RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III–transcribed RNA intermediate (original) (raw)
Main
The immune system must trigger an arsenal of defense measures to combat invading microbes. The innate immune system is the first line of defense1 and functions to control infection directly and relay signals to the adaptive immune system. Several classes of germline-encoded pattern-recognition receptors have been linked to innate defense. These include the Toll-like receptors (TLRs)2, the C-type lectin receptors3, the RIG-like helicases4, members of the Nod-like receptor family5 and cytosolic DNA sensors6,7,8. Individual pattern-recognition receptors recognize microbial products (also called 'pathogen-associated molecular patterns') from bacteria, viruses, fungi and parasites and trigger signaling pathways that regulate genes encoding molecules of the immune response. These include proinflammatory cytokines such as tumor necrosis factor (TNF), interleukin 1β and type I interferons1,5. Accumulating evidence shows that in addition to sensing microbial products, many of these same sensors detect 'danger' signals (or danger-associated molecular patterns) that are released from damaged or dying cells9.
A common theme in the recognition of pathogen-associated molecular patterns is the sensing of non-self nucleic acids. Viruses, for example, are sensed almost exclusively via their nucleic acid genomes or as a result of their replicative or transcriptional activity10. In the cytosol, RIG-I and Mda5 (encoded by Ddx58 and Ifih1, respectively) discriminate between different classes of RNA viruses11,12. RIG-I senses the nascent 5′-triphosphate moiety of viral genomes or virus-derived transcripts of negative-sense single-stranded RNA (ssRNA) viruses, whereas Mda5 is activated by long double-stranded RNA (dsRNA), a typical intermediate of the replication of positive-sense ssRNA viruses13,14. RIG-I also detects short blunt-ended dsRNA15. Both RIG-I and Mda5 engage the mitochondrial adaptor protein IPS-1 (also known as MAVS, Cardif or VISA)16,17,18,19. IPS-1 subsequently triggers 'downstream' signaling and activation of the kinase IKKα-IKKβ–transcription factor NF-κB pathway or the kinase TBK1–transcription factor IRF3 pathway, which promote the transcription of genes encoding inflammatory cytokines and type I interferon, respectively.
DNA is also a potent activator of innate immunity. In plasmacytoid dendritic cells (DCs), CpG DNA engages TLR9 to turn on transcription of the gene encoding interferon-α (IFN-α). A second DNA-sensing pathway elicits activation of the TBK1-IRF3 signaling pathway and transcription of genes encoding IFN-α and IFN-β, although the underlying mechanisms responsible for these last events are unclear. A candidate sensor, DAI (encoded by Zbp1)7, has been shown to bind synthetic double-stranded DNA (dsDNA) and activate TBK1 and IRF3 to promote the transcription of interferon genes. Knockdown experiments have indicated that DAI is involved in sensing cytosolic DNA in some cell lines7. However, DAI-deficient embryonic fibroblasts and macrophages respond normally to cytosolic DNA20, and DAI-deficient mice mount normal adaptive immune responses, which indicates possible redundancy with additional sensors. A second DNA sensor, AIM2 (absent in melanoma 2), has also been identified. AIM2 binds dsDNA and the adaptor protein ASC to form a caspase-1-activating inflammasome. However, AIM2 does not regulate the transcription of genes encoding type I interferon8,21,22,23.
It is likely that in most cell types, DNA viruses trigger the transcription of genes encoding type I interferons through TLR-independent DNA-sensing mechanisms. Although type I interferons are best studied in antiviral immunity, evidence of the involvement of these cytokines in bacterial, fungal and parasitic infection has also emerged. Francisella tularensis24, Streptococcus agalactiae25, Listeria monocytogenes26,27, Mycobacterium tuberculosis28, Legionella pneumophila29, Brucella abortus30 and Trypanosomai cruzi31 all trigger type I interferon by what seems to be a TBK1- and IRF3-dependent pathway. Neither the host sensor nor the microbial ligands that trigger these responses have been clearly identified, although DNA has been suggested as the likely ligand25,27. Endogenous DNA molecules generated during autoimmunity are also probably sensed by this pathway. For example, macrophages from DNase II–deficient mice, which fail to digest DNA from engulfed apoptotic cells, show robust production of type I interferon and inflammatory cytokines through IRF3 activation, which leads to lethal anemia and chronic arthritis32. Defining the molecular mechanisms responsible for sensing DNA and the induction of type I interferons and inflammatory cytokines therefore may aid in understanding and treating infectious as well as autoimmune diseases.
Here we identify a novel DNA-sensing pathway involving RIG-I. AT-rich dsDNA served as a template for RNA polymerase III, which normally functions to transcribe 5_S_ rRNA, tRNA and other small noncoding RNA through specific promoter regions. AT-rich dsDNA was transcribed by RNA polymerase III into dsRNA with a 5′-triphosphate moiety; this process converted AT-rich DNA into a RIG-I ligand. Moreover, we show that Epstein-Barr virus (EBV)-encoded RNAs (EBERs) was also transcribed by RNA polymerase III and then activated RIG-I and the transcription of genes encoding type I interferons. The RNA polymerase III–RIG-I pathway seems to be functional in both human and mouse cells, but in mouse cells, this pathway seems to be redundant with additional DNA-sensing mechanisms.
Note: Supplementary information is available on the Nature Immunology website.
Results
Poly(dA:dT) triggers type I interferons through RIG-I
To study the function of known RNA sensors in the detection of the negative-strand ssRNA paramyxovirus Sendai virus (SEV), we transfected human 293T embryonic kidney cells with small interfering RNA (siRNA) targeting TLR3, RIG-I, Mda5 or IPS-1. We used the dsDNA mimetic poly(dA:dT) as a control stimulus, as it has been shown to signal independently of these sensors in the mouse system33,34. As expected, SEV-mediated induction of type I interferon was completely abolished in 293T cells that had been treated with siRNA targeting RIG-I or IPS-1. Unexpectedly, poly(dA:dT)-triggered production of type I interferon was also abolished in 293T cells expressing RIG-I- or IPS-1-specific siRNA (Fig. 1a). Induction of type I interferon triggered by overexpression of IRF1 was unaffected by knockdown of RIG-I or IPS-1 (Fig. 1a). Dominant negative RIG-I constructs or targeting of IPS-1 with the hepatitis C virus–derived protease NS3-4A19 in 293T cells also completely abolished poly(dA:dT)-triggered induction of type I interferon (Supplementary Fig. 1a). Similar findings have been reported before5. We found that poly(dA:dT) also activated NF-κB-dependent gene transcription through RIG-I (Supplementary Fig. 1b).
Figure 1: Poly(dA:dT) triggers the induction of type I interferon via RIG-I in human cells.
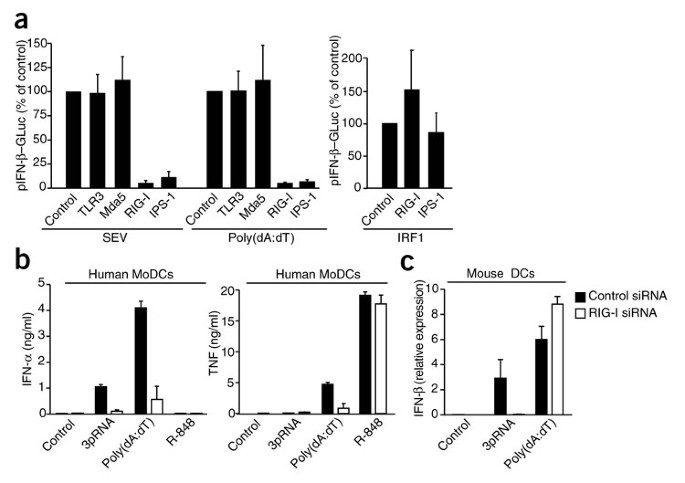
(a) Activity of an IFN-β reporter plasmid (pIFN-β–GLuc) in 293T cells transfected for 48 h with siRNA targeting TLR3, RIG-I, Mda5 or IPS-1 (three per gene, each tested in triplicate), then transfected with the reporter plasmid in conjunction with poly(dA:dT) or SEV and assessed after an additional of 24 h; results are normalized to results obtained with control siRNA, set as 100%. Right, IFN-β promoter transactivation in response to overexpression of IRF1 in 293T cells in which RIG-I or IPS1 was silenced. (b) Enzyme-linked immunosorbent assay (ELISA) of the production of IFN-α or TNF by MoDCs transfected by electroporation with control or RIG-I-specific siRNA, then stimulated with 3pRNA, poly(dA:dT) or R-848 48 h after electroporation and assessed 24 h later. (c) Real-time PCR analysis of IFN-β induction in mouse bone marrow–derived DCs mock transfected (Control) or transfected by electroporation with control or RIG-I-specific siRNA, then stimulated 48 h after electroporation and assessed 5 h later; results are presented relative to the expression of Hprt1 (encoding hypoxanthine guanine phosphoribosyl transferase). Data are from one representative of two (a) or three (b,c) experiments (mean and s.e.m.).
In primary human monocyte-derived DCs (MoDCs), RIG-I-specific siRNA also resulted in much lower type I interferon and TNF responses after poly(dA:dT) stimulation (Fig. 1b). TNF production in response to the TLR7-TLR8 ligand R-848 was unaffected, however. In agreement with published reports, mouse DCs or macrophages genetically deficient in IPS-1 (data not shown) or transfected by electroporation with siRNA targeting RIG-I were still responsive to poly(dA:dT) in terms of production of type I interferon (Fig. 1c). Together, these results indicate that RIG-I and IPS-1 are critical for poly(dA:dT)-triggered induction of type I interferon in human cells but are dispensable for these responses in cells of the mouse immune system.
Poly(dA:dT) activates RIG-I through an RNA intermediate
We next sought to understand how RIG-I, an RNA sensor, could respond to dsDNA. We first compared the kinetics of the interferon response after treatment of human peripheral blood mononuclear cells (PBMCs) with the true RIG-I stimulus 5′-triphosphate RNA (3pRNA) and transfected poly(dA:dT). PBMCs responded with slower kinetics to poly(dA:dT) than to 3pRNA (Fig. 2a), which suggested that poly(dA:dT) may activate RIG-I indirectly through the formation of an endogenous, secondary RNA ligand for RIG-I. To address that possibility, we isolated RNA from poly(dA:dT)-transfected 293T cells and tested its ability to stimulate the production of type I interferon in human PBMCs. We pretreated PBMCs with chloroquine to inhibit TLR9-dependent induction of type I interferon (Supplementary Fig. 2a) and then transfected the cells with RNA isolated from poly(dA:dT)-transfected or untransfected cells. Indeed, RNA derived from poly(dA:dT)-transfected 293T cells induced the production of type I interferon in PBMCs, but RNA derived from untransfected cells did not (Fig. 2b). The magnitude of the interferon response was similar to that obtained by SEV infection (Fig. 2b). This response was unaffected by treatment of the 293T cell–derived RNA with DNase I (Supplementary Fig. 2b), which indicated that the response was DNA independent. The induction of a stimulatory RNA species by poly(dA:dT) occurred within 4 h of transfection and peaked 9 h after delivery of poly(dA:dT) (Fig. 2c). The ability of poly(dA:dT) to generate a stimulatory RNA ligand was not restricted to 293T cells, as RNA derived from primary cells (for example, human monocytes) stimulated with poly(dA:dT) induced type I interferon when transfected into human PBMCs (Fig. 2d). RNA derived from mouse L929 fibroblasts (Fig. 2d) or mouse embryonic fibroblasts (MEFs; data not shown) transfected with poly(dA:dT) also potently induced the production of type I interferon, which indicated that this mechanism was conserved across humans and mice. Knockdown of RIG-I in human MoDCs or mouse DCs resulted in a much lower type I interferon response triggered by RNA derived from poly(dA:dT)-transfected cells (Fig. 2e). We obtained similar findings with dominant negative RIG-I or the hepatitis C virus protease NS3-4A in 293T cells (Supplementary Fig. 2c). Together, these results indicate that transfection of poly(dA:dT) led to the generation of RNA species in both human and mouse cells, which activated RIG-I.
Figure 2: Poly(dA:dT) triggers the formation of endogenous RIG-I-stimulatory RNA.
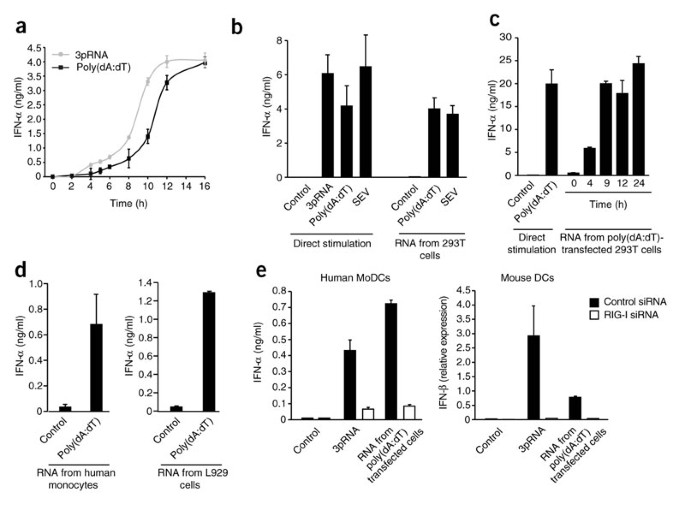
(a) ELISA of IFN-α production by human PBMCs transfected with 3pRNA or poly(dA:dT). (b) ELISA of IFN-α production by chloroquine-treated PBMCs transfected with RNA isolated from 293T cells stimulated with SEV or poly(dA:dT) (right) or PBMCs stimulated directly with 3pRNA, poly(dA:dT) or SEV (left), assessed 24 h after stimulation. (c) ELISA of IFN-α production by chloroquine-treated PBMCs stimulated with RNA isolated from 293T cells 0, 4, 9, 12 or 24 h after poly(dA:dT) transfection, assessed 24 h after stimulation. Far left, directly transfected poly(dA:dT) serves as a positive control; Control, mock-transfected cells (negative control). (d) ELISA of IFN-α production by PBMCs stimulated as described in c with RNA purified from poly(dA:dT)-transfected monocytes and L929 cells. (e) ELISA of IFN-α production (left) and real-time PCR analysis of IFN-β mRNA production (right) by human MoDCs or mouse DCs transfected by electroporation with control or RIG-I-specific siRNA and, after 48 h, stimulated with 3pRNA or RNA from poly(dA:dT)-transfected 293T cells, then assessed 24 h later (IFN-α) or 5 h later (IFN-β; presented relative to Hprt1 expression). Data are from one representative of two (a,c,d), three (e) or four (b) experiments (mean and s.e.m.).
Having established that transfection of poly(dA:dT) led to the generation of an endogenous RNA ligand for the RIG-I pathway, we next determined whether dsDNA from other sources also triggered the RIG-I pathway by generating an RNA intermediate. We generated dsDNA molecules of various lengths and random sequences by PCR and tested their ability to trigger the production of type I interferon in 293T cells and in primary human PBMCs. We selected dsDNA molecules in the size range of the poly(dA:dT) used for our studies (∼20–250 nucleotides; Fig. 3a). Notably, whereas poly(dA:dT) triggered type I interferon responses in both PBMCs and 293T cells, PCR-generated dsDNA failed to trigger this response in 293T cells (Fig. 3b). In contrast, we found a size-dependent interferon response to dsDNA in PBMCs (Fig. 3a and Supplementary Fig. 3). Contrary to the results we obtained with poly(dA:dT), the interferon response to PCR-generated dsDNA was not abolished in human MoDCs in which RIG-I was silenced (data not shown). In agreement with those findings, RNA derived from 293T cells transfected with poly(dA:dT) induced an interferon response, whereas RNA generated from PCR-generated dsDNA failed to do so (Fig. 3c). Collectively, these results indicate that in human cells, at least two independent receptor systems exist to sense dsDNA: an indirect RIG-I pathway for poly(dA:dT), and a second RIG-I-independent pathway for random long dsDNA molecules. Both of these pathways are independent of TLR9.
Figure 3: Non-poly(dA:dT) dsDNA triggers the induction of type I interferon through an RNA-independent pathway.
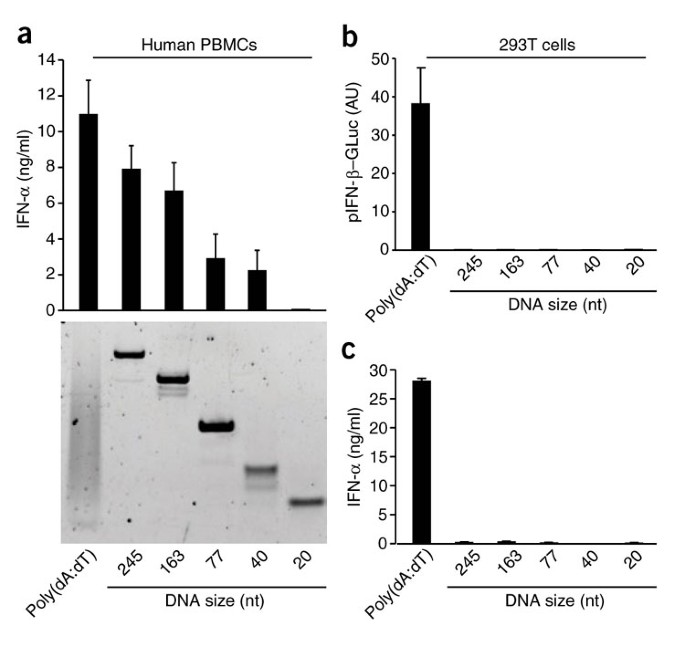
(a,b) ELISA (a) or IFN-β reporter analysis (b) of the induction of type I interferon in chloroquine-blocked PBMCs or 293T cells transfected with dsDNA of various lengths (derived from pcDNA3 by PCR) and assessed 24 h after stimulation. Below (a), nondenaturing PAGE. AU, arbitrary units. (c) ELISA of IFN-α production by chloroquine-blocked PBMCs transfected for 24 h with RNA from 293T cells that had been transfected with dsDNA of various lengths. Data are from one representative of four (a,c) or three (b) experiments (mean and s.e.m.).
Characterization of the RNA intermediate
To further characterize the poly(dA:dT)-dependent RNA species, we isolated and fractionated RNA from 293T cells that had been transfected with poly(dA:dT). By using different ethanol concentrations during the binding of RNA to a silica column, we were able to crudely fractionate RNA into different sizes. We then tested the ability of these RNA fractions to trigger interferon. The small RNA fraction (less than 200 nucleotides in length) was sufficient to induce type I interferon (Fig. 4a). The antiviral endoribonuclease RNase L, which is activated by 2′,5′-linked oligoadenylates derived from 2′,5′-oligoadenylate synthetase (OAS), produces small RNA-cleavage products from self RNA that can initiate interferon production through the RIG-I–IPS-1 pathway. Small RNA molecules of less than 200 nucleotides isolated from RNase L–activated cells have been shown to contain this stimulatory RNA species36. We therefore examined the function of the OAS–RNase L system in the interferon response to poly(dA:dT). Transient overexpression of OAS1, OAS2, OAS3 or RNase L did not alter poly(dA:dT)-induced induction of type I interferon (data not shown). Furthermore, overexpression of dominant negative RNase L mutants (data not shown) or targeting of RNase L, OAS1, OAS2, OAS3 or OASL with siRNA (Fig. 4b) did not inhibit poly(dA:dT)- or SEV-triggered type I interferon responses. Finally, RNA isolated from poly(dA:dT)-transfected wild-type or Rnasel −/− MEFs cells induced similar production of type I interferon (Fig. 4c). Thus, poly(dA:dT) transfection induces the formation of stimulatory small RNA independently of OAS and RNase L.
Figure 4: Poly(dA:dT) triggers the formation of a small stimulatory RNA species independently of the OAS–RNase L pathway.
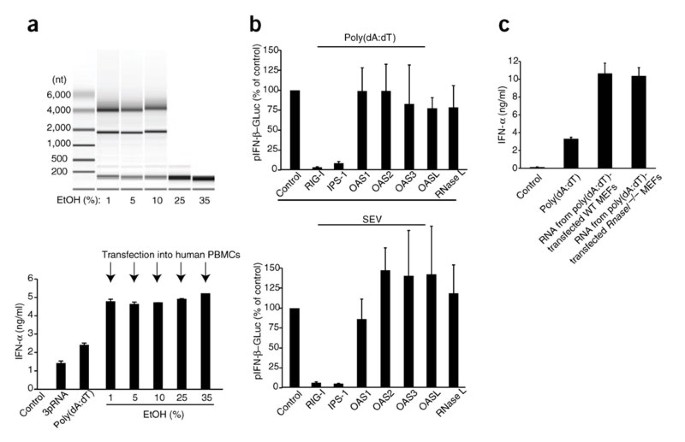
(a) Agilent RNA 6000 Pico Chip analysis of RNA isolated from poly(dA:dT)-transfected 293T cells and fractionated with various concentrations of ethanol during the initial binding step of the RNA in a silica matrix–based spin-column purification system (top). Below, induction of IFN-α by the RNA in chloroquine-treated PBMCs; direct stimulation with 3pRNA or poly(dA:dT) serves as a positive control. (b) IFN-β reporter activity in 293T cells transfected for 48 h with siRNA targeting various genes (horizontal axes; three per gene, each tested in triplicate), then transfected with an IFN-β reporter plasmid in conjunction with poly(dA:dT) (top) or SEV (bottom) and assessed after an additional 24 h; results are normalized to those of control siRNA, set as 100%. (c) ELISA of IFN-α production by chloroquine-treated PBMCs transfected with RNA isolated from poly(dA:dT)-transfected wild-type (WT) or Rnasel −/− MEFs. Far left, direct transfection of poly(dA:dT) serves as a control. Data are from one representative of four (a) or two (b,c) experiments (mean and s.e.m.).
To define the features of the poly(dA:dT)-triggered RNA species important for the interferon response, we took advantage of several RNA-modifying enzymes. To determine if phosphate groups were important features of the RNA, we treated poly(dA:dT) itself, poly(dA:dT)-triggered RNA and _in vitro_–transcribed 3pRNA with alkaline phosphatase to remove any 5′ or 3′ phosphates that might be present. Treatment of 3pRNA and poly(dA:dT)-triggered RNA with alkaline phosphatase considerably diminished their stimulatory activity, whereas poly(dA:dT) was unaffected by alkaline phosphatase treatment (Supplementary Fig. 4). In addition, RNA 5′-polyphosphatase, an enzyme that specifically removes the phosphate group in the γ-position and β-position of 5′-triphosphate or 5′-diphosphate RNA, completely abolished the induction of type I interferon by _in vitro_–transcribed 3pRNA and poly(dA:dT)-triggered RNA (Fig. 5a). Treating the stimulatory RNA with RNase III, an enzyme that degrades long dsRNA into short dsRNA molecules, also abolished the stimulatory ability (Fig. 5b). Of note, the _in vitro_–transcribed RNA species were completely sensitive to treatment with RNase III, in agreement with the finding that _in vitro_–transcribed RNA critically requires dsRNA conformation for its RIG-I-stimulatory activity37. Consistent with those observations, RNase T1, an endoribonuclease that degrades ssRNA at guanosine residues, did not inhibit the activity of the poly(dA:dT)-triggered RNA species or the _in vitro_–transcribed RNA (data not shown). However, when we used RNase T1 in conditions that denature dsRNA, the _in vitro_–transcribed RNA was rendered completely inactive, whereas the poly(dA:dT)-triggered RNA species was not affected (Fig. 5c). This was notable, as in denaturing conditions, RNase T1 treatment almost completely degraded the RNA from poly(dA:dT)-transfected cells (Fig. 5c, bottom). In contrast, the endoribonuclease RNase A, which cleaves both ssRNA and dsRNA at uridine and cytidine residues in low-salt conditions, led to complete degradation and loss of activity of both _in vitro_–transcribed RNA and poly(dA:dT)-triggered RNA (Fig. 5d). As RNase T1 cleaves ssRNA specifically after guanosine residues, we hypothesized that the stimulatory RNA of interest was devoid of guanosine, at least at critical residues required for the stimulatory activity. Indeed, an _in vitro_–transcribed RNA molecule that consisted only of alternating uridine and adenosine bases was not affected by RNase T1 treatment in denaturing conditions and was still active in terms of induction of type I interferon (Supplementary Fig. 5). From these results, we conclude that transfection of poly(dA:dT) triggers the formation of an endogenous, double-stranded, 3pRNA molecule that is devoid of guanosine.
Figure 5: Poly(dA:dT)-induced RNA is a 5′-triphosphate dsRNA species devoid of guanosine.
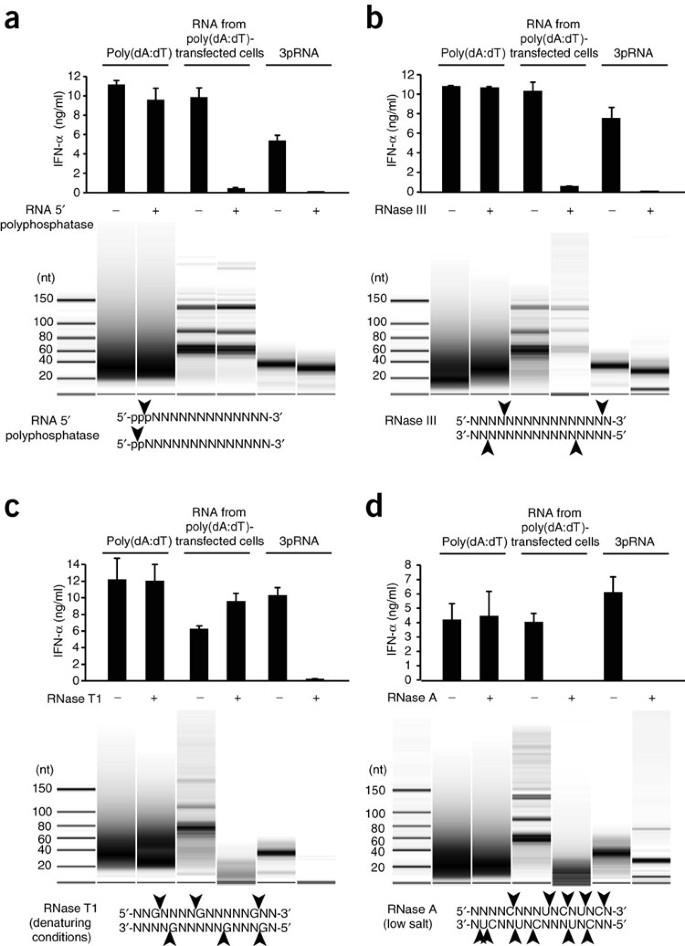
ELISA of IFN-α production by chloroquine-treated PBMCs transfected with RNA isolated from poly(dA:dT)-transfected 293T cells and treated with RNA 5′ polyphosphatase (a), RNase III (b), RNase T1 in denaturing conditions (c) or RNase A at a low salt concentration (d), or with poly(dA:dT) or 3pRNA treated the same way, and assessed 24 h later (top). Below, analysis of nucleic acids on an Agilent small RNA chip (bottom, enzymatic activity of enzymes). Data are from one representative of two (a,d) or four (b,c) experiments (mean and s.e.m.).
RNA polymerase III in type I interferon induction
The results reported above led us to hypothesize that poly(dA:dT) might itself serve as a template for the transcription of a 5′-triphosphate poly(rA:rU) molecule by a DNA-dependent RNA polymerase. Indeed, poly(dA:dT) has, for example, been used to study promoter-independent transcription by RNA polymerase III (refs. 38,39). Additionally, it has been shown that poly(dA:dT) is transcribed to poly(rA:rU) by RNA polymerase III (ref. 40). We therefore tested the possibility that poly(dA:dT) was transcribed in cells after transfection. In vitro_–transcribed poly(rA:rU) turned out to be an unsuitable template for specific RT-PCR amplification because of its homopolymeric nature (data not shown). We therefore constructed a synthetic homopolymeric dA:dT template consisting of 35 'units' of dA:dT (AT) plus an additional tail of 30 nucleotides (30N) containing a specific primer-binding site (AT+30N). Transfection of this synthetic template triggered a type I interferon response in PBMCs and also led to the generation of a stimulatory RNA species, albeit to a lower extent than that obtained with poly(dA:dT) (Supplementary Fig. 6a,b). Thus, we anticipated that this synthetic poly(dA:dT) construct would be transcribed together with the 30N downstream primer-binding site, which allowed us to use it as a specific tag for a 3′ rapid amplification of cDNA ends (RACE; Supplementary Fig. 7). Indeed, after transfection of this template into 293T cells, we were able to detect an RNA transcript containing the specific 3′ RACE tag (Fig. 6a), which indicated that the AT+30N dsDNA had been transcribed through its 30N 3′ end. We also assessed 5_S rRNA, an established RNA polymerase III transcript, by the 3′ RACE PCR method. In addition, we analyzed transcription of β2-microglobulin as an RNA polymerase II–dependent control (Fig. 6a). We obtained similar results when we transfected primary mouse DCs (Supplementary Fig. 6c), which indicated that this phenomenon was also operational in cells that responded to poly(dA:dT) in a RIG-I-independent way. Using the same template but with a poly(T) stretch separating the poly(dA:dT) portion from the 30N tag (AT+6T+30N) resulted in much less transcription of the 30N tag (Fig. 6b), which indicated that the polymerase activity was terminated before this part of the template. The fact that RNA polymerase III terminates transcription at poly(T) stretches additionally indicates involvement of RNA polymerase III in transcription of the AT+30N template. To address this possibility further, we took advantage of a specific inhibitor of RNA polymerase III, ML-60218. We treated 293T cells with ML-60218 and subsequently transfected the cells with AT+30N. Consistent with the involvement of RNA polymerase III in mediating transcription of the AT+30N template, ML-60218 inhibited transcription of 5_S_ rRNA and AT+30N DNA in a dose-dependent way (Fig. 6c).
Figure 6: RNA polymerase III transcribes AT-rich DNA and is required for poly(dA:dT)-mediated induction of type I interferon.
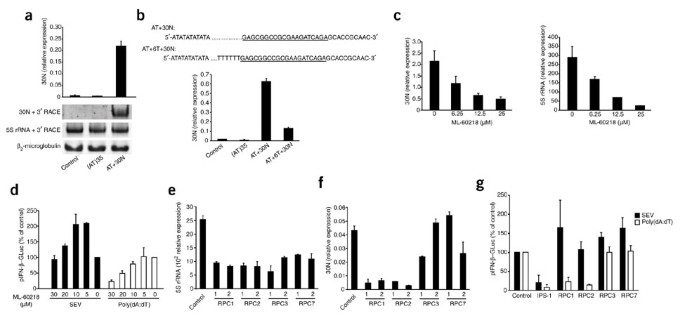
(a) Real-time PCR analysis (as in Supplementary Fig. 7) of expression of the 30N tag in 293T cells left untreated or transfected with the dsDNA template (AT)35 or AT+30N, assessed 8 h after transfection and presented relative to the expression of B2M (encoding β2-microglobulin). Below, standard PCR and PAGE of expression of 5_S_ rRNA, the 30N tag or β2-microglobulin. (b) Expression of the 30N tag in 293T cells left untreated (Control) or transfected with (AT)35, AT+30N or AT+6T+30N dsDNA, assessed 8 h after transfection and presented relative to B2M expression. Top, AT+30N and AT+6T+30N sequences, with the primer-binding sites for PCR underlined (dotted lines indicate additonal AT sequence). (c) Expression of the 30N tag and 5_S_ rRNA in 293T cells treated for 2 h with ML-60218 (concentration, horizontal axes) and transfected with AT+30N dsDNA and then assessed 16 h later; results are presented relative to B2M expression. (d) IFN-β reporter activity in 293T cells treated for 2 h with ML-60218 and then transfected with an IFN-β reporter plasmid in conjunction with poly(dA:dT) or SEV and assessed after 24 h. (e,f) Transcription of 5_S_ rRNA (e) and AT+30N (f) in 293T cells transfected for 48 h with two individual siRNA molecules (1 or 2) targeting genes encoding RPC1, RPC2, RPC3 or RPC7, then transfected with AT+30N and assessed after an additional 16 h; results are presented relative to B2M expression. (g) IFN-β reporter activity in 293T cells transfected for 48 h with siRNA specific for IPS-1, RPC1, RPC2, RPC3 or RPC7, then transfected with an IFN-β reporter plasmid in conjunction with poly(dA:dT) or SEV and assessed after an additional 24 h; results are presented relative to results obtained with control siRNA, set as 100%. Data are from one representative of three (a,b,g), two (c,e,f) or four (d) experiments (mean and s.e.m.).
To address the functional consequences of the inhibition of RNA polymerase III on the induction of type I interferon, we treated 293T cells with ML-60218 and then challenged these cells with poly(dA:dT) or SEV. SEV-triggered induction of type I interferon was enhanced at low concentrations of ML-60218 but was suppressed at higher concentrations (Fig. 6d). In contrast, poly(dA:dT)-mediated induction of type I interferon was blocked by ML-60218 in a dose-dependent way at concentrations that did not affect cell viability (data not shown). We also used RNA-mediated interference to 'knock down' essential subunits of the RNA polymerase III transcription apparatus to further study the function of RNA polymerase III in this response. We targeted RPC1 and RPC2 (encoded by Polr3a and Polr3b, respectively), the two core subunits that form the polymerase active center of RNA polymerase III, as well as RPC3 and RPC7 (encoded by Polr3c and Polr3g, respectively), which are not essential for elongation and termination during transcription with RNA polymerase III but are required for promoter-directed transcription initiation41 (Supplementary Fig. 8). As expected, transcription of 5S rRNA was affected by knockdown of RPC1, RPC2, RPC3 or RPC7 (Fig. 6e). However, whereas the core subunits RPC1 and RPC2 were essential for transcription of AT+30N, the RPC3 and RPC7 subunits were dispensable (Fig. 6f). In line with those data, induction of type I interferon triggered by poly(dA:dT) was critically dependent on RPC1 and RPC2 but not on RPC3 or RPC7 (Fig. 6g). SEV-mediated induction of type I interferon was unaffected by knockdown of any of these components (Fig. 6g). Thus, we conclude that promoter-independent transcription of poly(dA:dT) by RNA polymerase III leads to the formation of 3pRNA that in turn activates RIG-I.
RNA polymerase III–transcribed EBER RNA activates RIG-I
To address the physiological relevance of the RNA polymerase III pathway in antiviral host defenses, we examined the function of RNA polymerase III in the regulation of type I interferon responses to a DNA virus. We chose EBV because it encodes small EBER molecules that are transcribed by RNA polymerase III in very large amounts. EBER molecules are nonpolyadenylated, untranslated RNA molecules 167 nucleotides (EBER-1) or 172 nucleotides (EBER-2) in length42 and are the most abundant viral transcripts in cells with latent EBV infection43. Notably, EBV-immortalized lymphoblastoid cell lines and EBV-positive Burkitt lymphoma cell lines produce type I interferons in resting conditions44,45. To determine if pattern-recognition receptors are activated constitutively in Burkitt lymphoma cells and to define the involvement of RNA polymerase III in controlling the constitutive production of type I interferons, we treated the EBV-positive human Burkitt lymphoma Mutu III cell line with the RNA polymerase III inhibitor ML-60218. Inhibition of RNA polymerase III blocked IFN-α production in Mutu III cells in a dose-dependent way (Fig. 7a). The defect in interferon production correlated with repression of the expression of EBER-1, EBER-2 and 5S rRNA in these cells (Fig. 7b). To directly examine the ability of EBER to activate the RIG-I pathway, we cloned the entire EBER gene locus from EBV and transiently overexpressed this construct in interferon-primed 293T cells. Overexpression of EBER molecules led to substantial induction of type I interferon, albeit to a lower extent than did poly(dA:dT) (Fig. 7c). Expression of the loci for EBER-1 and EBER-2 separately showed that each EBER molecule could trigger type I interferon responses independently, although EBER-1 was slightly more active at high concentrations (Fig. 7c). Notably, when we expressed a dsDNA template encoding a blunt-ended version of EBER-1 RNA, we found more induction of type I interferon (Supplementary Fig. 9). This is in line with the finding that RIG-I favors fully blunt-ended 5′-triphosphate dsRNA37. As expected, expression of EBER RNA in 293T cells was suppressed by inhibition of RNA polymerase III activity, whereas RNA polymerase II–driven transcription of cyclophilin B was not affected (Fig. 7d). In addition, inhibition of RNA polymerase III or expression of RIG-I- or IPS-1-specific siRNA led to less EBER-induced type I interferon (Fig. 7e,f). Collectively, these results indicate that EBER molecules are transcribed by RNA polymerase III and trigger the induction of type I interferon through RIG-I. In line with those findings, we found that certain synthetic RNA polymerase III genes that encode blunt-ended dsRNA, such as short hairpin RNA, were potent triggers of RIG-I activation (Supplementary Fig. 10 and data not shown). These findings furthermore establish RIG-I as an important checkpoint in the detection of non-self RNA polymerase III transcripts.
Figure 7: EBER moleucles are transcribed by RNA polymerase III and trigger activation of RIG-I.
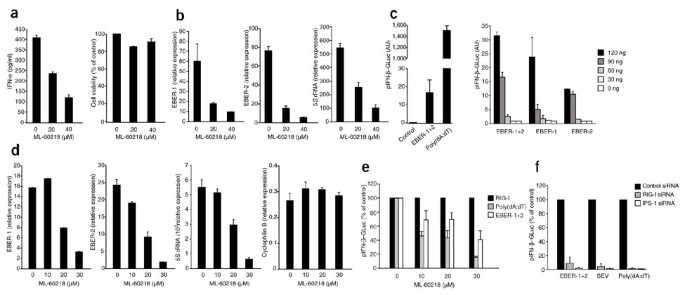
(a,b) ELISA of IFN-α production (a, left), calcein AM analysis of cell viability (a, right) and real-time PCR analysis of the production of EBER-1, EBER-2 and 5_S_ (b) of Mutu III cells treated for 24 h with ML-60218, presented relative to the expression of B2M. (c) IFN-β reporter activity in interferon-primed 293T cells transfected for 24 h with the genomic locus encoding EBER-1 and EBER-2 (EBER-1+2) or poly(dA:dT) (120 ng) (left) or decreasing amounts of the genomic locus encoding EBER-1 and EBER-2 or the portions encoding either EBER-1 or EBER-2 (right), together with an IFN-β reporter plasmid. Left, green fluorescent protein construct serves as a negative control (left). (d) Transcription of EBER-1, EBER-2, 5_S_ rRNA and cyclophilin B in interferon-primed 293T cells treated for 2 h with ML-60218 and transfected for 24 h with the genomic locus encoding EBER-1 and EBER-2, presented relative to the expression of B2M. (e) Transactivation of the IFN-β promoter in interferon-primed 293T cells treated as described in d and transfected with the genomic locus encoding EBER-1 and EBER-2 or with poly(dA:dT) or RIG-I (120 ng); results are presented relative to those obtained with the positive control RIG-I, set as 100%. (f) IFN-β reporter activity in 293T cells transfected for 48 h with two individual siRNA molecules targeting RIG-I or IPS-1 or control siRNA, then transfected with an IFN-β reporter plasmid in conjunction with the genomic locus encoding EBER-1 and EBER-2 or with poly(dA:dT) or SEV and assessed after an additional 24 h. Data are from one representative of three (a,e), two (b,d,f) or four (c) experiments (mean and s.e.m.).
Discussion
Here we have demonstrated a novel mechanism for the sensing of AT-rich dsDNA. The synthetic dsDNA mimetic poly(dA:dT) was converted by host RNA polymerase III into a 3pRNA intermediate, which was in turn recognized by RIG-I. RNA polymerase III–mediated conversion of poly(dA:dT) occurred in mouse and human cells, but at least one additional as-yet-undefined sensing mechanism operates in cells of the mouse immune system; this pathway responds to poly(dA:dT) in an RNA-independent way, presumably as a result of sensing DNA directly. These observations are consistent with published reports showing that mouse cells lacking RIG-I or IPS-1 still produce type I interferons in response to poly(dA:dT)33,34. This matter is further complicated by the fact that in the human system, at least one additional DNA-sensing mechanism exists to recognize dsDNA independently of a RIG-I-stimulatory RNA intermediate. Poly(dA:dT) induced the formation of the RIG-I-activating RNA species, but other dsDNA moleules that were not poly(dA:dT)-homopolymeric in nature failed to do so, despite being active when directly transfected into PBMCs. Nevertheless, our data clearly demonstrate that AT-rich DNA transcribed by RNA polymerase III is sensed by RIG-I in the human system in a nonredundant way.
Poly(dA:dT) has been used to study RNA polymerase III–mediated promoter-independent transcription38,39,40. This nonspecific transcriptional activity requires the core polymerase complex of RNA polymerase III, which includes RPC1 and RPC2, yet is independent of subunits used by RNA polymerase III for recruitment to specific promoter sites (RPC3, RPC6 and RPC7)41. It is unclear at present if RNA polymerase III has transcriptional activity independent of its characterized promoter sites in vivo, as no studies have addressed the RNA polymerase III transcriptome at a global cellular level in an unbiased way. It is plausible that RNA polymerase III also transcribes DNA templates from endogenous sources in a promoter-independent way. Such a mechanism could function as an innate defense strategy by tagging DNA as an RNA intermediate to be detected by RIG-I. Poly(dA:dT) or AT-rich DNA is a 'peculiar' template for RNA polymerase III and thereby indirectly for RIG-I for two main reasons. First, AT-rich sequences might be particularly suited for promoter-independent transcription by RNA polymerase III because of their propensity to form dsDNA regions with low helix stability, which may be particularly accessible as initiation sites for polymerases. However, other DNA templates have also been used to study promoter-independent transcription by RNA polymerase III and thus it is possible that non-AT DNA is also transcribed by RNA polymerase III in a promoter-independent way. Second, and probably more importantly, homopolymeric AT-rich DNA is also transcribed into a homopolymeric RNA molecule that has the propensity to form a complete RNA duplex. RIG-I strongly favors blunt-ended dsRNA over ssRNA or incompletely annealed dsRNA for binding and subsequent activity37. Although 5′-triphosphate ssRNA that is not self-complementary is equally active in terms of RIG-I activation when annealed to a complementary ssRNA strand, it shows little or no activity as a single-stranded molecule. Poly(rA:rU) RNA, in contrast, is completely self-complementary and thus has a strong propensity to form dsRNA with complete blunt-end formation, thereby making it ideally suited to be recognized by RIG-I.
Studying the function of host RNA polymerase III–dependent transcription of pathogen-derived DNA in innate immune defense will be important yet technically challenging. During acute infection with DNA viruses, it is likely that several DNA-sensing pathways are triggered in the nucleus or in the cytosol simultaneously, which makes it difficult to study the contribution of a single sensor. Here we focused on cells that were latently infected with EBV and thus contained viral DNA in the nucleus in steady-state conditions. EBV EBER molecules are transcribed in large amounts by RNA polymerase III in latently infected, EBV-positive cells43. In these cells, we found an RNA polymerase III–dependent activation of the induction of type I interferon that coincided with EBER RNA expression. Ectopic expression of the EBER locus also induced type I interferon by a mechanism dependent on RIG-I and IPS-1. Published work has also indicated EBER RNAs are endogenous triggers for RIG-I (ref. 46). We speculate that other herpes viruses also encode RNA polymerase III transcripts that can trigger RIG-I activation. For example, the genomes of some γ-herpesviruses contain variable numbers of internal repeats that are highly conserved across different viruses. In this context, it is noteworthy that a highly repetitive region in the genome of murine γ-herpesviruses 68 triggers type I interferon responses47.
Notably, published work has linked the RIG-like helicase–IPS-1 pathway to the detection of pathogens that replicate in the cytosol but are not RNA viruses. For example, induction of type I interferon triggered by L. pneumophila29 or vaccinia virus48 is strongly attenuated in cells devoid of IPS-1. It is unclear whether the activation of RIG-like helicases in these cases is due to pathogen-dependent RNA polymerase activity or host-derived RNA generated by an RNA polymerase III–dependent mechanism. Future studies that allow discrimination between host-derived and pathogen-derived RIG-I ligands could help clarify these issues.
Methods
Reagents.
Poly(dA:dT) and chloroquine were from Sigma-Aldrich. ML-60218 was from Calbiochem. Human interleukin 4 and granulocyte-macrophage colony-stimulating factor were from ImmunoTools. RNase T1, alkaline phosphatase and DNase I were from Fermentas. RNase III, RNase A, RNA 5′ polyphosphatase and poly(A) polymerase were from Epicentre. The synthetic imidazoquinoline resiquimod (R-848) and CpG oligonucleotide 2216 were from Invivogen.
Cell isolation and culture.
Human PBMCs were isolated from whole blood of healthy volunteers by density-gradient centrifugation (Biochrom). Red blood cells were lysed with red blood cell lysis buffer (Sigma). Human monocytes were isolated from PBMCs with anti-CD14 paramagnetic beads (Miltenyi Biotec) and were differentiated for 6 d into MoDCs in the presence of interleukin 4 (800 U/ml) and granulocyte-macrophage colony-stimulating factor (800 U/ml). Mouse bone marrow was cultured for 6 d with 5% (vol/vol) supernatant from J558L cells containing granulocyte-macrophage colony-stimulating factor for the generation of bone marrow–derived DCs. Primary cells were cultured in RPMI medium supplemented with L-glutamine, sodium pyruvate, 10% (vol/vol) FCS (all from Invitrogen) and ciprofloxacin (Bayer Schering Pharma). The 293T cells were cultured in DMEM (Invitrogen) with the same additives described above. Rnasel −/− MEFs and their respective controls were provided by R. Silverman. Cell viability was assessed by staining with the fluorescent cell viability indicator calcein AM as described8. Experiments involving human and mouse materials were approved by the institutional review board of the University Hospital of the University of Bonn and the University of Massachusetts Medical School.
RNA preparation.
If not indicated otherwise, DNA-transfected or SEV-infected 293T cells (2.4 × 105 cells per 12 wells) were collected 16 h after stimulation. The Mini RNA Isolation II kit (Zymo Research) was used for RNA isolation with the following modifications. Cells were lysed with TRIzol (Invitrogen), followed by one round of chloroform extraction. For total RNA extraction, the solution obtained was mixed with 0.7 volumes of ethanol and applied to the separation column and, after washing, column-bound RNA was eluted. For enrichment of small RNA molecules, 0.2 volumes of ethanol were added to the chloroform-extracted solution and applied to the separation column and the flow-through was collected. That fraction was then mixed with 1.0 volume of ethanol and 'passed' to a second column. After washing, column-bound RNA was eluted. For some experiments, the amount of ethanol used in the first binding step was varied to obtain flow-through fractions of different sizes.
Enzymatic reactions.
RNA or poly(dA:dT) (2 μg) was treated with alkaline phosphatase (100 U/ml), RNA 5′-polyphosphatase (1,000 U/ml), RNase A (50 μg/ml), RNase III (100 U/ml) or DNase I (1,000 U/ml to 100 U/ml) in the corresponding buffer solution in a volume of 10 μl for 30 min (RNA 5′ polyphosphatase) or 1 h (alkaline phosphatase, RNase A, RNase III or DNase I) at 37 °C. Samples were digested for 30 min at 55 °C with RNase T1 in denaturing conditions in 6 M urea and 17.8 mM sodium citrate, pH 3.5, and 0.9 mM EDTA.
Cell stimulation.
For reporter studies, if not indicated otherwise, 293T cells (2 × 104 cells per 96 wells) were transfected with 50 ng IFN-β promoter reporter plasmid pIFN-β–GLuc in conjunction with 50 ng poly(dA:dT) or other nucleic acids using Lipofectamine 2000 (Invitrogen). In some cases, 50 ng expression plasmid (pCMV-hsIRF1, pCMV-hsTBK1, pEF-BOS-RIG-I, pEF-BOS-RIG-IΔCard, pME18-NS3-4A WT or pME18-NS3-4A S139A) was transfected while the total amount of DNA was kept at 200 ng with pcDNA3 as a 'stuffer' plasmid. In some experiments, 293T cells were primed overnight with IFN-β (10 U/ml). Transactivation of the IFN-β promoter was measured 24 h after stimulation in an EnVision 2104 Multilabel Reader (Perkin Elmer). Human PBMCs, human MoDCs and mouse bone marrow–derived DCs (2 × 105 cells per 96 wells) were preincubated with chloroquine (2000 ng/ml), followed by transfection with 200 ng of DNA or RNA using Lipofectamine 2000. Sendai Virus (Cantell strain; Charles River Laboratories) was used at a concentration of 300 hemagglutinating units per ml.
3′ RACE.
For 3′ RACE analysis, isolated RNA was 'tailed' with poly(A) polymerase and subsequently reverse-transcribed with the RevertAid First Strand cDNA Synthesis kit (Fermentas) with the 3-RACE-RT primer (5′-CTATAGGCGCGCCACCGGTGTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTVN-3′). The cDNA was subsequently amplified by conventional or real-time PCR with a transcript-specific forward primer and the 3-RT-PCR reverse primer (Supplementary Table 1).
Quantitative real-time PCR.
After cDNA was synthesized from total RNA with the RevertAid First Strand cDNA Synthesis kit (Fermentas), a Roche LC480 with Maxima SYBR Green qPCR Master Mix (Fermentas) was used for quantitative RT-PCR analysis. The specificity of amplification was assessed for each sample by melting-curve and gel analysis. Standard curve analysis was used for relative quantification. Mouse quantification data are presented relative to Hprt1 expression; human data are presented relative to B2M expression. A detailed list of all primer sequences is in Supplementary Table 1.
RNA analysis.
Enriched small cellular RNA molecules and _in vitro_–transcribed RNA molecules were monitored with the Small RNA kit or RNA 6000 Pico Chip kit with an Agilent Bioanalyzer 2100 (Agilent Technologies).
ELISA.
Human IFN-α (Bender Med Systems) and TNF (BD Biosciences) were assessed with commercial ELISA kits according to the manufacturer's instructions.
_In vitro_–transcribed RNA and PCR-generated dsDNA.
In vitro transcription was done as described14 (templates, Supplementary Table 2). The 20-, 40-, 77-, 163-, 245-nucleotide dsDNA molecules and EBER dsDNA templates were generated by PCR with pCDNA3 or EBV genomic DNA as the template (primers, Supplementary Table 3).
RNA-mediated interference.
First, siRNA was reverse-transfected at a concentration of 25 nM into 293T cells (1 × 104 cells per 96 wells) with 0.5 μl Lipofectamine. Then, 48 h after transfection, cells were stimulated and, after an additional period of 24 h, luciferase activity was assessed. Electroporation of human MoDCs and mouse bone marrow–derived DCs was done as described49 (siRNA sequences, Supplementary Table 4).
References
- Kawai, T. & Akira, S. Toll-like receptor and RIG-I-like receptor signaling. Ann. NY Acad. Sci. 1143, 1–20 (2008).
Article CAS Google Scholar - Takeda, K. & Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 17, 1–14 (2005).
Article CAS Google Scholar - Huysamen, C. & Brown, G.D. The fungal pattern recognition receptor, Dectin-1, and the associated cluster of C-type lectin-like receptors. FEMS Microbiol. Lett. 290, 121–128 (2009).
Article CAS Google Scholar - Yoneyama, M. & Fujita, T. RIG-I family RNA helicases: cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 18, 545–551 (2007).
Article CAS Google Scholar - Meylan, E., Tschopp, J. & Karin, M. Intracellular pattern recognition receptors in the host response. Nature 442, 39–44 (2006).
Article CAS Google Scholar - Takaoka, A. & Taniguchi, T. Cytosolic DNA recognition for triggering innate immune responses. Adv. Drug Deliv. Rev. 60, 847–857 (2008).
Article CAS Google Scholar - Takaoka, A. et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505 (2007).
Article CAS Google Scholar - Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Article CAS Google Scholar - Kono, H. & Rock, K.L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289 (2008).
Article CAS Google Scholar - Takeuchi, O. & Akira, S. Innate immunity to virus infection. Immunol. Rev. 227, 75–86 (2009).
Article CAS Google Scholar - Takeuchi, O. & Akira, S. MDA5/RIG-I and virus recognition. Curr. Opin. Immunol. 20, 17–22 (2008).
Article CAS Google Scholar - Kato, H. et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 (2006).
Article CAS Google Scholar - Pichlmair, A. et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314, 997–1001 (2006).
Article CAS Google Scholar - Hornung, V. et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 (2006).
Article Google Scholar - Kato, H. et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205, 1601–1610 (2008).
Article CAS Google Scholar - Kawai, T. et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6, 981–988 (2005).
Article CAS Google Scholar - Seth, R.B., Sun, L., Ea, C.K. & Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122, 669–682 (2005).
Article CAS Google Scholar - Xu, L.G. et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 19, 727–740 (2005).
Article CAS Google Scholar - Meylan, E. et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172 (2005).
Article CAS Google Scholar - Ishii, K.J. et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature 451, 725–729 (2008).
Article CAS Google Scholar - Burckstummer, T. et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10, 266–272 (2009).
Article Google Scholar - Fernandes-Alnemri, T., Yu, J.W., Datta, P., Wu, J. & Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 (2009).
Article CAS Google Scholar - Roberts, T.L. et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060 (2009).
Article CAS Google Scholar - Henry, T., Brotcke, A., Weiss, D.S., Thompson, L.J. & Monack, D.M. Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987–994 (2007).
Article CAS Google Scholar - Charrel-Dennis, M. et al. TLR-independent type I interferon induction in response to an extracellular bacterial pathogen via intracellular recognition of its DNA. Cell Host Microbe 4, 543–554 (2008).
Article CAS Google Scholar - O'Riordan, M., Yi, C.H., Gonzales, R., Lee, K.D. & Portnoy, D.A. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. USA 99, 13861–13866 (2002).
Article CAS Google Scholar - Stetson, D.B. & Medzhitov, R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity 24, 93–103 (2006).
Article CAS Google Scholar - Stanley, S.A., Johndrow, J.E., Manzanillo, P. & Cox, J.S. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J. Immunol. 178, 3143–3152 (2007).
Article CAS Google Scholar - Opitz, B. et al. Legionella pneumophila induces IFNβ in lung epithelial cells via IPS-1 and IRF3, which also control bacterial replication. J. Biol. Chem. 281, 36173–36179 (2006).
Article CAS Google Scholar - Roux, C.M. et al. Brucella requires a functional type IV secretion system to elicit innate immune responses in mice. Cell. Microbiol. 9, 1851–1869 (2007).
Article CAS Google Scholar - Chessler, A.D., Ferreira, L.R., Chang, T.H., Fitzgerald, K.A. & Burleigh, B.A. A novel IFN regulatory factor 3-dependent pathway activated by trypanosomes triggers IFN-β in macrophages and fibroblasts. J. Immunol. 181, 7917–7924 (2008).
Article CAS Google Scholar - Yoshida, H., Okabe, Y., Kawane, K., Fukuyama, H. & Nagata, S. Lethal anemia caused by interferon-β produced in mouse embryos carrying undigested DNA. Nat. Immunol. 6, 49–56 (2005).
Article CAS Google Scholar - Sun, Q. et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 24, 633–642 (2006).
Article CAS Google Scholar - Kumar, H. et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 203, 1795–1803 (2006).
Article CAS Google Scholar - Cheng, G., Zhong, J., Chung, J. & Chisari, F.V. Double-stranded DNA and double-stranded RNA induce a common antiviral signaling pathway in human cells. Proc. Natl. Acad. Sci. USA 104, 9035–9040 (2007).
Article CAS Google Scholar - Malathi, K., Dong, B., Gale, M., Jr & Silverman, R.H. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448, 816–819 (2007).
Article CAS Google Scholar - Schlee, M. et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity published online, doi:10.1016/j.immuni.2009.05.008 (2 July 2009).
- Huet, J., Riva, M., Sentenac, A. & Fromageot, P. Yeast RNA polymerase C and its subunits. Specific antibodies as structural and functional probes. J. Biol. Chem. 260, 15304–15310 (1985).
CAS PubMed Google Scholar - Zaros, C. & Thuriaux, P. Rpc25, a conserved RNA polymerase III subunit, is critical for transcription initiation. Mol. Microbiol. 55, 104–114 (2005).
Article CAS Google Scholar - Thuillier, V., Brun, I., Sentenac, A. & Werner, M. Mutations in the α-amanitin conserved domain of the largest subunit of yeast RNA polymerase III affect pausing, RNA cleavage and transcriptional transitions. EMBO J. 15, 618–629 (1996).
Article CAS Google Scholar - Wang, Z. & Roeder, R.G. Three human RNA polymerase III-specific subunits form a subcomplex with a selective function in specific transcription initiation. Genes Dev. 11, 1315–1326 (1997).
Article CAS Google Scholar - Rosa, M.D., Gottlieb, E., Lerner, M.R. & Steitz, J.A. Striking similarities are exhibited by two small Epstein-Barr virus-encoded ribonucleic acids and the adenovirus-associated ribonucleic acids VAI and VAII. Mol. Cell. Biol. 1, 785–796 (1981).
Article CAS Google Scholar - Lerner, M.R., Andrews, N.C., Miller, G. & Steitz, J.A. Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 78, 805–809 (1981).
Article CAS Google Scholar - Schlee, M., Schuhmacher, M., Holzel, M., Laux, G. & Bornkamm, G.W. c-Myc impairs immunogenicity of human B cells. Adv. Cancer Res. 97, 167–188 (2007).
Article CAS Google Scholar - Schlee, M. et al. C-myc activation impairs the NF-κB and the interferon response: implications for the pathogenesis of Burkitt's lymphoma. Int. J. Cancer 120, 1387–1395 (2007).
Article CAS Google Scholar - Samanta, M., Iwakiri, D., Kanda, T., Imaizumi, T. & Takada, K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 25, 4207–4214 (2006).
Article CAS Google Scholar - Sanchez, D.J. et al. A repetitive region of γ-herpesvirus genomic DNA is a ligand for induction of type I interferon. J. Virol. 82, 2208–2217 (2008).
Article CAS Google Scholar - Deng, L. et al. Vaccinia virus subverts a mitochondrial antiviral signaling protein-dependent innate immune response in keratinocytes through its double-stranded RNA binding protein, E3. J. Virol. 82, 10735–10746 (2008).
Article CAS Google Scholar - Ablasser, A. et al. Selection of molecular structure and delivery of RNA oligonucleotides to activate TLR7 versus TLR8 and to induce high amounts of IL-12p70 in primary human monocytes. J. Immunol. 182, 6824–6833 (2009).
Article CAS Google Scholar
Acknowledgements
We thank R. Silverman (Lerner Research Institute, Cleveland Clinic) for Rnasel −/− MEFs and their respective controls; and G. Bornkamm and M. Schlee for discussions. Supported by the Deutsche Forschungsgemeinschaft (Ho2783/2-1 V.H.) and the US National Institutes of Health (AI-065483 to E.L.; AI-067497 to K.A.F.; AI-083713 to K.A.F. and E.L.).
Author information
Author notes
- Andrea Ablasser, Franz Bauernfeind, Katherine A Fitzgerald and Veit Hornung: These authors contributed equally to this work.
Authors and Affiliations
- Institute for Clinical Chemistry and Pharmacology, University of Bonn, Bonn, Germany
Andrea Ablasser, Franz Bauernfeind, Gunther Hartmann & Veit Hornung - Division of Infectious Diseases and Immunology, University of Massachusetts Medical School, Worcester, Massachusetts, USA
Eicke Latz & Katherine A Fitzgerald
Authors
- Andrea Ablasser
You can also search for this author inPubMed Google Scholar - Franz Bauernfeind
You can also search for this author inPubMed Google Scholar - Gunther Hartmann
You can also search for this author inPubMed Google Scholar - Eicke Latz
You can also search for this author inPubMed Google Scholar - Katherine A Fitzgerald
You can also search for this author inPubMed Google Scholar - Veit Hornung
You can also search for this author inPubMed Google Scholar
Contributions
A.A., F.B. and V.H. did the experiments; V.H. conceived of and K.A.F. and V.H. supervised the research; G.H. and E.L. provided technical support; E.L., K.A.F. and V.H. sponsored the research; and K.A.F. and V.H. prepared the manuscript.
Corresponding author
Correspondence toVeit Hornung.
Supplementary information
Rights and permissions
About this article
Cite this article
Ablasser, A., Bauernfeind, F., Hartmann, G. et al. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III–transcribed RNA intermediate.Nat Immunol 10, 1065–1072 (2009). https://doi.org/10.1038/ni.1779
- Received: 06 June 2009
- Accepted: 13 July 2009
- Published: 16 July 2009
- Issue Date: October 2009
- DOI: https://doi.org/10.1038/ni.1779