Intense and highly localized gene conversion activity in human meiotic crossover hot spots (original) (raw)
Main
Meiotic gene conversion in humans is defined as the recombinational transfer of information between alleles or loci without crossover. (This definition, though widely used, is not necessarily congruent with conversion classically defined by non-2:2 meiotic segregation ratios and does not include crossovers accompanied by conversion.) Homogenization events in multigene and dispersed repeat sequence families and in palindromic DNA3,4,5, as well as the direct detection of de novo conversion events in families6,10 and in sperm11,12, provide extensive evidence for interlocus conversion. Indirect evidence for interallelic conversion comes from seemingly anomalous low levels of linkage disequilibrium seen between some closely linked markers13 and from patchwork patterns of allelic diversity14,15 (though such patchworks could arise from sequential crossovers). Sperm analysis at the HLA-DPB1 locus has provided direct evidence that true interallelic conversions do occur, though at low frequency16. The relationship between crossover and conversion in humans, however, is unclear.
The high-resolution definition of human crossover hot spots by sperm typing8,9,15 has allowed us to investigate the connection between crossover and interallelic conversion without crossover. We chose the hot spot _DNA_3 located in the MHC for analysis8, because of its intense crossover activity and the availability of sperm donors with multiple single nucleotide polymorphism (SNP) heterozygosities needed to monitor recombination events. Our initial approach was similar to that used to detect conversion in HLA-DPB1 (ref. 16; Fig. 1a). We amplified pools of sperm DNA by PCR using allele-specific primers outside the hot spot to amplify one haplotype (Fig. 1b), then typed them with allele-specific oligonucleotides (ASOs) to see whether any pools contained markers from the other haplotype. This strategy simultaneously detects crossovers and any conversions that affect one or more markers (Fig. 1c). Analysis of two individuals (man 1 and man 2) for reciprocal recombination events identified intense conversion activity, but only at markers very close to the center of the crossover hot spot (Fig. 2). The frequency of conversion at the most active marker (AB12) was 1.3–3.4 × 10−3 per sperm, higher than the rate of crossover (0.9–1.2 × 10−3). The ratio of conversions to crossovers did not vary substantially between the four separate analyses (orientation A, B recombinants analyzed in both men; Fisher's exact test, P = 0.05). All conversions were simple, involving the transfer of a contiguous block of markers between haplotypes, and short, with the longest involving coconversion of markers AB12, AB5, AB13 and AB14 in man 1 (minimum tract length = 300 bp, maximum = 1,091 bp). Conversion rates declined rapidly with distance and defined a very steep gradient of gene conversion activity extending in each direction from the center of the hot spot.
Figure 1: Detecting gene conversion events in sperm DNA.
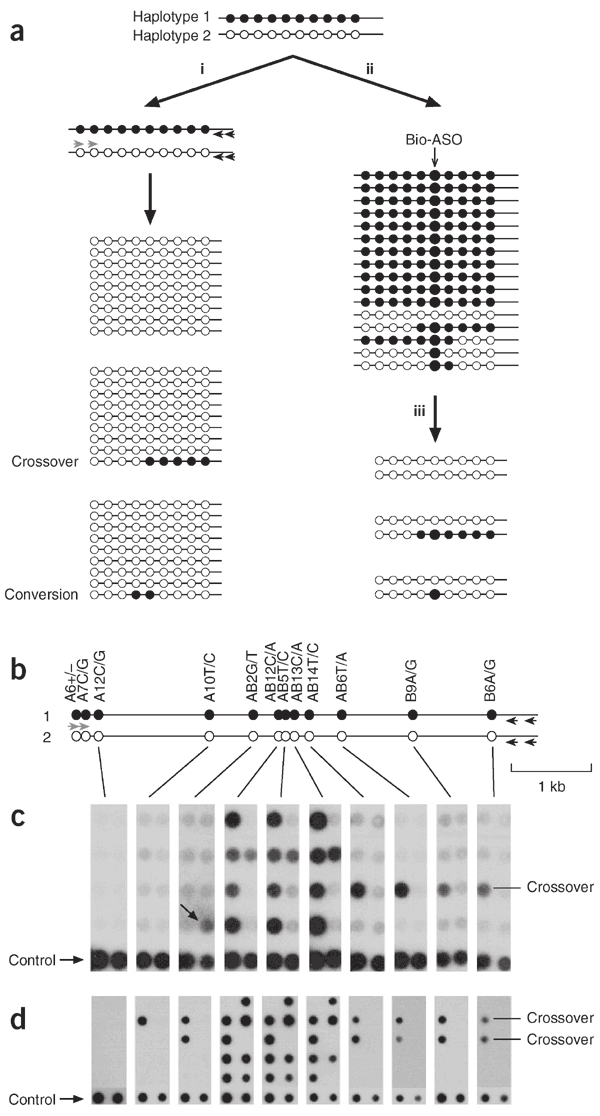
(a) Methods for detecting recombinants in a man carrying multiple SNP heterozygosities (black, white circles). (i) Screening without selection. Pools of sperm DNA are subjected to two rounds of nested PCR using allele-specific primers directed to haplotype 2 (gray arrows) and universal (not allele-specific) primers (black arrows). PCR products in each pool will be from haplotype 2, with the occasional presence of marker(s) from haplotype 1 signaling the presence of a recombinant molecule. (ii) Screening after selection by hybridization17 with a biotinylated ASO (Bio-ASO) specific for haplotype 1. The recovered single-stranded DNA is largely depleted in haplotype 2 and in any other molecules carrying the white allele at the selected SNP. (iii) Multiple aliquots of the enriched DNA are analyzed by allele-specific PCR, as described for screening without selection (i). Enrichment reduces the complexity of the sperm DNA pools, facilitating recombinant detection, but only recombinants exchanged at the selected SNP will be recovered. (b) Detecting sperm recombinants at hot spot _DNA_3: progenitor haplotypes in the individual tested, with the first and second allele (e.g., A6+ and A6−) corresponding to white and black circles, respectively. The hot spot is centered near SNP AB12. (c) Detection of recombinants without selection. Aliquots of sperm DNA each containing 100 amplifiable molecules of each haplotype were amplified using primers specific to haplotype 2, and PCR products were dot-blot hybridized with ASOs from haplotype 1, along with control PCR products (1:100 ratio of haplotype 1:haplotype 2). In the eight PCRs shown, one crossover molecule was detected, exchanged between markers AB2 and AB12, as well as four conversions affecting SNPs AB12, AB5 and AB13 and one putative conversion affecting AB2 alone (arrow). (d) Detection of recombinants after selection of sperm DNA with a biotinylated ASO specific for AB12A and AB5C (these SNPs are only 2 bp apart). Enriched DNA pools containing 300 molecules of haplotype 1 and about 2 remaining molecules of haplotype 2 were analyzed as described in c. Control contains a 1:1 ratio of haplotype 1:haplotype 2 PCR products. Two crossovers were detected in the ten PCRs shown, exchanged as predicted upstream of AB12 and AB5, as well as five conversion affecting AB12, AB5 and AB13 and one conversion involving only AB12 and AB5. Note the improvement in signal-to-noise ratio compared with c.
Figure 2: Gradients of gene conversion in hot spot _DNA_3.
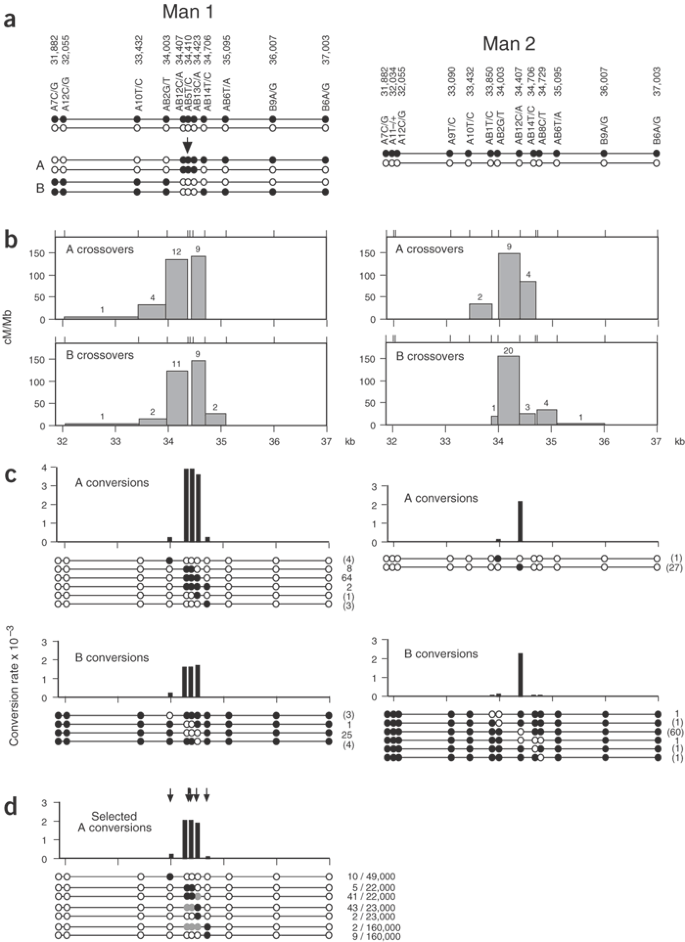
Sperm DNAs from man 1 and man 2 were assayed for reciprocal (A,B) crossovers and conversions as in Figure 1c,d. (a) Progenitor haplotypes in each man, with SNP coordinates taken from ref. 8. Illustrative examples of type A and B crossovers and conversions are shown for man 1. The numbers of amplifiable progenitor molecules of each haplotype screened for recombinants were as follows: man 1, 22,000 (A) and 22,000 (B); man 2, 14,000 (A) and 33,000 (B). (b) Crossover activity. The numbers of crossovers mapping to each interval (shown above each bar) were used to estimate the recombination efficiency in cM per Mb. (c) Conversion activity. The Poisson-corrected numbers of each type of convertant seen are shown, together with the deduced conversion rate per SNP site. Single-site conversions could not be verified, and numbers (in brackets) are therefore provisional. (d) Conversions detected after selection of sperm DNA from man 1 using four different biotinylated ASOs (AB2T, AB12A/AB5C, AB13A, AB14C; arrows). Conversions affecting SNPs AB12 or AB5 alone would not be detected by this approach, but these sites are only 2 bp apart and such conversions will be rare (∼4% of all conversions) and were not seen in the unselected sperm DNA conversions. The number of enriched molecules screened from each selection, and the Poisson-corrected numbers of convertants detected, are given at right. Selected markers in conversion molecules are shown in black, and unselected coconverted markers in gray. Crossover molecules detected in these surveys (Supplementary Table 2 online) agreed in number and distribution with those expected from the crossover distributions shown in b.
Defining the shape of this conversion gradient depends critically on the reliable detection of single-site conversion events. Detecting such events is difficult, particularly for SNPs with a substantial PCR misincorporation rate. We therefore used DNA enrichment by allele-specific hybridization17 to reduce the complexity of sperm DNA before conversion analysis (Fig. 1a). This improves the signal-to-noise ratio and allows much larger pools of sperm DNA to be surveyed for recombinants. Analysis of recombinants in man 1 by this method (Figs. 1d and 2d) confirmed the authenticity of single-site conversions and the shape of the gradient. We also applied this approach to analyzing sperm and blood DNA from a third man heterozygous with respect to the central marker AB12 (data not shown). We detected 19 AB12 conversions among 16,000 sperm molecules but no recombinants in 65,000 blood molecules. This is further evidence for the authenticity of sperm conversions and is consistent with these being products of meiotic recombination.
To determine whether this pattern of conversion occurs in other crossover hot spots, we analyzed hot spot _DMB_2 in the MHC8. This hot spot is much less active than _DNA_3 (crossover rate of 5 × 10−5 in the man analyzed), necessitating the use of DNA enrichment to analyze conversions, but is abundant in SNPs near the center of the hot spot. The pattern of recombination was very similar to that seen in hot spot _DNA_3, with detectable conversions and crossovers occurring at similar relative rates and with what appeared to be a very steep gradient of gene conversion activity in the hot spot (Fig. 3). Again, conversion tracts were short, with most conversions involving the transfer of a single SNP between haplotypes. We also analyzed an intense hot spot in the gene SHOX in the pseudoautosomal pairing region PAR1 on the sex chromosomes9. The crossover rate at this hot spot is much higher than at _DMB_2 (3.7 × 10−3 per sperm) but the low SNP density and lack of markers near the center of the hot spot again necessitated the use of enrichment. Only the marker closest to the center of the hot spot (5053G/C, located about 200 bp 5′ from the center) showed a substantial conversion rate, at about 30% of the crossover rate (Fig. 3f). In contrast, marker 5543C/G located about 300 bp 3′ to the hot-spot center yielded no detectable convertants. These data are again consistent with short conversion tracts and a steep gradient of conversion activity extending from the hot-spot center but were insufficient to establish maximum conversion rates at the hot-spot center and the shape of the gradient.
Figure 3: Gene conversion activity in the _DMB_2 and SHOX hot spots.
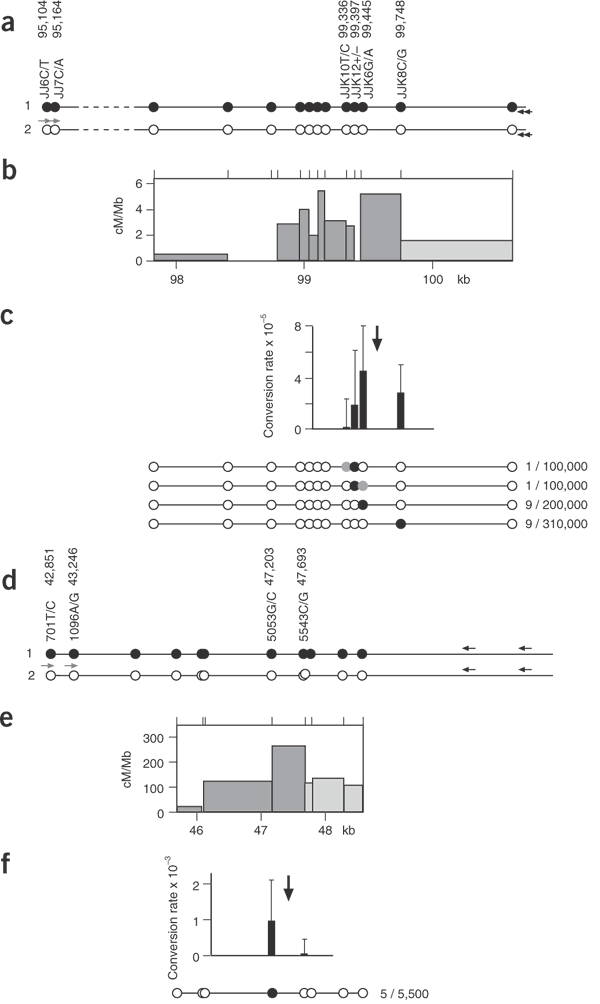
(a) _DMB_2 haplotypes in the man analyzed, with SNP coordinates taken from ref. 8. Sperm DNA was enriched using four different biotinylated ASOs (JJK10C, JJK12−, JJK6A, JJK8G) specific for haplotype 1 and spanning the center of the hot spot. Purified DNA was amplified by PCR using allele-specific primers for haplotype 2 (JJ6C, JJ7C located 4.4 kb upstream of the center of the hot spot) and crossovers and conversions detected as in Figure 1d. (b) _DMB_2 crossover activity determined from the 45 crossovers detected in the four assays; 26 of these mapped to the adjacent hot spot _DMB_1 located in the dashed region indicated in a (data not shown). Crossovers in the last interval (pale gray) were lost after enrichment and were therefore estimated from previous _DMB_2 data8. (c) _DMB_2 conversion rates, with 95% confidence intervals, and structures of convertants detected (black circle, selected marker; gray circle, coconverted marker). Enrichment with JJK10C yielded eight crossovers but no conversions in 130,000 molecules. The approximate location of the peak of crossover activity is marked with an arrow. Full details of the conversion and crossover data are provided in Supplementary Table 3 online. (d) Recombination in the SHOX hot spot; haplotypes in the man analyzed, with coordinates taken from ref. 9. Sperm DNA was enriched using biotinylated ASO 5053C or 5543G,which flank the peak crossover interval, and purified DNA was amplified by PCR using allele-specific primers for 701T and 1096A. (e) SHOX crossover activity estimated by a standard crossover assay9 and from the 32 crossovers detected in the two enriched DNA pools. The crossover rate is 3.7 × 10−3 per sperm. (f) SHOX conversion activity. Twenty-four crossovers but no convertants were detected among the 9,400 molecules enriched for 5543G. The arrow marks the approximate center of the crossover hot spot.
If recombination-initiating lesions, such as double-strand breaks18, occur at a single site in the hot spot, then all conversion tracts arising by subsequent resection and repair should share a common region of overlap corresponding to the initiation site. There is no unique region of overlap at either _DNA_3 or _DMB_2, however; for example, the double-site conversion in _DMB_2 at JJK10/12 and the single site conversions at JJK8 (Fig. 3c) cannot, under a simple double-strand break–repair model, arise from initiations at the same site. It therefore follows that although hot spots probably contain recombination initiation sites, these sites are probably diffused over a localized zone of activity.
Even with the extensive data at hot spot _DNA_3 and the abundance of markers, it is not possible to determine accurately the lengths of conversion tracts (Fig. 4a). But we could estimate these variables by simulating gene conversions under a range of models in which the locations and lengths of conversion tracts are varied and comparing the simulated conversion distributions with those observed experimentally (Fig. 2d and Supplementary Note online). Irrespective of the model used, the center of the conversion initiation zone coincided with the peak of crossover activity in the hot spot (Fig. 4b and Supplementary Table 1 online). This strongly suggests that crossovers and conversions arise from the same initiating lesions. All models similarly suggest that initiating lesions are diffused over a zone of ∼400–500 bp, similar to the spread of meiotic double-strand breaks observed in yeast hot spots19. The mean length of conversion tracts is less certain but probably lies in the range of 55–290 bp (Fig. 4a). Conversion tract length will also affect the proportion of conversions that alter haplotype and are thus experimentally detectable. This proportion, estimated at 16% and 75% for the shortest and longest permissible mean tracts, respectively, suggests that the ratio of conversions to crossovers observed at hot spot _DNA_3 (2.7:1, averaged over all assays in both men; Fig. 2) is probably an underestimate and is more likely ∼4–15:1. This implies that somewhere between 80% and 94% of recombinations at hot spot _DNA_3 are resolved as conversions rather than crossovers.
Figure 4: Conversion tract lengths and breakpoint distributions in hot spot _DNA_3.
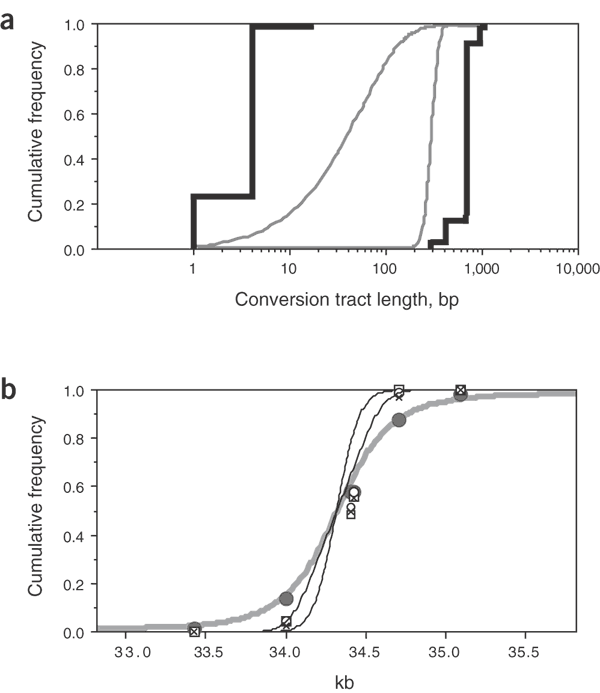
(a) The cumulative frequencies of minimum and maximum conversion tract lengths (black lines) determined from conversions detected by biotinylated ASO enrichment (Fig. 2d). The considerable spread between minimum and maximum lengths reflects marker spacing in this region. Continuous gray lines show the two most extreme distributions determined by simulation that are compatible with the observed conversion data (models 4 and 6 in Supplementary Table 1 online; model 4 assumes that conversion tracts have no minimum length and extend unidirectionally from sites of initiation with a fixed probability per base traversed that the tract terminates, whereas model 6 assumes that conversion tract lengths follow a normal distribution); these distributions have mean tract lengths of 55 and 290 bp, respectively. (b) Cumulative frequency distributions of breakpoints across hot spot _DNA_3. The distribution of crossover breakpoints in man 1 (gray circles) determined from 304 sperm crossovers is compared with the distribution of detectable conversion breakpoints in man 1 (crosses, unselected A conversions; squares, unselected B conversions; open circles, selected A conversions; Fig. 2c,d), as well as with all conversions, including those that do not affect any SNP sites, under the two extreme conversion models (black lines, each established from 100,000 simulations; the distribution with shorter conversion tracts gives the steeper curve). Inclusion of missing conversions, which would preferentially locate to the interval of 34.0–34.4 kb, shifts the conversion breakpoint distribution in the 5′ direction. The crossover and conversion distributions are significantly different (χ2 [3 d.f.] = 303, P < < 0.001). The thick gray line shows the least-squares best-fit distribution for crossovers assuming an identical distribution of initiating events; this distribution fits well with the observed crossover distribution (χ2 [5 d.f.] = 8.1, P = 0.15) and gives a mean length of conversion tracts accompanying crossover of about 460 bp. d.f., degrees of freedom.
Crossover breakpoints are more broadly distributed across hot spot _DNA_3 than are conversion breakpoints (Fig. 4b). The same seems to be true for hot spot _DMB_2 (Fig. 3b,c). This suggests that conversion tracts accompanying crossover20 are longer (mean length = ∼460 bp for _DNA_3) than conversion tracts without crossover (mean length = 55–290 bp). Thus, although crossovers and conversions seem to be initiated at the same sites, conversions may be generated by an alternative downstream processing pathway, such as synthesis-dependent strand annealing, as occurs in yeast21. Alternatively, the difference between crossover and conversion distributions could result from a quality control checkpoint at which short interactions between homologs are aborted as conversions and only longer interactions are processed into crossovers.
The conversion activity in human crossover hot spots provides strong evidence that they represent sites of crossover initiation, rather than resolution after migration from a distal initiation site, and is consistent with crossover asymmetry evidence most readily explained under the hot-spot initiation model20. The short conversion tracts seen in human hot spots are similar to those reported in a mouse hot spot22 and seem similar in length to interallelic sperm conversions seen in HLA-DPB1 (ref. 16) as well as conversions between loci3,6,7. This suggests that similar mechanisms operate in interallelic and interlocus gene conversion. But the germline specificity of hot spot _DNA_3 conversions contrasts with evidence for high-frequency interlocus conversions reported not only in sperm but also in blood DNA12. Bidirectional gene conversion gradients associated with human crossover hot spots are similar to those seen in Drosophila23 and yeast24, but seem to be much steeper and have shorter conversion tracts. There is little evidence for kilobase-long conversion tracts often seen in yeast25; we have never seen such tracts in human hot spots, and evidence for large scale conversion at the human CYP21A2 locus has now been discredited26. To our knowledge, the only reasonably clear evidence for long conversion tracts in humans comes from rare germline reversions of triplet repeat expansions27.
There is considerable current interest in establishing haplotype maps of the human genome and in understanding how such processes as recombination can influence haplotype structures28. Approaches include the exploration of models of crossover distribution that can account for observed haplotype block structures29,30. What is missing from this modeling is the inclusion of gene conversion as an additional process that can lead to haplotype diversification. The present work provides the first step in determining basic parameters of meiotic gene conversion in the human genome and suggests that interallelic conversion in hot spots has the potential to profoundly affect haplotype diversity, but only at an extremely localized level, in the hot spots themselves.
Methods
DNA preparation.
We collected, with approval from the Leicestershire Health Authority Research Ethics Committee, semen and blood samples with informed consent from UK men of north European descent, including volunteers and men attending fertility clinics, and prepared DNA as described previously under conditions designed to minimize the risk of contamination15. We digested DNA with _Hin_dIII, _Hin_dIII and _Blp_I, or _Nru_I and _Bsp_HI for hot spots _DNA_3, _DMB_2 or SHOX, respectively, to release the target region on a DNA fragment 7.0, 6.6 or 9.4 kb long, respectively. We then carried out DNA enrichment and recombination analysis.
DNA enrichment.
Enrichment of genomic DNA by allele-specific hybridization is described in detail elsewhere17. Briefly, we mixed 1–16 μg digested genomic DNA (depending on conversion rate and the efficiency of enrichment) at 100–200 μg ml−1 with 0.38 μM HPLC-purified 5′-tribiotinylated ASO (18 nt long, with the allele-specific base 11 nt from the 5′ end; Eurogentec) plus 1.5 μM competitor ASO complementary to the other SNP allele in hybridization buffer (45 mM Tris-HCl (pH 8.8), 11 mM ammonium sulfate, 4.5 mM MgCl2, 6.7 mM 2-mercaptoethanol, 4.4 μM EDTA and 2 μg ml−1 single-stranded high-molecular-weight herring sperm DNA as carrier). DNA was denatured at 96 °C for 75 s and then annealed for 5 min at 35–50 °C (depending on the GC content of the biotinylated ASO). We captured hybrids by adding Dynabeads M-280 Streptavidin (Dynal Biotech) to a final concentration of 9 mg ml−1 and incubating for 10 min at the annealing temperature with gentle mixing. Beads were magnetically separated and then washed with hybridization buffer at the hybridization temperature followed by elution buffer (0.14× hybridization buffer, 5 μg ml−1 single-stranded herring DNA) at room temperature. We released single-stranded target DNA by denaturation from the biotinylated ASO–bead complex using a further incubation in elution buffer for 2 min at 65 °C, or at 80 °C for biotinylated ASOs with >60% GC content. We recovered additional target by adding biotinylated ASO to 0.38 μM to the unbound DNA and carrying out two to three additional rounds of extraction. Two of the biotinylated ASOs (JJK10C, JJK12−) gave relatively poor enrichment (13- and 7-fold respectively), and so we subjected the recovered single-stranded DNA to a second cycle of purification17. We determined yields of recovered DNA by long PCR amplification across the entire target region and comparing PCR product yields with those obtained from decreasing inputs of unfractionated DNA. We verified yields by Poisson analysis of limiting single molecule dilutions of enriched single-stranded DNA15. We estimated the degree of purification using allele-specific PCR to determine the ratio of each haplotype in the enriched DNA17. Yields varied from 10% to 65%, depending on the SNP being selected and whether one or two cycles of enrichment were done, with purifications of 40-fold to 300-fold. Further details on the biotinylated ASOs used are provided elsewhere17.
Detection of recombinant molecules.
To detect recombinants in unfractionated genomic DNA, we amplified multiple aliquots of digested sperm DNA, each containing 50–100 amplifiable molecules of each progenitor haplotype (0.6–1.2 ng DNA), using one distal allele-specific primer and one universal primer. Long PCR conditions were described previously, with annealing temperatures optimized for each allele-specific primer to ensure good efficiency and excellent allele-specificity8,9,15. Each 10-μl PCR was amplified for 23 cycles then diluted to 0.2 ml with water. We used 0.5 μl of each diluted primary PCR to seed a 15-μl secondary PCR containing nested allele-specific and universal primers. These secondary PCRs were amplified for 36 cycles, using optimized annealing temperatures, and analyzed by dot-blot hybridization with 32P-labeled ASOs as described previously15. We analyzed recombinants in enriched DNA using the same method, but with larger DNA inputs (up to 3,000 molecules of the selected haplotype per PCR, depending on recombination rate and the degree of enrichment, but with no more than 50 molecules of the other contaminating haplotype per PCR).
Validation of multisite recombinants.
All primary PCR products from unfractionated sperm DNA showing crossover or conversion molecules were further analyzed using allele-specific primers to specifically amplify conversion or crossover molecules. We then typed these PCR products by ASO hybridization. We used this approach to validate the structure of recombinant molecules containing switches at more than one SNP site and to identify pools of sperm containing more than one recombinant (e.g., two different conversions or a conversion and a crossover molecule). We give further details in Supplementary Note online. We corrected the final inventory of crossover and conversion molecules across all PCRs by Poisson analysis for instances of two or more identical recombinant molecules being present in the same PCR15; these corrections were modest, resulting in at most only a 1.3-fold increase in the number of a given class of recombinant.
Analysis of conversion distributions.
We estimated conversion parameters by maximum likelihood analysis, comparing simulated sperm conversions with biotinylated ASO–selected conversion data from hot spot _DNA_3. We explored a range of conversion models, including conversion tracts extending unidirectionally or bidirectionally from a site of initiation with a fixed probability per base encountered that the tract terminated, as well as models with tracts of a minimum fixed length or with normally distributed lengths. We also explored the effects of different shapes of the initiation zone. For each model, we systematically varied the initiation rate (the proportion of sperm carrying conversions), the center and width of the zone of initiation, and the parameters controlling conversion tract length. For each combination of variables, we simulated sperm conversion events until 20,000 conversions affecting at least one SNP site were accumulated. Each conversion tract was classified according to which, if any, SNP markers had been exchanged between haplotypes. We used the frequency of each class of convertant to estimate the likelihood of obtaining the observed numbers of conversion-positive and conversion-negative PCRs in each biotinylated ASO enrichment. We combined these probabilities over the four different enrichments to give the overall likelihood of obtaining the entire observed data set for a given combination of conversion variables. We then identified the combination of variables that maximized this likelihood (Supplementary Fig. 1 online) and rejected any model that gave a maximum likelihood <1/100 of that seen in the best models. Details of procedures and results are given in Supplementary Note online.
Note: Supplementary information is available on the Nature Genetics website.
References
- Clarke, S.H., Claflin, J.L. & Rudikoff, S. Polymorphism in immunoglobulin heavy chains suggesting gene conversion. Proc. Natl. Acad. Sci. USA 79, 3280–3284 (1982).
Article CAS PubMed PubMed Central Google Scholar - Jinks-Robertson, S. & Petes, T.D. High-frequency meiotic gene conversion between repeated genes on nonhomologous chromosomes in yeast. Proc. Natl. Acad. Sci. USA 82, 3350–3354 (1985).
Article CAS PubMed PubMed Central Google Scholar - Powers, P.A. & Smithies, O. Short gene conversions in the human fetal globin gene region: a by-product of chromosome pairing during meiosis? Genetics 112, 343–358 (1986).
CAS PubMed PubMed Central Google Scholar - Ogasawara, K. et al. Recombination and gene conversion-like events may contribute to ABO gene diversity causing various phenotypes. Immunogenetics 53, 190–199 (2001).
Article CAS PubMed Google Scholar - Rozen, S. et al. Abundant gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 423, 873–876 (2003).
Article CAS PubMed Google Scholar - Collier, S., Tassabehji, M., Sinnott, P. & Strachan, T. A de novo pathological point mutation at the 21-hydroxylase locus: implications for gene conversion in the human genome. Nat. Genet. 3, 260–265 (1993).
Article CAS PubMed Google Scholar - Watnick, T.J., Gandolph, M.A., Weber, H., Neumann, H.P. & Germino, G.G. Gene conversion is a likely cause of mutation in PKD1. Hum. Mol. Genet. 7, 1239–1243 (1998).
Article CAS PubMed Google Scholar - Jeffreys, A.J., Kauppi, L. & Neumann, R. Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat. Genet. 29, 217–222 (2001).
Article CAS PubMed Google Scholar - May, C.A., Shone, A.C., Kalaydjieva, L., Sajantila, A. & Jeffreys, A.J. Crossover clustering and rapid decay of linkage disequilibrium in the Xp/Yp pseudoautosomal gene SHOX. Nat. Genet. 31, 272–275 (2002).
Article CAS PubMed Google Scholar - Jaatinen, T., Eholuoto, M., Laitinen, T. & Lokki, M.L. Characterization of a de novo conversion in human complement C4 gene producing a C4B5-like protein. J. Immunol. 168, 5652–5658 (2002).
Article CAS PubMed Google Scholar - Huang, M.M., Erlich, H.A., Goodman, M.F. & Arnheim, N. Analysis of mutational changes at the HLA locus in single human sperm. Hum. Mutat. 6, 303–310 (1995).
Article CAS PubMed Google Scholar - Tusie-Luna, M.T. & White, P.C. Gene conversions and unequal crossovers between CYP21 (steroid 21-hydroxylase gene) and CYP21P involve different mechanisms. Proc. Natl. Acad. Sci. USA 92, 10796–10800 (1995).
Article CAS PubMed PubMed Central Google Scholar - Ardlie, K. et al. Lower-than-expected linkage disequilibrium between tightly linked markers in humans suggests a role for gene conversion. Am. J. Hum. Genet. 69, 582–589 (2001).
Article CAS PubMed PubMed Central Google Scholar - Erlich, H.A. & Gyllensten, U.B. Shared epitopes among HLA class II alleles: gene conversion, common ancestry and balancing selection. Immunol. Today 12, 411–414 (1991).
Article CAS PubMed Google Scholar - Jeffreys, A.J., Ritchie, A. & Neumann, R. High-resolution analysis of haplotype diversity and meiotic crossover in the human TAP2 recombination hotspot. Hum. Mol. Genet. 9, 725–733 (2000).
Article CAS PubMed Google Scholar - Zangenberg, G., Huang, M.M., Arnheim, N. & Erlich, H. New HLA-DPB1 alleles generated by interallelic gene conversion detected by analysis of sperm. Nat. Genet. 10, 407–414 (1995).
Article CAS PubMed Google Scholar - Jeffreys, A.J. & May, C.A. DNA enrichment by allele-specific hybridisation (DEASH): a novel method for haplotyping and for detecting low frequency base substitutional variants and recombinant DNA molecules. Genome Res. 13, 2316–2324 (2003).
Article CAS PubMed PubMed Central Google Scholar - Szostak, J.W., Orr-Weaver, T.L., Rothstein, R.J. & Stahl, F.W. The double-strand-break repair model for recombination. Cell 33, 25–35 (1983).
Article CAS PubMed Google Scholar - Keeney, S. Mechanism and control of meiotic recombination initiation. Curr. Top. Dev. Biol. 52, 1–53 (2001).
Article CAS PubMed Google Scholar - Jeffreys, A.J. & Neumann, R. Reciprocal crossover asymmetry and meiotic drive in a human recombination hot spot. Nat. Genet. 31, 267–271 (2002).
Article CAS PubMed Google Scholar - Paques, F. & Haber, J.E. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 63, 349–404 (1999).
CAS PubMed PubMed Central Google Scholar - Guillon, H. & De Massy, B. An initiation site for meiotic crossing-over and gene conversion in the mouse. Nat. Genet. 32, 296–299 (2002).
Article CAS PubMed Google Scholar - Hilliker, A.J. et al. Meiotic gene conversion tract length distribution within the rosy locus of Drosophila melanogaster. Genetics 137, 1019–1026 (1994).
CAS PubMed PubMed Central Google Scholar - Schultes, N.P. & Szostak, J.W. Decreasing gradients of gene conversion on both sides of the initiation site for meiotic recombination at the ARG4 locus in yeast. Genetics 126, 813–822 (1990).
CAS PubMed PubMed Central Google Scholar - Borts, R.H. & Haber, J.E. Length and distribution of meiotic gene conversion tracts and crossovers in Saccharomyces cerevisiae. Genetics 123, 69–80 (1989).
CAS PubMed PubMed Central Google Scholar - Koppens, P.F., Hoogenboezem, T. & Degenhart, H.J. Carriership of a defective tenascin-X gene in steroid 21-hydroxylase deficiency patients: TNXB -TNXA hybrids in apparent large-scale gene conversions. Hum. Mol. Genet. 11, 2581–2590 (2002).
Article CAS PubMed Google Scholar - Losekoot, M. et al. Prenatal diagnosis of the fragile X syndrome: loss of mutation owing to a double recombinant or gene conversion event at the FMR1 locus. J. Med. Genet. 34, 924–926 (1997).
Article CAS PubMed PubMed Central Google Scholar - Wall, J.D. & Pritchard, J.K. Haplotype blocks and linkage disequilibrium in the human genome. Nat. Rev. Genet. 4, 587–597 (2003).
Article CAS PubMed Google Scholar - Gabriel, S.B. et al. The structure of haplotype blocks in the human genome. Science 296, 2225–2229 (2002).
Article CAS PubMed Google Scholar - Wang, N., Akey, J.M., Zhang, K., Chakraborty, R. & Jin, L. Distribution of recombination crossovers and the origin of haplotype blocks: the interplay of population history, recombination, and mutation. Am. J. Hum. Genet. 71, 1227–1234 (2002).
Article CAS PubMed PubMed Central Google Scholar
Acknowledgements
We thank J. Blower and volunteers for providing semen and blood samples, S. Mistry for oligonucleotide synthesis and colleagues for helpful discussions. This work was supported by grants to A.J.J. from the Medical Research Council and the Royal Society.
Author information
Authors and Affiliations
- Department of Genetics, University of Leicester, Leicester, LE1 7RH, UK
Alec J Jeffreys & Celia A May
Authors
- Alec J Jeffreys
You can also search for this author inPubMed Google Scholar - Celia A May
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toAlec J Jeffreys.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Jeffreys, A., May, C. Intense and highly localized gene conversion activity in human meiotic crossover hot spots.Nat Genet 36, 151–156 (2004). https://doi.org/10.1038/ng1287
- Received: 03 September 2003
- Accepted: 04 December 2003
- Published: 04 January 2004
- Issue Date: 01 February 2004
- DOI: https://doi.org/10.1038/ng1287