Primary culture of Caenorhabditis elegans developing embryo cells for electrophysiological, cell biological and molecular studies (original) (raw)
Introduction
Genetic model organisms provide a number of powerful experimental advantages for defining the genes and genetic pathways involved in biological processes. The nematode C. elegans is a particularly attractive model system1,2. C. elegans is well suited for mutagenesis and forward genetic analysis and has a fully sequenced and well-annotated genome. Gene expression in nematodes is relatively easy and economical to manipulate using RNA interference (RNAi), knockout and transgenesis. Genomic sequence and many other biological data on this organism are assembled in readily accessible public databases and numerous reagents including mutant worm strains and cosmid and yeast artificial chromosome clones spanning the genome are freely available through public resources.
Despite these many experimental advantages, the small size of C. elegans and most of its somatic cells, and the presence of a tough, pressurized cuticle surrounding the animal have limited access for cell-specific physiological and molecular studies. A C. elegans cell culture system would provide direct access to individual cell types for functional and molecular analyses. Early attempts at large-scale culture of C. elegans embryonic cells were described by Bloom3 and the feasibility of culturing differentiated worm neurons was demonstrated. However, Bloom noted significant problems with cell survival, attachment of cells to the growth substrate, cell differentiation and reproducibility of the methods. Initial attempts to patch-clamp cultured cells were unsuccessful. Buechner et al.4 also reported that cultured C. elegans embryonic cells undergo morphological differentiation resembling neurons and muscle cells.
Bloom's studies led to the widely held belief in the field that C. elegans cells could not be cultured reliably in vitro. However, we have recently developed a robust approach for the large-scale culture of cells isolated from developing worm embryos5,6. Analysis of gene expression patterns and cell-type frequency suggests that in vitro embryo cell cultures recapitulate the developmental characteristics of L1 larvae. The protocol described here provides step-by-step details for culturing worms, isolating eggs and removing eggshells, dissociating embryos into single cells, placing embryo cells in culture and in vitro RNAi methods. We also describe fluorescence-activated cell sorting (FACS) methods that allow enrichment of specific cell types and video-enhanced microscopy methods required for electrophysiological analyses of cultured cells. Limitations of the method are the same as those of any other primary cell culture technique and include the loss of critical cell–cell interactions and factors that may be required for proper cell development and differentiation. Cultured C. elegans embryo cells are, however, suitable for a wide range of cell physiological and cell biological applications including studies of signal transduction, ion channel physiology and gene expression.
Materials
Reagents
- Bacto Agar (Becton, Dickinson and Co., catalog no. 214010)
- Bacto Peptone (Becton, Dickinson and Co., catalog no. 211677)
- Bacto Tryptone (Becton, Dickinson and Co., catalog no. 211705)
- Bacto Yeast Extract (Becton, Dickinson and Co., catalog no. 212750)
- Cholesterol (Sigma Chemical Co., catalog no. C3045)
- NA22 Escherichia coli (Caenorhabditis Genetics Center)
- Chlorox bleach
- Chitinase (Sigma, catalog no. C6137 or C7809)
- L-15 cell culture medium (Invitrogen, catalog no. 21083-027)
- Heat-inactivated fetal bovine serum (Invitrogen, catalog no. 10082-139)
- Penicillin-streptomycin (Invitrogen, catalog no. 15140-122)
- Peanut lectin (Sigma, catalog no. L0881)
- Sucrose
- NaCl
- KCl
- CaCl2
- MgCl2
- HEPES
- NaOH
- MgSO4
- KH2PO4
- Ethanol
- Milli-Q or equivalent water
- Poly-L-lysine (Sigma, catalog no. P1274)
- Propidium iodide (Molecular Probes, catalog no. P-1304)
- Commercially available fluorescent beads for FACS machine calibration (BD Biosciences and Duke Scientific Corporation)
- Concentrated nitric acid
Equipment
- Microcentrifuge (e.g., Eppendorf centrifuge 5415R)
- Osmometer (e.g., Wescore Vapro vapor pressure osmometer 5520)
- Tabletop centrifuge with swinging bucket rotor (e.g., Thermo IEC Centra-CL2)
- Laminar flow hood
- Inverted microscope with × 10 and × 20 objectives
- Hemocytometer
- Cell culture incubator (optional)
- 1–200 μl siliconized pipette tips (VWR International Inc., catalog no. 53503-794)
- Sterile 18 gauge needles (Becton Dickinson and Co., catalog no. 305196)
- Sterile 3 ml syringes (Becton Dickinson and Co., catalog no. 309585)
- 5.0 μm Durapore filters (Millipore Corporation, catalog no. SLSV025LS)
- Mat Tek dishes (35 mm diameter sterile plastic Petri dishes with a no. 0 15 mm diameter glass coverslip glued onto the bottom; Mat Tek Corp., model no. P35G-0-14-C)
- 12 mm diameter glass coverslips (Fisher Scientific, catalog no. 12-545-80)
- 3.5″ × 8″ sterilization pouches (VWR International, catalog no. 11213-237)
- Nunc four-well culture dishes (VWR International, catalog no. 62407-068)
- Lab-Tek chamber slides (Nalge Nunc International)
- Model R-26G bath chamber (Warner Instrument Corp.)
Reagent setup
- 2 × YT medium Dissolve 16 g Bacto Tryptone, 10 g Bacto Yeast Extract and 5 g NaCl in 1 liter of Milli-Q water. Adjust pH to 7.0 with NaOH and autoclave. Medium can be stored for several weeks at room temperature (21–22 °C).
- Nematode growth medium agar plates Dissolve 3 g NaCl, 2.5 g Bacto Peptone and 17 g Bacto Agar in 1 liter of Milli-Q water and autoclave. After cooling to 55 °C, under sterile conditions add the following solutions in the given order, while swirling: 1 ml of cholesterol solution (5 mg cholesterol per ml ethanol), 1 ml of 1 M CaCl2 solution, 1 ml of 1 M MgSO4 solution and 25 ml of 1 M KH2PO4 (pH 6) solution. Pour liquid agar medium into 10 cm Petri dishes.
Critical
CaCl2, MgSO4 and KH2PO4 stock solutions should be sterile-filtered before use.
- Enriched peptone agar plates Dissolve 1.2 g NaCl, 20 g Bacto Peptone and 25 g Bacto Agar in 1 liter of Milli-Q water and autoclave. After cooling to 55 °C, under sterile conditions add the following solutions in the given order, while swirling: 1 ml of cholesterol solution (5 mg cholesterol per ml ethanol), 1 ml of 1 M MgSO4 solution and 25 ml of 1 M KH2PO4 (pH 6) solution. Pour liquid agar medium into 10 cm Petri dishes.
- Egg isolation solution Mix together 1 ml fresh Chlorox bleach, 0.25 ml 10 N NaOH and 3.75 ml sterile H2O.
Critical
This solution must be made fresh before each use. If you store bleach in smaller volumes on the bench, the container should be light tight. Replace bleach stored in smaller containers at least once a week.
- Egg buffer Contains 118 mM NaCl, 48 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 25 mM HEPES, pH 7.3 and should have an osmolality of 340 mOsm.
Critical
The osmolality of egg buffer should be measured with an osmometer and must be 340 ± 5 mOsm for cell viability.
- Chitinase solution Contains 1 U chitinase per ml sterile egg buffer.
Critical
Sigma chitinase no. C7809 is more expensive, but works faster than chitinase no. C6137.
Critical
Prepare chitinase solution under aseptic conditions in ice-cold egg buffer. Store aliquots at −80 °C and thaw immediately before use.
- Cell culture medium L-15 cell culture medium without phenol red, containing 10% (v/v) heat-inactivated fetal bovine serum, 50 U ml−1 penicillin and 50 μg ml−1 streptomycin.
Critical
The osmolality of L-15 culture medium varies from lot to lot. It is therefore important to measure the osmolality of each bottle of medium and adjust to 340 ± 5 mOsm by addition of an appropriate amount of sucrose. Sterile-filter the medium after osmolality is adjusted.
- Peanut lectin solution Dissolve peanut lectin in its original vial. Add sterile water to fill the vial and store it overnight at 4 °C. Transfer this material aseptically to a sterile 50 ml tube and rinse the vial with additional measured volumes of sterile water to ensure full recovery of the lectin and a final peanut lectin concentration of 0.5 mg ml−1. Mix well and transfer 1 ml aliquots into sterile Eppendorf tubes. Store aliquots at 4 °C. The aliquots remain usable for many months.
Critical
Stored aliquots will occasionally contain small amounts of insoluble lectin. Leave this material in the bottom of the tube when coating culture vessels. Attempting to redissolve this material promotes cell clumping.
Critical
Peanut lectin solutions cannot be filter-sterilized or autoclaved. Sterilize small bottles of peanut lectin using gamma irradiation if possible. The dosage and time of exposure vary with the gamma source. Discuss the proper sterilization procedure with the person who oversees your institution's gamma irradiation facility. Alternatively, lectin-coated culture vessels can be UV-irradiated in a laminar flow hood for several hours to overnight. The actual length of irradiation time will depend on the age of the UV lamp.
- Poly- L -lysine solution Prepare poly-L-lysine in sterile water to a final concentration of 40 μg ml−1 and sterilize by sterile filtration.
Equipment setup
- Acid-washing glass coverslips Glass coverslips must be acid-washed prior to coating with lectin or poly-L-lysine. Acid-washing removes manufacturing residues, dirt and/or fingerprints and is necessary to maximize cell attachment, differentiation and survival. In a fume hood, drop 100–200 coverslips individually into 50 ml of concentrated nitric acid in a 100 ml beaker and allow them to soak overnight. Decant the nitric acid into an appropriate waste container. Wash the coverslips in the beaker several times to remove the majority of the nitric acid. To ensure complete removal of the acid, remove single coverslips with forceps and dip them into a series of three beakers containing 400–500 ml of Milli-Q water. Lay coverslips on clean Kim Wipes to dry. When dry, place coverslips into sterilization pouches and autoclave. Store opened pouches under sterile conditions.
Caution
Use all standard laboratory safety procedures when handling and disposing of concentrated nitric acid.
Critical
It is essential to remove completely all nitric acid from the coverslips. Residual acid will harm or kill cells.
- Coating culture vessels with peanut lectin In a laminar flow hood, aseptically pipette a small volume of the peanut lectin solution onto the glass substrate so that it covers about 80% of the surface. Incubate the substrate with the lectin solution for 10–20 min and then remove completely by aspiration. Discard the lectin solution or, alternatively, you can pipette the solution onto another growth substrate to conserve the lectin. If the peanut lectin was not sterilized by gamma irradiation, the lectin-coated growth substrates should be sterilized by UV irradiation. Peanut lectin-coated culture vessels and coverslips can be stored for weeks under sterile conditions.
Critical
It is essential to completely remove the lectin solution. Excess lectin on the growth substrate causes cell clumping. Make sure to remove any lectin solution that may have crept underneath glass coverslips.
- Coating culture vessels with poly- L -lysine for FACS experiments Cover the glass growth substrate with poly-L-lysine solution for 1 h at room temperature. Aspirate the solution, wash three times with sterile water and air-dry.
Critical
Excess poly-L-lysine can be toxic to cells. It is therefore important to aspirate completely the poly-L-lysine solution from the culture vessel and to thoroughly wash the vessel after coating.
- Preparation of enriched peptone agar plates seeded with NA22 E. coli When seeded onto enriched peptone agar plates, these bacteria form a thick lawn that will support the growth of large numbers of gravid adult worms required for the preparation of isolated embryo cells; spread each enriched peptone agar plate with 0.4 ml of NA22 E. coli cultured overnight (12–16 h) in 2 × YT medium at 37 °C with shaking at 250 r.p.m. After the liquid has absorbed into the agar, invert the plate and then incubate overnight at 37 °C.
Procedure
Synchronization of worm cultures
Timing approximately 45–60 min
- 1
It is essential to synchronize worm cultures in order to obtain large numbers of gravid adult animals for subsequent embryo cell isolation and culture. To synchronize cultures, isolate eggs as described below (Steps 8–17) and resuspend in 100–200 μl of sterile water and seed onto a nematode growth medium agar plate without bacteria. - 2
Allow the eggs to hatch by incubating the plate at 16–24 °C for 12–16 h. Without food, worms will arrest at the L1 larval stage.
Critical Step
Do not incubate wild-type worms for longer than 16 h. With longer incubations, worms burrow into the agar, which reduces the final yield.
3. 3
After hatching, rinse the L1 larvae off the plate into a 15 ml conical tube with sterile water.
4. 4
Pellet the worms by centrifugation at ∼350_g_ for 3 min at room temperature.
5. 5
Remove the liquid and repeat the wash and pellet step a second time to eliminate dauer pheromone that may have accumulated during the starvation period. Using first a plastic transfer pipette and finishing with a 100 μl pipettor, remove as much of the final wash liquid as possible to create a dense pellet of larvae.
6. 6
Using a siliconized pipette tip, pipette 8 μl of larvae onto each 10 cm enriched peptone agar plate seeded with NA22 E. coli.
7. 7
Grow larvae into young gravid adults. The time to reach this end point varies with temperature, worm strain, etc. For N2 worms, the incubation temperatures and times listed in Table 1 work well.
Table 1 Time required for growing N2 wild-type L1 larvae at various temperatures to the gravid adult stage.
Full size table
Egg isolation
Timing approximately 45 min
- 8
Wash adult worms off agar growth plates with water into a 15 ml conical tube and pellet by centrifugation at ∼350_g_ for 3 min in a swinging bucket rotor in a tabletop centrifuge at room temperature. - 9
Wash pelleted worms with water 1–3 × until the supernatant is clear of bacteria. Recentrifuge the worms as described in Step 8 after each wash. If you are using multiple plates of worms, the washing and lysis steps will be more efficient if you also use multiple 15 ml tubes. - 10
After the last wash, lyse the worm pellet(s) by adding to each 15 ml conical tube 5 ml of freshly prepared egg isolation solution. Rock worms gently by hand during the lysis. The progress of the lysis reaction is monitored with a dissecting microscope by viewing 5–10 μl of the worm suspension pipetted onto a glass slide. The lysis reaction should be stopped when ∼50% of the worms are lysed.
Critical Step
Lysis time is critical. Under no conditions should the lysis reaction exceed 5 min.
4. 11
Stop the lysis reaction by filling the tube with egg buffer and immediately pellet the eggs and lysed worms by centrifugation at ∼350_g_ for 3 min at room temperature.
5. 12
Remove the supernatant using a sterile plastic transfer pipette and then wash 3 × with 10–12 ml of egg buffer. Repellet the lysed material after each wash by centrifuging at ∼350_g_ for 3 min at room temperature.
Critical Step
Make sure the pellet is completely resuspended in the egg buffer during each wash.
6. 13
After the last centrifugation, carefully remove the buffer by sterile plastic transfer pipette.
7. 14
Resuspend the pelleted eggs and lysed worms in 5 ml of sterile water and then add 5 ml of a sterile 60% (w/v) sucrose stock. Mix this solution well by shaking vigorously.
8. 15
Separate the eggs from lysed worms and other debris by centrifugation at ∼350_g_ for 4 min at room temperature using a swinging bucket rotor in a tabletop centrifuge; eggs will float in the sucrose solution and collect at the solution meniscus and just below the meniscus.
9. 16
Using a sterile plastic transfer pipette, transfer the eggs at the meniscus into sterile 15 ml conical tubes. Eggs that stick to the tube can be recovered by gently washing down the sides with a small volume of the sucrose solution and then removing them with a plastic transfer pipette.
Critical Step
You must use a plastic pipette to remove eggs as they will stick to glass.
Critical Step
Collect no more than 3–4 ml of the egg/sucrose suspension in each tube.
10. 17
To remove the sucrose and debris, fill each tube containing the isolated eggs with 10–12 ml of sterile water and repellet by centrifuging at ∼350_g_ for 3 min at room temperature. Using first a plastic transfer pipette and finishing with a 100 μl pipettor, remove as much of the final wash liquid as possible to recover a dense egg pellet.
Critical Step
When isolating eggs for establishing synchronized worm cultures, it is essential to remove debris produced during the worm lysis. If not removed, hatched larvae may feed on this material and fail to arrest.
Critical Step
After egg isolation, all subsequent steps are carried out in a laminar flow hood under sterile conditions.
Troubleshooting
Preparation of dissociated embryo cells
Timing approximately 50–110 min
- 18
Using a pipettor, add 100 μl of chitinase solution to the egg pellet in the 15 ml tube. Resuspend the eggs and then transfer them into a new sterile Eppendorf tube. To ensure maximal egg recovery, rinse the 15 ml tube and pipette tip 4 × using 100 μl of additional chitinase solution. Transfer each 100 μl aliquot of chitinase solution into the same sterile Eppendorf tube. The final volume of chitinase solution in the tube should be 500 μl. Mix the egg suspension in the Eppendorf tube by rocking gently at room temperature for 20–80 min. - 19
When ≥80% of the eggshells have been digested, add 800 μl of L-15 cell culture medium to the Eppendorf tube. The progress of eggshell digestion should be monitored by viewing samples of the egg suspension under a microscope at × 10–20 magnification. Figure 1 shows examples of the appearance of an isolated egg preparation before and during chitinase digestion.
Figure 1: Appearance of isolated C. elegans embryos during chitinase digestion of the eggshell.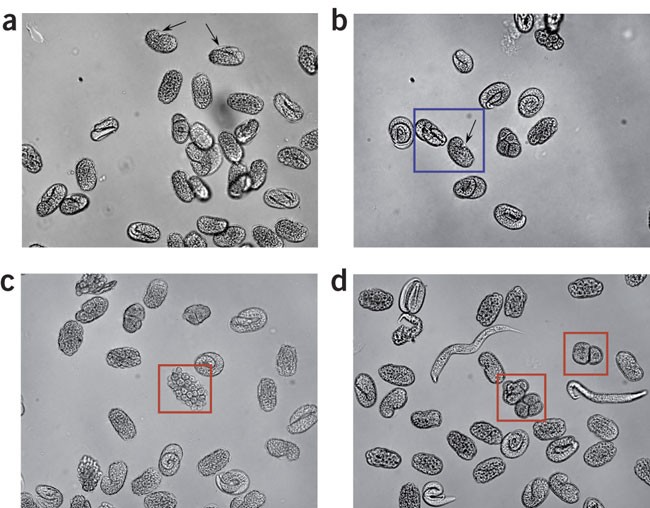
(a) Isolated embryos before exposure to chitinase. The eggshell is readily apparent (arrows). Also note the relatively uniform elongated oval shape of the eggshell-encased embryos. (b) Isolated embryos during early stages of chitinase digestion. Embryos in the blue box have a largely intact eggshell. The eggshell is clearly visible (arrow) and the embryos retain their elongated oval shape. The other embryos in the field still have an intact eggshell; however, their eggshell is weakening, as evidenced by the loss of the elongated oval shape and rounding of the embryos. (c,d) Isolated embryos during late stages of chitinase digestion. Eggshell has been completely digested away from embryos in red boxes. In c, the outlined embryo shows a “grape cluster”-like appearance, which is indicative of complete loss of the eggshell. Presence of free larvae (d) in the preparation is also indicative of eggshell digestion. Embryos were imaged at × 20 using an inverted microscope and conventional bright-field optics.
Full size image
Critical Step
Note that each lot of chitinase tends to vary in potency. Consequently, incubation times will have to be assessed for each lot.
3. 20
Pellet the eggs by centrifugation at ∼900_g_ for 3 min at 4 °C in a microcentrifuge.
4. 21
Carefully remove the supernatant and add 800 μl of fresh L-15 cell culture medium.
5. 22
Gently dissociate the cells by repeatedly pipetting the cell suspension against the side of the Eppendorf tube using a 100–1,000 μl pipettor. Monitor the degree of dissociation by periodically placing an ∼10 μl drop of the suspension on a microscope slide and viewing at 20 ×. Continue the dissociation until at least 50% of the cellular material in the visual field is single cells. The preparation will also contain some intact eggs, undissociated embryos, clumps of cells and hatched larvae (see Fig. 2a).
Figure 2: Appearance of embryo cells during various isolation steps.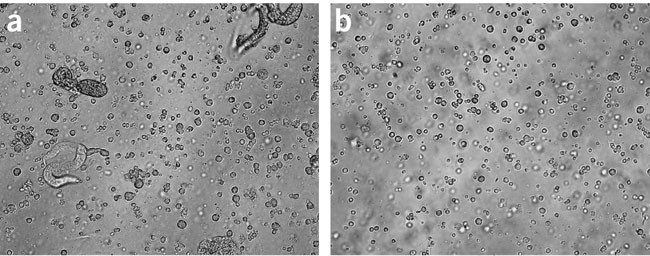
(a) Embryo cells immediately following dissociation step. The majority of material in the preparation is single cells. Intact eggs, hatched larvae, cell clumps and debris are also present. (b) Filtered embryo cells. Preparations were imaged at × 20 using an inverted microscope and conventional bright-field optics.
Full size image
Critical Step
Use of excessive force during the dissociation step will damage the cells. It should be stressed that one should not attempt to prepare a homogeneous population of dissociated cells. In our experience, doing so typically causes excessive cell damage.
6. 23
Pellet the dissociated cell suspension by centrifugation at ∼900_g_ and 4 °C for 3 min.
7. 24
Remove the supernatant, which may be somewhat cloudy, and resuspend the pellet in 500 μl of L-15 cell culture medium.
Critical Step
It is important to remove as much of the supernatant as possible before resuspending the cells in L-15 medium. The fine debris in the supernatant tends to clog the filter used in the filtration step described below.
Filtration of dissociated embryo cells
Timing approximately 15–20 min
- 25
Hatched larvae in the cultures will eat isolated embryo cells. Consequently, cell suspensions must be filtered to remove these larvae as well as cell clumps. Attach a sterile 18 gauge needle to a sterile 3 ml syringe and draw 1 ml of L-15 medium into the syringe. - 26
Draw the cell suspension from Step 24 slowly into the syringe.
Critical Step
Draw the cell suspension up slowly enough so that it does not mix with the L-15 medium in the syringe.
3. 27
Keeping the syringe held vertically, remove the needle and secure a 5.0 μm Durapore filter to the syringe. Force the cell suspension and L-15 medium through the filter with “medium” pressure into a sterile Eppendorf tube.
Critical Step
Excessive force during the filtration step can damage the cells. Use of too little force will reduce cell yield.
4. 28
To maximize the yield of single cells, rinse the filter with additional L-15 medium as follows: remove the filter unit from the syringe and attach a new, sterile needle. Draw an additional 1.0–1.5 ml of L-15 medium into the syringe, remove the needle and replace with the filter. Force the L-15 medium through the filter into a second sterile Eppendorf tube.
5. 29
Pellet the cells by centrifugation of both Eppendorf tubes (from Steps 27 and 28) at ∼900_g_ and 4 °C for 3 min.
6. 30
Remove the supernatants and resuspend the cells in 25–200 μl of L-15 cell culture medium. The actual volume depends on the size of the cell pellet. Small pellets should be resuspended in small volumes of medium.
7. 31
After resuspension, pool the two samples. Figure 2b shows a field of cells after filtration.
Troubleshooting
Setting up cultures
Timing approximately 30–150 min
- 32
Prepare appropriate dilutions of the cell suspension in L-15 medium and determine cell density using a hemocytometer. Useful counting dilutions range from 1:10 to 1:200. For our experimental purposes, we count only “large” and “medium” cells and ignore any small cells.
Critical Step
Plating density depends on the nature of the experiments that are going to be performed. For patch-clamp and optical experiments, we find that a seeding density of ∼230,000 cells per cm2 is optimal. In addition, we have used seeding densities of 8,000,000 cells per well of a one-well chamber slide for western analyses and 800,000 cells per well of a four-well chamber slide for immunocytochemistry.
2. 33
Plate cells onto lectin-coated glass growth substrates using the following methods, according to the type of culture vessel used: use option A for 12 mm diameter glass coverslips and Mat Tek dishes, and option B for chamber slides:
- A
Plating cells onto 12 mm diameter glass coverslips or Mat Tek dishes- i
For 12 mm diameter glass coverslips, place single coverslips into 35 mm diameter Petri dishes. - ii
Pipette 75–100 μl of cell suspension onto the center of each 12 mm coverslip or the center of the Mat Tek dish coverslip and allow the cells to settle and attach for a minimum of 2 h. - iii
Add an additional 2 ml of L-15 medium to each Petri or Mat Tek dish.
- i
- B
Plating cells onto chamber slides- i
Add 2.5 ml of cell suspension to a one-well chamber slide or 600 μl of cell suspension to each well of a four-well chamber slide. - ii
A cell settling period is not needed for chamber slides and they can be placed in the incubator immediately after seeding.
Critical Step
The type of culture vessel used will depend on the type of experiments being performed. For electrophysiology experiments, we culture cells on 12 mm diameter acid-washed glass coverslips. For high-resolution differential interference contrast (DIC) and fluorescence microscopy studies, we usually culture cells in Mat Tek dishes. If larger numbers of cells are needed, we have used one- and four-well Lab-Tek chamber slides.
Critical Step
Isolated embryo cells must adhere tightly to the growth substrate for differentiation to occur and cells do not attach well to coated or uncoated plastic growth substrates. Peanut lectin coating of glass growth substrates is therefore used to promote cell adhesion (see EQUIPMENT SETUP).
Troubleshooting - i
- 34
Keep cultures in small sealed Tupperware containers lined with wet paper towels during seeding and throughout the culture period to prevent evaporation of culture medium. Keep Tupperware containers in a humidified incubator at room temperature and ambient air.
Critical Step
Prevention of culture medium evaporation is critical. As a general rule, we do not use cultures that are older than 5 days. If cultures are kept longer than this, it may be necessary to replace the culture medium periodically.
4. 35
To carry out optional applications of cell cultures such as RNAi, FACS or patch-clamp electrophysiology, see Box 1, 2 or 3, respectively, for procedures.
Troubleshooting
Troubleshooting advice can be found in Table 2.
Table 2 Troubleshooting table.
Timing
Steps 1–7, synchronization of worm cultures: approximately 45–60 min
Steps 8–17, egg isolation: approximately 45 min
Steps 18–24, preparation of dissociated embryo cells: approximately 50–110 min
Steps 25–31, filtration of dissociated embryo cells: approximately 15–20 min
Steps 32–34, setting up cultures: approximately 30–150 min
Anticipated results
Embryo cells terminally differentiate within 24 h after isolation (see Fig. 3) and exhibit developmental characteristics and gene expression patterns that resemble those of the L1 larva. Our limited studies suggest that postembryonic cell differentiation does not occur in embryo cell cultures6. Identification of specific cell types in vitro is greatly facilitated by culturing cells from transgenic worm strains expressing cell-specific GFP reporters (see Figs. 3b,c, 4 and 5). It is also possible to identify certain cell types by specific morphological features. Cell survival is excellent for 2–3 weeks6. However, it should be stressed that these are primary cultures and they may dedifferentiate with time. We typically prepare cells weekly and do not use cultures for physiology experiments that are older than 5 days.
Figure 3: Morphological and GFP reporter expression characteristics in cultured terminally differentiated C. elegans embryo cells.
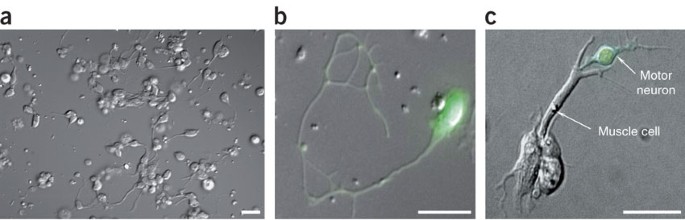
(a) DIC image of a typical culture of differentiated C. elegans embryonic cells 4 days after isolation and plating. Neurons and muscle cells are the most predominant cell type in the visual field. Scale bar, 5 μm. (b) Combined DIC and fluorescence micrograph of a _unc-119_∷GFP-expressing neuron. unc-119 encodes a conserved metazoan protein that may function in signaling pathways that control axonal patterning. (c) Combined DIC and fluorescence micrograph of a _unc-4_∷GFP-expressing cholinergic motor neuron physically interacting with a body wall muscle cell. unc-4 encodes a homeodomain transcription factor. Scale bars, 10 μm (b,c). Reproduced from Christensen et al.6 with permission of Elsevier, copyright 2002.
Figure 4: Whole-cell and cell-attached currents recorded from C. elegans mechanosensory neurons.
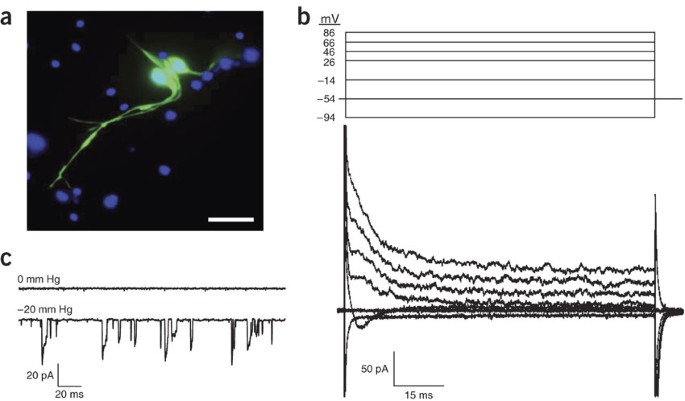
(a) Fluorescent micrograph of cultured mechanosensory neurons expressing _mec-4_∷GFP. mec-4 encodes an amiloride-sensitive Na+ channel or “degenerin” protein20. Cell nuclei are labeled with DAPI and shown in blue. Scale bar, 10 μm. (b) Whole-cell currents in a cultured mechanosensory neuron. Currents were elicited by clamping membrane voltage from −94 to 86 mV. (c) Cell-attached patch currents recorded from a mechanosensory neuron. Holding potential is −60 mV. Channel activity is not detected in the absence of mechanical force (0 mm Hg). Application of suction (−20 mm Hg) to the patch pipette activates inward currents. The currents rapidly and repeatedly activate and inactivate throughout the recording period. x and y axes in scale bars are milliseconds (ms) and picoamperes (pA), respectively.
Figure 5: Patch-clamp electrophysiology and intracellular Ca2+ imaging in cultured C. elegans intestinal cells.
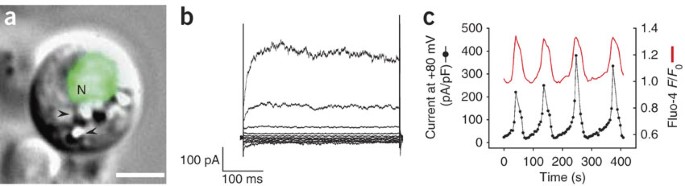
(a) Combined fluorescence and DIC micrographs of a cultured intestinal cell expressing the intestine-specific reporter _elt-2_∷GFP (ref. 33) in the cell nucleus (labeled N). elt-2 encodes a GATA transcription factor. Scale bar, 2.5 μm. Arrowheads denote refractile granules that are a characteristic feature of the worm intestine. (b) Whole-cell ORCa channel currents recorded from a cultured intestinal cell. Pipette was buffered with 10 mM 1,2-bis(_o_-aminophenoxy)ethane-N,N,_N_′,_N_′-tetraacetic acid (BAPTA). Currents were elicited by stepping membrane voltage from −100 to +100 mV in 20 mV steps from a holding potential of 0 mV. x and y axes in scale bars are milliseconds (ms) and picoamperes (pA), respectively. a and b reproduced from Estevez et al.11 with permission of The Rockefeller University Press, copyright 2003. (c) Simultaneous measurement of ORCa channel current and intracellular Ca2+ in a cultured intestinal cell. Pipette solution contained 1 mM BAPTA and 30 μM fluo-4, a Ca2+-sensitive fluorescent probe. Fluo-4 fluorescence intensity changes are plotted as the ratio of F/_F_0, where _F_0 is the fluorescence intensity measured at time 0. Current density on the y axis is reported as picoamperes/picofarad (pA/pF) and was recorded at a membrane potential of +80 millivolts (mV). Reproduced from Estevez and Strange15 with permission of Blackwell Publishing, copyright 2005.
Except for their small size, cultured embryo cells present no serious challenges for patch-clamp studies. C. elegans cell culture has been exploited recently to characterize ion channel and transporter function and regulation5,6,11,12,13,14,15,16,17,18,19. Figure 4 shows examples of whole-cell and cell-attached patch currents recorded from cultured C. elegans mechanosensory neurons expressing _mec-4_∷GFP (see Fig. 4a). mec-4 encodes an amiloride-sensitive Na+ channel or “degenerin” protein20. The whole-cell currents shown (see Fig. 4b) are largely due to the activity of K+ and Ca2+ channels (M. Christensen & K. Strange, unpublished observations) and resemble those recorded from C. elegans mechanosensory neurons in vivo21. Inward currents activated by application of suction to the patch pipette are observed in cell-attached patches (see Fig. 4c). The channels carrying these currents activate and inactivate rapidly and repeatedly in the presence of constant pipette suction. Channel inactivation observed in the presence of continuous mechanical force is referred to as “adaptation” and allows mechanoreceptors to respond to transient and dynamic stimuli22. Rapid adaptation has also been observed in whole-cell mechanoreceptor currents recorded in vivo from worm mechanosensory neurons21.
Cultured embryo cells are also well suited for studies using optical probes of signal transduction and ion activity. Cells can be cultured from worms expressing cell-specific genetically encoded fluorescent indicators23,24 or cells can be loaded in vitro with fluorescent dyes. Figure 5 shows an example of simultaneous measurement of whole-cell current and intracellular Ca2+ in a cultured C. elegans intestinal cell (see Fig. 5a). These cells express an outwardly rectifying Ca2+ (ORCa) channel (see Fig. 5b) (see ref. 11). When cells are patch-clamped with solutions containing low concentrations of Ca2+ buffers, channel activity oscillates. Oscillating channel activity in turn gives rise to intracellular Ca2+ oscillations (see Fig. 5c) (see ref. 15). Such oscillating channel activity may play an important role in driving intestinal Ca2+ oscillations that control the C. elegans defecation rhythm25,26,27.
Gene expression can be silenced by RNAi in C. elegans cell culture simply by adding double-stranded RNA (dsRNA) to the culture medium6,28. Figure 6a shows an example of a whole-cell current in a cultured worm intestinal cell that is activated by depletion of Ca2+ from endoplasmic reticulum (ER) Ca2+ stores11. This store-operated Ca2+ channel or SOCC requires the function of the ER transmembrane protein STIM-1 (ref. 28) and the plasma membrane protein ORAI-1 (ref. 29). Addition of stim-1 or orai-1 dsRNA to the culture medium for 2–3 days inhibits SOCC activation >90% (see Fig. 6) (see refs. 28,29).
Figure 6: Effect of stim-1 RNAi on whole-cell SOCC currents recorded from cultured C. elegans intestinal cells.
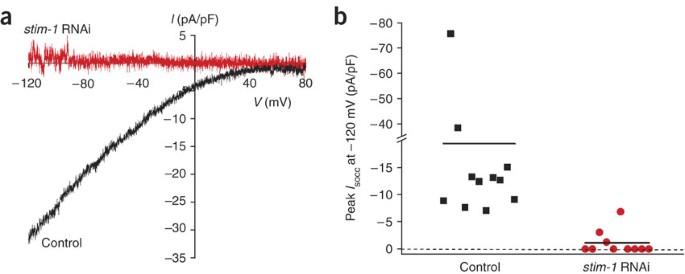
(a) Current-to-voltage relationships of whole-cell Ca2+ currents detected in cultured C. elegans intestinal cells after depletion of ER Ca2+ stores. Currents are peak currents recorded 5–6 min after store depletion was induced. Calcium stores were depleted by dialyzing cells with a pipette solution containing 10 μM IP3, 10 mM BAPTA and a free Ca2+ concentration of ∼18 nM. SOCC current fails to activate in cells treated with stim-1 dsRNA for 2–3 days. The y axis is current (I) reported as picoamperes/picofarad (pA/pF). The x axis is membrane voltage (V) expressed as millivolts (mV). (b) Effect of stim-1 dsRNA on peak SOCC current measured 5 min after obtaining whole cell access. Solid lines are the mean currents for the cells shown. Reproduced from Yan et al.28 with permission of The Rockefeller University Press, copyright 2006.
Fluorescence-activated sorting of C. elegans cultured cells has to date been used to enrich mechanosensory9, motor30,31, and thermosensory and olfactory neurons32 expressing cell-specific GFP reporters. Isolation of cell populations with purities for these neurons of 40–90% has been reported. These enriched cell populations have in turn been used for cell-specific gene expression studies9,30,31,32.
References
- Barr, M.M. Super models. Physiol. Genomics 13, 15–24 (2003).
Article CAS Google Scholar - Strange, K. From genes to integrative physiology: ion channel and transporter biology in Caenorhabditis elegans . Physiol. Rev. 83, 377–415 (2003).
Article CAS Google Scholar - Bloom, L. Genetic and molecular analysis of genes required for axon outgrowth in Caenorhabditis elegans 1–412 (Massachusetts Institute of Technology, Boston, MA, 1993).
Google Scholar - Buechner, M., Hall, D.H., Bhatt, H. & Hedgecock, E.M. Cystic canal mutants in Caenorhabditis elegans are defective in the apical membrane domain of the renal (excretory) cell. Dev. Biol. 214, 227–241 (1999).
Article CAS Google Scholar - Christensen, M. & Strange, K. Developmental regulation of a novel outwardly rectifying mechanosensitive anion channel in Caenorhabditis elegans . J. Biol. Chem. 276, 45024–45030 (2001).
Article CAS Google Scholar - Christensen, M. et al. A primary culture system for functional analysis of C. elegans neurons and muscle cells. Neuron 33, 503–514 (2002).
Article CAS Google Scholar - Grishok, A. RNAi mechanisms in Caenorhabditis elegans . FEBS Lett. 579, 5932–5939 (2005).
Article CAS Google Scholar - Kamath, R.S. & Ahringer, J. Genome-wide RNAi screening in Caenorhabditis elegans . Methods 30, 313–321 (2003).
Article CAS Google Scholar - Zhang, Y. et al. Identification of genes expressed in C. elegans touch receptor neurons. Nature 418, 331–335 (2002).
Article CAS Google Scholar - The Axon CNS Guide, A Laboratory Guide to Electrophysiology and Biophysics (Molecular Devices Corporation, Union City, CA, 2006).
- Estevez, A.Y., Roberts, R.K. & Strange, K. Identification of store-independent and store-operated Ca2+ conductances in Caenorhabditis elegans intestinal epithelial cells. J. Gen. Physiol. 122, 207–223 (2003).
Article CAS Google Scholar - Yuan, A. et al. The sodium-activated potassium channel is encoded by a member of the Slo gene family. Neuron 37, 765–773 (2003).
Article CAS Google Scholar - Carvelli, L., McDonald, P.W., Blakely, R.D. & DeFelice, L.J. Dopamine transporters depolarize neurons by a channel mechanism. Proc. Natl. Acad. Sci. USA 101, 16046–16051 (2004).
Article CAS Google Scholar - Park, K.H., Hernandez, L., Cai, S.Q., Wang, Y. & Sesti, F. A family of K+ channel ancillary subunits regulate taste sensitivity in Caenorhabditis elegans . J. Biol. Chem. 280, 21893–21899 (2005).
Article CAS Google Scholar - Estevez, A.Y. & Strange, K. Calcium feedback mechanisms regulate oscillatory activity of a TRP-like Ca2+ conductance in C. elegans intestinal cells. J. Physiol. 567, 239–251 (2005).
Article CAS Google Scholar - Bianchi, L. et al. The neurotoxic MEC-4(d) DEG/ENaC sodium channel conducts calcium: implications for necrosis initiation. Nat. Neurosci. 7, 1337–1344 (2004).
Article CAS Google Scholar - Suzuki, H. et al. In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron 39, 1005–1017 (2003).
Article CAS Google Scholar - Teramoto, T., Lambie, E.J. & Iwasaki, K. Differential regulation of TRPM channels governs electrolyte homeostasis in the C. elegans intestine. Cell Metab. 1, 343–354 (2005).
Article CAS Google Scholar - Mullen, G.P. et al. The Caenorhabditis elegans snf-11 gene encodes a sodium-dependent GABA transporter required for clearance of synaptic GABA. Mol. Biol. Cell 17, 3021–3030 (2006).
Article CAS Google Scholar - Goodman, M.B. & Schwarz, E.M. Transducing touch in Caenorhabditis elegans . Annu. Rev. Physiol. 65, 429–452 (2003).
Article CAS Google Scholar - O'Hagan, R., Chalfie, M. & Goodman, M.B. The MEC-4 DEG/ENaC channel of Caenorhabditis elegans touch receptor neurons transduces mechanical signals. Nat. Neurosci. 8, 43–50 (2005).
Article CAS Google Scholar - Hamill, O.P. & Martinac, B. Molecular basis of mechanotransduction in living cells. Physiol. Rev. 81, 685–740 (2001).
Article CAS Google Scholar - Frokjaer-Jensen, C. et al. Effects of voltage-gated calcium channel subunit genes on calcium influx in cultured C. elegans mechanosensory neurons. J. Neurobiol. 66, 1125–1139 (2006).
Article CAS Google Scholar - Suzuki, H. et al. In vivo imaging of C. elegans mechanosensory neurons demonstrates a specific role for the MEC-4 channel in the process of gentle touch sensation. Neuron 39, 1005–1017 (2003).
Article CAS Google Scholar - Dal Santo, P., Logan, M.A., Chisholm, A.D. & Jorgensen, E.M. The inositol trisphosphate receptor regulates a 50-second behavioral rhythm in C. elegans . Cell 98, 757–767 (1999).
Article CAS Google Scholar - Espelt, M.V., Estevez, A.Y., Yin, X. & Strange, K. Oscillatory Ca2+ signaling in the isolated Caenorhabditis elegans intestine: role of the inositol-1,4,5-trisphosphate receptor and phospholipases C β and γ. J. Gen. Physiol. 126, 379–392 (2005).
Article CAS Google Scholar - Teramoto, T. & Iwasaki, K. Intestinal calcium waves coordinate a behavioral motor program in C. elegans . Cell Calcium 40, 319–327 (2006).
Article CAS Google Scholar - Yan, X. et al. Function of a STIM1 homologue in C. elegans: evidence that store-operated Ca2+ entry is not essential for oscillatory Ca2+ signaling and ER Ca2+ homeostasis. J. Gen. Physiol. 128, 459 (2006).
Article Google Scholar - Lorin-Nebel, C., Xing, J., Yan, X. & Strange, K. CRAC channel activity in C. elegans is mediated by Orai1 and STIM1 homologs and is essential for ovulation and fertility. J. Physiol. 580, 67–85 (2007).
Article CAS Google Scholar - Cinar, H., Keles, S. & Jin, Y. Expression profiling of GABAergic motor neurons in Caenorhabditis elegans . Curr. Biol. 15, 340–346 (2005).
Article CAS Google Scholar - Fox, R.M. et al. A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genomics 6, 42 (2005).
Article Google Scholar - Colosimo, M.E. et al. Identification of thermosensory and olfactory neuron-specific genes via expression profiling of single neuron types. Curr. Biol. 14, 2245–2251 (2004).
Article CAS Google Scholar - Fukushige, T., Hawkins, M.G. & McGhee, J.D. The GATA-factor elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev. Biol. 198, 286–302 (1998).
CAS PubMed Google Scholar
Acknowledgements
This work was supported by NIH R01 grants DK51610, DK61168 and GM74229 to K.S.
Author information
Authors and Affiliations
- Departments of Anesthesiology, Molecular Physiology and Biophysics, and Pharmacology, Vanderbilt University Medical Center, Nashville, 37232, Tennessee, USA
Kevin Strange, Michael Christensen & Rebecca Morrison
Authors
- Kevin Strange
You can also search for this author inPubMed Google Scholar - Michael Christensen
You can also search for this author inPubMed Google Scholar - Rebecca Morrison
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toKevin Strange.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Strange, K., Christensen, M. & Morrison, R. Primary culture of Caenorhabditis elegans developing embryo cells for electrophysiological, cell biological and molecular studies.Nat Protoc 2, 1003–1012 (2007). https://doi.org/10.1038/nprot.2007.143
- Published: 19 April 2007
- Issue Date: April 2007
- DOI: https://doi.org/10.1038/nprot.2007.143