3-dependent and Notch-independent, whereas the delayed induction is Notch-dependent, and mediated by Smad3 and Erk-induced Jagged1 expression 164.
Notch signaling has also been shown to function in the TGF-β-mediated EMT that occurs during endocardial cushion formation. Activation of Notch1 was shown to induce Snail1 expression in mice and Snail2 expression in chicks, which then lead to repression of VE-cadherin expression and promote endocardial cell migration and invasion into the cardiac jelly. Mutations in the Notch pathway components Notch1 or RBPJK cause reduction in TGF-β2 and Snail1 expression, accompanied by persistent VE-cadherin expression 165, 166.
Perspectives
The phenotypic changes during EMT – the loss of epithelial cell-cell junctions, gain of stress fibers and invasive properties – are shared among diverse cell types, but the molecular underpinnings often diverge among tissues from different origins. This multi-tasking is achieved by TGF-β proteins through a complex network of effectors. TGF-β proteins activate both Smad and non-Smad signals, which crosstalk with various signal transduction pathways at multiple levels to provide context-dependent outcomes. Hence, context-specific effects can be generated by distinct strength and duration of Smad and non-Smad pathway activation, changes in levels of interacting protein partners and availability of repressors/activators. Although our understanding of the molecular mechanism of EMT has significantly advanced during the past decade, much work is needed to define the transcriptional regulatory networks and the key target genes that drive EMT in a context-specific way.
References
- Akhurst R . TGF-β signaling in epithelial-mesenchymal transition and invasion and metastasis. In: Derynck R, Miyazono K, eds. The TGF-beta family. Cold Spring Harbor Laboratory Press: New York 2007:939–964.
Google Scholar - Tsukita S, Furuse M, Itoh M . Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2001; 2:285–293.
Article CAS PubMed Google Scholar - Assemat E, Bazellieres E, Pallesi-Pocachard E, Le Bivic A, Massey-Harroche D . Polarity complex proteins. Biochim Biophys Acta 2008; 1778:614–630.
Article CAS PubMed Google Scholar - Martin-Belmonte F, Mostov K . Regulation of cell polarity during epithelial morphogenesis. Curr Opin Cell Biol 2008; 20:227–234.
Article CAS PubMed Google Scholar - Niessen CM, Gottardi CJ . Molecular components of the adherens junction. Biochim Biophys Acta 2008; 1778:562–571.
Article CAS PubMed PubMed Central Google Scholar - Wheelock MJ, Johnson KR . Cadherins as modulators of cellular phenotype. Annu Rev Cell Dev Biol 2003; 19:207–235.
Article CAS PubMed Google Scholar - Llorens A, Rodrigo I, Lopez-Barcons L, et al. Down-regulation of E-cadherin in mouse skin carcinoma cells enhances a migratory and invasive phenotype linked to matrix metalloproteinase-9 gelatinase expression. Lab Invest 1998; 78:1131–1142.
CAS PubMed Google Scholar - Navarro P, Lozano E, Cano A . Expression of E-or P-cadherin is not sufficient to modify the morphology and the tumorigenic behavior of murine spindle carcinoma cells. Possible involvement of plakoglobin. J Cell Sci 1993; 105:923–934.
CAS PubMed Google Scholar - Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004; 117:927–939.
Article CAS PubMed Google Scholar - Ansieau S, Bastid J, Doreau A, et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008; 14:79–89.
Article CAS PubMed Google Scholar - De Craene B, Gilbert B, Stove C, et al. The transcription factor snail induces tumor cell invasion through modulation of the epithelial cell differentiation program. Cancer Res 2005; 65:6237–6244.
Article CAS PubMed Google Scholar - Ohkubo T, Ozawa M . The transcription factor Snail downregulates the tight junction components independently of E-cadherin downregulation. J Cell Sci 2004; 117:1675–1685.
Article CAS PubMed Google Scholar - Garrod D, Chidgey M . Desmosome structure, composition and function. Biochim Biophys Acta 2008; 1778:572–587.
Article CAS PubMed Google Scholar - Yin T, Green KJ . Regulation of desmosome assembly and adhesion. Semin Cell Dev Biol 2004; 15:665–677.
Article CAS PubMed Google Scholar - Cano A, Perez-Moreno MA, Rodrigo I, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2000; 2:76–83.
Article CAS PubMed Google Scholar - Savagner P, Yamada KM, Thiery JP . The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol 1997; 137:1403–1419.
Article CAS PubMed PubMed Central Google Scholar - Miettinen PJ, Ebner R, Lopez AR, Derynck R . TGF-β-induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol 1994; 127:2021–2036.
Article CAS PubMed Google Scholar - Imamichi Y, Menke A . Signaling pathways involved in collagen-induced disruption of the E-cadherin complex during epithelial-mesenchymal transition. Cells Tissues Organs 2007; 185:180–190.
Article CAS PubMed Google Scholar - Zhao Y, Min C, Vora S, et al. The lysyl oxidase pro-peptide attenuates fibronectin-mediated activation of FAK and p130CAS in breast cancer cells. J Biol Chem 2008 Nov 21. doi: 10.1074/jbc.M802612200.
Article CAS PubMed Google Scholar - Frame MC, Inman GJ . NCAM is at the heart of reciprocal regulation of E-cadherinand integrin-mediated adhesions via signaling modulation. Dev Cell 2008; 15:494–496.
Article CAS PubMed Google Scholar - Shirakihara T, Saitoh M, Miyazono K . Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-β. Mol Biol Cell 2007; 18:3533–3544.
Article CAS PubMed PubMed Central Google Scholar - Deckers M, van Dinther M, Buijs J, et al. The tumor suppressor Smad4 is required for transforming growth factor β-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res 2006; 66:2202–2209.
Article CAS PubMed Google Scholar - Cavallaro U, Christofori G . Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer 2004; 4:118–132.
Article CAS PubMed Google Scholar - Grunert S, Jechlinger M, Beug H . Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol 2003; 4:657–665.
Article CAS PubMed Google Scholar - Moustakas A, Heldin CH . Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci 2007; 98:1512–1520.
Article CAS PubMed Google Scholar - Peinado H, Olmeda D, Cano A . Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer 2007; 7:415–428.
Article CAS PubMed Google Scholar - Piek E, Moustakas A, Kurisaki A, Heldin CH, ten Dijke P . TGF-β type I receptor/ALK-5 and Smad proteins mediate epithelial to mesenchymal transdifferentiation in NMuMG breast epithelial cells. J Cell Sci 1999; 112:4557–4568.
CAS PubMed Google Scholar - Valcourt U, Kowanetz M, Niimi H, Heldin CH, Moustakas A . TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol Biol Cell 2005; 16:1987–2002.
Article CAS PubMed PubMed Central Google Scholar - Shook D, Keller R . Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mech Dev 2003; 120:1351–1383.
Article CAS PubMed Google Scholar - Kishigami S, Mishina Y . BMP signaling and early embryonic patterning. Cytokine Growth Factor Rev 2005; 16:265–278.
Article CAS PubMed Google Scholar - Lee JM, Dedhar S, Kalluri R, Thompson EW . The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 2006; 172:973–981.
Article CAS PubMed PubMed Central Google Scholar - Nakajima Y, Yamagishi T, Hokari S, Nakamura H . Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-β and bone morphogenetic protein (BMP). Anat Rec 2000; 258:119–127.
Article CAS PubMed Google Scholar - Mercado-Pimentel ME, Runyan RB . Multiple transforming growth factor-β isoforms and receptors function during epithelial-mesenchymal cell transformation in the embryonic heart. Cells Tissues Organs 2007; 185:146–156.
Article CAS PubMed Google Scholar - Azhar M, Schultz Jel J, Grupp I, et al. Transforming growth factor β in cardiovascular development and function. Cytokine Growth Factor Rev 2003; 14:391–407.
Article CAS PubMed PubMed Central Google Scholar - Pelton RW, Hogan BL, Miller DA, Moses HL . Differential expression of genes encoding TGFs β1, β2, and β3 during murine palate formation. Dev Biol 1990; 141:456–460.
Article CAS PubMed Google Scholar - Nawshad A, LaGamba D, Hay ED . Transforming growth factor β (TGFβ) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch Oral Biol 2004; 49:675–689.
Article CAS PubMed Google Scholar - Klattig J, Englert C . The Müllerian duct: recent insights into its development and regression. Sex Dev 2007; 1:271–278.
Article CAS PubMed Google Scholar - Hugo H, Ackland ML, Blick T, et al. Epithelial-mesenchymal and mesenchymal-epithelial transitions in carcinoma progression. J Cell Physiol 2007; 213:374–383.
Article CAS PubMed Google Scholar - Cui W, Fowlis DJ, Bryson S, et al. TGFβ1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell 1996; 86:531–542.
Article CAS PubMed Google Scholar - Derynck R, Akhurst RJ, Balmain A . TGF-β signaling in tumor suppression and cancer progression. Nat Genet 2001; 29:117–129.
Article CAS PubMed Google Scholar - Iwano M, Plieth D, Danoff TM, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 2002; 110:341–350.
Article CAS PubMed PubMed Central Google Scholar - Schnaper HW, Hayashida T, Hubchak SC, Poncelet AC . TGF-β signal transduction and mesangial cell fibrogenesis. Am J Physiol Renal Physiol 2003; 284:F243–F252.
Article CAS PubMed Google Scholar - Willis BC, Borok Z . TGF-β-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol 2007; 293:L525–L534.
Article CAS PubMed Google Scholar - Gressner AM, Weiskirchen R, Breitkopf K, Dooley S . Roles of TGF-β in hepatic fibrosis. Front Biosci 2002; 7:d793–d807.
Article CAS PubMed Google Scholar - Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 2007; 13:952–961.
Article CAS PubMed Google Scholar - Feng XH, Derynck R . Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol 2005; 21:659–693.
Article CAS PubMed Google Scholar - Portella G, Cumming SA, Liddell J, et al. Transforming growth factor β is essential for spindle cell conversion of mouse skin carcinoma in vivo: implications for tumor invasion. Cell Growth Differ 1998; 9:393–404.
CAS PubMed Google Scholar - Oft M, Heider KH, Beug H . TGFβ signaling is necessary for carcinoma cell invasiveness and metastasis. Curr Biol 1998; 8:1243–1252.
Article CAS PubMed Google Scholar - Han G, Lu SL, Li AG, et al. Distinct mechanisms of TGF-β1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. J Clin Invest 2005; 115:1714–1723.
Article CAS PubMed PubMed Central Google Scholar - Lamouille S, Derynck R . Cell size and invasion in TGF-β-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 2007; 178:437–451.
Article CAS PubMed PubMed Central Google Scholar - Eger A, Stockinger A, Park J, et al. β-Catenin and TGFβ signalling cooperate to maintain a mesenchymal phenotype after FosER-induced epithelial to mesenchymal transition. Oncogene 2004; 23:2672–2680.
Article CAS PubMed Google Scholar - Mercado-Pimentel ME, Hubbard AD, Runyan RB . Endoglin and Alk5 regulate epithelial-mesenchymal transformation during cardiac valve formation. Dev Biol 2007; 304:420–432.
Article CAS PubMed Google Scholar - Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A . Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest 2003; 112:1486–1494.
Article CAS PubMed PubMed Central Google Scholar - Ashcroft GS, Yang X, Glick AB, et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol 1999; 1:260–266.
Article CAS PubMed Google Scholar - Hoot KE, Lighthall J, Han G, et al. Keratinocyte-specific Smad2 ablation results in increased epithelial-mesenchymal transition during skin cancer formation and progression. J Clin Invest 2008; 118:2722–2732.
CAS PubMed PubMed Central Google Scholar - Ju W, Ogawa A, Heyer J, et al. Deletion of Smad2 in mouse liver reveals novel functions in hepatocyte growth and differentiation. Mol Cell Biol 2006; 26:654–667.
Article CAS PubMed PubMed Central Google Scholar - Oft M, Akhurst RJ, Balmain A . Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat Cell Biol 2002; 4:487–494.
Article CAS PubMed Google Scholar - Takano S, Kanai F, Jazag A, et al. Smad4 is essential for down-regulation of E-cadherin induced by TGF-β in pancreatic cancer cell line PANC-1. J Biochem 2007; 141:345–351.
Article CAS PubMed Google Scholar - Kaimori A, Potter J, Kaimori JY, et al. Transforming growth factor-β1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in vitro. J Biol Chem 2007; 282:22089–22101.
Article CAS PubMed Google Scholar - Bardeesy N, Cheng KH, Berger JH, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 2006; 20:3130–3146.
Article CAS PubMed PubMed Central Google Scholar - Desgrosellier JS, Mundell NA, McDonnell MA, Moses HL, Barnett JV . Activin receptor-like kinase 2 and Smad6 regulate epithelial-mesenchymal transformation during cardiac valve formation. Dev Biol 2005; 280:201–210.
Article CAS PubMed Google Scholar - Armstrong EJ, Bischoff J . Heart valve development: endothelial cell signaling and differentiation. Circ Res 2004; 95:459–470.
Article CAS PubMed PubMed Central Google Scholar - Saika S, Ikeda K, Yamanaka O, et al. Transient adenoviral gene transfer of Smad7 prevents injury-induced epithelial-mesenchymal transition of lens epithelium in mice. Lab Invest 2004; 84:1259–1270.
Article CAS PubMed Google Scholar - Xu GP, Li QQ, Cao XX, et al. The fffect of TGF-β1 and SMAD7 gene transfer on the phenotypic changes of rat alveolar epithelial cells. Cell Mol Biol Lett 2007; 12:457–472.
Article CAS PubMed PubMed Central Google Scholar - Dooley S, Hamzavi J, Ciuclan L, et al. Hepatocyte-specific Smad7 expression attenuates TGF-β-mediated fibrogenesis and protects against liver damage. Gastroenterology 2008; 135:642–659.
Article CAS PubMed Google Scholar - Zavadil J, Böttinger EP . TGF-β and epithelial-to-mesenchymal transitions. Oncogene 2005; 24:5764–5774.
Article CAS PubMed Google Scholar - Nieto MA . The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol 2002; 3:155–166.
Article CAS PubMed Google Scholar - Barrallo-Gimeno A, Nieto MA . The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development 2005; 132:3151–3161.
Article CAS PubMed Google Scholar - Batlle E, Sancho E, Franci C, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2000; 2:84–89.
Article CAS PubMed Google Scholar - Hemavathy K, Guru SC, Harris J, Chen JD, Ip YT . Human Slug is a repressor that localizes to sites of active transcription. Mol Cell Biol 2000; 20:5087–5095.
Article CAS PubMed PubMed Central Google Scholar - Jamora C, Lee P, Kocieniewski P, et al. A signaling pathway involving TGF-β2 and snail in hair follicle morphogenesis. PLoS Biol 2005; 3:e11.
Article CAS PubMed Google Scholar - Martinez-Alvarez C, Blanco MJ, Perez R, et al. Snail family members and cell survival in physiological and pathological cleft palates. Dev Biol 2004; 265:207–218.
Article CAS PubMed Google Scholar - Yanez-Mo M, Lara-Pezzi E, Selgas R, et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N Engl J Med 2003; 348:403–413.
Article PubMed Google Scholar - Peinado H, Quintanilla M, Cano A . Transforming growth factor β1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. J Biol Chem 2003; 278:21113–21123.
Article CAS PubMed Google Scholar - Romano LA, Runyan RB . Slug is an essential target of TGFβ2 signaling in the developing chicken heart. Dev Biol 2000; 223:91–102.
Article CAS PubMed Google Scholar - Cho HJ, Baek KE, Saika S, Jeong MJ, Yoo J . Snail is required for transforming growth factor-β-induced epithelial-mesenchymal transition by activating PI3 kinase/Akt signal pathway. Biochem Biophys Res Commun 2007; 353:337–343.
Article CAS PubMed Google Scholar - Morita T, Mayanagi T, Sobue K . Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J Cell Biol 2007; 179:1027–1042.
Article CAS PubMed PubMed Central Google Scholar - Grotegut S, von Schweinitz D, Christofori G, Lehembre F . Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J 2006; 25:3534–3545.
Article CAS PubMed PubMed Central Google Scholar - Olmeda D, Jorda M, Peinado H, Fabra A, Cano A . Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene 2007; 26:1862–1874.
Article CAS PubMed Google Scholar - Bolos V, Peinado H, Perez-Moreno MA, et al. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci 2003; 116:499–511.
Article CAS PubMed Google Scholar - Hajra KM, Chen DY, Fearon ER . The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res 2002; 62:1613–1618.
CAS PubMed Google Scholar - Carver EA, Jiang R, Lan Y, Oram KF, Gridley T . The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol Cell Biol 2001; 21:8184–8188.
Article CAS PubMed PubMed Central Google Scholar - Nieto MA, Sargent MG, Wilkinson DG, Cooke J . Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science 1994; 264:835–839.
Article CAS PubMed Google Scholar - Ikenouchi J, Matsuda M, Furuse M, Tsukita S . Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J Cell Sci 2003; 116:1959–1967.
Article CAS PubMed Google Scholar - Kajita M, McClinic KN, Wade PA . Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol 2004; 24:7559–7566.
Article CAS PubMed PubMed Central Google Scholar - Wang Z, Wade P, Mandell KJ, et al. Raf 1 represses expression of the tight junction protein occludin via activation of the zinc-finger transcription factor slug. Oncogene 2007; 26:1222–1230.
Article CAS PubMed Google Scholar - Kurrey NK, K A, Bapat SA . Snail and Slug are major determinants of ovarian cancer invasiveness at the transcription level. Gynecol Oncol 2005; 97:155–165.
Article CAS PubMed Google Scholar - Whiteman EL, Liu CJ, Fearon ER, Margolis B . The transcription factor snail represses Crumbs3 expression and disrupts apico-basal polarity complexes. Oncogene 2008; 27:3875–3879.
Article CAS PubMed PubMed Central Google Scholar - Guaita S, Puig I, Franci C, et al. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J Biol Chem 2002; 277:39209–39216.
Article CAS PubMed Google Scholar - Moreno-Bueno G, Cubillo E, Sarrio D, et al. Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer Res 2006; 66:9543–9556.
Article CAS PubMed Google Scholar - Boutet A, De Frutos CA, Maxwell PH, et al. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J 2006; 25:5603–5613.
Article CAS PubMed PubMed Central Google Scholar - Boutet A, Esteban MA, Maxwell PH, Nieto MA . Reactivation of Snail genes in renal fibrosis and carcinomas: a process of reversed embryogenesis? Cell Cycle 2007; 6:638–642.
Article CAS PubMed Google Scholar - Cicchini C, Filippini D, Coen S, et al. Snail controls differentiation of hepatocytes by repressing HNF4α expression. J Cell Physiol 2006; 209:230–238.
Article CAS PubMed Google Scholar - Hattrup CL, Gendler SJ . Structure and function of the cell surface (tethered) mucins. Annu Rev Physiol 2008; 70:431–457.
Article CAS PubMed Google Scholar - Taki M, Verschueren K, Yokoyama K, Nagayama M, Kamata N . Involvement of Ets-1 transcription factor in inducing matrix metalloproteinase-2 expression by epithelial-mesenchymal transition in human squamous carcinoma cells. Int J Oncol 2006; 28:487–496.
CAS PubMed Google Scholar - Jorda M, Olmeda D, Vinyals A, et al. Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J Cell Sci 2005; 118:3371–3385.
Article CAS PubMed Google Scholar - Postigo AA, Depp JL, Taylor JJ, Kroll KL . Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J 2003; 22:2453–2462.
Article CAS PubMed PubMed Central Google Scholar - Postigo AA . Opposing functions of ZEB proteins in the regulation of the TGFβ/BMP signaling pathway. EMBO J 2003; 22:2443–2452.
Article CAS PubMed PubMed Central Google Scholar - Nishimura G, Manabe I, Tsushima K, et al. DeltaEF1 mediates TGF-β signaling in vascular smooth muscle cell differentiation. Dev Cell 2006; 11:93–104.
Article CAS PubMed Google Scholar - Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10:593–601.
Article CAS PubMed Google Scholar - Park SM, Gaur AB, Lengyel E, Peter ME . The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 2008; 22:894–907.
Article CAS PubMed PubMed Central Google Scholar - Korpal M, Lee ES, Hu G, Kang Y . The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem 2008; 283:14910–14914.
Article CAS PubMed PubMed Central Google Scholar - Long J, Zuo D, Park M . Pc2-mediated sumoylation of Smad-interacting protein 1 attenuates transcriptional repression of E-cadherin. J Biol Chem 2005; 280:35477–35489.
Article CAS PubMed Google Scholar - Comijn J, Berx G, Vermassen P, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 2001; 7:1267–1278.
Article CAS PubMed Google Scholar - Grooteclaes ML, Frisch SM . Evidence for a function of CtBP in epithelial gene regulation and anoikis. Oncogene 2000; 19:3823–3828.
Article CAS PubMed Google Scholar - Eger A, Aigner K, Sonderegger S, et al. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005; 24:2375–2385.
Article CAS PubMed Google Scholar - Vandewalle C, Comijn J, De Craene B, et al. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res 2005; 33:6566–6578.
Article CAS PubMed PubMed Central Google Scholar - Bindels S, Mestdagt M, Vandewalle C, et al. Regulation of vimentin by SIP1 in human epithelial breast tumor cells. Oncogene 2006; 25:4975–4985.
Article CAS PubMed Google Scholar - Spaderna S, Schmalhofer O, Wahlbuhl M, et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res 2008; 68:537–544.
Article CAS PubMed Google Scholar - Aigner K, Dampier B, Descovich L, et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 2007; 26:6979–6988.
Article CAS PubMed PubMed Central Google Scholar - Massari ME, Murre C . Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol 2000; 20:429–440.
Article CAS PubMed PubMed Central Google Scholar - Perez-Moreno MA, Locascio A, Rodrigo I, et al. A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem 2001; 276:27424–27431.
Article CAS PubMed Google Scholar - Kondo M, Cubillo E, Tobiume K, et al. A role for Id in the regulation of TGF-β-induced epithelial-mesenchymal transdifferentiation. Cell Death Differ 2004; 11:1092–1101.
Article CAS PubMed Google Scholar - Kowanetz M, Valcourt U, Bergstrom R, Heldin CH, Moustakas A . Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol Cell Biol 2004; 24:4241–4254.
Article CAS PubMed PubMed Central Google Scholar - Kang Y, Chen CR, Massagué J . A self-enabling TGFβ response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell 2003; 11:915–926.
Article CAS PubMed Google Scholar - Chen ZF, Behringer RR . Twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev 1995; 9:686–699.
Article CAS PubMed Google Scholar - el Ghouzzi V, Le Merrer M, Perrin-Schmitt F, et al. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nature Genetics 1997; 15:42–46.
Article CAS PubMed Google Scholar - Howard TD, Paznekas WA, Green ED, et al. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nature Genetics 1997; 15:36–41.
Article PubMed Google Scholar - Yang J, Mani SA, Weinberg RA . Exploring a new twist on tumor metastasis. Cancer Res 2006; 66:4549–4552.
Article CAS PubMed Google Scholar - Thuault S, Valcourt U, Petersen M, et al. Transforming growth factor-β employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol 2006; 174:175–183.
Article CAS PubMed PubMed Central Google Scholar - Derynck R, Zhang YE . Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003; 425:577–584.
Article CAS PubMed Google Scholar - Kretzschmar M, Doody J, Timokhina I, Massagué J . A mechanism of repression of TGFβ/ Smad signaling by oncogenic Ras. Genes Dev 1999; 13:804–816.
Article CAS PubMed PubMed Central Google Scholar - Funaba M, Zimmerman CM, Mathews LS . Modulation of Smad2-mediated signaling by extracellular signal-regulated kinase. J Biol Chem 2002; 277:41361–41368.
Article CAS PubMed Google Scholar - Lehmann K, Janda E, Pierreux CE, et al. Raf induces TGFβ production while blocking its apoptotic but not invasive responses: a mechanism leading to increased malignancy in epithelial cells. Genes Dev 2000; 14:2610–2622.
Article CAS PubMed PubMed Central Google Scholar - Davies M, Robinson M, Smith E, et al. Induction of an epithelial to mesenchymal transition in human immortal and malignant keratinocytes by TGF-β1 involves MAPK, Smad and AP-1 signalling pathways. J Cell Biochem 2005; 95:918–931.
Article CAS PubMed Google Scholar - Lee MK, Pardoux C, Hall MC, et al. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J 2007; 26:3957–3967.
Article CAS PubMed PubMed Central Google Scholar - Galliher AJ, Schiemann WP . Src phosphorylates Tyr284 in TGF-β type II receptor and regulates TGF-β stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res 2007; 67:3752–3758.
Article CAS PubMed Google Scholar - Grande M, Franzén A, Karlsson JO, et al. Transforming growth factor-β and epidermal growth factor synergistically stimulate epithelial to mesenchymal transition (EMT) through a MEK-dependent mechanism in primary cultured pig thyrocytes. J Cell Sci 2002; 115:4227–4236.
Article CAS PubMed Google Scholar - Janda E, Lehmann K, Killisch I, et al. Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol 2002; 156:299–313.
Article CAS PubMed PubMed Central Google Scholar - Uttamsingh S, Bao X, Nguyen KT, et al. Synergistic effect between EGF and TGF-β1 in inducing oncogenic properties of intestinal epithelial cells. Oncogene 2008; 27:2626–2634.
Article CAS PubMed Google Scholar - Xie L, Law BK, Chytil AM, et al. Activation of the Erk pathway is required for TGF-β1-induced EMT in vitro. Neoplasia 2004; 6:603–610.
Article CAS PubMed PubMed Central Google Scholar - Schmidt CR, Gi YJ, Patel TA, et al. E-cadherin is regulated by the transcriptional repressor SLUG during Ras-mediated transformation of intestinal epithelial cells. Surgery 2005; 138:306–312.
Article PubMed Google Scholar - Marchetti A, Colletti M, Cozzolino AM, et al. ERK5/MAPK is activated by TGFβ in hepatocytes and required for the GSK-3β-mediated Snail protein stabilization. Cell Signal 2008; 20:2113–2118.
Article CAS PubMed Google Scholar - Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL . p38 mitogen-activated protein kinase is required for TGFβ-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci 2002; 115:3193–3206.
CAS PubMed Google Scholar - Yu L, Hébert MC, Zhang YE . TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. EMBO J 2002; 21:3749–3759.
Article CAS PubMed PubMed Central Google Scholar - Sano Y, Harada J, Tashiro S, et al. ATF-2 is a common nuclear target of Smad and TAK1 pathways in transforming growth factor-signaling. J Biol Chem 1999; 274:8949–8957.
Article CAS PubMed Google Scholar - Santibanez JF . JNK mediates TGF-β1-induced epithelial mesenchymal transdifferentiation of mouse transformed keratinocytes. FEBS Lett 2006; 580:5385–5391.
Article CAS PubMed Google Scholar - Alcorn JF, Guala AS, van der Velden J, et al. Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal transition induced by TGF-β1. J Cell Sci 2008; 121:1036–1045.
Article CAS PubMed Google Scholar - Liu Q, Mao H, Nie J, et al. Transforming growth factor β1 induces epithelial-mesenchymal transition by activating the JNK-Smad3 pathway in rat peritoneal mesothelial cells. Perit Dial Int 2008; 28 Suppl 3:S88–S95.
CAS PubMed Google Scholar - Han S, Ritzenthaler JD, Sitaraman SV, Roman J. Fibronectin increases matrix metalloproteinase 9 expression through activation of c-Fos via extracellular-regulated kinase and phosphatidylinositol 3-kinase pathways in human lung carcinoma cells. J Biol Chem 2006; 281:29614–29624.
Article CAS PubMed Google Scholar - Wang W, Zhou G, Hu M, Yao Z, Tan TH . Activation of the hematopoietic progenitor kinase-1 (HPK1)-dependent, Stress activated c-Jun N-terminal Kinase (JNK) pathway by transforming growth factor-β (TGF-β)-activated Kinase (TAK1), a kinase mediator of TGF-β signal transduction. J Biol Chem 1997; 272:22771–22775.
Article CAS PubMed Google Scholar - Delaney JR, Mlodzik M . TGF-β activated kinase-1: new insights into the diverse roles of TAK1 in development and immunity. Cell Cycle 2006; 5:2852–2855.
Article CAS PubMed Google Scholar - Yamashita M, Fatyol K, Jin C, et al. TRAF6 mediates Smad-indepent activation of JNK and p38 by TGF-β. Mol Cell 2008; 31:918–924.
Article CAS PubMed PubMed Central Google Scholar - Sorrentino A, Thakur N, Grimsby S, et al. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol 2008; 10:1199–1207.
Article CAS PubMed Google Scholar - Tavares AL, Mercado-Pimentel ME, Runyan RB, Kitten GT . TGF-β-mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Dev Dyn 2006; 235:1589–1598.
Article CAS PubMed Google Scholar - Cho HJ, Yoo J . Rho activation is required for transforming growth factor-β-induced epithelial-mesenchymal transition in lens epithelial cells. Cell Biol Int 2007; 31:1225–1230.
Article CAS PubMed Google Scholar - Pellegrini S, Mellor H . Actin stress fibres. J Cell Sci 2007; 120:3491–3499.
Article CAS Google Scholar - Vardouli L, Moustakas A, Stournaras C . LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-β. J Biol Chem 2005; 280:11448–11457.
Article CAS PubMed Google Scholar - Bhowmick NA, Ghiassi M, Bakin A, et al. Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell 2001; 12:27–36.
Article CAS PubMed PubMed Central Google Scholar - Ozdamar B, Bose R, Barrios-Rodiles M, et al. Regulation of the polarity protein Par6 by TGFβ receptors controls epithelial cell plasticity. Science 2005; 307:1603–1609.
Article CAS PubMed Google Scholar - Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL . Phosphatidylinositol 3-kinase function is required for transforming growth factor-β-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem 2000; 275:36803–36810.
Article CAS PubMed Google Scholar - Lee YI, Kwon YJ, Joo CK . Integrin-linked kinase function is required for transforming growth factor β-mediated epithelial to mesenchymal transition. Biochem Biophys Res Commun 2004; 316:997–1001.
Article CAS PubMed Google Scholar - Rodriguez-Barbero A, Dorado F, Velasco S, et al. TGF-β1 induces COX-2 expression and PGE2 synthesis through MAPK and PI3K pathways in human mesangial cells. Kidney Int 2006; 70:901–909.
Article CAS PubMed Google Scholar - Lien SC, Usami S, Chien S, Chiu JJ . Phosphatidylinositol 3-kinase/Akt pathway is involved in transforming growth factor-β1-induced phenotypic modulation of 10T1/2 cells to smooth muscle cells. Cell Signal 2006; 18:1270–1278.
Article CAS PubMed Google Scholar - Lin CC, Chiang LL, Lin CH, et al. Transforming growth factor-β1 stimulates heme oxygenase-1 expression via the PI3K/Akt and NF-κB pathways in human lung epithelial cells. Eur J Pharmacol 2007; 560:101–109.
Article CAS PubMed Google Scholar - Yeh YY, Chiao CC, Kuo WY, et al. TGF-β1 increases motility and αvβ3 integrin up-regulation via PI3K, Akt and NF-κB-dependent pathway in human chondrosarcoma cells. Biochem Pharmacol 2008; 75:1292–1301.
Article CAS PubMed Google Scholar - Yi JY, Shin I, Arteaga CL . Type I transforming growth factor β receptor binds to and activates phosphatidylinositol 3-kinase. J Biol Chem 2005; 280:10870–10876.
Article CAS PubMed Google Scholar - Sarbassov DD, Ali SM, Sabatini DM . Growing roles for the mTOR pathway. Curr Opin Cell Biol 2005; 17:596–603.
Article CAS PubMed Google Scholar - Kattla JJ, Carew RM, Heljic M, Godson C, Brazil DP . Protein kinase B/Akt activity is involved in renal TGF-β1-driven epithelial-mesenchymal transition in vitro and in vivo. Am J Physiol Renal Physiol 2008; 295:F215–225.
Article CAS PubMed PubMed Central Google Scholar - Pon YL, Zhou HY, Cheung AN, Ngan HY, Wong AS . p70 S6 kinase promotes epithelial to mesenchymal transition through snail induction in ovarian cancer cells. Cancer Res 2008; 68:6524–6532.
Article CAS PubMed Google Scholar - Nelson WJ, Nusse R . Convergence of Wnt, β-catenin, and cadherin pathways. Science 2004; 303:1483–1487.
Article CAS PubMed PubMed Central Google Scholar - Liebner S, Cattelino A, Gallini R, et al. β-catenin is required for endothelial-mesenchymal transformation during heart cushion development in the mouse. J Cell Biol 2004; 166:359–367.
Article CAS PubMed PubMed Central Google Scholar - Nawshad A, Medici D, Liu CC, Hay ED . TGFβ3 inhibits E-cadherin gene expression in palate medial-edge epithelial cells through a Smad2-Smad4-LEF1 transcription complex. J Cell Sci 2007; 120:1646–1653.
Article CAS PubMed Google Scholar - Zavadil J, Cermak L, Soto-Nieves N, Böttinger EP . Integration of TGF-β/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J 2004; 23:1155–1165.
Article CAS PubMed PubMed Central Google Scholar - Grego-Bessa J, Diez J, Timmerman L, de la Pompa JL . Notch and epithelial-mesenchyme transition in development and tumor progression: another turn of the screw. Cell Cycle 2004; 3:718–721.
Article CAS PubMed Google Scholar - Timmerman LA, Grego-Bessa J, Raya A, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev 2004; 18:99–115.
Article CAS PubMed PubMed Central Google Scholar
Author information
Authors and Affiliations
- Department of Cell and Tissue Biology, Programs in Cell Biology and Developmental Biology, University of California-San Francisco, San Francisco, CA, USA
Jian Xu, Samy Lamouille & Rik Derynck
Authors
- Jian Xu
- Samy Lamouille
- Rik Derynck
Corresponding author
Correspondence toRik Derynck.
Rights and permissions
About this article
Cite this article
Xu, J., Lamouille, S. & Derynck, R. TGF-β-induced epithelial to mesenchymal transition.Cell Res 19, 156–172 (2009). https://doi.org/10.1038/cr.2009.5
- Published: 20 January 2009
- Issue Date: February 2009
- DOI: https://doi.org/10.1038/cr.2009.5