Estimating the selective effects of heterozygous protein-truncating variants from human exome data (original) (raw)
References
- Mukai, T., Chigusa, S.I., Mettler, L.E. & Crow, J.F. Mutation rate and dominance of genes affecting viability in Drosophila melanogaster. Genetics 72, 335–355 (1972).
Article CAS PubMed PubMed Central Google Scholar - Deng, H.W. & Lynch, M. Estimation of deleterious-mutation parameters in natural populations. Genetics 144, 349–360 (1996).
Article CAS PubMed PubMed Central Google Scholar - Wang, T. et al. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015).
Article CAS PubMed PubMed Central Google Scholar - Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Article CAS PubMed PubMed Central Google Scholar - Williamson, S.H. et al. Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc. Natl. Acad. Sci. USA 102, 7882–7887 (2005).
Article CAS PubMed PubMed Central Google Scholar - Boyko, A.R. et al. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 4, e1000083 (2008).
Article PubMed PubMed Central CAS Google Scholar - Kryukov, G.V., Pennacchio, L.A. & Sunyaev, S.R. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am. J. Hum. Genet. 80, 727–739 (2007).
Article CAS PubMed PubMed Central Google Scholar - Kryukov, G.V., Shpunt, A., Stamatoyannopoulos, J.A. & Sunyaev, S.R. Power of deep, all-exon resequencing for discovery of human trait genes. Proc. Natl. Acad. Sci. USA 106, 3871–3876 (2009).
Article CAS PubMed PubMed Central Google Scholar - Eyre-Walker, A. & Keightley, P.D. The distribution of fitness effects of new mutations. Nat. Rev. Genet. 8, 610–618 (2007).
Article CAS PubMed Google Scholar - Do, R. et al. No evidence that selection has been less effective at removing deleterious mutations in Europeans than in Africans. Nat. Genet. 47, 126–131 (2015).
Article CAS PubMed PubMed Central Google Scholar - Fu, W., Gittelman, R.M., Bamshad, M.J. & Akey, J.M. Characteristics of neutral and deleterious protein-coding variation among individuals and populations. Am. J. Hum. Genet. 95, 421–436 (2014).
Article CAS PubMed PubMed Central Google Scholar - Lohmueller, K.E. The distribution of deleterious genetic variation in human populations. Curr. Opin. Genet. Dev. 29, 139–146 (2014).
Article CAS PubMed Google Scholar - Gravel, S. When is selection effective? Genetics 203, 451–462 (2016).
Article CAS PubMed PubMed Central Google Scholar - Williamson, S., Fledel-Alon, A. & Bustamante, C.D. Population genetics of polymorphism and divergence for diploid selection models with arbitrary dominance. Genetics 168, 463–475 (2004).
Article PubMed PubMed Central Google Scholar - Balick, D.J., Do, R., Cassa, C.A., Reich, D. & Sunyaev, S.R. Dominance of deleterious alleles controls the response to a population bottleneck. PLoS Genet. 11, e1005436 (2015).
Article PubMed PubMed Central CAS Google Scholar - Simons, Y.B., Turchin, M.C., Pritchard, J.K. & Sella, G. The deleterious mutation load is insensitive to recent population history. Nat. Genet. 46, 220–224 (2014).
Article CAS PubMed PubMed Central Google Scholar - MacArthur, D.G. et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 335, 823–828 (2012).
Article CAS PubMed PubMed Central Google Scholar - Samocha, K.E. et al. A framework for the interpretation of de novo mutation in human disease. Nat. Genet. 46, 944–950 (2014).
Article CAS PubMed PubMed Central Google Scholar - Francioli, L.C. et al. Genome-wide patterns and properties of de novo mutations in humans. Nat. Genet. 47, 822–826 (2015).
Article CAS PubMed PubMed Central Google Scholar - Solomon, B.D., Nguyen, A.-D., Bear, K.A. & Wolfsberg, T.G. Clinical genomic database. Proc. Natl. Acad. Sci. USA 110, 9851–9855 (2013).
Article CAS PubMed PubMed Central Google Scholar - Yang, Y. et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 312, 1870–1879 (2014).
Article CAS PubMed PubMed Central Google Scholar - Lee, H. et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 312, 1880–1887 (2014).
Article PubMed PubMed Central CAS Google Scholar - Saleheen, D. et al. Human knockouts in a cohort with a high rate of consanguinity. Preprint at bioRxiv http://dx.doi.org/10.1101/031518 (2015).
- Koscielny, G. et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 42, D802–D809 (2014).
Article CAS PubMed Google Scholar - Georgi, B., Voight, B.F. & Buc´an, M. From mouse to human: evolutionary genomics analysis of human orthologs of essential genes. PLoS Genet. 9, e1003484 (2013).
Article CAS PubMed PubMed Central Google Scholar - Roessler, E. et al. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat. Genet. 14, 357–360 (1996).
Article CAS PubMed Google Scholar - Kang, S., Graham, J.M., Olney, A.H. & Biesecker, L.G. GLI3 frameshift mutations cause autosomal dominant Pallister–Hall syndrome. Nat. Genet. 15, 266–268 (1997).
Article CAS PubMed Google Scholar - Vortkamp, A., Gessler, M. & Grzeschik, K.H. GLI3 zinc-finger gene interrupted by translocations in Greig syndrome families. Nature 352, 539–540 (1991).
Article CAS PubMed Google Scholar - Wild, A. et al. Point mutations in human GLI3 cause Greig syndrome. Hum. Mol. Genet. 6, 1979–1984 (1997).
Article CAS PubMed Google Scholar - Roessler, E. et al. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc. Natl. Acad. Sci. USA 100, 13424–13429 (2003).
Article CAS PubMed PubMed Central Google Scholar - Chiang, C. et al. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407–413 (1996).
Article CAS PubMed Google Scholar - Hui, C.C. & Joyner, A.L. A mouse model of Greig cephalopolysyndactyly syndrome: the extra-toes J mutation contains an intragenic deletion of the Gli3 gene. Nat. Genet. 3, 241–246 (1993).
Article CAS PubMed Google Scholar - Mo, R. et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 124, 113–123 (1997).
Article CAS PubMed Google Scholar - Huang, D.W., Sherman, B.T. & Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2008).
Article CAS Google Scholar - Seidman, J.G. & Seidman, C. Transcription factor haploinsufficiency: when half a loaf is not enough. J. Clin. Invest. 109, 451–455 (2002).
Article CAS PubMed PubMed Central Google Scholar - NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 41, D8–D20 (2013).
- Raychaudhuri, S. et al. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 5, e1000534 (2009).
Article PubMed PubMed Central CAS Google Scholar - Agrawal, A.F. & Whitlock, M.C. Inferences about the distribution of dominance drawn from yeast gene knockout data. Genetics 187, 553–566 (2011).
Article CAS PubMed PubMed Central Google Scholar - Simmons, M.J. & Crow, J.F. Mutations affecting fitness in Drosophila populations. Annu. Rev. Genet. 11, 49–78 (1977).
Article CAS PubMed Google Scholar - Wright, S. Evolution in Mendelian populations. Bull. Math. Biol. 52, 241–295 (1990).
Article CAS PubMed Google Scholar - Petrovski, S. et al. The intolerance of regulatory sequence to genetic variation predicts gene dosage sensitivity. PLoS Genet. 11, e1005492 (2015).
Article PubMed PubMed Central CAS Google Scholar - Kiezun, A. et al. Exome sequencing and the genetic basis of complex traits. Nat. Genet. 44, 623–630 (2012).
Article CAS PubMed PubMed Central Google Scholar - Li, W.H. & Nei, M. Total number of individuals affected by a single deleterious mutation in a finite population. Am. J. Hum. Genet. 24, 667–679 (1972).
CAS PubMed PubMed Central Google Scholar - Li, W.H. The first arrival time and mean age of a deleterious mutant gene in a finite population. Am. J. Hum. Genet. 27, 274–286 (1975).
CAS PubMed PubMed Central Google Scholar - Maruyama, T. The age of a rare mutant gene in a large population. Am. J. Hum. Genet. 26, 669–673 (1974).
CAS PubMed PubMed Central Google Scholar - Maruyama, T. The age of an allele in a finite population. Genet. Res. 23, 137–143 (1974).
Article CAS PubMed Google Scholar - Messer, P.W. SLiM: simulating evolution with selection and linkage. Genetics 194, 1037–1039 (2013).
Article PubMed PubMed Central Google Scholar - Tennessen, J.A. et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69 (2012).
Article CAS PubMed PubMed Central Google Scholar - Wang, S.R. et al. Simulation of Finnish population history, guided by empirical genetic data, to assess power of rare-variant tests in Finland. Am. J. Hum. Genet. 94, 710–720 (2014).
Article CAS PubMed PubMed Central Google Scholar - Huttlin, E.L. et al. The BioPlex Network: a systematic exploration of the human interactome. Cell 162, 425–440 (2015).
Article CAS PubMed PubMed Central Google Scholar - Ayadi, A. et al. Mouse large-scale phenotyping initiatives: overview of the European Mouse Disease Clinic (EUMODIC) and of the Wellcome Trust Sanger Institute Mouse Genetics Project. Mamm. Genome 23, 600–610 (2012).
Article PubMed PubMed Central Google Scholar
Acknowledgements
We thank I. Adzhubei, K. Karczewski, E. Minikel, and A. Kondrashov for helpful advice. This work was supported by US National Institutes of Health (NIH) grants HG007229 (C.A.C.), GM078598 (S.R.S., D.M.J., D.J.B.), and MH101244 (S.R.S., D.W.).
Author information
Author notes
- Christopher A Cassa, Donate Weghorn, Daniel J Balick and Daniel M Jordan: These authors contributed equally to this work.
Authors and Affiliations
- Division of Genetics, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, USA
Christopher A Cassa, Donate Weghorn, Daniel J Balick, David Nusinow & Shamil R Sunyaev - Program in Medical and Population Genetics, Broad Institute, Cambridge, Massachusetts, USA
Christopher A Cassa, Daniel G MacArthur, Mark J Daly & Shamil R Sunyaev - Department of Genetic and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, New York, USA
Daniel M Jordan - Analytic and Translational Genetics Unit, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts, USA
Kaitlin E Samocha, Anne O'Donnell-Luria, Daniel G MacArthur & Mark J Daly - Program in Biological and Biomedical Sciences, Harvard Medical School, Boston, Massachusetts, USA
Kaitlin E Samocha - Division of Genetics and Genomics, Boston Children's Hospital, Boston, Massachusetts, USA
Anne O'Donnell-Luria - Center for Developmental Biology and Regenerative Medicine, Seattle Children's Research Institute, Seattle, Washington, USA
David R Beier - Department of Pediatrics, University of Washington School of Medicine, Seattle, Washington, USA
David R Beier
Authors
- Christopher A Cassa
You can also search for this author inPubMed Google Scholar - Donate Weghorn
You can also search for this author inPubMed Google Scholar - Daniel J Balick
You can also search for this author inPubMed Google Scholar - Daniel M Jordan
You can also search for this author inPubMed Google Scholar - David Nusinow
You can also search for this author inPubMed Google Scholar - Kaitlin E Samocha
You can also search for this author inPubMed Google Scholar - Anne O'Donnell-Luria
You can also search for this author inPubMed Google Scholar - Daniel G MacArthur
You can also search for this author inPubMed Google Scholar - Mark J Daly
You can also search for this author inPubMed Google Scholar - David R Beier
You can also search for this author inPubMed Google Scholar - Shamil R Sunyaev
You can also search for this author inPubMed Google Scholar
Contributions
Overall concept and approach conceived and developed by C.A.C., D.R.B., and S.R.S. Implementation, data analysis, and interpretation conducted by D.W.,C.A.C., D.J.B., D.M.J., and D.N. Data sets and advice were provided by D.G.M., M.J.D., K.E.S., and A.O'D.-L. The article was written by C.A.C. and S.R.S. with contributions from D.W. and D.J.B. All authors read and discussed the manuscript.
Corresponding authors
Correspondence toDavid R Beier or Shamil R Sunyaev.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Population genetics simulations of model assumptions.
To validate the assumption that estimates of selection can be made under mutation-selection balance independent of demography or population size for variants under sufficiently strong selection ( Methods ), we used SLiM 2.0 to conduct forward population genetics simulations. We compare the theoretical mutation load (defined as the sum of PTV allele frequencies calculated as _U/s_het) with the simulated mutation load in four groups (African, Non-Finnish European, Finnish, and Combined). The combined group includes pooled site frequency spectra from African, Non-Finnish European, and Finnish populations in proportions represented in the EXAC dataset for _s_het ∈ {-5x10-2,-5x10-3,-5x10-4,-5x10-5,-5x10-6} from left to right on the x-axis. μ = 2x10-8, each gene is 100 base pairs, and U = 2x10-6 for all simulations. Plotted points are mean values across 10,000 replicates. The simulations support our assumption of mutation-selection balance (with no appreciable effect from drift) in the strong selection regime (|_s_het| > 1x10-3), which appears to be appropriate for PTVs even in case of the Finnish population that underwent a recent bottleneck and a subsequent population expansion.
Supplementary Figure 2 ROC curve for mode of inheritance gene classifier.
We train a Naïve Bayes classifier to predict the mode of inheritance in a set of solved clinical exome sequencing cases from Baylor College of Medicine (N=283 cases) and UCLA (N=176 cases). Using data from UCLA as the training dataset, we are able to cross-predict the mode of inheritance in separately ascertained Baylor cases with classification accuracy of 88.0%, sensitivity of 86.1%, specificity of 90.2%, and an AUC of 0.931. Genes that were related to diagnosis in both clinics (overlapping genes) were removed from the larger Baylor set.
Supplementary Figure 3 Association of _s_hetQUOTE _s_het estimates with known disease genes.
Proportion of genes listed to have a disease association in the Human Gene Mutation Database, and number of disease associations related to each gene in OMIM MorbidMap, in each _s_het decile. Each bin is expected to contain 10% of all covered genes, ordered from greatest to smallest _s_het values, in bins 1 through 10, respectively.
Supplementary Figure 4 Enrichment in germline cancer predisposition genes.
In a large screen of germline cancer predisposition genes in the Pediatric Cancer Genome Project (PCGP), the enrichment of variants in pediatric cancer cases is measured over individuals in ExAC. Genes with greater enrichment of variants in cancer cases over ExAC are correlated with higher selection coefficients. Data are separated by _s_het bins on a log scale. Box plots range from 25th-75th percentile values and whiskers include 1.5 times the interquartile range.
Supplementary Figure 5 Enrichments of _s_het in de novo variants from autism spectrum disorder (ASD) case and control trios.
In a set of de novo ASD case (N=2,939) and control (N=1,429) trios, _s_het estimates can help discriminate between all protein-coding variants, protein-truncating variants (including all frameshift, nonsense, and essential splice site variants), and individually for nonsense, frameshift, and missense variants which are predicted to be PolyPhen-2 damaging. Box plots range from 25th-75th percentile values and whiskers include 1.5 times the interquartile range.
Supplementary Figure 6 Association of _s_het estimates with PubMed gene score.
[a] The average PubMed gene score is calculated by _s_het decile. Estimates of selection (_s_het) are positively correlated with the average PubMed gene score. Each bin contains 10% of all covered genes, ordered from greatest to smallest _s_het values, in bins 1 through 10, respectively. [b] The PubMed gene score is significantly positively correlated with the (p<0.0001) using a logarithmic model (y=4.557*log(_s_het)+44.449) with R2=0.00409.
Supplementary Figure 7 Most and least published genes from top _s_het decile.
The proportion of annotations related to genes with the fewest and most publications in Entrez Gene. From the set of genes under the strongest selection (top 10% of _s_het values), we create two sets of 250 genes. The first set of genes has the fewest publications associated with each gene, as defined by our PubMed gene score ( Methods ), and the second set has the greatest number of associated publications. Between the two groups, we compare the _s_het values, number of protein-protein interactions, viability of orthologous mouse knockouts (IMPC), and cell essentiality assays (KBM-7 CRISPR score and Gene Trap Score). These results suggest that the genes in the least published set are similar to those in the most published set, and are also potentially important developmental genes.
Supplementary Figure 8 Relationship between gene mutation rate and selection.
Relationship between the estimate of local mutation rate, U, and the naïve estimator for heterozygous selection against PTVs, ν/n=NU/n, for all 17,199 genes. Light green dots represent genes with ◯ =_n/N_>0.001 (1,201), which we omit in the inference of the distribution of P (_s_het). Light gray dots are used genes with _n_>0 (14,274), while dark blue dots correspond to those with n_=0 (1,724). The latter were assigned a fixed selection coefficient estimate of 1 for illustration purposes. We computed the mean U in logarithmic bins of ν/n for the range 0.00003<ν/n≤_0.012, and for the last bin from all genes with _ν/n>_0.012, including those with _n_=0 (large gray dots). Error bars denote s.e.m. The slight positive correlation between U and selection strength motivates the division of the data set into terciles of U and separate estimation of the parameters of the distribution of selection coefficients in each.
Supplementary Figure 9 Fit to the observed distribution of PTV counts.
Fitted distribution P(n) (black dots) from maximum likelihood fit to the observed distribution Q(n) (histogram) of PTV counts n across 15,998 considered genes divided into terciles according to mutation rate U, assuming _s_het_~_IG
Supplementary Figure 10 Inferred distribution of fitness effects for heterozygous loss of gene function in non-Finnish Europeans.
We separately repeated the inference procedure for _P (s_het) using data from a single population group, Non-Finnish Europeans (NFE, N=33,370, as annotated by ExAC), and generated a corresponding set of _s_het estimates. The inferred parameters are very similar to those from the larger sample. Estimates of parameters from maximum likelihood fit to the observed distribution of PTV counts n across genes with X =n/N<0.001 in the set of non-Finnish Europeans (16,279 genes), assuming _s_het_~_IG(α,β) in terciles of the mutation rate U. Parameter estimates are (α1,β1) = (0.093, 0.0068), (α2,β2) = (0.046, 0.0110), and (α3,β3) = (0.078, 0.0183), and shown is the mixture distribution of the three components with equal weights.
Supplementary Figure 11 Inferred distribution of fitness effects for heterozygous loss of gene function when excluding Finnish individuals.
We re-generated estimates of the distribution of heterozygous selection coefficients _s_het using the set of PTVs from all individuals in ExAC (N=60,706) and the set that excludes all Finnish individuals (N=57,399), using ExAC version 0.3.1 with LOFTEE annotations. Estimates of parameters from maximum likelihood fit to the observed distribution of PTV counts n across genes with 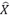 =n/N<0.001, assuming s_het_~IG(α,β). We find no substantial difference in the estimation of the prior for the distribution of selection coefficients in the ExAC sample that excludes Finnish individuals.
=n/N<0.001, assuming s_het_~IG(α,β). We find no substantial difference in the estimation of the prior for the distribution of selection coefficients in the ExAC sample that excludes Finnish individuals.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–11, Supplementary Table 3 and Supplementary Note. (PDF 3379 kb)
Supplementary Table 1
Distribution of _s_het estimates. We provide _s_het estimates in Supplementary Table 1. This file includes the mean of the posterior distribution Eq. 7 for each gene as well as the upper and lower 95% credibility intervals for each gene estimate. Credibility intervals have precision of 10-3 where _s_het > 0.005 and 10-5 otherwise. (XLSX 1814 kb)
Supplementary Table 2
Predicted mode of inheritance for each gene. For each gene, we generate a probability of mode of inheritance (either autosomal dominant or autosomal recessive). Estimates are generated using a logistic regression, trained on the full set of labeled case examples from two clinical exome sequencing programs (Baylor and UCLA)21,22. These estimates are applicable for interpretation of genes in cases that are similarly ascertained as these two clinical exome sequencing programs. (XLSX 579 kb)
Supplementary Table 4
Most published and least published genes from top _s_het decile.Full annotations for the PubMed Score in the top _s_het decile for the top 250 and bottom 250 PubMed genes scores. From the set of genes under the strongest selection (top 10% of _s_het values), we create two sets of 250 genes. We then annotated these lists with the results from neutrally-ascertained screens of gene importance and gene essentiality. We summarize these screens using a heuristic score. (XLSX 60 kb)
Supplementary Table 5
Functional analysis terms from DAVID. We include the results of GO term enrichment screening from DAVID that reach Bonferroni corrected significance in genes with _s_het > 0.15, _s_het > 0.25 and _s_het > 0.5. (XLSX 185 kb)
Supplementary Table 6
Functional analysis clusters from DAVID. We include the results of functional cluster enrichment screening from DAVID that reach Bonferroni corrected significance in genes with _s_het > 0.15, _s_het > 0.25 and _s_het > 0.5. (XLSX 198 kb)
Rights and permissions
About this article
Cite this article
Cassa, C., Weghorn, D., Balick, D. et al. Estimating the selective effects of heterozygous protein-truncating variants from human exome data.Nat Genet 49, 806–810 (2017). https://doi.org/10.1038/ng.3831
- Received: 13 September 2016
- Accepted: 07 March 2017
- Published: 03 April 2017
- Issue Date: May 2017
- DOI: https://doi.org/10.1038/ng.3831