Novel evidence that crosstalk between the complement, coagulation and fibrinolysis proteolytic cascades is involved in mobilization of hematopoietic stem/progenitor cells (HSPCs) (original) (raw)
Introduction
Hematopoietic stem/progenitor cells (HSPCs) express the chemokine receptor CXCR4 and the very late antigen-4 receptor (VLA-4, also known as α4β1 integrin) on their surface and are retained in bone marrow (BM) niches by interaction of these receptors with their respective ligands, α-chemokine stromal-derived growth factor-1 (SDF-1) and vascular adhesion molecule-1 (VCAM-1, also known as CD106) that are expressed by cells in the BM microenvironment (for example, osteoblasts and fibroblasts).1, 2, 3, 4, 5
HSPCs residing in BM are released from their niches and circulate under steady-state conditions at detectable levels in the peripheral blood (PB), and their number increases in response to (1) systemic or local inflammation, (2) strenuous exercise, (3) stress, (4) tissue/organ injury and (5) pharmacological agents.6, 7, 8 All these processes involve activation of the complement cascade (ComC), and mice deficient in a downstream component of ComC, complement protein 5 (C5), are very poor mobilizers.9 This has been explained by demonstration that the C5 cleavage fragment C5a, which is activated in BM sinusoids, is crucial for egress of granulocytes and monocytes from the BM and that these cells pave the way for HSPCs through the PB–BM barrier during mobilization.9 At the same time, C5a activates granulocytes and monocytes to release several proteolytic enzymes in the BM microenvironment that attenuate SDF-1–CXCR4 and VLA4–VCAM-1 retention signals in BM niches.4, 9
It is also known that activation of the ComC, similar to coagulation cascade (CoaC) and fibrynolytic cascade (FibC), is based on sequential activation of proteolytic proenzymes from the top to the bottom of the cascade.10, 11 Therefore, the lack of upstream C3 should theoretically affect generation of ComC-generated C5 convertase, a proteolytic enzyme activating C5. Surprisingly, C3−/− mice are easy mobilizers12 that suggests that other proteolytic enzymes in blood plasma substitute for ComC-generated C5 convertase. To explain how C5 can be activated during the mobilization process even when C3 is missing, we hypothesized that other proteases that are products of the activated CoaC and FibC compensate for the lack of proteolytic activity of ComC-derived C5 convertase. In support of this hypothesis, it has been demonstrated that both CoaC and FibC have vigorous crosstalk with ComC during some innate immunity-mediated responses.10 It has also been demonstrated that thrombin (Dr T Lapidot, personal communication) or plasmin administration13 may enhance mobilization of HSPCs, suggesting a role for these enzymes in the mobilization process. To support this further, G-CSF-induced mobilization of HSPCs was facilitated in plasminogen activator inhibitor-1- and plasmin inhibitor-α2 antiplasmin-deficient mice.13 Moreover, both membrane-anchored plasminogen activator, urokinase receptor14 and cleaved form of soluble urokinase receptor15 have been implicated in HSPCs mobilization.
In our experiments, mobilization was evaluated in C3-deficient mice (C3−/−) and normal wild-type (WT) littermates mobilized by granulocyte colony-stimulating factor (G-CSF) or the CXCR4 receptor antagonist AMD3100 in the presence or absence of refludan (a direct inhibitor of thrombin) and tranexamic acid (an inhibitor of plasminogen activation). In parallel, we measured the activation of all three cascades by detecting the level of C5a in PB and measuring prothrombin time(PT) and activated partial thromboplastin time (APTT) as well as the concentrations of thrombin/antithrombin and plasmin/antiplasmin complexes.
The data presented in this work demonstrate for the first time the existence of vigorous crosstalk between all three evolutionarily ancient proteolytic enzyme cascades, the ComC, CoaC and FibC, in the process of mobilizing HSPCs. We observed that G-CSF-induced mobilization of HSPCs was significantly reduced in normally easy-mobilizing C3−/− mice when the mice were treated with refludan (a CoaC inhibitor) or tranexamic acid (an FibC inhibitor) and that this reduction correlated with significant inhibition of C5 activation/cleavage. Significantly, we also observed that inhibitors of the CoaC and FibC had a negative effect on mobilization of HSPCs in normal WT animals. Our observations of crosstalk between the ComC, CoaC and FibC better explain the effect of thrombin and plasmin on stem cell mobilization and may lead to the development of more efficient mobilization strategies in poor mobilizers and mobilized patients who are treated with drugs that interfere with CoaC/FibC.
Materials and methods
Animals
Mobilization experiments were performed on 6–8-week-old C57B1/6 C3−/− and WT C57Bl/6 C3+/+ age- and sex-matched littermates (breeding colony purchased from The Jackson Laboratory, Bar Harbor, ME, USA). C57B1/6 and C3−/− mice were identified by screening for serum C3 protein levels (using radial immunodiffusion), and before the experiment, PCR was used to confirm the genotype of the C3−/− mice. Animal studies were approved by the Animal Care and Use Committee of the University of Louisville (Louisville, KY, USA).
Mobilization studies
Mice were mobilized by G-CSF (6 days, 250 μg/kg, subcutaneously) or AMD3100 (5 mg/kg subcutaneous) in the absence or presence of refludan, a direct inhibitor of thrombin (5 mg/kg/day, divided in two doses; Bayer, Whippany, NJ, USA) or tranexamic acid, an inhibitor of plasminogen activation (20 mg/mice/day; Sigma-Aldrich, MO, USA). Control mice were injected with vehicle, refludan or tranexamic acid only. At 6 h after the last G-CSF injection or 1 h after AMD3100 injection, PB was obtained from the vena cava (with a 25-gauge needle and 1 ml syringe containing 250 U heparin).
Peripheral blood parameter counts
To obtain leukocyte counts, blood samples were collected from the retro-orbital plexus of the mice into microvette EDTA-coated tubes (Sarstedt Inc., Newton, NC, USA) and run within 3 h of collection on a HemaVet 950 hematology analyzer (Drew Scientific Inc., Oxford, CT, USA; http://www.drew-scientific.com).
Fluorescence-activated cell sorting analysis of SKL cells
The following monoclonal antibodies were employed to stain Sca-1+/c-Kit+/Lin– (SKL cells): biotin-conjugated rat anti-mouse Ly-6A/E (Sca-1, clone E13–161.7), streptavidin–phycoerythrin (PE)–Cy5-conjugated anti-mouse c-Kit (clone 2B8) and lineage markers anti-mouse CD45R/B220–PE (clone RA3–6B2), anti-mouse TCRab–PE (clone H57–597), anti-mouse TCRγζ–PE (clone GL3), anti-mouse CD11b–PE (clone M1/70), anti-mouse Ter119–PE (clone TER-119) and anti-mouse Gr-1–PE (clone RB6–8C5), as described.4 All monoclonal antibodies were added at saturating concentrations, and the cells were then incubated for 30 min on ice, washed twice, resuspended in RPMI-1640+2% fetal bovine serum, and analyzed with an LSR II flow cytometer (BD, San Diego, CA, USA).
Enumeration of the number of colony-forming unit-granulocyte/macrophage (CFU-GM) mobilized into PB
After PB red blood cell lysis (BD Pharm Lyse Buffer, San Jose, CA, USA), nucleated cells were washed, counted and 1 × 106 cells were resuspended in 10% culture medium with 90% human methylcellulose base media supplemented with 25 ng/ml recombinant murine GM-CSF and 10 ng/ml recombinant murine IL-3 (PeproTech, Rocky Hill, NJ, USA). After 1 week of culture, the numbers of CFU-GM colonies were scored using an inverted microscope (Olympus, Center Valley, PA, USA).
Evaluation of HSPC mobilization
For evaluation of circulating CFU-GM and Sca-1+/c-Kit+/Lin− (SKL) cells, the following formulas were used: (number of white blood cells (WBCs) × number of CFU-GM colonies)/number of WBCs plated=number of CFU-GM per μl of PB; and (number of WBCs × number of SKL cells)/number of gated WBCs=number of SKL cells per μl of PB.
Plasma concentration of C5b-C9 (MAC complex)
The concentration of C5b-C9 was measured by employing the commercially available, highly sensitive enzyme-linked immunosorbent assay (ELISA) kit K-ASSAY (Kamiya Biomedical Company, Seattle, WA, USA), according to the manufacturer’s protocol. For analysis, PB from C3−/− mice was collected on day 6 of G-CSF-induced mobilization by retro-orbital plexus bleeding into cold microvette EDTA-coated tubes (Sarstedt Inc.). Subsequently, blood was centrifuged at 2000 g for 20 min in 4 °C to obtain plasma.
Activation of coagulation cascade
PT and APTT were evaluated by employing coagulometry. For measurement of CoaC components, PB from C57Bl/6 mice was collected on day 6 after G-CSF- or 1 h AMD3100-induced mobilization into tubes with 3.2% sodium citrate (ratio: 1:9). Subsequently, blood was centrifuged for 10 min at 700 g at 4 °C to obtain plasma. PT and APTT were measured within 4 h of blood collection. For PT evaluation, THROMBOPLASTIN PT-S (Cormay, Lomianki, Poland) was warmed to 37 °C, and 100 μl was added to 0.05 ml of serum. The time of appearance of a clot was measured using a coagulometer. For APTT evaluation, to 0.05 ml of serum, 0.05 ml of APTT-P-REAGENT (Cormay) was added, and after 5 min of incubation (37 °C), calcium chloride (warmed to 37 °C) was added. The time of appearance of a clot was evaluated using a coagulometer. Thrombin/antithrombin complexes were measured by employing ELISA assay, according to the manufacturer’s protocols (USCN Life Science, Wuhan, China).
Activation of fibrinolysis cascade
Plasmin/antiplasmin complexes was measured by employing ELISA assay, according to the manufacturer’s protocols (USCN Life Science). Plasma was collected in the same way, as reported above.
Statistical analysis
Arithmetic means and s.d. were calculated using Excel. Statistical significance was defined as P<0.05. Data were analyzed using Student’s _t_-test for unpaired samples (Excel), and ANOVA was performed using STATISTICA 10.0 (StatSoft, Krakow, Poland).
Results
G-CSF and AMD3100 mobilization is impaired in C3−/− mice in the presence of refludan and/or tranexamic acid
We have already reported that inhibition of the CoaC by refludan impairs G-CSF-induced mobilization of HSPCs in C3−/− mice.16 In this report, we show that, in addition to inhibiting the CoaC, inhibition of the FibC has a similar effect. Moreover, inhibition of both these proteolytic cascades at the same time even more significantly affects the numbers of mobilized WBCs, SKL and CFU-GM circulating in PB (Figure 1a). We report here, also for the first time, that refludan and tranexamic acid have similar negative effects on AMD3100-induced mobilization of HSPCs in C3−/− mice (Figure 1b). These data support the hypothesis that both thrombin and plasmin in fact possess C5 convertase-like activity.10
Figure 1
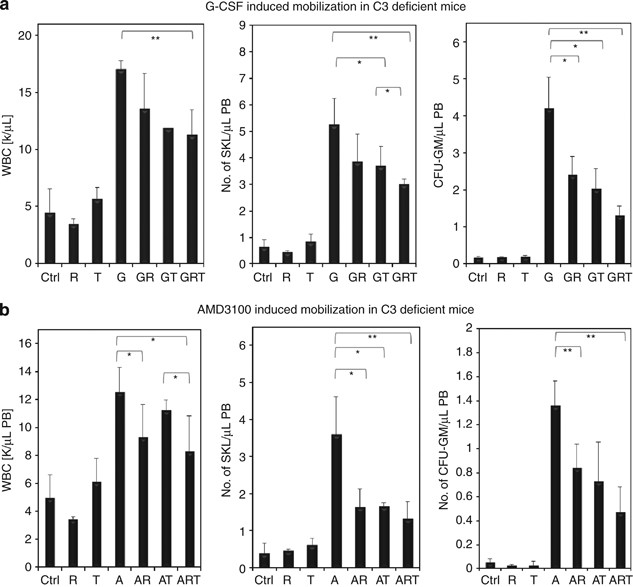
Effect of inhibition of CoaC and FibC on mobilization of HSPCs in C3-deficient mice. C3−/− mice were mobilized for 6 days with (a) G-CSF (250 μg/kg subcutaneously (s.c.) per day; _n_=6 mice per group) or with (b) AMD3100 (5 mg/kg s.c.) in the presence or absence of refludan (administered daily for 6 days, 5 mg/kg intraperitoneally (i.p.)) or tranexamic acid (administered for 6 days, 20 mg/mice/day s.c.). The numbers of circulating leukocytes (left panels), SKL cells (middle panels) and CFU-GM progenitors (right panels) per μl of PB are shown. A, AMD3100 only; A+R, AMD3100+refludan; A+R+T, AMD3100+refludan+tranexamic acid; A+T, AMD3100+tranexamic acid; Ctrl, control; G, G-CSF only; G+R+T, G-CSF+refludan+tranexamic acid; G+T, G-CSF+tranexamic acid; R, refludan only; T, tranexamic acid only. *P<0.05; **P<0.01. These are representative results from three independent experiments for G-CSF and two independent experiments for AMD3100.
Nevertheless, as is shown in Figure 1, mobilization was not completely inhibited after simultaneous inhibition of both CoaC and FibC that implies that some residual activity of both thrombin and plasmin may be present, or some other plasma proteases partially compensate for thrombin and plasmin activity.17 It also shows that, when employed alone, neither refludan nor tranexamic acid affects the pool of circulating HSPCs.
Administration of refludan and/or tranexamic acid also impairs stem cell mobilization in normal WT littermates
In this study, we also present data showing that inhibition of both CoaC and FibC has a negative effect on mobilization of HSPCs after either G-CSF- (Figure 2a) or AMD3100-induced (Figure 2b) mobilization. This effect was particularly visible in G-CSF-induced mobilization when refludan and tranexamic acid were employed together. However, in AMD3100-induced mobilization, the addition of tranexamic acid to refludan did not significantly increase the effect of refludan alone in WT animals (Figure 2b).
Figure 2
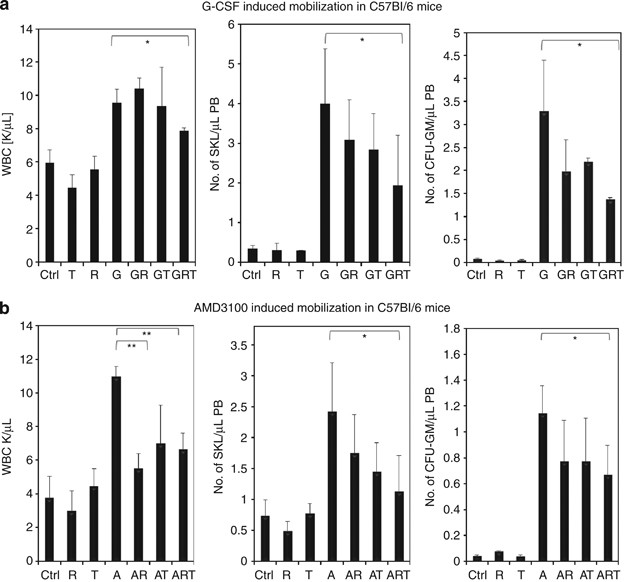
Effect of inhibition of CoaC and FibC on mobilization of HSPCs in WT mice. WT mice were mobilized for 6 days with (a) G-CSF (250 μg/kg subcutaneously (s.c.) per day, _n_=6 mice per group) or with (b) AMD3100 (5 mg/kg s.c.) in the presence or absence of refludan (administered daily for 6 days, 5 mg/kg intraperitoneally (i.p.)) or tranexamic acid (administered for 6 days, 20 mg/mice/day s.c.). The numbers of circulating leukocytes (left panels), SKL cells (middle panels) and CFU-GM progenitors (right panels) per μl of PB are shown. A, AMD3100 only; A+R, AMD3100+refludan; A+R+T, AMD3100+refludan+tranexamic acid; A+T, AMD3100+tranexamic acid; Ctrl, control; G, G-CSF only; G+R+T, G-CSF+refludan+tranexamic acid; G+T, G-CSF+tranexamic acid; R, refludan only; T, tranexamic acid only. *P<0.05; **P<0.005. These are combined results from three independent experiments for G-CSF and two independent experiments for AMD3100.
As described in the past,12 we observed that in response to G-CSF administration, C3−/− mice mobilize greater numbers of HSPCs into PB (Figure 1a) than WT littermates (Figure 2a) that suggests that C3 cleavage fragments promote CXCR4-dependent retention of HSPCs in BM.4, 12 Interestingly, when we employed the CXCR4 antagonist AMD3100 in our current study, this difference in mobilization was not apparent (Figure 1b vs Figure 2b). Again, when employed alone, neither refludan nor tranexamic acid affected the pool of circulating HSPCs.
All three evolutionarily ancient proteolytic cascades are activated during G-CSF- and AMD3100-induced mobilization
In support of our in vivo mobilization data, in the presence or absence of CoaC and FibC inhibitors, we directly measured activation of both cascades in the PB of mice mobilized by G-CSF or AMD3100.
Figure 3 demonstrates that administration of G-CSF (Figure 3A) or AMD3100 (Figure 3B) significantly decreases both PT and APTT and AMD3100 significantly increases thrombin/antithrombin that indicates activation of the CoaC. G-CSF administration also increased the concentration of thrombin/antithrombin complexes in PB, although this increase was not significant. At the same time, as shown in Figure 3A, AMD3100 significantly activated the FibC, as seen by an increase in plasmin/antiplasmin concentration. Finally, activation of the ComC during G-CSF- and AMD-3100-induced mobilization, as previously reported, was inhibited after administration of refludan and/or tranexamic acid (Figure 3C).
Figure 3
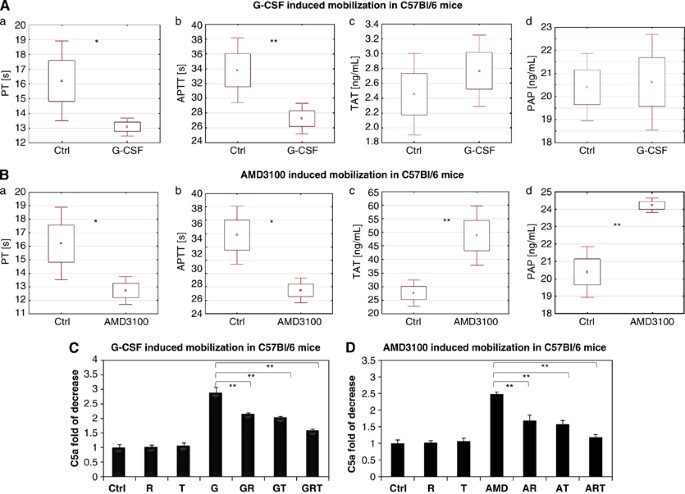
Evidence for activation of CoaC, FibC and ComC during mobilization of HSPCs. (A and B) Activation of the CoaC in WT mice mobilized by G-CSF or AMD3100, as measured by decrease of prothrombin time (PT) (a), APTT (b; *P<0.05, **P<0.01) and by the increase in serum level of thrombin/antithrombin (TAT) complexes (c; *P<0.005). (d) Activation of the FibC in WT mice mobilized by G-CSF or AMD3100 as measured by the increase in serum level of and plasmin/antiplasmin (PAP) complexes. *P<0.005. These are combined results from two independent experiments (_n_=10 mice/group). (C and D) Activation of ComC as measured by increase in concentration of C5a cleavage fragments in PB (control values in nonmobilized mice were assumed to be 1.0). These are combined results from two independent experiments (_n_=6 mice/group). *P<0.005.
Discussion
The mechanisms that regulate mobilization of HSPCs are still not well understood, and a relatively large number of patients, particularly those pretreated by chemotherapy, are poor mobilizers in response to G-CSF.7, 18, 19, 20
It has been reported that administration of G-CSF or AMD3100 induces a highly proteolytic microenvironment in BM because of several proteolytic enzymes released by activated granulocytes and monocytes.21, 22 Thus, it is widely accepted that the first step in the release of HSPCs from their niches in BM into PB is related to attenuation of the interaction between the major homing/retention receptors (CXCR4 and VLA-4) expressed on the surface of HSPCs with their corresponding ligands (SDF-1 and VCAM-1) in BM niches.1, 2, 3, 4, 5 This could be achieved by the action of the above-mentioned proteolytic enzymes or blockade with specific receptor-blocking molecules (for example, AMD3100). However, the role of BM-expressed proteolytic enzymes, such as neutrophil elastase, cathepsin-G or metalloproteinases,17 has been challenged by findings that knockout of several of these enzymes has only a negligible effect on the mobilization process,17 and this also suggests the existence of compensatory mechanisms involving other proteolytic enzymes. We hypothesize that proteases generated in the activation of CoaC and FibC may play a role here. In fact, it has been suggested that CoaC and FibC play a role in the release of HSPCs from BM;13, 14, 15 however, the molecular mechanisms behind this phenomenon are still not fully understood.
It is well known that three evolutionarily ancient proteolytic enzyme cascades, ComC, CoaC and FibC, are activated in PB during several events in which HSPCs are released from the BM into PB (for example, inflammation or strenuous exercise). All of these cascades also show circadian rhythms in activation because of a drop in blood pH during deep sleep at night and are most likely responsible for circadian changes in HSPC trafficking in PB.23, 24 Therefore, in addition to circadian changes in the tonus of the vegetative nervous system, circadian activation of the ComC, CoaC and FibC is most likely also involved in the circadian release of HSPCs from BM into PB.6 What is also important, the three cascades show a vigorous crosstalk because of mutual cross-activation by proteolytic enzymes released, for example, during inflammation.10
We previously reported on the importance of the ComC in G-CSF- and AMD3100-induced mobilization.4 We have also shown that mice defective in C3, which is a proximal component of the ComC, are easy mobilizers.12 We have explained this phenomenon by a lack of C3 cleavage and C3a formation that increases because of a so-called priming effect, CXCR4–SDF-1 axis-mediated retention signals for HSPCs in BM, thus preventing uncontrolled release of these cells into PB. In contrast to C3−/− animals, mice defective in C5, which is a distal component of the ComC, have a profound defect in mobilization of HSPCs.9 This observation is explained by the fact that the C5 cleavage fragment (C5a) generated in the BM microenvironment inside the BM–PB barrier is responsible for augmenting the release of proteolytic enzymes from myeloid cells that attenuate SDF-1–CXCR4 and VLA4–VCAM-1 retention signals in BM niches. Furthermore, C5a generated in BM sinusoids is a potent chemoattractant for granulocytes and monocytes, the first cells to egress from the BM and thus permeabilize the BM–PB barrier, facilitating subsequent egress of HSPCs that express proteolytic enzymes at an ∼100-fold lower level.4, 9 In addition, another C5 cleavage fragment, C5b, is involved in formation of C5b-C9 (membrane attack complex) that may increase the PB level of sphingosine-1-phosphate,25 a crucial direct chemoattractant in PB directing egress of HSPCs. Thus, the failure of C5-deficient mice to generate both C5a and membrane attack complex explains why these animals are poor mobilizers.9
Taking into consideration that C3 is necessary for generation of C5 convertase, an enzyme that cleaves C5 during activation of ComC, one would expect that C3-deficient mice, similar to C5-deficient animals, would be poor mobilizers. However, as mentioned above, this is not the case. To address why C3−/− mice, despite with a defect in generation of C5 convertase, are easy mobilizers, we considered the involvement of alternative proteolytic pathways that provide C5 convertase-like activity and thus cleave/activate C5 in C3−/− mice. Therefore we focused on the CoaC and FibC, knowing that both thrombin and plasminogen may possess C5 convertase-like activity, as demonstrated in some experimental settings.10 In our previous report we showed that inhibition of CoaC by refludan negatively affects G-CSF-induced mobilization in C3−/− animals, suggesting the involvement of the CoaC in this process.16 In further support of this interpretation, as mentioned above thrombin and plasmin have been shown to mobilize HSPCs when administered in vivo. To our knowledge, we have demonstrated for the first time that administration of both G-CSF and AMD3100 activate CoaC, ComC and FibC at the same time and shed more light on the role of CoaC and FibC in the mobilization process. Specifically, based on the observation that C5 cleavage is crucial for egress of HSPCs from BM,9 we show here that both thrombin, as a product of the activated CoaC, and plasmin, as a product of the activated FibC, provide C5 convertase-like activity to facilitate C5 cleavage as a crucial step in HSPC mobilization (Figure 4).
Figure 4
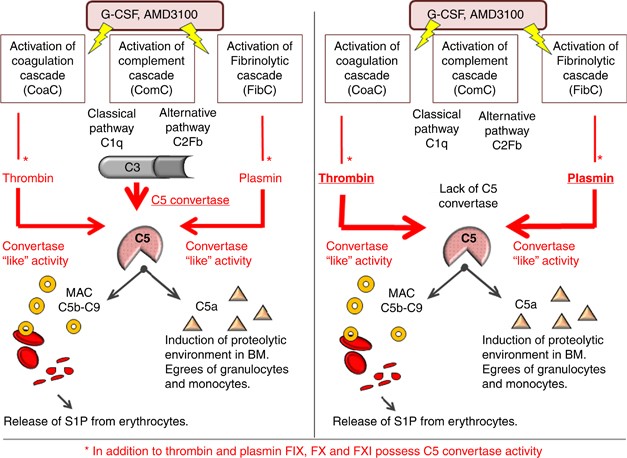
Postulated crosstalk between ComC, CoaCand FibC in the mobilization of HSPCs that is explained by each providing C5 convertase activity. C5 cleavage and release of C5a and C5b is crucial for egress of HSPCs from BM.9 (Left panel) G-CSF and AMD3100 activate ComC, CoaC and FibC that leads to generation of C5 convertase (via the ComC), C5 convertase-like activity by thrombin (via the CoaC) and C5a convertase-like activity by plasmin (via the FibC). (Right panel) C5 convertase-like activity because of activation of CoaC and generation of thrombin as well as activation of the FibC and generation of plasmin that compensates for the lack of C3 and thus the lack of ‘classical’ C5 convertase in C3−/− mice. This substitution enables efficient mobilization of HSPCs in C3−/− mice, despite a defect in activation of the proximal (upstream of C5) part of the ComC. * indicates involvement of other potential pathways. To support this, factors IX, X and XI also possess C5 convertase activity.10
Thus, these results confirm that C5 can be activated/cleaved in a thrombin- and plasmin-dependent manner10 in the absence of classical C3-dependent, ComC-generated C5 convertase—and thus both thrombin and plasmin may provide ‘C5-convertase-like’ activity during mobilization of HSPCs. We employed C3−/− animals that are easy mobilizers as a model of this effect, although our data also support thrombin and plasmin providing C5-convertase-like activity in G-CSF- and AMD3100-mobilized WT animals. However, as inhibition of both proteases did not completely inhibit egress of HSPCs from BM, one has to consider the involvement of other components of both CoaC and FibC in providing C5 convertase-like activity. In support of this conclusion, C5 can also be cleaved by several other proteases released from granulocytes and monocytes unrelated to the CoaC and ComC, such as cathepsin, matrix metalloproteinase (MMP)-2 and MMP-9.17
In conclusion, the data presented in this work demonstrate, for the first time, the existence of crosstalk between three evolutionarily ancient proteolytic cascades, ComC, CoaC and FibC, in the mobilization of HSPCs by providing C5 convertase activity. Further study is needed to see whether proteolytic enzymes from those cascades also penetrate the blood–BM barrier and affect CXCR4–SDF-1- and VLA-4–VCAM-1-mediated retention signals for HSPCs. It is very likely that proteolytic activity of these cascades compensates for the lack of granulocyte/monocyte-derived proteases in mobilization studies in animals with deficiencies in these enzymes. Finally, as all these cascades are activated late at night because of the drop of pH in PB, more studies are needed to clarify their involvement in the circadian release of HSPCs from the BM into PB. Finally, these observations are relevant to the mobilization of the patients who are treated with drugs that potentially interfere with CoaC/FibC.
References
- Lévesque JP, Helwani FM, Winkler IG . The endosteal ‘osteoblastic’ niche and its role in hematopoietic stem cell homing and mobilization. Leukemia 2010; 24: 1979–1992.
Article Google Scholar - Bonig H, Papayannopoulou T . Hematopoietic stem cell mobilization: updated conceptual renditions. Leukemia 2013; 27: 24–31.
Article CAS Google Scholar - Lapidot T, Kollet O . The brain-bone-blood triad: traffic lights for stem-cell homing and mobilization. Hematology Am Soc Hematol Educ Program 2010; 2010: 1–6.
Article Google Scholar - Ratajczak MZ, Kim CH, Wojakowski W, Janowska-Wieczorek A, Kucia M, Ratajczak J . Innate immunity as orchestrator of stem cell mobilization. Leukemia 2010; 24: 1667–1675.
Article CAS Google Scholar - Doan PL, Chute JP . The vascular niche: home for normal and malignant hematopoietic stem cells. Leukemia 2012; 26: 54–62.
Article CAS Google Scholar - Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA et al. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006; 124: 407–421.
Article CAS Google Scholar - Hoggatt J, Pelus LM . Hematopoietic stem cell mobilization with agents other than G-CSF. Methods Mol Biol 2012; 904: 49–67.
CAS PubMed Google Scholar - Ratajczak MZ, Kim CH, Abdel-Latif A, Schneider G, Kucia M, Morris AJ et al. A novel perspective on stem cell homing and mobilization: review on bioactive lipids as potent chemoattractants and cationic peptides as underappreciated modulators of responsiveness to SDF-1 gradients. Leukemia 2012; 26: 63–72.
Article CAS Google Scholar - Lee HM, Wu W, Wysoczynski M, Liu R, Zuba-Surma EK, Kucia M et al. Impaired mobilization of hematopoietic stem/progenitor cells in C5-deficient mice supports the pivotal involvement of innate immunity in this process and reveals novel promobilization effects of granulocytes. Leukemia 2009; 23: 2052–2062.
Article CAS Google Scholar - Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B et al. Molecular intercommunication between the complement and coagulation systems. J Immunol 2010; 185: 5628–5636.
Article CAS Google Scholar - Patthy L . Evolution of blood coagulation and fibrinolysis. Blood Coagul Fibrinolysis 1990; 2: 153–166.
Google Scholar - Ratajczak J, Reca R, Kucia M, Majka M, Allendorf DJ, Baran JT et al. Mobilization studies in mice deficient in either C3 or C3a receptor (C3aR) reveal a novel role for complement in retention of hematopoietic stem/progenitor cells in bone marrow. Blood 2004; 103: 2071–2078.
Article CAS Google Scholar - Tjwa M, Janssens S, Carmeliet P . Plasmin therapy enhances mobilization of HPCs after G-CSF. Blood 2008; 112: 4048–4050.
Article CAS Google Scholar - Tjwa M, Sidenius N, Moura R, Jansen S, Theunissen K, Andolfo A et al. Membrane-anchored uPAR regulates the proliferation, marrow pool size, engraftment, and mobilization of mouse hematopoietic stem/progenitor cells. J Clin Invest 2009; 119: 1008–1018.
CAS PubMed PubMed Central Google Scholar - Selleri C, Montuori N, Ricci P, Visconte V, Baiano A, Carriero MV et al. In vivo activity of the cleaved form of soluble urokinase receptor: a new hematopoietic stem/progenitor cell mobilizer. Cancer Res 2006; 66: 10885–10890.
Article CAS Google Scholar - Borkowska S, Suszynska M, Wysoczynski M, Ratajczak MZ . Mobilization studies in C3-deficient mice unravel the involvement of a novel crosstalk between the coagulation and complement cascades in mobilization of hematopoietic stem/progenitor cells. Leukemia 2013; 27: 1928–1930.
Article CAS Google Scholar - Levesque JP, Liu F, Simmons PJ, Betsuyaku T, Senior RM, Pham C et al. Characterization of hematopoietic progenitor mobilization in protease-deficient mice. Blood 2004; 104: 65–72.
Article CAS Google Scholar - Karpova D, Dauber K, Spohn G, Chudziak D, Wiercinska E, Schulz M et al. The novel CXCR4 antagonist POL5551 mobilizes hematopoietic stem and progenitor cells with greater efficiency than Plerixafor. Leukemia 2013; 27: 2322–2331.
Article CAS Google Scholar - Motabi IH, DiPersio JF . Advances in stem cell mobilization. Blood Rev 2012; 26: 267–278.
Article CAS Google Scholar - Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med 2005; 201: 1307–1318.
Article CAS Google Scholar - Winkler IG, Pettit AR, Raggatt LJ, Jacobsen RN, Forristal CE, Barbier V et al. Hematopoietic stem cell mobilizing agents G-CSF, cyclophosphamide or AMD3100 have distinct mechanisms of action on bone marrow HSC niches and bone formation. Leukemia 2012; 26: 1594–1601.
Article CAS Google Scholar - Lévesque JP, Hendy J, Takamatsu Y, Williams B, Winkler IG, Simmons PJ . Mobilization by either cyclophosphamide or granulocyte colony-stimulating factor transforms the bone marrow into a highly proteolytic environment. Exp Hematol 2002; 30: 440–449.
Article Google Scholar - Reis ES, Lange T, Köhl G, Herrmann A, Tschulakow AV, Naujoks J et al. Sleep and circadian rhythm regulate circulating complement factors and immunoregulatory properties of C5a. Brain Behav Immun 2011; 25: 1416–1426.
Article CAS Google Scholar - Wolk R, Gami AS, Garcia-Touchard A, Somers VK . Sleep and cardiovascular disease. Curr Probl Cardiol 2005; 30: 625–662.
Article Google Scholar - Ratajczak MZ, Lee H, Wysoczynski M, Wan W, Marlicz W, Laughlin MJ et al. Novel insight into stem cell mobilization-plasma sphingosine-1-phosphate is a major chemoattractant that directs the egress of hematopoietic stem progenitor cells from the bone marrow and its level in peripheral blood increases during mobilization due to activation of complement cascade/membrane attack complex. Leukemia 2010; 24: 976–985.
Article CAS Google Scholar
Acknowledgements
This work was supported by the NIH Grants 2R01 DK074720 and R01HL112788, the Stella and Henry Endowment and the Maestro Grant 2011/02/A/NZ4/00035 to MZR.
Author information
Authors and Affiliations
- Stem Cell Institute at James Graham Brown Cancer Center, University of Louisville, Louisville, KY, USA
S Borkowska, M Suszynska, K Mierzejewska, A Ismail, M Budkowska, D Salata, B Dolegowska, M Kucia, J Ratajczak & M Z Ratajczak - Department of Physiology, Pomeranian Medical University, Szczecin, Poland
S Borkowska, M Suszynska, K Mierzejewska, A Ismail, M Budkowska, D Salata, B Dolegowska, M Kucia, J Ratajczak & M Z Ratajczak
Authors
- S Borkowska
You can also search for this author inPubMed Google Scholar - M Suszynska
You can also search for this author inPubMed Google Scholar - K Mierzejewska
You can also search for this author inPubMed Google Scholar - A Ismail
You can also search for this author inPubMed Google Scholar - M Budkowska
You can also search for this author inPubMed Google Scholar - D Salata
You can also search for this author inPubMed Google Scholar - B Dolegowska
You can also search for this author inPubMed Google Scholar - M Kucia
You can also search for this author inPubMed Google Scholar - J Ratajczak
You can also search for this author inPubMed Google Scholar - M Z Ratajczak
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toM Z Ratajczak.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Borkowska, S., Suszynska, M., Mierzejewska, K. et al. Novel evidence that crosstalk between the complement, coagulation and fibrinolysis proteolytic cascades is involved in mobilization of hematopoietic stem/progenitor cells (HSPCs).Leukemia 28, 2148–2154 (2014). https://doi.org/10.1038/leu.2014.115
- Received: 08 January 2014
- Revised: 24 February 2014
- Accepted: 13 March 2014
- Published: 26 March 2014
- Issue Date: November 2014
- DOI: https://doi.org/10.1038/leu.2014.115