Gene Expression Profiling of Depression and Suicide in Human Prefrontal Cortex (original) (raw)
INTRODUCTION
Major depression is a disorder of the central nervous system with a pathophysiology involving neocortex. Genetics and environmental factors contribute to the development of mood disorders (Nestler et al, 2002), whereas the diathesis for suicide has independent genetic and clinical risk factors (Mann et al, 1999). No gene mutations or specific molecular and/or neural disruptions have been identified as causative factors for mood disorders or suicide, although some candidate genes have been hypothesized and changes have been reported in several neurotransmitter systems (Mann et al, 2001). For subjects hospitalized with major depression, the lifetime mortality due to suicide reaches 15%. Conversely 60% of suicide cases are associated with mood disorders (Mann, 1998). Post-mortem and in vivo studies of monoaminergic systems have identified abnormalities in serotonergic function, independently correlated with mood disorders and suicidal behavior (Arango et al, 1995; Drevets et al, 2000; Malison et al, 1998; Mann et al, 2000; Sargent et al, 2000), however, serotonergic dysfunctions are likely to represent only part of the pathophysiology of mood disorders and suicide. Moreover, the diagnosis of mood disorders is based on variable sets of symptoms, and subtypes have been proposed based on patterns of symptoms, such as major depression, bipolar disorder involving manic episodes, dysthymia, a milder chronic form of the disease, or melancholic, reactive, psychotic, and atypical depressions (Nestler et al, 2002). However, it is not known whether they represent biological variants of a single family of mood disorders.
Biological studies of mood disorders have most frequently focused on the serotonergic and noradrenergic systems partly due to the fact that they are targets of effective antidepressant treatments. More recently a hypothesis for a cellular neurobiology of depression and antidepressant action emphasized the modulation of neuroplasticity and cellular resilience (Duman et al, 1997; Manji et al, 2001). These distinct lines of experimental research have in fact converged, in that serotonergic and adrenergic antidepressant treatments increase neurogenesis in rodent hippocampus (Malberg et al, 2000), and promote cellular resilience by increasing cell survival factors such as the neurotrophic factor BDNF (Manji et al, 2001). Valproate and lithium, two mood stabilizers with possible antidepressant effects, achieve similar results, possibly by blocking glycogen synthase kinase (GSK3), an enzyme involved in the modulation of proneurodegenerative agents (Manji et al, 1999), although other mechanisms have been suggested (Williams et al, 2002). Consistent with these findings is the post-mortem report of less neuropil in prefrontal cortex of subjects (Rajkowska et al, 1999). Other pathological findings may include a decrease in neuronal density in suicide victims (Arango et al, 2002). The prefrontal cortex is involved in processing of emotion, and implicated in mood disorders (Mann et al, 2001; Robinson et al, 1984). The ventral prefrontal cortex contributes to rational decision-making, controls aggressive/impulsive behaviors (Damasio et al, 1994), and is altered in suicide and aggression (Mann et al, 2001). In vivo imaging studies have also demonstrated altered prefrontal cortex function in depression, with mostly reduced activity and decreased cortical volume (Bremner et al, 2002; Drevets et al, 1997; Mayberg et al, 1999). Treatment of depression may reverse these prefrontal cortical deficits (Goodwin et al, 1993; Nobler et al, 1994), while tryptophan depletion-induced relapse of depressive episode correlates with decreased prefrontal cortical activity (Bremner et al, 1997). All these approaches suffer from the limitation that they explore just a small fraction of the neurotransmitter systems in the brain. An unbiased system for screening many neurotransmitter and other biological systems may provide a more complete picture of the pathobiology of mood disorders.
DNA microarray technology involves large-scale monitoring of relative differences in RNA abundance between samples, which are assumed a priori to represent changes in function. Such approaches have already yielded insights into mechanisms of diseases and biological functions, including cancer, diabetes, and development. As neuropsychiatric disorders have lacked the precise clinical and pathological classification of the cancer field, studies have scarcely begun to address molecular variability in diseases, such as schizophrenia (Hakak et al, 2001; Mirnics et al, 2000; Vawter et al, 2001), alcoholism (Lewohl et al, 2000), and to a lesser extend in bipolar disorder (Bezchlibnyk et al, 2001). For instance, based on large-scale gene expression profiles and functional classification of related genes, dysregulation of presynaptic (Mirnics et al, 2000), metabolic (Middleton et al, 2002), and myelination-related functions (Hakak et al, 2001) have been suggested in the prefrontal cortex of schizophrenic patients.
In this report, by combining large-scale gene expression approaches, bioinformatic analysis, and careful selection of a relatively large cohort of clinical cases, we have investigated molecular and cellular pathways in two areas of the prefrontal cortex, a brain region that may be responsible for dysfunctions in depression and suicidal behavior. We have applied analytical methods to our gene expression database to formally test several hypotheses of depression and suicide, including, first, molecular changes at the level of single or groups of genes, second, the existence of subgroups of patients or disease subtypes, and third, the possibility of common biological pathways being affected in the disease process.
METHODS
Clinical Samples
Brain samples were obtained from the brain collection of the Human Neurobiology Core, Sylvio Conte Center for the Neuroscience of Mental Disorders, at the New York State Psychiatric Institute and Columbia University. All subjects and controls were psychiatrically characterized by psychological autopsies, using our previously validated psychological autopsy method (Kelly and Mann, 1996). Depressed–suicide victims (_n_=19) were selected according to the following criteria: death by violent method, lifetime diagnosis of a major depressive episode (DSM-III-R), absence of psychotropic or illegal drugs on toxicological screens, sudden death without prolonged agonal state or protracted medical illness, and matched with a control group on the basis of sex, age, post-mortem interval, and race. Controls (_n_=19) did not have an Axis I psychiatric disorder, were drug-free and died suddenly without a prolonged agonal period from causes other than suicide. Due to the potentially confounding effect of pharmacological treatments in post-mortem studies, only brain samples from psychotropic medication-free cases were investigated. Other drug exposures were minimal (analgesic (acetominophen) in one case; anesthetic (lidocaine) in two controls; antihypertensive (papaverine) in one control; antihystamine (diphenhydramine) in one case and one control; benzodiazepine in one case and one control; alcohol in three controls). Peripheral and brain toxicological screens ruled out psychotropic medication just prior to death. For psychological autopsies, axis I (SCID I) and axis II diagnoses were obtained using structured clinical interviews. Interviews were done with spouses, close relatives or friends by a PhD psychologist. Axis I and II diagnoses were based on SCID I and II structured clinical interviews, DSM III-R criteria and a consensus conference with a psychiatrist (JJM). Demographic, childhood history, treatment history, and family health care history were assessed systematically using the Columbia Baseline Demographic scale. As a group, subjects did not differ statistically from controls on age (subjects, 44.6+21.2 years; controls, 44.5+21.5 years; mean+SD), sex ratio (subjects and controls: 75% males) and post-mortem delay (subjects, 16.5+8.0 h (4–27); controls 18.5+7.3 h (6–33); mean+SD (range)). Prefrontal cortex Brodmann area 9 (BA9) and B47 were dissected from frozen brain sections that had been transferred from −80 to −20°C for 2 h prior to gray matter sampling.
U133A Oligonucleotide DNA Microarrays
Total RNA was extracted by the guanidine thiocyanate method using the TRIZOL protocol (Invitrogen, Carlsbad, CA) and cleaned with Rneasy microcolumns (QIAGEN GmbH, Germany). The RNA purity and integrity were assessed by optical densitometry, gel electrophoresis, and subsequent array parameters. Microarray samples were prepared according to the Affymetrix protocol (http://www.affymetrix.com/support/). In brief, 10 μg of total RNA was reverse-transcribed and converted into double-stranded cDNA. A biotinylated complementary RNA (cRNA) was then transcribed in vivo, using a RNA polymerase T7 promoter site, which had been introduced during the reverse-transcription of RNA into cDNA. After fragmentation into pieces of 50–200 bases long, 15 μg of labeled cRNA sample was hybridized onto oligonucleotide U133A microarrays, using standard protocols with the Affymetrix microarray oven and fluidics station. Approximately 22 000 annotated genes and well-characterized expressed sequence tag (EST) clusters are represented on U133A arrays. Microarray quality control parameters were as follows: noise (RawQ) less than 5, background signal less than 100 (250 targeted intensity for array scaling), consistent number of genes detected as present across arrays, consistent scale factors, Actin and GAPDH 5′/3′ signal ratios less than 3, and consistent detection of BioB and BioC hybridization spiked controls. Based on these criteria, 36 arrays were retained for further analysis in BA9 and 33 in BA47. Details of procedure and microarray quality control parameters are described elsewhere (Galfalvy et al, 2003). Probeset signal intensities were extracted with the robust multi-array average (RMA) algorithm (Irizarry et al, 2003), which can be found in the R package affy that can be downloaded from the Bioconductor project website (http://www.bioconductor.org). Pairwise comparisons were performed with Microarray Suite 5.0. This software assigns a reliable significance level for differential expression between two samples hybridized on separate arrays that is based on multiple probe level data information, and is therefore well suited for matched-pair experimental design.
Statistics
To test for a disease effect that manifests itself in a shift in the intensity of individual genes, RMA-extracted gene intensities for the depression-suicide and control groups were first compared using gene-by-gene _t_-tests. The resulting _p_-values were adjusted for multiple testing using the Benjamini–Hochberg procedure for controlling the false discovery rate. Based on sex-chromosome-linked genes as internal controls, we have previously demonstrated that this approach is well suited to separate true differences from false positive results, with no evidence for false negative results (Galfalvy et al, 2003). As the false discovery rate is a function of the number of genes tested, _p_-values threshold adjustments were diminished by excluding transcripts with very low variability and reducing the number of genes being tested (11 838 instead of 22 000). Next, taking into consideration that a number of genes are affected by aging, gene-by-gene analysis of variance and analysis of covariance models were used to obtain _p_-values for the age-adjusted disease effect. The _p_-values were adjusted with the same Benjamini–Hochberg procedure mentioned above. Possible interactions between age and disease, and gender and disease, were explored by analysis of variance models with interactions.
An alternative hypothesis regarding the nature of the disease is that it affects the regulation of groups of genes. One way to test this hypothesis was to compare the variability of gene intensities between control and depressed–suicide groups. This could also be viewed as a simple test for heterogeneity in the disease group. Gene-by-gene variance tests were performed, and the resulting _p_-values were adjusted for multiple comparisons. Note, however, that this variance tests relies heavily on the assumption that the distributions of gene intensities in the two groups are normal, an assumption that is violated for at least some of the genes. Gene regulation can be considered at the level of individual genes or groups of genes. Testing the hypothesis that the disease manifests itself as a breakdown of the coregulation of gene groups is difficult, since the gene groups themselves, let alone the nature of the coregulation, are not well defined. As a starting point, we selected a simplified way of looking at coregulation by reducing it to the second-order relationships, as represented by the correlation matrix of gene intensities. Studying the differences in gene coregulation between subject and control groups was reduced in this step to comparing the first two principal components of the correlation matrices for these two groups. Using the distance between the principal components for depressed–suicide victims and controls as test statistics, a permutation test was performed. In a permutation test the samples' group labels (‘subject’ or ‘control’) have been randomly permuted and the tests statistics computed for each permutation. At the end, the ‘observed’ test statistic was compared to the set of ‘permuted’ statistics, and a _p_-value computed as the proportion of permuted test statistics larger than the observed value.
Genes and Sample Pattern Discovery
Genes and samples subgroup analysis was performed with Genes@Work, a pattern discovery algorithm developed by the IBM Functional Genomics group (Califano et al, 2000) and downloadable from http://www.research.ibm.com/FunGen/index.htm. In large genomic databases, the potential number of gene and/or sample patterns of expression profile is exponential. A solution to identifying relevant patterns was proposed, based on a modified supervised learning algorithm. It couples a complex, nonlinear similarity metric, which maximizes the probability of discovering discriminative gene expression patterns, and a pattern discovery algorithm that discovers all statistically significant gene expression patterns in the phenotype set. Statistical significance is evaluated based on the probability of a pattern to occur by chance in the control set. In addition to identifying patterns, it highlights the relevant marker genes, their expression range in the phenotype, and the independent patterns that relate them. Under present conditions, reliable differences in gene expression within subgroups as small as four to five subjects, and/or affecting as little as eight genes were determined to be the lower analytical threshold for subgroup detection.
Functional Class Scoring Analysis
Instead of analyzing genes one at a time, the gene functional class scoring (Pavlidis et al, 2002) gives scores to classes or groups of genes, identified by a common gene ontology (GO, Ashburner et al, 2000) annotation. The input scoring for each gene in subject samples was the statistical distance for this gene transcript level to the expression level of control samples (gene score). Genes present in less than 10% of arrays or with very low variability were not retained for analysis. The class scoring reflects how the distribution of gene scores differs in this class from that which would be expected by chance. GO terms were only considered if between 4 and 150 unique members were assayed for that class in the sample. 1970 GO classes were retained. Classes with high scores tend to have numerous genes with good individual scores. The ‘experiment score’ method described by Pavlidis et al (2002) was applied to the data with some modifications. Briefly, a raw score for each set of genes with a GO term in common is calculated as the mean of the negative log of the gene scores for all genes in the class. When a gene is represented more than once, only the best score is counted. This raw score is transformed into a _p_-value by comparing the score to an empirically determined distribution of the raw scores. This distribution is obtained by randomly generating gene classes of the same size as the class being tested; this is repeated 100 000 times to generate the distribution of scores expected if high gene scores are not concentrated in the class. This method was applied to each sample from the disease group. To identify gene classes (GO terms) associated with depression/suicide, scores for each GO term were ranked for each disease sample. The data were assessed by examining the median and mean ranking for each class across the individual depressed–suicide victims.
Real-time PCR
Small PCR products (100–200 base pairs) were amplified in quadruplets on an Opticon real-time PCR machine (MJ Research, Waltham, MA), using universal PCR conditions (65C to 59C touch-down, followed by 35 cycles (15″at 95°C, 10″ at 59°C and 10″ at 72°C)). A measure of 150 pg of cDNA was amplified in 20 μl reactions (0.3X Sybr-green, 3 mM MgCl2, 200 μM dNTPs, 200 μM primers, 0.5 U Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA)). Primer-dimers were assessed by amplifying primers without cDNA. Results were calculated as relative signal intensity compared to actin. RNA levels for over 15 genes were independently quantified in most samples.
RESULTS
Prefrontal Cortex Oligonucleotide Microarray Samples
Large-scale gene expression was investigated in BA9 and BA47, in the dorsolateral and ventral prefrontal cortex, respectively, between suicide–depressed cases (_n_=19) and controls (_n_=19). Independent analyses of experimental and demographic parameters indicated no effect of post-mortem interval or brain pH on gene expression, but revealed an age effect involving many genes (Erraji-Benchekroun et al, 2003) and a highly specific sex effect restricted to a few genes on sex chromosomes. For instance, signal differences as low as 20% were robustly detected for Y-chromosome-linked genes between male (_n_=30) and female (_n_=8) samples (Galfalvy et al, 2003). Based on these results, only age was considered as a covariate for the analysis of disease effect on gene expression. Three main hypotheses for disease effect on gene expression were tested.
Hypothesis 1: Do Depressed–Suicide Victims have Common Identifiable Changes in Gene Expression?
For analytical purposes, a simple assumption to seek gene expression changes that correlated with the disease was to consider depressed–suicide subjects and controls as two independent groups. Unpaired _t_-test between subjects and controls, or paired _t_-test analysis between age-matched samples were adjusted for multiple testing to control the false discovery rate (Reiner et al, 2003). No single gene was identified in correlation with depression and suicide by this approach. Due to the large sample size, many uncorrected gene _p_-value were in the 10e−2–10e−6 range. A power analysis for the unpaired _t_-test indicated that a two-fold change in a gene with medium intersubject expression variability would be almost certain to be detected. A 40% change could still be detected with 70% probability at this level of variability, while chances of detecting similar changes for highly variable genes are lower. Therefore, it is important to account for other known or suspected sources of intersubject variation in the gene expression levels while looking for the disease effect. Statistical tests including age and/or sex as covariates also did not identify differences in transcript levels between subjects and controls. We also individually examined genes with the smallest unadjusted _p_-values and did not detect any evidence or trends towards differential expression between controls and depressed–suicide victims.
Several gene products and biological systems that have been suggested to be involved in putative mechanisms of depression and suicide were examined individually (Table 1, also Supplementary Material, Table S1). Expression levels for most genes were highly similar between the two experimental groups, as observed by group or age-matched pairwise comparisons. Due to the large sample size and multiple statistical testing, the false discovery rate, as controlled for in this study, typically places statistical threshold for significance in large-scale microarray studies in the 10e−4–10e−6 range (see Methods). A few statistical _p_-values in our list of candidate genes reached the 10e−3 range (ie 5-HT1E receptor in Table 1, see also Table S1), with nevertheless overall minimal fold change, marginal variability and no reproducibility by other comparable analyses (ie paired _t_-test and age-adjusted _p_-value, Tables 1 and S1). Microarray signal intensities for the 5-HT1A receptor, a gene product previously shown to be altered in both suicide and depression, were low, probably representing background signal (Figure 1a), therefore we independently measured mRNA levels for this receptor by quantitative real-time PCR on cDNAs obtained from the original RNA samples (Figure 1d). No difference was observed between depressed–suicide victims and control samples. Overall, real-time PCR analysis agreed well with microarray results over several genes independently tested (Figure 1c) and confirmed the lack of group differences in transcript level for selected genes (Table 1; Figure 1d).
Table 1 Candidate Gene Expression Levels in Depressed Suicide Victims Vs Controls
Figure 1
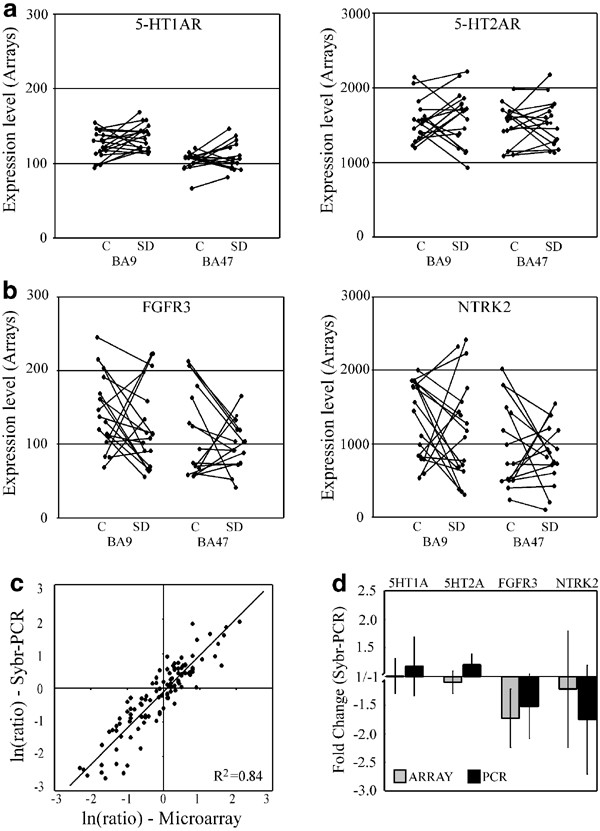
Microarray and real-time PCR mRNA quantification for selected serotonin and neurotrophic receptors in depressed–suicide victims vs controls. Microarray expression levels between depressed–suicide victims and age-matched controls (black lines) are displayed for 5-HT1A and 5-HT2A serotonin receptors (a), and for fibroblast growth factor receptor 3 (FGFR3) and neurotrophic tyrosine kinase receptor 2 (NTRK2, trkB) (b). Note the difference in scales, reflecting different baseline expression across genes. FGFR3 and NTRK2 demonstrated high variability in transcript levels. (c) Real-time PCR (Sybr-PCR) mRNA quantification. Overall, PCR analysis agreed well with microarray results. For genes selected with significant differences in microarray signals across several experimental paired samples, 94% of individual pairwise comparisons with over two-fold expression level differences were confirmed by real-time PCR and 82% in the 1.4–2-fold range. Results from five genes (FGFR3, GPRC5B, MAOA, NTRK2 and MET1A), each quantified in 10–15 paired samples between two brain areas, were combined in this graph (_R_2, coefficient of correlation). (d) Average fold change differences in gene expression levels between depressed–suicide victims and controls, quantified by microarray and real-time PCR. The large range of microarray expression levels for FGFR3 and NTRK2 is also reflected in high standard error across samples, as measured by real-time PCR. No group differences reached statistical levels, whether transcript levels were assessed by microarray or Sybr-PCR.
Most genes display some degree of coregulation of their transcriptional activity, representing the presence of shared regulatory elements, but also numerous and mostly unknown biological interactions. Thus, to extend the hypothesis of single genes being affected by suicide and depression, we tested the hypothesis that the disease affected either the extent of coregulation between genes, or the grouping of coregulated genes within the control and subject groups. A principal component analysis tested the covariance matrices of the gene intensities for depressed–suicide victims and controls separately. The first principal components, representing the main aspects of genes coregulation were compared. If the covariance structures were different between controls and suicides, it would be reflected in differences between components, which in turn would tend to separate controls from suicides on a graphical representation (Figure 2). A permutation test to control for multiple testing demonstrated that it was not the case, as illustrated by the lack of suicide/control sample spatial discrimination in Figure 2a.
Figure 2
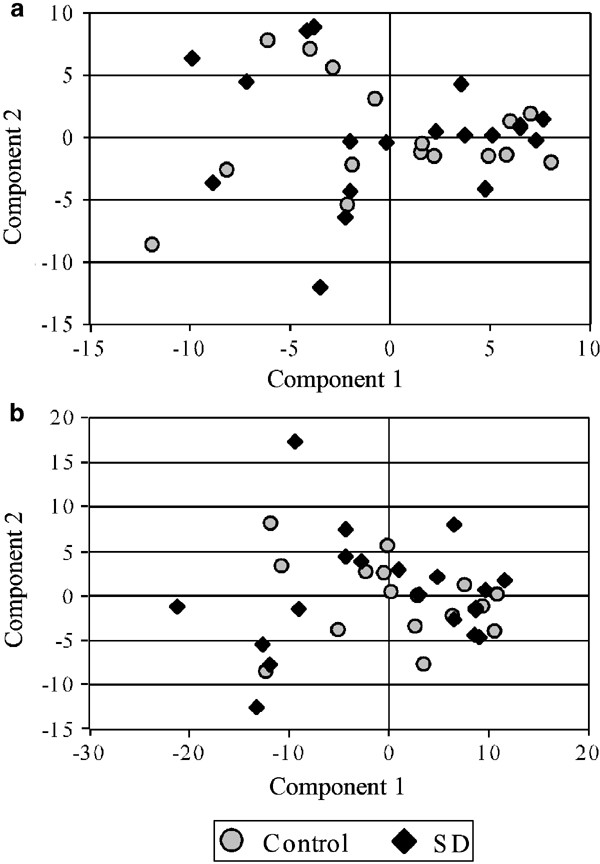
Disease subgroup analysis by principal component analysis. Based on robust coefficients of variation, genes with the highest differences in variability in the depressed–suicide group compared to the control group were selected for principal component analysis. The first two scores for each subjects (components 1 and 2) explain most of the variability in gene expression for this subject. Scores (X_–_Y axis) are standardized to mean 0 and variance proportional to the respective eigenvalue. On a two-dimensional scatterplot, samples are distributed according to similarities in their correlation matrices for these genes. Spatial subgrouping of samples typically represent putative underlying biological differences between the subgroups. No statistical subgroups were identified, as represented here by the lack of spatial segregation of even small number of suicide–depressed patients from the rest of the samples. Similar results were obtained with 200, 1000, and 5000 most variable genes selected, as well as with serotonergic- and adrenergic-related genes. Displayed are results for analyses based on 200 genes selected (a) and on 121 serotonergic- and adrenergic-related genes (b) in BA47. Comparable graphs were obtained in BA9.
As the serotonergic and noradrenergic systems have been repeatedly implicated in the neuropathology and/or therapeutic treatment of mood disorders, we further examined a subgroup of genes coding for most serotonin and norepinephrine receptors and their direct signal transduction partners. A total of 121 genes were selected based on function and reliable detection of expression levels (see Supplementary Material, Table S3). The expression levels of this group of genes were investigated by principal component analysis for the presence of more complex regulatory changes in markers of monoaminergic function. Focusing on a smaller number of genes increased the sensitivity to detect any change in coregulation within this subset of genes. No differences were observed between depressed–suicide victims and controls (Figure 2b).
In summary, the analysis of expression levels of more than 22 000 genes and ESTs in orbital (BA47) and dorsolateral (BA9) prefrontal cortex of depressed–suicide victims did not identify changes in transcript levels compared to control samples.
Hypothesis 2: Is the Clinical Heterogeneity of Mood Disorders Reflected in Biological Subgroups?
To determine whether gene expression differences could identify the presence of subgroups of depressed–suicide victims, which may reflect distinct biological subgroups, underlying one common disease phenotype, two independent approaches were undertaken, based, first, on microarray technology-derived pairwise comparisons of samples, and second, on systematic analysis of statistically significant patterns of gene expression.
A pairwise analysis identified a group of 231 genes with significant differences in RNA levels in at least 50% of the age-matched paired samples (see Supplementary Material, Table S2). These genes displayed high variability, significant levels of coregulation and identified apparent subgroups of depressed–suicide samples based on similarities in expression levels across numerous genes (see Supplementary Material, Figure S1a). However, these changes were not restricted to the patient group only, but were also evident in the control samples (Figure S1b). These results were confirmed by a separate test of analysis of variance of gene expression levels across all samples, indicating that more variable genes were not confined to a specific experimental group (see Methods).
To further seek patterns of gene expression that may correlate with subgroups of subjects, or subtypes of disease, an independent analysis of sample and gene subgroups was undertaken using a novel pattern discovery algorithm which maximizes the probability of discovering all statistically significant gene expression patterns in a dataset (Califano et al, 2000). This approach is especially well suited to study complex phenotypes, which may be composed of unknown subphenotypes or subgroups (see Methods). Between our sample size and the number of genes tested, reliable differences in gene expression within subgroups as small as four to five subjects, and/or affecting as little as eight genes would be detected. Analyzing all samples together easily identified groups of samples corresponding to sex differences, as well as other independent subgroups of samples with coregulated changes in gene expression (G Stolovitzky, J Lepre, and E Sibille, unpublished results). These subgroups confirmed the comprehensive and discriminative power of this analytical approach to detect unknown sample subgroups within a complex gene expression database. However, no patterns of gene expression were identified that correlated with subgroups of suicide–depressed samples vs controls.
Taken together, several lines of analysis provided no evidence in our cohort of samples for the presence of subgroups of depressed–suicide victims with distinct changes in gene expression.
Hypothesis 3: Are Common Biological Functions Affected in Suicide and Depression?
Correlated changes in gene expression levels across gene families, metabolic pathways or functionally related genes may result in common pathological end points, although attained through disruptions of closely related, but not necessarily identical genes within each individual depressed–suicide subject. To formally test this hypothesis, genes were organized according to the GO classification, based on biological processes, cellular components, and molecular functions (Ashburner et al, 2000). The cumulated effect of genes belonging to a single gene group was scored as described in Pavlidis et al (2002), as a measurement of a putative disease effect on this function within each subject (see Methods). Finally, gene families were ranked according to their cumulated effect across all depressed–suicide samples and examined individually (Table 2 and Supplementary Material, Table S4). No specific biological or functional gene group emerged as being consistently affected in most psychiatric subjects. Two overlapping gene families (epidermal growth factor (GO:0005006) and enzyme-linked receptor protein signaling pathway (GO:0007167)) were present in the top-ranked gene groups, mostly due to a moderate effect in a small group of samples (Table 2). Plotting gene expression changes across all psychiatric samples for this functional group (199 genes; see Supplementary Material, Table S5) revealed a similar, but attenuated, bimodal distribution of differential expression that had been detected by age-matched pairwise comparisons in both suicide–depressed and control subjects (see hypothesis 2, Figure S1). For instance, variability in expression levels for genes coding for the neurotrophic tyrosine kinase receptor, type 2 (trkB), or the fibroblast growth factor receptor 3 contributed to the overall score for the GO : 0007167 gene family (Table S5). However, these genes also demonstrated similar high variability in expression levels in the control and patient subgroups (Figure 2b, c).
Table 2 Functional Genomic Analysis in Depression and Suicide
Taken together, a genome-wide functional analysis did not reveal common disease-related dysregulation within functional gene groups or biological pathways. Identified functional changes were not specific to depressed–suicide victims.
DISCUSSION
In this report, we simultaneously measured expression levels for thousands of genes in post-mortem dorsolateral and ventral prefrontal cortex of subjects with a history of major depression who died by suicide and in nonpsychiatric controls who died from other causes. Using a battery of analytical approaches, we tested our genomic database for evidence supporting several hypotheses of the disease. Within the limits in analytical power of this relatively large study, we found no evidence for differences in gene expression that correlated with major depression and suicide. Specifically, there was no effect or trend towards altered gene expression at the level of single genes or groups of genes. There was no indication of stratification in the disease group, ruling out the presence of patient or disease subtypes with important molecular pathologies. Furthermore, using a genome-wide functional analysis, we found no evidence for common biological functions being affected across patients. The lack of detectable differences in gene expression in depression and suicide raised several experimental and biological issues that we discuss here.
Microarray studies of complex tissues, such as the brain, are technically challenging and may suffer from numerous limitations that would have reduced our chances to detect gene expression differences that correlated with depression and suicide. First and foremost is the fact that changes in gene expression in a small number of neurons are very likely to be attenuated in the overall RNA pool that was extracted from our gray matter samples. For instance, hypothesizing that depression affected a neural network consisting of only 10% of the neuronal population of a specific brain area, changes in expression levels as large as a 10-fold magnitude within these neurons would be reduced to less than 10% between BA9 and BA47 samples and would therefore be missed. In this report, we have attempted to enrich our samples in gray vs white matter and have concentrated on delimited brain areas (BA9 and BA47), however, our negative results still cannot rule out that dilution of signal differences due to tissue cellular heterogeneity may have masked a real disease effect. Importantly, these limits in detection sensitivity due to attenuation of signal differences in complex tissues also affect all analytical procedures that were described here. A more appropriate method to increase the probability of detecting rare changes in transcript levels would be to microdissect specific cellular layers or neuronal population to limit the dilution of putative disease-related changes in signal levels. However, signal amplification protocols associated with these procedures are less quantitative, and in the absence of candidate systems or identified disease markers to select these cells, this approach is currently difficult to implement.
Despite these limitations, an analysis of gene content on U133A microarrays used in this study indicated that numerous brain-specific functions were reliably monitored in our samples, through consistent expression levels of genes involved in neuronal structure (ie receptors, trophic, and transcription factors) and function (synaptic transmission). The fact that real-time PCR quantification of mRNA levels highly correlated with microarray data, and that consistent differences in signal intensities as low as 20% could be statistically identified for Y-chromosome-linked genes (Galfalvy et al, 2003), underscores the sensitivity of the genomic approach to reliably monitor changes in transcript levels between samples, and indicates that, if present, differences of that magnitude would have been detected between our experimental samples. Alternatively, genes involved in the pathology of depression and suicide may not have been represented on the U133A arrays that were used in this study. An exhaustive search for disease-related changes in gene expression will need to include other microarray platforms, with complementary gene coverage. Alternate methods to measure RNA levels, such as serial analysis of gene expression (SAGE, Velculescu et al, 1995) may be more comprehensive in gene coverage, but are technically demanding for studies including large number of samples. As the characterization of genomic DNA sequences and gene annotations improve, future generation of microarrays will be able to cover a larger biological spectrum, including more genes and a better knowledge of gene structures, including splice variants and other RNA modifications.
In view of a legitimate concern that depression is a heterogeneous disease, we have attempted to generate a homogeneous cohort of subjects through careful selection of clinical cases. In particular the combination of suicide by violent means with episodes of major depression probably already selected for a more consistent putative pathology. Our samples were devoid of the possible confounding effects of other comorbid psychiatric disorders, recent psychotropic drug treatments, or prolonged agonal phases. Although the toxicological screen makes it unlikely that there was a psychotropic drug exposure immediately prior to death, the recent taking of drugs with short half-lives is still possible and could have introduced heterogeneity into the study. Normal intersubject gene expression variability was investigated separately by seeking genes whose expression correlated with either experimental or demographic parameters. Only a restricted effect of sex on sex-chromosome genes (Galfalvy et al, 2003) and a widespread effect of age (Erraji-Benchekroun et al, 2003) were identified. Others parameters had at best a marginal effect on very few genes, although we cannot exclude that additional unknown parameters that are linked to post-mortem conditions may have masked putative disease effects. Most genes, but not all, displayed tight control on their expression levels across the different subjects in this study. For genes displaying low- to medium high variability of expression levels in the control group, even a 40% change due to disease could be detected with a reasonable probability by _t_-test, and the discovery of a two-fold change would be almost certain. For highly variable genes, the probability to detect a change in expression level is much lower, so it becomes important to account for other known sources of variation while testing for a disease effect. Accordingly, an analysis of covariance methods with age and gender as covariates of the disease was applied, but did not identify any changes that correlated with the disease.
The combination of a relatively homogeneous psychiatric population, of a substantial sample size for neuropsychiatric genomic analysis (patients, _n_=19; controls, _n_=19), and of the investigation of two closely related prefrontal cortical areas, increased our analytical power to detect putative disease-related changes in gene expression. Within the above-described limits in detection and analysis, the absence of any correlated increase or decrease in expression levels between depressed–suicide victims and controls is surprising and may reflect redundant homeostatic controls over gene expression levels and the heterogeneity of the disorder, in addition to the anatomical and cellular complexities of the brain tissue. Depression is a heterogeneous disease but it is not known whether biological variants exist. Such disease subtypes may manifest as correlated changes in gene expression within subgroups of patients and not in controls. Using several analytical approaches, we found no evidence supporting such biological subgroups in this study. Some level of stratification was observed in the genomic database; however, subgroups of samples overlapped between subjects and controls and did not correspond to any known clinical or demographic parameters. In particular, coregulated changes in transcripts levels were not restricted to the patient group only, but were also evident in the control samples. The existence of a group of coregulated genes under less-strict transcriptional regulatory control, which contained trophic factors (trkB, FGFR2, FGFR3), cystoskeletal, and elements of synaptic transmission (see Supplementary Material, Figure S1b, c), was intriguing and suggested the presence of a pool of genes with potential for rapid structural adaptation in the prefrontal cortex, regardless of disease status. Importantly, the detection of such subgroups highlights the potential of our combined analytical approaches to discover putative biological disease subgroups, had large disease-related differences been present. On the other hand, if the heterogeneity of the disease were higher than suspected, that is, if most depressed subjects varied from each other's, our combination of analytical approaches would not have detected it. Future studies may need more refined or new disease classifications for sample selection, but may also require microarray sample size closer to population genetic studies in order to discriminate between putative disease subgroups. For instance, a recent genomic study aimed at classifying breast cancer tumors, necessitated close to 300 patients to achieve adequate analytical power for a putative prognosis purpose (Van de Vijver et al, 2002).
Gene expression levels were also investigated in depressed–suicide victims for correlated changes within gene families and molecular pathways. This approach relies on the hypothesis that common pathological end points may be attained through disruptions of closely related genes or biochemical pathways, and thus does not require identical, but functionally related changes in gene expression between affected individuals. These approaches are exploratory by nature and rely on continuous improvements in gene annotations, but have proven successful at characterizing biological events through genomic studies of complex organs. Thus, the lack of common functional dysregulations in depression and suicide that is observed here may be due either to the absence of common biological mechanisms in the disease or to current limitations in annotation and understanding of gene interactions. This analysis may therefore need to be reassessed in the future, concurrently with improvements in human genome annotation.
Overall, this absence of changes in prefrontal cortex transcriptome is difficult to reconcile with the large body of evidence from post-mortem biochemical studies and in vivo functional analysis, which mostly support the notion of altered prefrontal cortical function in depression and suicide. For instance, altered or decreased neuronal and glial densities has been reported by our group and others in orbitofrontal cortex in depression (Arango et al, 2002; Rajkowska et al, 1999). Neuroimaging studies of brain structure and function in depression have consistently described a smaller volume, particularly in the prefrontal cortex (Bremner et al, 2002), and lower blood flow and metabolism, although there is some debate over the specific brain regions that are involved (Drevets, 1999; Mayberg et al, 1999). Moreover, altered pre- and postsynaptic markers of the serotonergic system have been consistently reported in depression and suicide. Downregulation of serotonin transporter-binding sites was mapped by quantitative autoradiography throughout the prefrontal cortex (Arango et al, 1995; Mann et al, 2000). Prefrontal cortex postsynaptic 5-HT1A receptor density, as measured by antagonists, was higher in most suicide victims (Arango et al, 1995), restricted to violent suicide (Matsubara et al, 1991) or not changed (Dillon et al, 1991), while lower 5-HT1A receptor agonist binding was reported in vivo in depressed patients by positron-emission tomography (Drevets et al, 2000; Sargent et al, 2000). Taken together, the prefrontal cortex is believed to be a crucial cortical area participating in a putative brain network involved in the expression and pathology of depression.
Within the analytical limits of this relatively large genomic study, the results presented here suggest that the pathology of suicide and depression may either be below the detection level of current genomic approaches, or that it does not manifest in altered transcriptional activity, at least in the two prefrontal cortical areas investigated. We did not reproduce changes in expression levels for several genes that had been previously associated with depression. Differences between studies may arise due to case selection, but may also be due to our relatively large sample size. It is also possible that the amplitude of changes that were reported in protein levels and/or functions are either blunted or not present at the level of messenger RNAs, due to differences in homeostatic controls between transcription and translation. Furthermore, post-transcriptional modifications of gene products, such as RNA splicing or editing, are potent regulators of cellular mechanisms, but are not assessed by current genomic studies. Again, it is also important to note that mRNA differences restricted to small number of key neurons involved in a putative neural network affected in depression, may be considerably diluted and indistinguishable in the overall RNA pool extracted from the whole brain area. On the other hand, the pathogenic processes that involve gene regulation may have been long past. Altered functions in depression and suicide, as measured by receptor binding or brain imaging may reflect changes in protein function, due to altered levels or post-translational modifications, independently of mRNA levels. Finally, neurons may not display cellular or structural changes, and yet demonstrate altered activity when challenged, due to altered inputs from distal neural network components. In turn, neuronal activity induces transcriptional activity, however, it is not known whether episodic events of altered neuronal activity in the prefrontal cortex may lead to long-term detectable changes in transcript levels. Accordingly, neural network dysfunctions may be more elusive to genomic analysis.
In conclusion, using genomic approaches, we have investigated molecular and cellular pathways in the human prefrontal cortex, which may be responsible for brain dysfunctions in depression and suicidal behavior. Within the limits of this study, a battery of analytical approaches found no changes in single gene expression levels, altered gene coregulation within patient subgroups, or altered gene expression in common biological pathways that correlated with depression and suicide. In view of the relative homogeneity in disease profile of our neuropsychiatric patient cohort and of the absence of major confounding effects, these results suggest a pathology in suicide and depression that does not manifest as large transcriptional changes. Future gene expression studies will need to enrich samples for potential markers of disease pathology, mostly by addressing the two key experimental issues of cellular/tissue heterogeneity and of disease classification. Alternatively, the molecular pathology of depression and suicide may be localized to other cortical and subcortical brain areas, or may be more associated with network changes, protein levels and/or functions rather than altered transcriptome in the prefrontal cortex.
References
- Arango V, Underwood MD, Gubbi AV, Mann JJ (1995). Localized alterations in pre- and postsynaptic serotonin binding sites in the ventrolateral prefrontal cortex of suicide victims. Brain Res 688: 121–133.
Article CAS Google Scholar - Arango V, Underwood MD, Mann JJ (2002). Serotonin brain circuits involved in major depression and suicide. Prog Brain Res 136: 443–453.
Article CAS Google Scholar - Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al (2000). Gene ontology: tool for the unification of biology. Gene Ontol Consortium Nat Genet 25: 25–29.
CAS Google Scholar - Bezchlibnyk YB, Wang JF, McQueen GM, Young LT (2001). Gene expression differences in bipolar disorder revealed by cDNA array analysis of post-mortem frontal cortex. J Neurochem 79: 826–834.
Article CAS Google Scholar - Bremner JD, Innis RB, Salomon RM, Staib LH, Ng CK, Miller HL et al (1997). Positron emission tomography measurement of cerebral metabolic correlates of tryptophan depletion-induced depressive relapse. Arch Gen Psychiatry 54: 364–374.
Article CAS Google Scholar - Bremner JD, Vythilingam M, Vermetten E, Nazeer A, Adil J, Khan S et al (2002). Reduced volume of orbitofrontal cortex in major depression. Biol Psychiatry 51: 273–279.
Article Google Scholar - Califano A, Stolovitzky G, Tu Y (2000). Analysis of gene expression microarrays for phenotype classification. Proc Int Conf Intell Systems Mol Biol 8: 75–85.
CAS Google Scholar - Damasio H, Grabowski T, Frank R, Galaburda AM, Damasio AR (1994). The return of Phineas Gage: clues about the brain from the skull of a famous patient. Science 264: 1102–1105.
Article CAS Google Scholar - Dillon KA, Gross-Isseroff R, Israeli M, Biegon A (1991). Autoradiographic analysis of serotonin 5-HT1A receptor binding in the human brain postmortem: effects of age and alcohol. Brain Res 554: 56–64.
Article CAS Google Scholar - Drevets WC (1999). Prefrontal cortical-amygdalar metabolism in major depression. Ann NY Acad Sci 877: 614–637.
Article CAS Google Scholar - Drevets WC, Frank E, Price JC, Kupfer DJ, Greer PJ, Mathis C (2000). Serotonin type-1A receptor imaging in depression. Nucl Med Biol 27: 499–507.
Article CAS Google Scholar - Drevets WC, Price JL, Simpson Jr JR, Todd RD, Reich T, Vannier M et al (1997). Subgenual prefrontal cortex abnormalities in mood disorders. Nature 386: 824–827.
Article CAS Google Scholar - Duman RS, Heninger GR, Nestler EJ (1997). A molecular and cellular theory of depression. Arch Gen Psychiatry 54: 597–606.
Article CAS Google Scholar - Erraji-Benchekroun L, Underwood MD, Arango V, Galfalvy H, Pavlidis P, Smyrniotopoulos P et al (2003). Extensive molecular aging in human prefrontal cortex. submitted.
- Galfalvy HC, Erraji-Benchekroun L, Smyrniotopoulos P, Pavlidis P, Ellis SP, Mann JJ et al (2003). Sex genes as internal controls for genomic analysis: a study in human brain. BMC Bioinformat 4: 37.
Article Google Scholar - Goodwin GM, Austin MP, Dougall N, Ross M, Murray C, O'Carroll RE et al (1993). State changes in brain activity shown by the uptake of 99mTc-exametazime with single photon emission tomography in major depression before and after treatment. J Affect Disord 29: 243–253.
Article CAS Google Scholar - Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD et al (2001). Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci USA 98: 4746–4751.
Article CAS Google Scholar - Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP (2003). Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31: e15.
Article Google Scholar - Kelly TM, Mann JJ (1996). Validity of DSM-III-R diagnosis by psychological autopsy: a comparison with clinician ante-mortem diagnosis. Acta Psychiatr Scand 94: 337–343.
Article CAS Google Scholar - Lewohl JM, Wang L, Miles MF, Zhang L, Dodd PR, Harris RA (2000). Gene expression in human alcoholism: microarray analysis of frontal cortex. Alcoholism: Clin Exp Res 24: 1873–1882.
Article CAS Google Scholar - Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20: 9104–9110.
Article CAS Google Scholar - Malison RT, Price LH, Berman R, van Dyck CH, Pelton GH, Carpenter L et al (1998). Reduced brain serotonin transporter availability in major depression as measured by [123I]-2 beta-carbomethoxy-3 beta-(4-iodophenyl)tropane and single photon emission computed tomography. Biol Psychiatry 44: 1090–1098.
Article CAS Google Scholar - Manji HK, Drevets WC, Charney DS (2001). The cellular neurobiology of depression. Nat Med 7: 541–547.
Article CAS Google Scholar - Manji HK, Moore GJ, Chen G (1999). Lithium at 50: have the neuroprotective effects of this unique cation been overlooked? Biol Psychiatry 46: 929–940.
Article CAS Google Scholar - Mann JJ (1998). The neurobiology of suicide. Nat Med 4: 25–30.
Article CAS Google Scholar - Mann JJ, Brent DA, Arango V (2001). The neurobiology and genetics of suicide and attempted suicide: a focus on the serotonergic system. Neuropsychopharmacology 24: 467–477.
Article CAS Google Scholar - Mann JJ, Huang YY, Underwood MD, Kassir SA, Oppenheim S, Kelly TM et al (2000). A serotonin transporter gene promoter polymorphism (5-HTTLPR) and prefrontal cortical binding in major depression and suicide. Arch Gen Psychiatry 57: 729–738.
Article CAS Google Scholar - Mann JJ, Waternaux C, Haas GL, Malone KM (1999). Toward a clinical model of suicidal behavior in psychiatric patients. Am J Psychiatry 156: 181–189.
CAS PubMed Google Scholar - Matsubara S, Arora RC, Meltzer HY (1991). Serotonergic measures in suicide brain: 5-HT1A binding sites in frontal cortex of suicide victims. J Neural Transm Gen Sect 85: 181–194.
Article CAS Google Scholar - Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA et al (1999). Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry 156: 675–682.
CAS Google Scholar - Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P (2002). Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci 22: 2718–2729.
Article CAS Google Scholar - Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P (2000). Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron 28: 53–67.
Article CAS Google Scholar - Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM (2002). Neurobiology of depression. Neuron 34: 13–25.
Article CAS Google Scholar - Nobler MS, Sackeim HA, Prohovnik I, Moeller JR, Mukherjee S, Schnur DB et al (1994). Regional cerebral blood flow in mood disorders, III. Treatment and clinical response. Arch Gen Psychiatry 51: 884–897.
Article CAS Google Scholar - Pavlidis P, Lewis DP, Noble WS (2002). Exploring gene expression data with class scores. Pac Symp Biocomput 7: 474–485.
Google Scholar - Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY et al (1999). Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry 45: 1085–1098.
Article CAS Google Scholar - Reiner A, Yekutieli D, Benjamini Y (2003). Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19: 368–375.
Article CAS Google Scholar - Robinson RG, Kubos KL, Starr LB, Rao K, Price TR (1984). Mood disorders in stroke patients. Importance of location of lesion. Brain 107(Part 1): 81–93.
Article Google Scholar - Sargent PA, Kjaer KH, Bench CJ, Rabiner EA, Messa C, Meyer J et al (2000). Brain serotonin1A receptor binding measured by positron emission tomography with [11C]WAY-100635: effects of depression and antidepressant treatment. Arch Gen Psychiatry 57: 174–180.
Article CAS Google Scholar - Van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW et al (2002). A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med 347: 1999–2009.
Article CAS Google Scholar - Vawter MP, Barrett T, Cheadle C, Sokolov BP, Wood III WH, Donovan DM et al (2001). Application of cDNA microarrays to examine gene expression differences in schizophrenia. Brain Res Bull 55: 641–650.
Article CAS Google Scholar - Velculescu VE, Zhang L, Vogelstein B, Kinzler KW (1995). Serial analysis of gene expression. Science 270: 484–487.
Article CAS Google Scholar - Williams RS, Cheng L, Mudge AW, Harwood AJ (2002). A common mechanism of action for three mood-stabilizing drugs. Nature 417: 292–295.
Article CAS Google Scholar
Acknowledgements
This work was supported by a NIMH Conte Center for the Neuroscience of Mental Disorders (MH62185 JJM, VA), NIMH-NRSA (ES), NIMH-K01 (MH067721 ES), NIMH R01 (MH40210 VA) and the American Foundation for Suicide Prevention (ES). We are thankful to Peggy Smyrniotopoulos for technical assistance, to Andrew J Dwork (MH64168) for providing additional samples and to Jie Qin for assistance with Gene Ontology annotations.
Author information
Authors and Affiliations
- Department of Psychiatry, Columbia University, New York, NY, USA
Etienne Sibille, Victoria Arango, Loubna Erraji-Benchekroun, Steve P Ellis & J John Mann - Department of Neuroscience, New York State Psychiatric Institute, New York, NY, USA
Etienne Sibille, Victoria Arango, Hanga C Galfalvy, Loubna Erraji-Benchekroun, Steve P Ellis & J John Mann - Department of Biomedical Informatics and Genome Center, Columbia University, New York, NY, USA
Paul Pavlidis
Authors
- Etienne Sibille
You can also search for this author inPubMed Google Scholar - Victoria Arango
You can also search for this author inPubMed Google Scholar - Hanga C Galfalvy
You can also search for this author inPubMed Google Scholar - Paul Pavlidis
You can also search for this author inPubMed Google Scholar - Loubna Erraji-Benchekroun
You can also search for this author inPubMed Google Scholar - Steve P Ellis
You can also search for this author inPubMed Google Scholar - J John Mann
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toEtienne Sibille.
Additional information
Supplementary Information accompanies the paper on Neuropsychopharmacology website (http://www.nature.com/npp)
Supplementary information
Supplementary File 1 (XLS 29 kb)
Supplementary File 2 (XLS 50 kb)
Supplementary File 3 (XLS 30 kb)
Supplementary File 4 (XLS 631 kb)
Supplementary File 5 (XLS 158 kb)
Supplementary File 6
Gene coregulation in prefrontal cortex. (DOC 3270 kb)
Based on Affymetrix MAS5.0 statistical threshold for differential expression in pairwise comparison, 231 genes were significantly decreased or increased in at least 50% of the subject-control pairs.
A) Using differences in expression level between depressed–suicide victims and controls, a clustering analysis identified high levels of coregulation between genes, as demonstrated by similarities in expression profiles along horizontal gene clusters (note the large cluster at the bottom of graph) and consistently identified two subgroups of patients (main two branches of vertical clusters).
B) Highly similar hierarchical clustering results were obtained when using the same set of 231 genes, but with expression level ratios between pairs of aged-matched control samples. Over 90% of the genes were distributed in highly similar horizontal clusters (i.e. bottom clusters in graphs A and B), indicating that the coregulation structure of these genes was not restricted to the depressed–suicide subgroup.
Similar results were obtained when variables genes were selected based on comparisons between control groups and plotted using aged-matched depressed–suicide paired values, confirming that more variable genes were not confined to a specific experimental group.
Logarithms of signal ratios between aged-matched cases and controls (a) or between controls (b) were normalized and mean-centered for genes and arrays. Vertical lines are individual arrays or samples, horizontal lines are genes. Red and green indicate increased or decreased expression levels. Results from both brain areas were combined in this figure. Similar results were obtained with both brain areas analyzed separately.
Rights and permissions
About this article
Cite this article
Sibille, E., Arango, V., Galfalvy, H. et al. Gene Expression Profiling of Depression and Suicide in Human Prefrontal Cortex.Neuropsychopharmacol 29, 351–361 (2004). https://doi.org/10.1038/sj.npp.1300335
- Received: 02 April 2003
- Revised: 18 September 2003
- Accepted: 22 September 2003
- Published: 05 November 2003
- Issue Date: 01 February 2004
- DOI: https://doi.org/10.1038/sj.npp.1300335