Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells (original) (raw)
Main
Viral infections of the airways are a major cause of morbidity and mortality. Since CD8+ T cells are central in host defense against such viral infections1,2, the ability to generate and maintain effective CD8+ T cell memory is a key objective for vaccine development, with potentially major effects on global health.
Memory CD8+ T cells have traditionally been categorized as central memory T cells (TCM cells) and effector memory T cells (TEM cells). These populations both recirculate in blood and tissues but differ in tissue distribution, immediate effector ability and ability to expand during secondary infection3. Published findings have shown that a subset of memory CD8+ T cells does not recirculate but is maintained at the site of infection4. These tissue-resident memory T cells (TRM cells) are required for optimal protection at mucosal sites such as the lungs, intestine and female reproductive tract5,6. The best-characterized TRM cells express the integrin αE (CD103) and the C-type lectin CD69, which promote localization to epithelia through interaction with E-cadherin5 and prevent homing to blood and lymph by interfering with activity of the receptor for the bioactive lipid S1P, respectively7. However, published results indicate that some TRM cells express neither CD69 nor CD103 but nonetheless exhibit TRM cell characteristics8,9,10. TRM cells keep recall infections from spreading into the lower airways and thereby curb the development of pathology1. This function requires swift responsiveness of these cells. One mechanism used by TRM cells is the production of interferon-γ (IFN-γ), which promotes antiviral resistance of the tissue and induces the production of chemokines to recruit auxiliary immune cells11. Remaining in tissues after clearance of infection, TRM cells do not have access to the niches in secondary lymphoid organs where circulating memory T cells receive their maintenance signals. How TRM cells are maintained in situ is not clear, although signaling via the receptor for interleukin 15 (IL-15) is apparently important8.
TRM cells in the lungs confer superior protective immunity relative to that provided by circulating T cells12,13,14. Vaccination strategies should therefore aim at eliciting stable antigen-specific TRM cell populations. The ability to design such strategies requires thorough understanding of these cells. Lung TRM cells have been characterized in mouse models8. However, it is not clear to what degree such data can be extrapolated to human T cells, given the considerable divergence between these species in genes encoding products associated with immunity15 and the differences in the time frame associated with immunological memory, spanning decades in humans, compared with a few years in mice16.
Here we analyzed the genetic programs of memory CD8+ T cell populations obtained from human lung-resection tissues. These populations had gene-expression signatures very different from those of blood-derived T cells. Lung TRM cells constitutively expressed deployment-ready mRNAs encoding effector molecules but also expressed many inhibitory regulators. These cells therefore seemed to be poised for prompt reactivity to pathogens but under tight control to limit impairment of the air-exchange function in the delicate lung mucosa. Furthermore, TRM cells exhibited an active Notch signaling signature, and activity of this pathway was required for their maintenance.
Results
Distinct transcriptional profile of lung TRM cells
To study lung-resident memory CD8+ T cells, we isolated CD45RA− T cells from paired samples of healthy lung tissue and peripheral blood from patients undergoing lung resection (Supplementary Fig. 1). Many memory CD8+ T cells from the lungs and some from the blood expressed CD103 (Fig. 1a,b). Whereas the latter nearly all coexpressed the costimulatory receptor CD27 (Fig. 1c) and thus resembled TCM cells, expression of CD27 was low or negative on both CD103+ memory T cells and CD103− memory T cells from lungs, characteristic of TEM cells (Fig. 1c). The CD103+ population from lungs almost universally expressed CD69, but 5–30% of the lung CD103− population lacked expression of this marker (Fig. 1d).
Figure 1: Distinct gene-expression program of lung TRM cells.
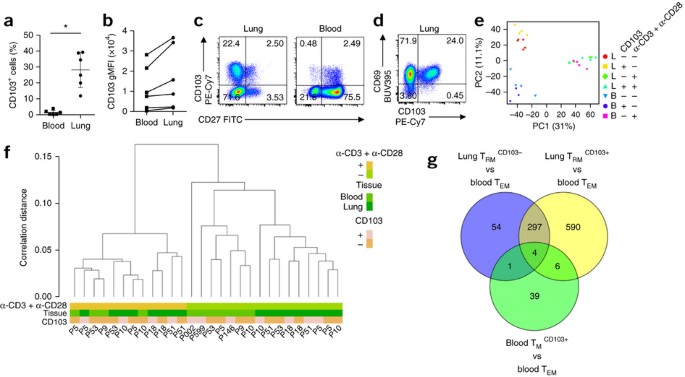
(a) Frequency of CD103-expressing memory CD8+ T cells in paired samples (n = 6) from the peripheral blood and lungs (horizontal axis), measured by flow cytometry. Each symbol represents an individual donor; small horizontal lines indicate the mean (± s.d.). (b) Geometric mean fluorescence intensity (gMFI) of CD103 on paired sets of CD103+CD8+ T cells derived from the peripheral blood and lungs (lines connect paired samples). (c) Flow cytometry (bi-exponential scales) analyzing the expression of CD103 and CD27 on CD8+ T cells derived from the lungs (left) and blood (right). Numbers in quadrants indicate percent cells in each throughout. (d) Flow cytometry analyzing the expression of CD69 and CD103 on lung-derived CD8+ T cells. (e) Principal-component analysis of global gene-expression data for CD8+ memory T cell subsets derived from the lungs (L) or blood (B) (n = 6 donors) that expressed CD103 (+) or not (−), assessed under resting conditions (−) or after stimulation (+) for 16 h with anti-CD3 plus anti-CD28 (α-CD3 + α-CD28) (key). Each symbol represents a cell population from an individual donor; PC1 and PC2 indicate principal components 1 and 2. (f) Hierarchical clustering of T cell populations as in e (key; horizontal axis); bottom ('P' and number), donor identification. (g) Quantification of genes with significantly differential expression in various comparisons (periphery) of T cell subsets (with a false-discovery rate (FDR) of <0.05), and overlap among those groups. *P < 0.001 (paired _t_-test). Data are representative of three experiments (a,b), nine independent experiments (c,d) or one experiment (e–g).
We determined by microarray the global gene-expression profiles of CD103+ and CD103− memory CD8+ T cell subsets from the lungs and blood by isolating mRNA directly ex vivo and after stimulation of cells with antibody to the invariant signaling protein CD3 (anti-CD3) and antibody to the co-receptor CD28 (anti-CD28). CD103− memory T cells from blood were additionally selected for lack of CD27 expression to obtain TEM cells (populations that included >95% CCR7− cells; Supplementary Fig. 1). The results of this microarray analysis were confirmed by quantitative PCR analysis of ten genes (Supplementary Fig. 2).
We used lung tissue from two types of patients: non-cancerous lobectomy tissue from patients with non-small cell lung carcinoma, and lung tissue from patients with chronic obstructive pulmonary disease (whose lungs were removed in preparation for lung transplantation). The results of samples from both sources were very similar (Supplementary Fig. 3). Principal-component analysis and hierarchical clustering revealed that the expression profiles of unstimulated cells segregated according to anatomical origin rather than by donor or donor type (Fig. 1e,f). CD103+ T cells from blood proved more similar to blood-derived CD103− TEM cells than to the CD103+ subset from lungs (Fig. 1e,f), which suggested that most blood-derived CD103+ T cells were not lung TRM cells that had mobilized into circulation. Also, the CD103− subset from lungs was more similar to the CD103+ lung subset than to either population from the blood (Fig. 1e,f). We therefore considered this CD103− CD8+ memory T cell population from lungs to be mostly tissue resident. We call the CD103+ lung population and CD103− lung population specifically 'TRM103+ cells' and 'TRM103− cells', respectively, here.
Given that blood TEM cells have been well characterized, we used their transcriptome as reference to which we compared the other transcriptomes. The greatest number of differentially expressed genes (897) appeared in the comparison between lung TRM103+ cells and blood TEM cells (Fig. 1g). Of those, 301 were shared with the lung TRM103− cell subset, while 596 were not (Fig. 1g). However, most of those 596 genes also did exhibit a similar expression pattern in the comparison between TRM103− cells and circulating TEM cells (Supplementary Fig. 4), but this result did not reach statistical significance. Only 10 genes showed significantly differential expression in the lung memory CD8+ T cell populations (Supplementary Fig. 4). Likewise, only a limited number of genes (50) had differential expression in the two blood-derived populations (Fig. 1g). We concluded that both lung-derived T cell subsets were distinct from blood memory CD8+ T cells and were related (but not identical) to each other. Because TRM103+ cells are the population that dwells in the epithelial barrier and are therefore most acutely involved in maintaining border integrity, we focused further analysis on this subset.
Top differentially expressed genes and gene sets
Comparison of the gene-expression patterns of lung TRM103+ cells with those of blood TEM cells yielded genes encoding the factors also observed in studies of mice8,17. Expression of ITGAE (which encodes CD103), CTLA4 (which encodes the immunomodulatory receptor CTLA-4) and KLRC1 (which encodes the inhibitory receptor NKG2A) was elevated in TRM103+ cells, whereas the expression of S1PR1 (which encodes the S1P receptor), SELL (which encodes the lymph-node-homing receptor CD62L) and KLRG1 (which encodes the activation marker KLRG1) was low (Fig. 2a,b). Among the top 100 genes with the greatest difference in expression in TRM103+ cells relative to that in blood TEM cells were those encoding heat-shock proteins (HSPA1A, HSPA7, HSPA2 and HSPD1), transcription factors (EGR2, FOSB, ATF3 and RBPJ), a chemokine (XCL1), a chemokine receptor (CXCR6), the ligand for the death receptor Fas (FASLG), anti-apoptotic factors (PHLDA1 and BIRC3), members of the tumor-necrosis factor (TNF) receptor signaling family (TRAF1 and TANK), an adhesion G-protein-coupled receptor (CD97) and interferon-γ (IFNG), all of which exhibited higher expression in TRM103+ cells than in blood TEM cells (Fig. 2c). Furthermore, TRM103+ cells 'preferentially' expressed several splice variants of sprouty 1 (encoded by SPRY1), which is an inhibitor of signaling via the T cell antigen receptor (TCR)18 (Fig. 2c and Supplementary Fig. 5). Genes with much lower expression in TRM103+ cells than in blood TEM cells included those encoding the transcription factor Kruppel-like factor 12 (KLF12) and granzyme M (GZMM) (Fig. 2c).
Figure 2: Top differentially expressed genes and gene sets in lung TRM cells.
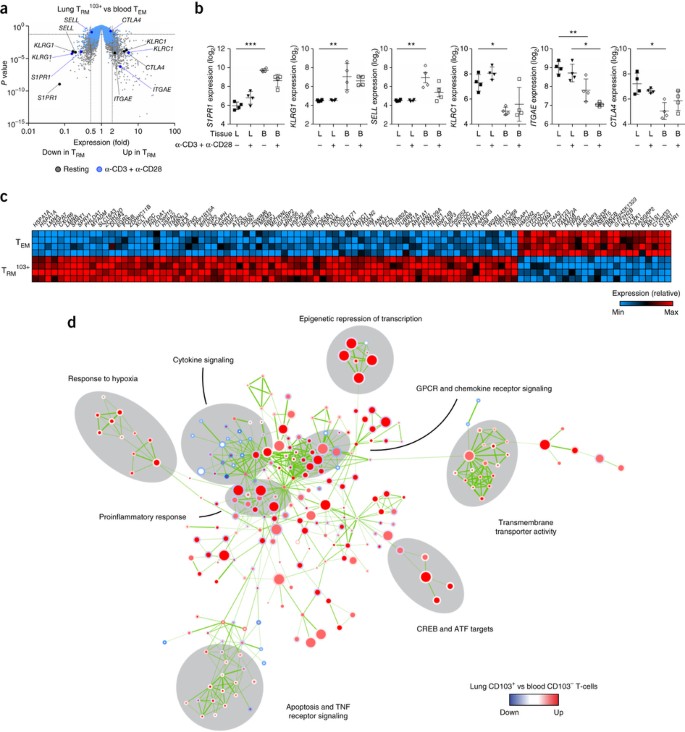
(a) Gene expression in lung TRM103+ cells versus that in peripheral-blood-derived TEM cells under resting conditions or after stimulation with anti-CD3 plus anti-CD28 (key), showing genes upregulated (Up in TRM; right) or downregulated (Down in TRM; left) in the TRM103+ cells relative to their expression in the TEM cells (dashed vertical lines indicate a 0.5-fold or 2-fold difference in expression or no difference (1-fold) in expression), plotted against P values (dashed horizontal line indicates P = 0.05). Each symbol represents an individual probe set (key TRM cell genes are labeled). (b) Expression (log2-transformed normalized values) of various genes (vertical axes) in T cell subsets derived from the lungs or blood and analyzed under resting conditions or after stimulation with anti-CD3 plus anti-CD28 (below plots). *FDR < 0.05, **FDR < 0.01 and ***FDR < 0.001. Each symbol represents an individual donor; small horizontal lines indicate the mean (± s.d.). (c) Expression of mRNA from the top 100 genes (above plot) with the greatest difference in expression (key) in lung TRM103+ cells relative to that in blood TEM cells (left margin), under resting conditions. (d) Gene-set–enrichment analysis of lung TRM103+ cells versus blood TEM cells: labels along periphery indicate prominent biological functional categories (grouped by gray ellipses); nodes represent gene sets (derived from the Molecular Signatures Database (MSigDB)) with differential expression in lung TRM103+ cells relative to that in blood-derived TEM cells; node color and intensity indicate degree of significance of enrichment, and node size is proportional to the number of genes in the set; circles represent comparison between resting samples (inner circles) or stimulated samples (outer circles). Data are representative of one experiment.
Gene-set–enrichment analysis with the CAMERA gene-set test procedure19 showed that major differences between TRM103+ cells and blood-derived TEM cells were associated with cytokine signaling, chemokine receptor signaling, active transport of ions and small molecules, apoptosis, TNF receptor signaling and the proinflammatory immune response (Fig. 2d and Supplementary Table 1). The gene set most highly enriched among the genes with differential expression between these two cell types indicated that TRM103+ cells exhibited a glucose-deprivation signature (Table 1), consistent with the lower glucose concentration in airway fluid than in blood20. Gene sets encoding products related to hypoxia were also enriched in this comparison (Fig. 2d and Supplementary Table 1). Correspondingly, lung TRM103+ cells had elevated expression of HIF1A mRNA (which encodes HIF-1α) and EPAS1 mRNA (which encodes HIF-2α) (Supplementary Fig. 5).
Table 1 Enriched gene sets
Chemokine and homing receptors
The distinct anatomical localization of lung TRM103+ cells required specific migratory properties, as reflected by enrichment for gene sets encoding chemokine receptors, among the genes with differential expression in lung TRM103+ cells relative to that in blood-derived TEM cells (Fig. 2d). Receptors that distinguished lung TRM103+ cells from circulating TEM cells included CXCR3, CXCR6, CCR5 and CCR6 (Fig. 3a,b). With the exception of CCR6, which could not be reliably detected anymore after collagenase treatment, elevated expression of these receptors was confirmed by flow cytometry (Supplementary Fig. 6). Of these, CCR6 also had high expression in CD103+ memory CD8+ T cells from the blood (Supplementary Fig. 5b) and was therefore not sufficient for localization to the lung epithelium. Furthermore, TRM103+ cells expressed little CX3CR1 (Fig. 3b), a chemokine receptor that mediates transmigration through endothelial layers21. Low expression of this receptor in TRM103+ cells fits with the hypothesis that these cells had reached their target destination and did not need to interact with endothelium.
Figure 3: Specific localization program of lung TRM103+ cells.
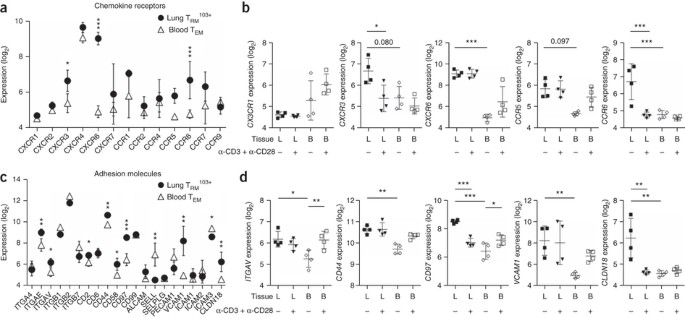
(a,c) Expression (log2-transformed normalized values) of genes (horizontal axes) encoding chemokine receptors (a) or adhesion molecules (c) in lung TRM103+ cells and blood TEM cells (keys) under resting conditions. (b,d) Expression (as in a) of genes (vertical axes) encoding chemokine receptors (b) or adhesion molecules (d) in lung- or blood-derived T cell subsets under resting conditions or after stimulation with anti-CD3 plus anti-CD28 (below plots). *FDR < 0.05, **FDR < 0.01 and ***FDR < 0.001 (other FDR values above bracketed comparisons). Each symbol represents an individual donor; small horizontal lines indicate the mean (± s.d.). Data are representative of one experiment.
The repertoire of adhesion molecules in lung TRM103+ cells was also different from that in blood TEM cells. Apart from their high expression and low expression, respectively, of ITGAE and SELL (Fig. 3c), lung TRM103+ cells had high expression of the adhesion-molecule-encoding genes ITGAV, CD44, CD97, VCAM1 and CLDN18 (Fig. 3d). Thus, the lung TRM103+ cells displayed patterns of chemokine receptors and adhesion molecules that clearly differed from those on circulating cells, consistent with their unique tissue localization.
Constitutive expression of effector-molecule-encoding genes
As a first line of defense, lung TRM cells must act without delay to prevent respiratory pathogens from establishing a foothold. Notably, lung TRM cells had constitutively high expression of mRNAs encoding effector molecules, such as granzyme B, IFN-γ and TNF, without the need for in vitro stimulation (Fig. 4a,b). The amounts of these transcripts in resting lung TRM103+ cells were as high as those reached in circulating TEM cells after activation via the TCR and CD28 (Fig. 4b). Notably, resting lung TRM103+ cells had little expression of these effector molecules at the protein level17 (Fig. 4c,d), unlike blood TEM cells, which, for example, clearly had granzyme B protein, despite their much lower constitutive expression of mRNA encoding granzyme B (Fig. 4b–d). Lung TRM cells thus maintained large amounts of mRNA without accumulating protein.
Figure 4: Rapid but strictly regulated effector function of lung TRM103+ cells.
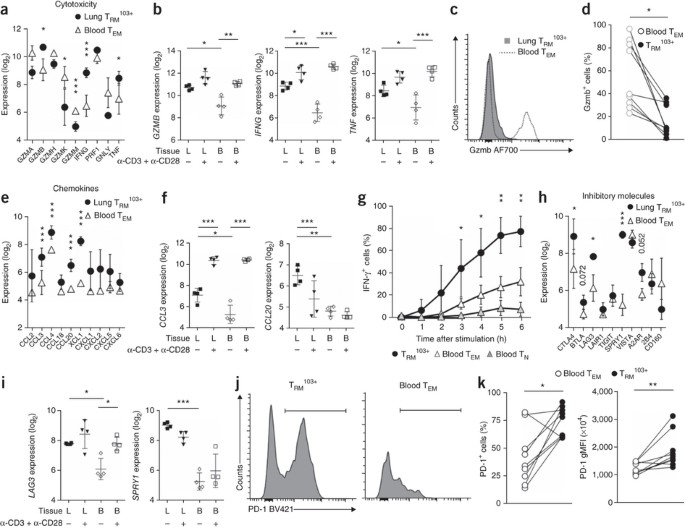
(a,e,h) Expression (log2-transformed normalized values) of genes (horizontal axes) encoding cytotoxic mediators (a), chemokines (e) or inhibitory molecules (h) in lung and blood T cell subsets (key) under resting conditions. (b,f,i) Expression (as in a) of various genes (horizontal axes) in lung- and blood-derived T cell subsets under resting conditions or after stimulation with anti-CD3 plus anti-CD28 (below plots). (c,d) Flow cytometry (c) and quantification (d) of granzyme B (Gzmb) in paired samples (n = 9) of lung TRM103+ cells and blood TEM cells (keys); lines (d) connect paired samples. (g) Frequency of IFN-γ+ T cells in unpaired samples of lung TRM103+ cells (n = 4 samples) and blood TEM cells or naive T cells (TN cells) (n = 2 samples each) stimulated for various times (horizontal axis) with PMA and ionomycin in the presence of brefeldin A. (j,k) Flow cytometry (j) and quantification (k) of PD-1 expression in paired samples (n = 9) of lung TRM103+ and blood TEM cells (key in k; lines connect paired samples). Each symbol (a,b,e,f,h,i) represents an individual donor; small horizontal lines indicate the mean (± s.d. in b,f,i). *FDR < 0.05, **FDR < 0.01 and ***FDR < 0.001 (a,b,e,f,h,i). *P < 0.05 and **P < 0.01 (paired _t_-test (c,d,k) or two-way analysis of variance (ANOVA) with the Bonferroni post-hoc test (g)). Data are representative of one experiments (a,b,e,f,h,i), nine experiments (c,d,j,k) or three experiments (g; mean ± s.d.).
An important function of TRM cell–derived IFN-γ is to induce the secretion of chemokines by epithelial cells11,22. We found that lung TRM103+ cells themselves also expressed genes encoding many chemokines (Fig. 4e,f), which might contribute to this sense-and-alarm function. Thus, lung TRM103+ cells had constitutively elevated expression of the chemokine-encoding genes CCL3, CCL4, C_CL20_ and XCL1 (Fig. 4e,f). Regulation varied among the chemokines. Expression of the gene encoding CCL3, an attractant for macrophages, CD4+ T cells and CD8+ T cells, was (further) induced by the activation via the TCR and CD28 on TRM103+ cells, whereas the expression of genes encoding other chemokines, such as CCL20, was hardly induced at all or was even reduced (Fig. 4e,f).
The expression of deployment-ready mRNAs encoding effector molecules could save precious time, which might make the difference between containment of microbes in the upper airways and full-blown infection. Indeed, lung TRM103+ cells secreted IFN-γ more rapidly than blood-derived T cells did after in vitro stimulation with the phorbol ester PMA and ionomycin (Fig. 4g), as found for murine airway CD8+ T cells23.
Despite such robust effector function, lung TRM103+ cells simultaneously expressed genes encoding inhibitory molecules, such as CTLA4, BTLA, LAG3, SPRY1 and the adenosine receptor A2AR (Fig. 4h,i). Furthermore, most of these cells were PD-1hi (Fig. 4j,k). Finally, lung TRM103+ cells had higher expression of transcription factors that inhibit effector function, such as TWIST1 and BACH2, than that of blood TEM cells24,25 (Supplementary Fig. 5). While seemingly at odds with the requisite vigilance of TRM103+ cells, expression of all these inhibitors might impose a degree of restraint to help prevent unnecessary loss of tissue integrity upon infection.
Transcription factors that define TRM cells
Given their constitutive expression of mRNAs encoding effector molecules, we expected that lung TRM103+ cells would have abundant expression of factors that drive the transcription of these mRNAs. However, two major regulators of such genes, T-bet and eomesodermin26, were not expressed in lung TRM103+ cells (Fig. 5a–c), as reported for mouse skin-resident T cells27. To search for candidates that might control constitutive expression of effector-molecule-encoding genes, we analyzed the expression patterns of transcription factors. We found that 58 transcription factors had differential expression in lung-derived T cells relative to that in circulating TEM cells (Fig. 5d). Many of these factors fell into interconnected clusters in a protein-association network; these included the activator protein AP-1, Notch1-RBPJ (RBPJ is also known as CSL) and NF-κB transcription-factor complexes (Fig. 5e). We found that 33 transcription factors had higher expression in lung-derived memory T cells than in circulating TEM cells (Fig. 5d). Among these were RUNX3, which might regulate CD103 expression28, as well as BATF and AHR, which regulate the expression of homing receptors and the maintenance of mouse TRM cells, respectively29,30 (Fig. 5d and Supplementary Fig. 5). The transcription factors Hobit (ZNF683) and BLIMP1 (encoded by PRDM1), identified as master regulators of TRM cells in mice31, were not expressed differentially by lung TRM103+ cells relative to their expression by circulating TEM cells, because the latter population, unlike their mouse counterpart31, also expressed Hobit and BLIMP1 (ref. 32) (Supplementary Fig. 5).
Figure 5: The transcription-factor circuitry of lung TRM103+ cells.

(a) Expression (log2-transformed normalized values) of genes encoding eomesodermin (EOMES) and T-bet (TBX21) in lung- and blood-derived T cell subsets under resting conditions or after stimulation with anti-CD3 plus anti-CD28 (below plots). Number above bracketed line indicates FDR value. (b,c) Flow cytometry (bi-exponential scale) (b) and quantification (c) of the expression of eomesodermin (Eomes) and T-bet in paired samples (n = 6) of lung TRM103+ cells and blood TEM cells (lines (c) connect paired samples). (d) Expression (key) of mRNA encoding 58 transcription factors (right margin) differentially expressed in lung TRM103+ cells versus blood TEM cells under resting conditions; brackets above indicate hierarchical clustering. (e) Network analysis of the 58 transcription factors expressed differentially by lung TRM103+ cells relative to their expression by blood TEM cells, showing the transcription-factor complexes of AP-1 (green), Notch1-RBPJ (blue) and NF-κB (yellow). (f) Differential gene expression by lung TRM103+ cells versus blood CD8+ T cells for all probes (black) and gene sets (keys) that are targets of HIF-1α (MSigDB gene sets M2513, M12299, M255 and M6189, combined; top) or Notch1 (MSigDB gene set M1869; bottom), plotted against P values. (g) Expression (as in a) of NOTCH1 and RBPJ in lung- and blood-derived T cell subsets under resting conditions or after stimulation with anti-CD3 plus anti-CD28 (below plots). *FDR < 0.05, **FDR < 0.01 and ***FDR < 0.001. (h) Quantitative PCR analysis of IFNG mRNA in lung TRM103+ cells and blood TEM cells (horizontal axis) after 4 h of rest or stimulation with plate-bound DLL1 and DLL4 (DLL) (key); results are normalized to those of mRNA encoding the mitochondrial ribosomal protein S18 and are presented relative to those obtained in the resting condition. Each symbol (a,c,g,h) represents an individual donor; small horizontal lines (a,g,h) indicate the mean (± s.d.). *P < 0.05 (paired _t_-test (c) or unpaired _t_-test (h)). Data are representative of one experiment (a,d–g), nine experiments (b,c), or two experiments with three independent donors (h).
Many of the transcription factors with high expression in lung TRM103+ cells are known drivers of effector function, including RUNX3, ETS1, EPAS1, IRF4, various members of the NF-κB family (NF-κB1, REL, NF-κB2, NFAT5 and RELA) and Notch1 (ref. 33). The idea that many of these transcription factors were biologically active in lung TRM cells was supported by gene-set–enrichment analysis of target genes through the use of CAMERA, in which we searched for transcription-factor 'fingerprints' within the regulatory regions of genes with higher expression in lung TRM cells than in blood TEM cells (Table 2). The most significantly enriched gene sets in this analysis were those that contain targets of the transcription factors HIF-1α and Notch1 (Fig. 5f and Supplementary Table 2), which are both promoters of T cell effector function34,35,36,37.
Table 2 Transcription factors with target gene sets showing significant enrichment
Notch is a cell-surface receptor that is cleaved by a γ-secretase after ligand-induced activation, which allows its intracellular domain to migrate to the nucleus. There, the intracellular domain of Notch acts as a transcriptional activator by associating with the DNA-binding factor CSL (encoded by RBPJ). RBPJ was among the genes expressed most differentially by lung TRM103+ cells relative to their expression by blood TEM cells; its mRNA was present at much higher levels in TRM103+ cells than in blood TEM cells (Fig. 5g). Expression of NOTCH1 was also high in lung TRM103+ cells (Fig. 5g). As Notch transactivates the gene encoding IFN-γ (Ifng) in mice38, we investigated whether activation of this pathway would elicit the expression of IFNG in human lung CD8+ TRM cells. Culture of lung TRM cells in vitro resulted in a decrease in the steady-state abundance of IFNG mRNA (data not shown), consistent with a requirement for external signal input. Stimulation of these cells with recombinant Delta-like ligands (DLLs), which activate Notch signaling, resulted in higher expression of IFNG mRNA than that of mock-stimulated control cells (Fig. 5h). Notably, this induction did not require stimulation through the TCR (Fig. 5h). These results suggested that Notch contributed to maintenance of constitutive IFNG expression in lung TRM cells.
Control of the number of TRM103+ cells by Notch
How TRM cells are maintained is not clear. Notch has been linked to the maintenance of CD4+ memory T cells39,40. As TRM103+ cells exhibited an active Notch signaling signature, we hypothesized that Notch might also serve to maintain TRM103+ cells in situ in the tissue. A core TRM103+ cell gene-expression signature has been determined in mouse T cells8. That signature was strongly conserved in human TRM103+ cells (Fig. 6a and Supplementary Fig. 7), which indicated that mice were a suitable model for determining the function of Notch in the TRM103+ cell subset. Indeed, like their human counterparts, mouse lung TRM cells (identified here on the basis of expression of CD69) also had higher expression of RBPJ than that of CD69− CD8 T cells from the lungs (Fig. 6b). Consistent with a possible function for the Notch pathway here, TRM103+ cells from mouse lungs exhibited surface expression of Notch molecules, especially of Notch2 (Supplementary Fig. 8a). Furthermore, ligands for Notch were expressed by cell types expected to make contact with lung TRM cells, including a subpopulation of CD31−CD326+ lung epithelial cells and CD11b+ lung dendritic cells (Supplementary Fig. 8b). The necessary components therefore seemed to be in place to activate Notch in TRM103+ cells in the lung tissue.
Figure 6: Notch controls the number of lung TRM103+ cells.
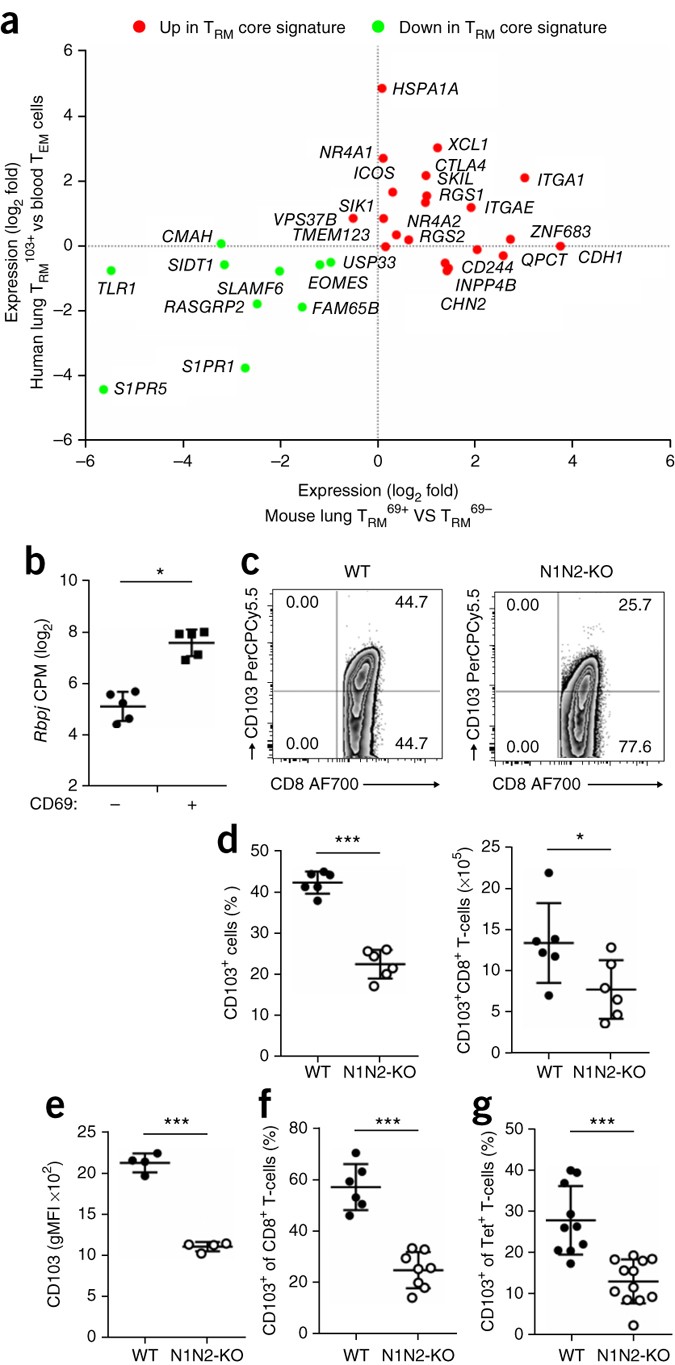
(a) Expression (log2 fold values) of a minimal core signature of TRM cells8 for human lung TRM103+ cells and mouse lung CD69+ CD8+ (TRM69+) cells (RNA sequencing of TRM69+ cells from the lungs of wild-type mice), including genes with higher (Up) or lower (Down) expression in mouse TRM cells than in splenic T cells8 (key). (b) Expression (log2 counts per million (CPM) normalized values) of Rbpj in paired samples (n = 5) of lung CD8+ CD69− T cells and CD69+ T cells (horizontal axis) under resting conditions. *FDR < 0.01. (c) CD103 expression by lung CD8+ T cells from _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre+ mice (N1N2-KO) and _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre− mice (WT), under steady-state conditions. (d) Frequency (left) and absolute number (right) of CD103-expressing (CD103+) lung CD8+ T cells (protected from labeling with anti-CD8β) obtained from mice as in c (n = 6 per group), assessed by flow cytometry. (e) Geometric mean fluorescence intensity of CD103 in CD103+ cells from mice as in c (n = 4 per genotype). (f) Frequency of CD103+ CD8+ T cells in the lungs of _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre+ mice (n = 8) and _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre− mice (n = 6) 10 d after intranasal infection with influenza virus strain HKx31. (g) Frequency of CD103-expressing (CD103+) CD8+ T cells specific for the influenza A virus NP(366–374) tetramer (Tet+) in _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre+ mice (n = 12) and _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre− mice (n = 10) 10 d after intranasal infection with HKx31. Each symbol (b,d,e–g) represents an individual donor; small horizontal lines indicate the mean (± s.d.). *P < 0.05, **P < 0.01 and ***P < 0.001 (unpaired _t_-test). Data are representative of one experiment (a), three experiments (b,g), three experiments with six independent stainings (c) or two experiments (d–f).
To study the function of Notch in lung TRM103+ cells, we inactivated Notch1 and Notch2, which often compensate for each other37, in both CD4+ T cells and CD8+ T cells. To this end, we crossed mice carrying _lox_P-flanked Notch1 and Notch2 alleles (_Notch1_fl/fl_Notch2_fl/fl) with mice with transgenic expression of Cre recombinase from the T cell–specific gene Cd4 (_Cd4_-Cre)36, a setting that does not overtly affect T cell development36. We intravenously injected fluorescence-labeled antibodies to CD8β into the resultant mice briefly before sacrificing them, to allow unequivocal discrimination between CD8+ T cells in blood (labeled with the antibody) and those that reside in the lung tissue (not labeled)41. Lungs from _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre+ mice had considerably fewer TRM103+ cells than did those from their _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre− littermates (Fig. 6c,d), although the overall frequency of CD8+ T cells in these lungs was similar in both groups of mice (Supplementary Fig. 8c). Furthermore, the average surface expression of CD103 by those remaining TRM103+ cells was also lower in _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre+ mice than in their _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre− littermates (Fig. 6c,e).
To study the role of Notch in antigen-specific TRM103+ cells, we infected mice intranasally with influenza A virus, strain HKx31 (H3N2). After 10 d, we sacrificed the mice and measured total CD8+ T cells as well as CD8+ T cells that bound the influenza-virus-specific tetramer of H-2Db and an epitope of amino acids 366–374 of influenza virus nucleocapsid protein (NP(366–374)). There was a distinctly lower frequency of the overall TRM103+ cell population (Fig. 6f) as well as of the H-2Db–NP(366–374)-tetramer-binding TRM103+ cell population (Fig. 6g), in the _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre+ mice than in their _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre− littermate controls. Thus, Notch controlled either the generation or the maintenance of TRM103+ cells in the lungs of mice.
Control of the maintenance of TRM103+ cells by Notch
The Notch signature in the TRM cell transcriptome suggested that this pathway is active in TRM cells in situ in the lungs, consistent with the hypothesis that Notch signaling has a role in maintenance of TRM cells. To investigate this, we first induced a population of lung TRM cells by intranasally infecting wild-type mice with influenza virus. After 35 d, we treated mice with γ-secretase inhibitor (GSI) or vehicle for 5 continuous days to block Notch signaling in already established TRM cells. Treatment with GSIs resulted in a significant decrease in TRM103+ cells in the total and H-2Db–NP(366–374)-tetramer-binding populations (Fig. 7a,b). These results demonstrated that Notch signaling was required for the persistence of TRM103+ cells.
Figure 7: Notch controls the maintenance of TRM103+ cells.
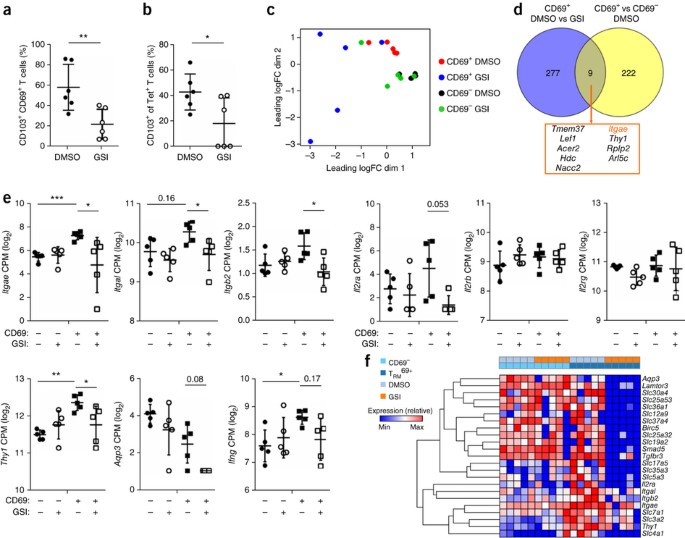
(a) Frequency of CD103+ CD8+ T cells among CD69+ T cells (protected from labeling by intravenously injected anti-CD8β) in wild-type mice (n = 5 per group) infected with influenza virus HKx31 and, 35 d later, treated for 5 d with the vehicle DMSO or GSI. (b) Frequency of CD103-expressing (CD103+) CD8+ T cells specific for the influenza A virus NP(366–374) tetramer (Tet+) in mice as in a. (c) Multi-dimensional scaling analysis (of RNA-sequencing results) of lung CD69+ and CD69− CD8+ T cell populations (key) sorted from mice infected with influenza virus HKx31 and, 35 d later, treated for 2 d with DMSO or GSI (key), presented as 'leading fold change' (logFC) in the first dimension (dim1) and second dimension (dim2), which represent the elements of the data that constitute the greatest difference between samples (indicative of the degree of sample relatedness). (d) Genes differentially expressed by CD69+ memory CD8+ T cells from the lungs of mice as in c treated with DMSO versus those treated with GSI (CD69+ DMSO vs GSI) and by CD69+ memory CD8+ T cells versus CD69− memory CD8+ T cells from DMSO-treated mice (CD69+ versus CD69− DMSO), and the overlap of those groups (middle and bottom). FDR < 0.05. (e) Expression (as in Fig. 6b) of genes (vertical axes) in lung T cell subsets (key) obtained from mice pre-infected with influenza virus and treated for 2 d with DMSO or GSI (key) as in c. *FDR < 0.05, **FDR < 0.01 and ***FDR < 0.001. (f) Expression (key) of mRNA from various genes (right margin) in CD69− memory CD8+ T cells (CD69−) or TRM69+ cells (TRM69+) from mice as in c (key, and above plot); brackets (left margin) indicate hierarchical clustering. Each symbol (a,b,e) represents an individual donor; small horizontal lines indicate the mean (± s.d.). *P < 0.05, **P < 0.01 and ***P < 0.001 (unpaired _t_-test (a,b)). Data are representative of two experiments (a,b) or one experiment (c–f).
To understand how Notch carries out that function, we performed whole-transcriptome analysis of memory T cell populations isolated from mice treated with GSI. To avoid a negative bias due to (possible) loss of CD103+ cells and consequent selection for the remaining cells not affected by GSI treatment, we assessed the gene-expression profiles of the total CD69+ population, in which the TRM103+ cell signature was readily detectable (Fig. 6a). Loss of TRM103+ cells after treatment with GSI should be reflected by a global reduction in the TRM103+ cell gene-expression signature within the transcriptome of CD69+ cells. We furthermore limited the treatment to 48 h to capture gene-expression changes before most of the TRM103+ cells were lost (Supplementary Fig. 8d). Multidimensional scaling analysis showed that GSI treatment prominently affected gene expression in CD69+ lung memory T cells but not in the CD69− population (Fig. 7c). Inhibition of Notch affected expression of only a few TRM cell–specific genes (Fig. 7d). Most notable among these was Itgae (Fig. 7e), consistent with the lower surface intensity of CD103 on TRM cells from _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre+ mice than on those from _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre− mice (Fig. 6e). Although Notch activation induced expression of IFNG in human TRM cells, expression of Ifng in mouse cells was not significantly inhibited, despite a downward trend (Fig. 7e).
The large majority of the 277 genes affected by GSI treatment did not belong to the TRM cell–specific transcriptome (Fig. 7d). These genes included known Notch targets, such as Il2ra (Fig. 7e). Expression of Il2rb and Il2rg (which encode the receptor for IL-15) was not affected by GSI treatment (Fig. 7e). This finding was important, as signaling via the IL-15 receptor has been linked to the maintenance of TRM cells8,27. Gene-set–enrichment analysis also revealed that GSI treatment affected processes such as signaling via the metabolic checkpoint complex mTORC1 and glycolysis (Table 3), which have previously been linked to Notch signaling in T cells37. Closer examination of specific genes showed that several genes encoding products in the integrin signaling pathway (Itgal, Itgb2 and Thy1) were downregulated, in addition to Itgae (Fig. 7e). Notable was also the downregulation of a large number of genes encoding transporters for amino acids (Slc36a1, Slc7a1 and Slc3a2), metabolites (Slc12a9, Slc30a4 and Slco4a1), trace elements and ions (Slc12a9, Slc30a4 and Slc4a1), and metabolites and nutrients (Slc37a4, Slc25a32, Slc19a2 and Aqp3) (Fig. 7f). These results suggested that a major function for Notch signaling is to control metabolic programs in TRM cells, a hypothesis further reinforced by the enrichment, within the Notch dependent transcriptome, for multiple gene sets associated with metabolism, including glycolysis, oxidative phosphorylation and fatty acid metabolism (Table 3).
Table 3 Enriched gene sets affected by GSI treatment
Discussion
The respiratory tract is a front line where immune cells must promptly ward off pathogens but avoid excessive damage to the delicate lung tissues. Our data have demonstrated specialized features of lung CD8+ TRM103+ cells that equip them for this balancing act. First among these was their eponymous anatomical localization, reflected by the distinct expression of genes encoding products associated with migration and adhesion. Expression of the chemokine receptor CCR6 stood out in TRM103+ cells. This receptor is responsive to CCL20 produced by lung epithelial cells and, in a potential positive feedback loop, by lung TRM cells themselves. The CCR6–CCL20 axis is, however, not restricted to the respiratory tract42. A receptor that might help determine lung tropism is CXCR6, as its ligand, CXCL16, has high expression by lung epithelial cells43. A second specialization was their constitutive expression of deployment-ready mRNAs encoding pro-inflammatory cytokines and cytotoxic mediators, which would presumably prevent delays required for transcription. Indeed, lung TRM cells rapidly produce IFN-γ after in vitro stimulation23. TRM cells recruit auxiliary immune cells via IFN-γ-mediated induction of chemokine production by epithelial cells11. Interestingly, human lung TRM103+ cells themselves expressed multiple chemokine-encoding genes, which would possibly allow more expeditious recruitment of auxiliary troops than would indirect production of chemokines by epithelial cells. Despite their constitutive mRNA expression, resting lung TRM103+ cells did not have the corresponding effector proteins17. This suggests that a mechanism exists to translate such mRNAs into proteins only when justified by TCR activation. The poised effector program in TRM103+ cells was accompanied by a gene-expression program associated with inhibition of T cell activation, including genes encoding inhibitory receptors (CTLA4, BTLA4 and KLRC1), suppressive transcription factors (BACH2 and TWIST1) and an inhibitor of TCR signaling (SPRY1). Although apparently at odds with the vigilance required of TRM103+ cells, these inhibitory modules might impose restraint to prevent immunopathology from excessive immunoreactivity.
How is constitutive expression of mRNAs encoding effector molecules maintained? Constitutive transcription probably contributes to this, independently of agonist-peptide-driven TCR stimulation. Although TRM cells expressed little T-bet and eomesodermin27, other regulators of genes encoding effector molecules (for example, NOTCH, HIF1A, IRF4 and NFKB) were active in these cells. Activity of HIF-1α, a transcription factor that operates in response to hypoxia, seems inconsistent with the high oxygen tension in the lungs. Although we considered that hypoxia during sample processing might have affected our results, we believe this is unlikely. First, such a signature was not observed in lung CD4+ TRM cells isolated in parallel (A.O., R.A.v.L. and P.H., data not shown). Furthermore, lung TRM103+ cells had higher expression of both HIF1A mRNA and HIF2A mRNA than that of blood TEM cells, whereas hypoxia controls this pathway via the (post-translational) stabilization of proteins of the HIF family. Indeed, transcription factors of the HIF family also operate in T cells under normoxic conditions, and their expression is induced by cytokines or extracellular ATP44,45.
Notch controls the effector differentiation and function of CD8+ T cells36,38. We found that stimulation with DLL ligands elicited expression of IFNG mRNA in human lung TRM cells. Nonetheless, short-term inhibition of Notch signaling in mice diminished the expression of Ifng mRNA only slightly. This finding suggests that these transcripts are very stable or that other transcription factors are sufficient to drive constitutive Ifng expression, at least in mice. Determination of the role of Notch in the control of effector-molecule-encoding genes during reinfection and in immunity to pathogens awaits the generation of a system that allows inducible elimination of this pathway only in TRM cells.
We found that Notch controlled the maintenance of TRM103+ cells by multiple parallel mechanisms. Notch controlled maximal expression of CD103, which presumably helps to anchor TRM103+ cells in epithelia5. Most of the TRM103+ cell–specific gene-expression program, however, was independent of Notch. Expression of the adhesion molecules LFA1 (encoded by Itgal and Itgb2) and the glycoprotein Thy1 depended on Notch, but the consequences of this are not currently clear. Notch signaling controls the persistence of circulating memory CD4+ T cells through the regulation of glucose metabolism40,46. However, the concentration of glucose is low in the lung mucosa20, as emphasized by the prominent expression of genes associated with glucose starvation in human lung TRM cells. Circulating memory CD8+ T cells rely on the import of glycerol, which can be catabolized via the glycolytic pathway and subsequent oxidative phosphorylation47. Both these metabolic pathways were affected by inhibition of Notch in mouse TRM cells, as was expression of the glycerol importer aquaporin-3. In addition, Notch regulated the expression of a series of other transporters for amino acids, trace elements and ions. Together these results suggest that Notch controls the maintenance of TRM103+ cells at least in part by regulating basic metabolic functions.
Apart from Notch, the transforming growth factor-β (TGF-β) pathway has an important role in TRM103+ cells8,48. Extensive crosstalk exists between these two pathways. TGF-β induces the expression of Notch ligands in epithelial cells, and the intracellular domain of Notch interacts with signal transducers of the Smad family, which are the effectors of the TGF-β pathway49,50. Secretion of TGF-β and expression of Notch ligands thus constitute an integrated mechanism by which the lung mucosal tissue actively maintains the T cell population that protects it from infectious assault.
There is growing appreciation of the clinical importance of tissue-specific T cell memory. We believe that apart from yielding novel insights into the biology of these cells, our study will serve as a valuable resource for further studies into local adaptive immune processes in lungs. Such studies will ultimately aid the development of strategies for vaccination against respiratory infections and possibly also for immunotherapy of cancers of the lung.
Methods
Subjects.
Material was collected from a total of six subjects (four male and two female). The median age of subjects was 58 years. Three patients underwent a lobectomy for a peripheral primary lung tumor, and three received lung transplantation because of end-stage pulmonary disease. Patients with a history of asthma or a recent (<4 weeks) lower respiratory tract infection at the time of inclusion in the study or in the recent past were excluded from the study. None of the patients received systemic corticosteroids, immunosuppressive therapy, chemotherapy or radiotherapy. All subjects were former smokers. Lobectomy patients were recruited from the Academic Medical Center and the Tergooi Hospitals. Two of the lobectomy patients had normal lung function and one had mild chronic obstructive pulmonary disease. All transplantation patients received a double lung transplant and were recruited from the University Medical Center Groningen. All patients gave written informed consent before inclusion in the study, and the study was approved by the Ethical Review Board (ERB) of the Academic Medical Center and the local ERBs of the other participating centers according to the Declaration of Helsinki. Unrelated buffy-coat donors were retrieved from Sanquin Blood Supply Foundation, Amsterdam, the Netherlands.
Mice.
_Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre+ mice and their _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre− littermates, all on C57BL/6/NCrl background were bred and housed in pathogen-free conditions at the Animal Resources Center of the Academic Medical Center (AMC, Amsterdam, the Netherlands). Mice (both male and female) were between 8 and 16 weeks of age at the start of the experiment. _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre+ mice and their _Notch1_fl/fl_Notch2_fl/fl_Cd4_-Cre− mice were housed together to avoid 'cage bias'. Lungs of these mice were cut into small pieces and digested with Collagen type 1 (1% v/w) for 1 h at 37 °C, followed by filtration and flow cytometry. Cells were stained with the relevant fluorochrome-conjugated mAbs for 30 min at 4 °C in PBS containing 0.5% BSA and 0.02% NaN3. For intracellular staining, cells were fixed and permeabilized using Cytofix/Cytoperm (BD Biosciences). Data acquisition and analysis was done on a LSRFortessa (Becton Dickinson) and FlowJo (Tree Star Inc.) software. To study CD103 expression on activated T cells in vivo, mice were intranasally infected with 100× 50% tissue culture effective dose (TCID50) of the H3N2 influenza A virus HKx31. Viral stocks were obtained by infection of MDCK or LLC-MK2 cells51. At 10 d after primary infection, or 8 d after secondary infection, 1 μg anti-CD8-PE (eBioscience, clone H35-17.2) was injected intravenously 8 min before the animals were sacrificed. In this way we were able to discriminate between circulating T cells and T cells shielded from labeling within the tissues. Influenza-virus-specific CD8+ T cells were enumerated using a different, noncompeting antibody CD8 (BD Biosciences, anti-CD8a, clone 53-6.7, dilution 1/200) and tetramers of H-2Db containing the influenza-A-virus-derived nucleocapsid protein peptide NP(366–374) (Sanquin Blood Supply Foundation). For interference of Notch signaling mice were treated with 5 mg/kg LY411575 (Sigma) in DMSO or DMSO control by intraperitoneal injection on a daily basis for up to 2 or 5 d. No weight loss or diarrhea was observed. All mice were used in accordance of institutional and national animal experimentation guidelines.
Isolation of mononuclear cells from peripheral blood and lung tissue.
Heparinized peripheral blood samples were obtained before or during the surgical procedure. Peripheral blood mononuclear cells (PBMCs) were isolated using standard density gradient techniques. Directly after lobectomy, a piece of peripheral lung tissue as far from the tumor as possible was cut off by a pathologist. Lung mononuclear cells (LMCs) were isolated from this tissue specimen as described52. In brief, tissue specimens (1 cm × 1 cm) were sliced with a McIlwain tissue chopper into pieces of 1 mm and incubated for 20 min in RPMI with 20 mM HEPES, pH 7.4, 15% FCS (FCS) and 50 U/ml DNAse type I (Sigma-Aldrich) while shaking at 37 °C. Tissue pieces were carefully dried with sterile gauze and were transferred to medium supplemented with collagenase type I 300 U/ml (Worthington). The material was incubated in this medium for 60 min while shaking at 37 °C. A cell suspension was obtained by grinding the tissue through a flow-through chamber. Mononuclear cells were isolated from the lung cell suspension by standard density gradient techniques. To exclude the possibility of contamination with peripheral blood, the erythrocyte counts were confirmed to be less than 5% of erythrocyte counts in the paired blood sample. Isolated cells were cryopreserved in liquid nitrogen for later analysis. Due to the low frequency of CD103+ T cells in peripheral blood, this subset was obtained from non-related buffy coat donors. Overnight T cell stimulation was performed by adding anti-CD3 plus anti-CD28 Dynabeads (Lifetechnologies) in a 1:1 ratio (c:b) for a period of 16 h. Intracellular cytokine production was measured by stimulation of prepared lymphocyte fractions with PMA (1 ng/ml, Life Technologies) and ionomycin (1 μM, Life Technologies) for up to 6 h in the presence of brefeldin A (5 μg/ml, Life Technologies). Then cells were labeled for markers of T cell subsets as described above and fixed and permeabilized (FoxP3 buffers kit, eBioscience), and intracellular IFN-γ (anti-IFN-γ PE; BioLegend, clone 4S.B3, dilution1/400) was measured by flow cytometry. For in vitro stimulation with Notch ligands, CD8+ T cells isolated by MACS (Miltenyi Biotec) were cultured for 4 h with recombinant sDLL1 and sDLL4 (5 μg/ml, Peprotech) and RNA was isolated. For analysis, a standard Student's _t_-test (unpaired) was applied with GraphPad Prism 6 software. P values of <0.05 were considered statistically significant.
Analysis and cell sorting by flow cytometry.
Human PBMCs or LMCs were incubated with the following antibodies: anti-CD103 PE-Cy7 (eBioscience, clone B-Ly7, dilution 1/200), anti-CD103 PE (eBioscience, clone B-Ly7, dilution 1/100), anti-CD27 FITC (Pelicluster, clone CLB-27/1, dilution 1/25), anti-CD45RA PerCP-Cy5 (eBioscience, clone HI100, dilution 1/200), anti-CD8 APC (BD, clone RPA-T8, dilution 1/100), anti-CD69 BUV395 (BD, clone FN50, dilution 1/50), anti-GZMB AF700 (BD, clone GB11, dilution 1/400), anti-PD1 (BioLegend, clone EH12.2H7, dilution 1/50), anti-anti-Tbet BV421 (BioLegend, clone 4B10, dilution 1/50), anti-EOMES eF660 (eBioscience, clone WD1928, dilution 1/400), anti-CXCR3 PE (R&D, clone 49801, dilution 1/10), anti-CCR5 PE (BD, clone 2D7, dilution 1/25), anti-CXCR6 AF647 (BioLegend, clone K041E5, 1/25) and anti-KLRG1 clone 13F12F2 (dilution 1/400, ref. 53). To prevent premature activation of T cells following staining with anti-CD3, this marker was left out of the phenotyping panel. Hence, NK cell contamination was prevented through the use of anti-CD16 PE (BD, clone 3G8, dilution 1/100) and anti-CD56 PE (BD, clone B159, dilution 1/100) as negative selection criteria. Cells used for transcriptome analysis were CD8+CD45RA−CD103+ or CD103− and negative for antibodies from the NK cell mix. Near-IR and Red LIFE/DEATH fixable dyes (ThermoFisher) were used to stain death cells. Cells were labeled according to manufacturer's instructions and were washed and analyzed in PBS containing 0.5% (w/v) bovine serum albumin (BSA). Cells were analyzed or sorted with a purity >99% using a LSR Fortessa (BD Biosciences) and FACSAria III cell sorter (BD Biosciences), respectively. Analysis was performed using FlowJo software (Tree Star Inc). For analysis of mouse TRM cells, the following antibodies were used: anti-CD8β AF700 (eBioscience, clone Ly-2, 53-6.7, dilution 1/200), anti-CD62L FITC (eBioscience, clone MEL-14, dilution 1/400), anti-CD44 BV785 (eBioscience, clone IM7, dilution 1/200), anti-CD69 eF450 (eBioscience, clone H1.2F3, dilution 1/200), anti-KLRG1 PECY7 (eBioscience, clone 2F1, 1/200), anti-CD103 PerCPCy5.5 (BD, clone M290, dilution 1/200), anti-CD4 Qd605 (Thermofisher, clone RM4-5, dilution 1/1000) anti-CD3 BV510 (BioLegend, clone 17A2, dilution 1/200), anti-DLL1 PE (eBioscience, clone HMD1-5, dilution 1/200), anti-Jagged1 PE (eBioscience, clone HMJ1-29, dilution 1/200), anti-Jagged2 PE (eBioscience, clone HMJ2-1, dilution 1/200), anti-Notch1 PE (BioLegend, clone HMN1-12, dilution 1/200), anti-Notch2 APC (BioLegend, clone HMN2-35, dilution 1/200). For statistical analysis of flow cytometry data, a two-way ANOVA was applied with GraphPad Prism 6 software. P values of <0.05 were considered statistically significant. Numerical analysis of human and mouse lung TRM cells within the post-sort fractions is demonstrated in Supplementary Table 3.
RNA isolation, amplification, labeling and hybridization.
RNA was isolated from 36 sorted cell samples (average of 83 × 103 cells per sample) with the Qiagen RNeasy Plus Micro kit according to manufacturer's instructions. Amplification, labeling, hybridization and data extraction were performed by ServiceXS (Leiden, the Netherlands). Hybridization was performed on the Whole Human Genome HT12-Microarrays (Illumina). The arrays were scanned using the Illumina iScan array scanner and the data retrieved using Illumina's GenomeStudio v2011.1 software. Eight microarray samples were excluded after hybridization, since their average signal was too low.
Microarray pre-processing and data-analysis.
Analyses were carried out with packages from Bioconductor in the statistical software package R (version 3.0.0). Normexp-by-control background correction, quantile normalization, and log2 transformation54 were performed on the Illumina sample and control probe profiles using the limma package (version 3.16.8). The arrayQualityMetrics package (version 3.16.0) was used to assess whether the microarray data were of good quality. Only probes detected (detection P value, <0.05) on at least one array were included in the differential expression analysis. Gene-wise linear models were fitted using the limma package. Differential gene expression between the different conditions was assessed via a moderated _t_-test. The resulting P values were corrected for multiple testing using the Benjamini-Hochberg false-discovery rate (FDR). The illuminaHumanv4.db package (version 1.18.0) was used to update the probe annotation provided by Illumina. Principal-component analysis was performed on unscaled data (function prcomp). The variance explained by the first two principal components was calculated as percentage of the total variance. Hierarchical clustering was done with Pearson correlation as distance measure and complete linkage as agglomeration method (function hclust). Gene set enrichment analysis was perfomed using CAMERA (limma package). CAMERA tests whether a set of genes is highly ranked relative to other genes in terms of differential expression, accounting for inter-gene correlation. CAMERA was applied using gene-set collections C2, C3, C5 and C7 from the Molecular Signatures Database (MSigDB v4.0) that contains information about curated, motif and gene ontology (GO) gene sets and immunological signatures. Low-quality probes that according to the updated probe annotation match repeat sequences, intergenic or intronic regions, or are unlikely to provide specific signal for any transcript were filtered out in the CAMERA analysis. In case multiple probes mapped to the same Entrez Gene ID according to the updated probe annotation, the probe with highest s.d. of its expression values was chosen. P values were calculated for each gene set for two alternative hypotheses (up or down). Gene-set–enrichment results were visualized using the EnrichmentMap Cytoscape plug-in. The enrichment map was generated including all gene sets with a P value < 0.02 and similarity cutoff value of 0.5. Singletons were removed to create the final gene-set interaction network. For network analysis, the STRING 9.1 functional protein interaction database was used55. Confidence view of known and predicted interactions between was applied. Edges represent functional associations; stronger associations are represented by thicker lines. Node colors indicate groups of proteins that are most related according to K-means clustering.
RNA-sequencing pre-processing and data-analysis.
RNA was isolated from 200–1,500 T cells. Samples were precipitated in 0.3 M sodium acetate (Sigma), 2 μg glycogen (Thermo Scientific) and 70% ethanol overnight at −20C. After washing in 70% ethanol, RNA pellets were dissolved in primer mix and incubated for 2 min at 70 °C and processed56. cDNA libraries were sequenced on an Illumina HiSeq 2500 using 50-bp paired-end sequencing. For analysis of paired end CEL-seq reads, sample-specific barcodes in the 'left' reads were used for barcode splitting with the FASTX-Toolkit (version 0.0.12), and the 'right' reads were aligned to the mouse genome (UCSC, mm10) with TopHat (version 2.1.0). All reads that aligned to (exonic regions of) genes annotated by Ensembl (release 78) were quantified using featureCounts (version 1.4.3-p1). Reads were normalized using edgeR57 (version 3.10.5) in R (version 3.2.2), and genes were removed that had less than 1 cpm in three or fewer samples. Multidimensional scaling (MDS) plots were made to detect outlier samples and two samples from one mouse were removed from further analysis. To find genes expressed differentially by CD69+ DMSO-treated cells relative to their expression by CD69− DMSO-treated cells, edgeR's GLM likelihood ratio test was used, taking variation into account caused by study design. To find genes expressed differentially by CD69+ GSI treated cells at day 2 relative to their expression by CD69− GSI treated cells at day 2, or by CD69+ GSI treated cells at day 2 relative to their expression by CD69+ DMSO treated cells at day 2, the GLM likelihood ratio test was used, taking variation caused by study design into account. FDR correction for multiple testing was used, and adjusted P values of <0.05 were considered significant.
Quantitative PCR.
Quantitative PCR analysis was performed in duplicates with an StepOnePlus Real-Time PCR System (Applied Biosystem) using Power SYBR Green (Applied Biosystem). Reaction mixtures were incubated for 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C, 1 min at 60 °C, and, finally, 15 s at 95 °C, 1 min 60 °C and 15 s at 95 °C. Gene expression was normalized by the S18 rRNA in each sample. The primers used are listed in Supplementary Table 4.
Statistical analysis.
All procedures were approved by the local Animal Ethics Committees. For pairwise comparisons, a standard two-sided Student's _t_-test (paired or unpaired when applicable), was applied with GraphPad Prism 6 software. P values of <0.05 were considered statistically significant. Gene-E v3.0.206 software, developed by the Broad Institute, was used to generate heat maps and perform hierarchical clustering. Values were converted to heat map colors using the mean and maximum values for each row. Spearman's rank correlation with average linkage was used for clustering. For statistical analysis of flow cytometry data, two-way ANOVA with the Bonferroni post-hoc test was applied with Graphpad Prsim 6 software.
Accession codes.
GEO: human mRNA data, GSE61397 (MIAME-compliant format); mouse mRNA data, GSE79774.
Accession codes
Primary accessions
Gene Expression Omnibus
Change history
28 November 2016
In the version of this article initially published, the word 'products' was misspelled (as 'prducts') in the final sentence of the third paragraph of the introduction; in Table 1, the third column was incorrectly labeled 'FDR' instead of the correct 'Genes in set'; the citation in the penultimate sentence of paragraph 1 of the third Results subsection (Chemokine and homing receptors) was incorrectly noted as a Supplementary Figure and should be cited as "(Fig. 3b)" instead; in Figure 5f, the P values defining the horizontal dashed lines were not visible and should be moved right for greater visibility, and each is P = 0.05; and in the legend to Figure 6b, 'CPM' was incorrectly defined as 'counts per minute' instead of the correct 'counts per million'. These errors have been corrected in the HTML and PDF versions of the article.
References
- Wu, T. et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol. 95, 215–224 (2014).
Article PubMed PubMed Central CAS Google Scholar - Turner, D.L. et al. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 7, 501–510 (2014).
Article CAS PubMed Google Scholar - Woodland, D.L. & Kohlmeier, J.E. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat. Rev. Immunol. 9, 153–161 (2009).
Article CAS PubMed Google Scholar - Sheridan, B.S. & Lefrançois, L. Regional and mucosal memory T cells. Nat. Immunol. 12, 485–491 (2011).
Article CAS PubMed PubMed Central Google Scholar - Sathaliyawala, T. et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 38, 187–197 (2013).
Article CAS PubMed Google Scholar - Park, C.O. & Kupper, T.S. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat. Med. 21, 688–697 (2015).
Article CAS PubMed PubMed Central Google Scholar - Skon, C.N. et al. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 14, 1285–1293 (2013).
Article CAS PubMed PubMed Central Google Scholar - Mackay, L.K. et al. The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 14, 1294–1301 (2013).
Article CAS PubMed Google Scholar - Sheridan, B.S. et al. Oral infection drives a distinct population of intestinal resident memory CD8+ T cells with enhanced protective function. Immunity 40, 747–757 (2014).
Article CAS PubMed PubMed Central Google Scholar - Steinert, E.M. et al. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161, 737–749 (2015).
Article CAS PubMed PubMed Central Google Scholar - Ariotti, S. et al. T cell memory. Skin-resident memory CD8 T cells trigger a state of tissue-wide pathogen alert. Science 346, 101–105 (2014).
Article CAS PubMed Google Scholar - Wilkinson, T.M. et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat. Med. 18, 274–280 (2012).
Article CAS PubMed Google Scholar - Sridhar, S. et al. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 19, 1305–1312 (2013).
Article CAS PubMed Google Scholar - Slütter, B., Pewe, L.L., Kaech, S.M. & Harty, J.T. Lung airway-surveilling CXCR3hi memory CD8+ T cells are critical for protection against influenza A virus. Immunity 39, 939–948 (2013).
Article PubMed CAS Google Scholar - Shay, T. et al. Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc. Natl. Acad. Sci. USA 110, 2946–2951 (2013).
Article CAS PubMed PubMed Central Google Scholar - den Braber, I. et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity 36, 288–297 (2012).
Article CAS PubMed Google Scholar - Piet, B. et al. CD8+ T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. J. Clin. Invest. 121, 2254–2263 (2011).
Article CAS PubMed PubMed Central Google Scholar - Akbulut, S. et al. Sprouty proteins inhibit receptor-mediated activation of phosphatidylinositol-specific phospholipase C. Mol. Biol. Cell 21, 3487–3496 (2010).
Article CAS PubMed PubMed Central Google Scholar - Wu, D. & Smyth, G.K. Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res. 40, e133 (2012).
Article CAS PubMed PubMed Central Google Scholar - Pezzulo, A.A. et al. Glucose depletion in the airway surface liquid is essential for sterility of the airways. PLoS One 6, e16166 (2011).
Article CAS PubMed PubMed Central Google Scholar - Jung, S. et al. Analysis of fractalkine receptor CX3CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 20, 4106–4114 (2000).
Article CAS PubMed PubMed Central Google Scholar - Schenkel, J.M., Fraser, K.A., Vezys, V. & Masopust, D. Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol. 14, 509–513 (2013).
Article CAS PubMed PubMed Central Google Scholar - McMaster, S.R., Wilson, J.J., Wang, H. & Kohlmeier, J.E. Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-γ production. J. Immunol. 195, 203–209 (2015).
Article CAS PubMed Google Scholar - Yang, M.H. et al. Direct regulation of TWIST by HIF-1a promotes metastasis. Nat. Cell Biol. 10, 295–305 (2008).
Article CAS PubMed Google Scholar - Roychoudhuri, R. et al. BACH2 represses effector programs to stabilize Treg-mediated immune homeostasis. Nature 498, 506–510 (2013).
Article CAS PubMed PubMed Central Google Scholar - Intlekofer, A.M. et al. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 6, 1236–1244 (2005).
Article CAS PubMed Google Scholar - Mackay, L.K. et al. T-box transcription factors combine with the cytokines TGF-β and IL-15 to control tissue-resident memory T cell fate. Immunity 43, 1101–1111 (2015).
Article CAS PubMed Google Scholar - Grueter, B. et al. Runx3 regulates integrin alpha E/CD103 and CD4 expression during development of CD4−/CD8+ T cells. J. Immunol. 175, 1694–1705 (2005).
Article CAS PubMed Google Scholar - Li, Y. et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 147, 629–640 (2011).
Article CAS PubMed Google Scholar - Wang, C. et al. BATF is required for normal expression of gut-homing receptors by T helper cells in response to retinoic acid. J. Exp. Med. 210, 475–489 (2013).
Article CAS PubMed PubMed Central Google Scholar - Mackay, L.K. et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463 (2016).
Article CAS PubMed Google Scholar - Vieira Braga, F.A. et al. Blimp-1 homolog Hobit identifies effector-type lymphocytes in humans. Eur. J. Immunol. 45, 2945–2958 (2015).
Article CAS PubMed Google Scholar - Kaech, S.M. & Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 12, 749–761 (2012).
Article CAS PubMed PubMed Central Google Scholar - Kuijk, L.M. et al. Notch controls generation and function of human effector CD8+ T cells. Blood 121, 2638–2646 (2013).
Article CAS PubMed Google Scholar - Doedens, A.L. et al. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol. 14, 1173–1182 (2013).
Article CAS PubMed PubMed Central Google Scholar - Backer, R.A. et al. A central role for Notch in effector CD8+ T cell differentiation. Nat. Immunol. 15, 1143–1151 (2014).
Article CAS PubMed PubMed Central Google Scholar - Amsen, D., Helbig, C. & Backer, R.A. Notch in T cell differentiation: all things considered. Trends Immunol. 36, 802–814 (2015).
Article CAS PubMed Google Scholar - Bailis, W. et al. Notch simultaneously orchestrates multiple helper T cell programs independently of cytokine signals. Immunity 39, 148–159 (2013).
Article CAS PubMed PubMed Central Google Scholar - Helbig, C. et al. Notch controls the magnitude of T helper cell responses by promoting cellular longevity. Proc. Natl. Acad. Sci. USA 109, 9041–9046 (2012).
Article CAS PubMed PubMed Central Google Scholar - Maekawa, Y. et al. Notch controls the survival of memory CD4+ T cells by regulating glucose uptake. Nat. Med. 21, 55–61 (2015).
Article CAS PubMed Google Scholar - Anderson, K.G. & Masopust, D. Editorial: Pulmonary resident memory CD8 T cells: here today, gone tomorrow. J. Leukoc. Biol. 95, 199–201 (2014).
Article PubMed CAS Google Scholar - Esplugues, E. et al. Control of TH17 cells occurs in the small intestine. Nature 475, 514–518 (2011).
Article CAS PubMed PubMed Central Google Scholar - Day, C., Patel, R., Guillen, C. & Wardlaw, A.J. The chemokine CXCL16 is highly and constitutively expressed by human bronchial epithelial cells. Exp. Lung Res. 35, 272–283 (2009).
Article CAS PubMed PubMed Central Google Scholar - Wang, R. et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882 (2011).
Article CAS PubMed PubMed Central Google Scholar - Mascanfroni, I.D. et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-α. Nat. Med. 21, 638–646 (2015).
Article CAS PubMed PubMed Central Google Scholar - Wang, W. et al. Notch receptor-ligand engagement maintains hematopoietic stem cell quiescence and niche retention. Stem Cells 33, 2280–2293 (2015).
Article CAS PubMed PubMed Central Google Scholar - Cui, G. et al. IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell 161, 750–761 (2015).
Article CAS PubMed PubMed Central Google Scholar - Zavadil, J., Cermak, L., Soto-Nieves, N. & Böttinger, E.P. Integration of TGF-β/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 23, 1155–1165 (2004).
Article CAS PubMed PubMed Central Google Scholar - Blokzijl, A. et al. Cross-talk between the Notch and TGF-β signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. J. Cell Biol. 163, 723–728 (2003).
Article CAS PubMed PubMed Central Google Scholar - Elyaman, W. et al. Notch receptors and Smad3 signaling cooperate in the induction of interleukin-9-producing T cells. Immunity 36, 623–634 (2012).
Article CAS PubMed PubMed Central Google Scholar - Bodewes, R. et al. Vaccination against human influenza A/H3N2 virus prevents the induction of heterosubtypic immunity against lethal infection with avian influenza A/H5N1 virus. PLoS One 4, e5538 (2009).
Article PubMed PubMed Central CAS Google Scholar - Holt, P.G. et al. Extraction of immune and inflammatory cells from human lung parenchyma: evaluation of an enzymatic digestion procedure. Clin. Exp. Immunol. 66, 188–200 (1986).
CAS PubMed PubMed Central Google Scholar - Marcolino, I. et al. Frequent expression of the natural killer cell receptor KLRG1 in human cord blood T cells: correlation with replicative history. Eur. J. Immunol. 34, 2672–2680 (2004).
Article CAS PubMed Google Scholar - Shi, W., Oshlack, A. & Smyth, G.K. Optimizing the noise versus bias trade-off for Illumina whole genome expression BeadChips. Nucleic Acids Res. 38, e204 (2010).
Article PubMed PubMed Central CAS Google Scholar - Snel, B., Lehmann, G., Bork, P. & Huynen, M.A. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 28, 3442–3444 (2000).
Article CAS PubMed PubMed Central Google Scholar - Grün, D. et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature 525, 251–255 (2015).
Article PubMed CAS Google Scholar - Robinson, M.D., McCarthy, D.J. & Smyth, G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Article CAS PubMed Google Scholar
Acknowledgements
We thank K.P. Gisbergen and I.J. ten Berge for discussions; E. Mul and T. Poplonski for assistance with flow cytometry sorting; J. Kloek, M. Jonker, E. Hendriks, J. Weening, H. Stel, E. Prins, G. Nossent and E. Verschuuren for help with obtaining patient material; A. ten Brinke (Sanquin, Amsterdam) for tetramer reagents; and R. Lutter, B.S. Dierdorp and T. Dekker for coordinating technical assistance. Supported by the Landsteiner Stichting voor Bloedtransfusie Research (1136 to R.A.W.v.L.), the Netherlands Asthma Foundation (3.2.06.020 to R.A.W.v.L.), the Alexander von Humboldt Foundation (R.S.) and the Netherlands organization for scientific research (NWO-ALW; to D.A.).
Author information
Author notes
- Christina Helbig, Ronald A Backer and Berber Piet: These authors contributed equally to this work.
- Perry D Moerland, Derk Amsen and René A W van Lier: These authors jointly directed this work.
Authors and Affiliations
- Department of Hematopoiesis, Sanquin Research and Landsteiner Laboratory, Amsterdam, the Netherlands
Pleun Hombrink, Christina Helbig, Ronald A Backer, Anna E Oja, Regina Stark, Giso Brasser, Derk Amsen & René A W van Lier - Department of Experimental Immunology, Academic Medical Center, Amsterdam, the Netherlands
Berber Piet - Department of Clinical Epidemiology, Biostatistics and Bioinformatics, Academic Medical Center, Amsterdam, the Netherlands
Aldo Jongejan & Perry D Moerland - Department of Respiratory Medicine, Academic Medical Center, Amsterdam, the Netherlands
René E Jonkers - Department of Blood Cell Research, Sanquin Research and Landsteiner Laboratory, Amsterdam, the Netherlands
Benjamin Nota - Hubrecht Institute-KNAW (Royal Netherlands Academy of Arts and Sciences), Utrecht, the Netherlands
Onur Basak & Hans C Clevers
Authors
- Pleun Hombrink
You can also search for this author inPubMed Google Scholar - Christina Helbig
You can also search for this author inPubMed Google Scholar - Ronald A Backer
You can also search for this author inPubMed Google Scholar - Berber Piet
You can also search for this author inPubMed Google Scholar - Anna E Oja
You can also search for this author inPubMed Google Scholar - Regina Stark
You can also search for this author inPubMed Google Scholar - Giso Brasser
You can also search for this author inPubMed Google Scholar - Aldo Jongejan
You can also search for this author inPubMed Google Scholar - René E Jonkers
You can also search for this author inPubMed Google Scholar - Benjamin Nota
You can also search for this author inPubMed Google Scholar - Onur Basak
You can also search for this author inPubMed Google Scholar - Hans C Clevers
You can also search for this author inPubMed Google Scholar - Perry D Moerland
You can also search for this author inPubMed Google Scholar - Derk Amsen
You can also search for this author inPubMed Google Scholar - René A W van Lier
You can also search for this author inPubMed Google Scholar
Contributions
P.H., C.H., H.C.C. and D.A. designed the experiments; P.H., C.H., R.A.B., B.P., A.E.O., R.S., G.B. and O.B. did the experiments; P.H., C.H., A.J., B.N. and P.D.M. analyzed the data; B.P. and R.E.J. contributed patient samples; R.A.W.v.L. initiated the research program and directed the study together with P.D.M. and D.A.; and P.H. and D.A. wrote the manuscript.
Corresponding author
Correspondence toDerk Amsen.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Flow cytometry showing the lung TRM cell–sorting strategy.
a. Pure cell populations of lung TRM103+ were sorted on basis of expression of CD8+ and CD103+ and lack of the NK markers CD16, CD56 and the CD45RA isoform. Note that lung derived T-cells lack expression of CD27. To prevent CD3 mediated activation of CD8+ T-cells, αCD3 mAb was excluded from the phenotyping panel. Data representative for 6 digested lung samples. b. Percentage of blood TEM cells that express CCR7 as analyzed by flow cytometry surface staining. c. Pure non-death mice lung TRM69+ were sorted on basis of expression of CD3 and CD8. T cells protected from injected CD8b PE were considered to be within the tissue. TRM69+ and their CD69– counterparts were sorted based on the expression of CD69 and a non-naïve phenotype. Data representative for 8 stainings. Numerical analysis of lung TRM cells within the post-sort fraction are provided in Supplementary Table 4.
Supplementary Figure 2 Validation of HT12 gene expression.
Quantitative PCR was used to validate microarray gene expression profiling for a set of 10 genes. Dotplots displaying the log2 transformed normalized expression of the depicted genes in lung (black), and blood (white) derived T-cell subsets under resting and stimulated conditions for the HT12 microarray (left). For PCR (right) the y-axis is shown as the relative expression to the mitochondrial ribosomal protein S18.
Supplementary Figure 3 Transcriptome comparison of lung T cells from chronic obstructive pulmonary disease versus lobectomy.
a. Heat map of 1078 probes that are differentially expressed between Lung TRM and Blood TEM. Indicated is whether material was derived from COPD or lobectomy patients. Selected are probes with an FDR≤0.05. b. Selection of 7 genes that are significantly differentially expressed between COPD and lobectomy patients (using an additive linear model with variables cell type and disease).
Supplementary Figure 4 Transcriptome comparison of lung TRM103+ cells versus lung TRM103– cells.
a. Scatter plot displaying genes that are differentially expressed between either the lung TRM103+ and blood TEM or the lung TRM103- and blood TEM under resting conditions with a FDR<0.05. Fold change is plotted as log2 scale. Genes defining TRM, including ITGAE, CTLA4, KLRC1, S1PR1, SELL and KLRG1 are depicted in red. b. Heat map of genes that are differentially expressed between lung TRM103+ and lung TRM103- under resting condition with a FDR<0.1. c. Dot plots displaying the log2 transformed normalized expression of the depicted genes in lung (TRM103+; square and TRM103-; circle), and blood (TEM; diamond) derived T-cell subsets under resting (black) and stimulated (white) conditions. (Mean ± SD)
Supplementary Figure 5 Gene expression in lung- and blood-derived T cell subsets.
a. Dot plots displaying the log2 transformed normalized expression of the depicted genes in lung (black), and blood (white) derived T-cell subsets under resting and stimulated conditions. b. Normalized expression of depicted genes in resting lung (L) and blood (B) CD103+ and CD103- cells (*FDR<0.05, **FDR<0.01, ***FDR<0.001, Mean ± SD)
Supplementary Figure 6 Chemokine receptor expression lung TRM103+ cells.
a. Representative examples for the surface expression of the chemokine receptors; CCR5, CXCR3 and CXCR6 by lung TRM103+. Histograms display overlay between paired lung TRM103+ (white) and blood TEM (black) T-cell subsets obtained under steady state conditions as analyzed by FACS. Displayed scales are in bi-exponential scale and cell counts are normalized to mode b. Histograms showing the geometric means of two independent paired lung TRM103+ and blood TEM samples. Data representative for 2 experiments.
Supplementary Figure 7 Comparison of the transcriptomes of human and mouse lung TRM cells.
a. Comparison of gene expression between human lung TRM103+ (vertical axis) and murine lung TRM69+ (horizontal axis). In Red; genes that are upregulated in the TRM core signature. In Green; genes that are downregulated in the TRM core signature. Representative examples for the expression of Notch1 and Notch2 by lung CD8+ T-cell subsets obtained from wild type mice under steady state conditions. b. Table showing prominent gene symbols and products resulting from the human lung TRM103+ and murine lung TRM69+ comparison above.
Supplementary Figure 8 Notch1 signaling in lung TRM cells.
a. Representative examples for the expression of Notch1 and Notch2 by lung CD8+ T-cell subsets obtained from wild type mice under steady state conditions. b. Representative example of Notch ligand Jagged1&2 and DLL1 expression by murine respiratory CD31-CD326+ epithelial cells and CD11b+ lung dendritic cells. Expression is compared to isotype control and data is plotted as geometric mean. Data representative for 2 experiments c. The percentage of CD8+ T-cells in naive N1/2ko versus wild type mice under steady state conditions. To specifically analyze T-cells present in lung tissue, mice were intravenously injected with fluorescently labeled antibodies to CD8a, 8 minutes before sacrifice. This allows unequivocal discrimination between CD8+ T-cells in blood (labeled with the antibody) and those that reside in the lung tissue (protected from label). d. The percentage of CD103+ of CD69+ TRM cell within the lung of wild type mice 40 days after intranasal infection with HKx31. Prior to sacrifice mice were treated with a daily dose of DMSO (black) or GSI (white) for a 2 days period. (Mean ± SD).
Supplementary information
Source data
Rights and permissions
About this article
Cite this article
Hombrink, P., Helbig, C., Backer, R. et al. Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells.Nat Immunol 17, 1467–1478 (2016). https://doi.org/10.1038/ni.3589
- Received: 09 June 2016
- Accepted: 21 September 2016
- Published: 24 October 2016
- Issue Date: December 2016
- DOI: https://doi.org/10.1038/ni.3589