p58IPK suppresses NLRP3 inflammasome activation and IL-1β production via inhibition of PKR in macrophages (original) (raw)
Introduction
The mammalian innate immune system recognizes and responds to different types of pathogens in order to protect the host through multiple signaling responses. The responses are initiated when pathogen-associated or damage-associated molecular patterns are recognized by specific cell surface receptors such as Toll-like receptors (TLRs) and C-type lectin receptors or cytoplasmic proteins including the retinoic acid-inducible gene (RIG)-I-like receptors and NOD-like receptors (NLRs)1. Upon engagement, these receptors activate a series of signaling pathways resulting in upregulation of genes encoding pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNFα), interleukin 1β (IL-1β) and interleukin 6 (IL-6). These molecules orchestrate the inflammatory response and play critical roles in inducing acute-phase proteins, modifying endothelial function and permeability, recruiting immune cells to injury site and regulating survival and cell death. While TNFα and IL-6 are regulated primarily at transcriptional and translational levels, production of IL-1β is regulated by a two-step mechanism. The first step is the expression of inactive IL-1β precursor (pro-IL-1β) induced by the TLR signaling. The second step is the cleavage of pro-IL-1β by activated caspase-12. The complex that activates caspase-1, called the inflammasome, is composed of NLRs, apoptosis-associated speck-like proteins containing caspase recruitment domains (ASC) and pro-caspase-1.
Inflammasome formation is triggered by a range of endogenous and exogenous substances that emerge during infections, tissue damage and metabolic imbalances. Once the protein complexes have formed, the inflammasomes activate caspase-1, which proteolytically activates the inactive IL-1β and IL‑18 precusors. Processed (active) IL-1β is one of the key inflammatory cytokines implicated in many human diseases including autoinflammatory syndromes3, diabetes4 and neurodegenerative disorder5. It has been shown to mediate pancreatic β cell destruction in type 1 diabetes and increases the risk for type 2 diabetes by inducing insulin resistance4. Inhibition of IL-1β signaling by IL-1 receptor antagonist IL-1RA or IL-1β antibody demonstrate encouraging therapeutic effect in type 2 diabetes6,7. Thus, understanding the regulatory mechanisms and identifying endogenous suppressors for IL-1β holds great promise for developing new treatment of inflammatory human diseases.
p58IPK is a multifunctional molecular chaperone that regulates protein homeostasis. When presenting in the cytosol p58IPK functions as an inhibitor of eIF-2 kinases to control protein translation8,9,10, while in the endoplasmic reticulum (ER) it acts as a co-chaperone and regulator of glucose-related protein 78 (GRP78) and participates in protein folding11. Among the eIF-2 kinases, double-stranded RNA dependent protein kinase (PKR) was the first being identified to bind to p58IPK and regulate protein translation during viral infections12. Apart from its regulatory role in protein translation, activation of PKR was found implicated critically in regulation of the expression and production of proinflammatory cytokines through governing multiple pathways mediated by mitogen activated protein kinases (MAPK)13,14, IκB kinase (IKK)15, IFN-β-promoter simulator 1 (IPS-1) signaling16 and transcriptional factors including NF-κB and, c-Jun and activating transcription factor 2 (ATF2)14,15,17. Recently, Lu and colleagues reported a novel role of PKR in coordinating the activation of inflammasomes and maturation of IL-1β18. Interestingly, this finding was challenged by another study showing that PKR is dispensable for inflammasome activation19. Nevertheless, the role of PKR and its regulation in macrophage activation and IL-1β production is intriguing.
Here we investigate the influence of p58IPK as an inhibitor of PKR on TLR4 signaling, NLRP3 inflammasomes and IL-1β production in macrophages. Using bone marrow derived macrophages (BMDM) from wild-type and p58IPK knockout mice, we demonstrate that p58IPK inhibits lipopolysaccharide (LPS)-induced activation of NF-κB and c-jun N-terminal kinase (JNK) and decreases the expression of pro-inflammatory cytokines TNFα, IL-1β and IL-6. Furthermore, we show that p58IPK suppresses NLRP3 inflammasome activation and reduces IL-1β production through inhibition of PKR.
Results
p58IPK regulates LPS-induced PKR, NF-κB and JNK activation in macrophages
Recent studies have implicated PKR in TLR4-dependent signaling in mouse cells20,21. To determine whether p58IPK as an inhibitor of PKR is involved in regulation of TLR4 pathway in macrophages, we isolated BMDM from WT and p58IPK knockout mice and exposed the cells to LPS to activate TLR4 signaling. We found that LPS treatment for 15 min induced markedly increased phosphorylation of PKR, NF-κB p65 and JNK in WT (p58IPK+/+) BMDM and to a significantly greater extent in p58IPK-deficient (p58IPK−/−) cells (Fig. 1A,B). Interestingly, LPS induced a less increase in the activation of p38 MAPK in p58IPK−/− BMDM compared to controls. To determine whether PKR is a key mediator of these changes, BMDM were pretreated with specific PKR inhibitor C13H8N4OS (C16). We found that inhibition of PKR largely abolished LPS-induced phosphorylation of NF-κB p65 and JNK but augmented p38 MAPK activation (Fig. 1C). These results indicate that the effect of p58IPK on TLR4 signaling is likely mediated by regulation of PKR.
Figure 1
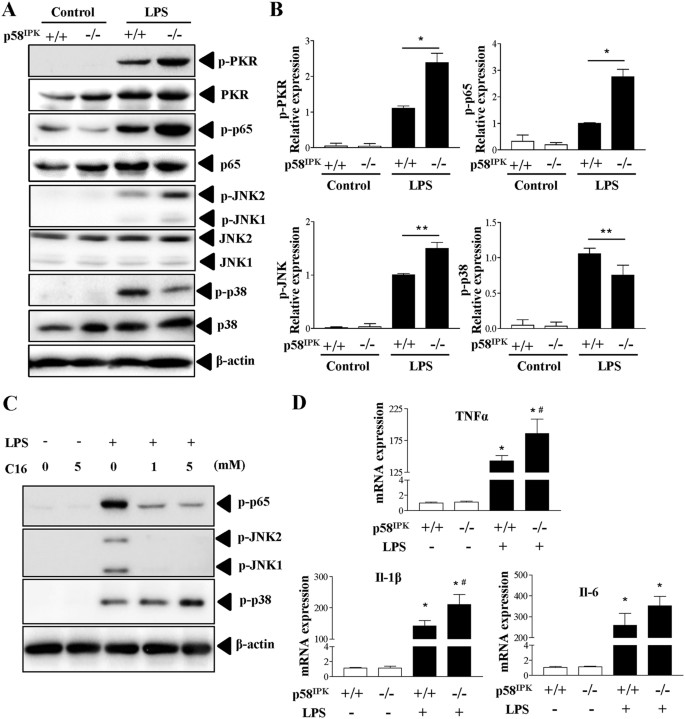
p58IPK regulates LPS-induced activation of PKR, NF-κB and JNK MAPK pathways.
(A,B) BMDM isolated from WT (p58IPK+/+) or p58IPK knockout (p58IPK−/−) mice were cultured and treated with LPS for 15 min. Phosphorylation of PKR, NF-Κb p65, JNK and p38 MAPK was evaluated by western blot analysis and quantified by densitometry. β-actin was used as a loading control. Data represent means ± SD from at least 3 independent experiments. *P < 0.05 and **P < 0.01. (C) BMDM from WT mice were pretreated with specific PKR inhibitor C-16 and then exposed to LPS or vehicle for 15 min. Whole cell lysates were subjected to western blot analysis. (D) BMDM from WT (p58IPK+/+) or p58IPK knockout (p58IPK−/−) mice were treated with LPS for 2 h. mRNA expressions of TNFα, IL-1β and IL-6 were determined by real-time qPCR and normalized by 18S. Data represent means ± SD of 3 independent experiments. *P < 0.05 vs. no LPS treatment, #p < 0.05 vs LPS-treated p58IPK+/+ cells.
Since TLR4 downstream transcription factors NF-κB and AP-1 critically regulate several proinflammatory genes, including TNFα22, IL-1β23 and IL-624, we evaluated the role of p58IPK in regulation of proinflammatory gene expression in LPS-treated macrophages. Compared to WT controls, LPS induced a significantly higher increase in TNFα and IL-1β expression in BMDM of p58IPK−/− (Fig. 1D). In contrast, p58IPK deletion led to a modestly higher increase in IL-6 expression, which, however, did not differ significantly from the WT control.
p58IPK deficiency exacerbates NLRP3 inflammasome activation
Next, we explored the role of p58IPK in the regulation of NLRP3 inflammasome activation in macrophages. To induce inflammasome activation, BMDM were primed with ultra-pure LPS and then stimulated with ATP. Inflammasome activation was evaluated by cleavage of caspase-1 and maturation of IL-1β. Compared to the WT control, p58IPK−/− macrophages show significantly higher levels of cleaved caspase-1 and mature IL-1β (Fig. 2A,B). The levels of pro-caspase-1 and pro-IL-1β as well as NLRP3, did not differ in p58IPK−/− and control macrophages.
Figure 2
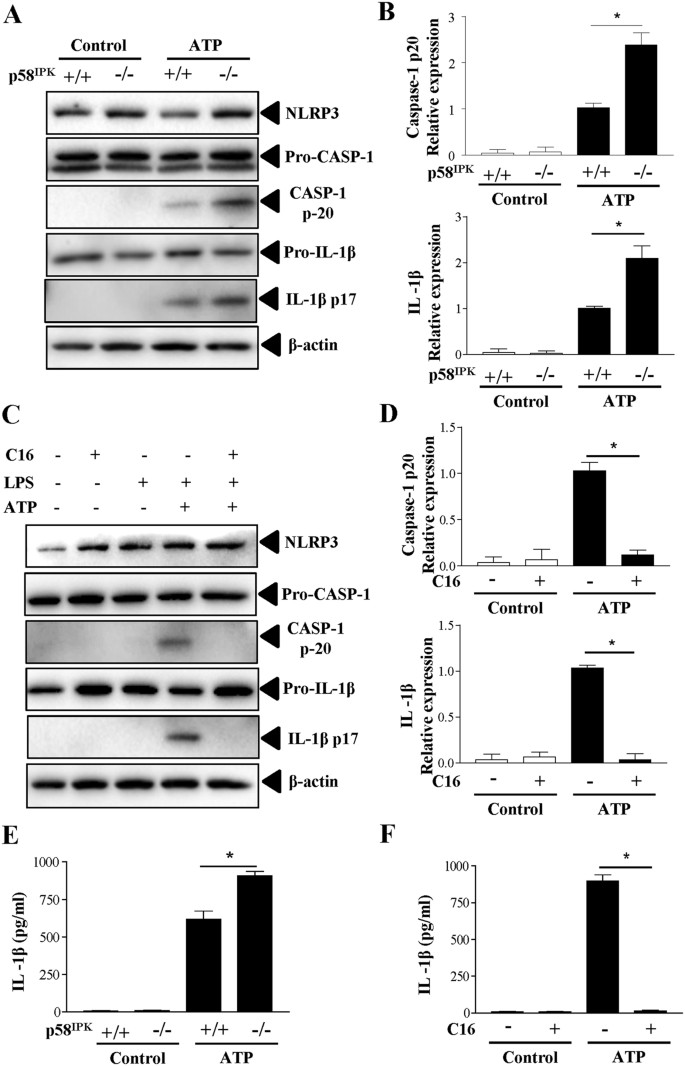
p58IPK deficiency exacerbates NLRP3 inflammasome activation via PKR.
(A,B) BMDM from WT (p58IPK+/+) or p58IPK knockout (p58IPK−/−) mice were primed with LPS (500 ng/ml) for 4 h and then stimulated with ATP (5 mM) for 1 h. Protein levels of NLRP3, pro- and cleaved capase-1 and IL-1β were determined by western blot analysis and quantified by densitometry. Data represent means ± SD of 3 independent experiments. *P < 0.01. (C,D) BMDM from p58IPK knockout (p58IPK−/−) mice were pretreated with C16 and then exposed to LPS and ATP to induce inflammasome activation. Whole cell lysates were assessed by western blot analysis. Protein levels were quantified by densitometry and normalized by β-actin. Data represent means ± SD of at least 3 independent experiments. *P < 0.01. (E,F) Levels of IL-1β secreted into the medium of p58IPK+/+ and p58IPK−/− BMDM cultures were measured by ELISA. Data represent means ± SD of at least 3 independent experiments. *P < 0.01.
PKR activation has been shown to play a crucial role in inflammasome activation18. To elucidate whether increased NLRP3 inflammasome activation in p58IPK−/− macrophages was attributed to enhanced PKR activation, we isolated BMDM from p58IPK−/− mice and pretreated the cells with PKR inhibitor C16 prior to inducing inflammasome activation. We found that inhibition of PKR completely blocked LPS-induced caspase-1 activation and IL-1β maturation (Fig. 2C,D). Furthermore, we measured the level of IL-1β secreted from macrophages into the media. Coincident with the changes in inflammasome activation, IL-1β secretion was significantly increased in p58IPK−/− BMDM and the increase in IL-1β secretion was completely abolished in cells pretreated with C16 (Fig. 2E,F).
Overexpression of p58IPK reduces NLRP3 inflammasome activation and IL-1β secretion
To further evaluate the effect of p58IPK on activation of NLRP3 inflammasomes, we overexpressed p58IPK by adenovirus (Ad-p58IPK) in BMDM isolated from WT mice. Adenovirus expressing β-galactosidase (Ad-LacZ) was used as a control. After 24 h, adenoviral transduced cells were primed with LPS and then stimulated with ATP to activate inflammasomes. As shown in Fig. 3A,B, overexpression of p58IPK significantly reduced caspase-1 activation and IL-1β maturation, suggesting an inhibition in inflammasome activation. Moreover, p58IPK overexpression drastically reduced IL-1β secretion from macrophages (Fig. 3C), further confirming an inhibitory role of p58IPK in inflammasome activation.
Figure 3
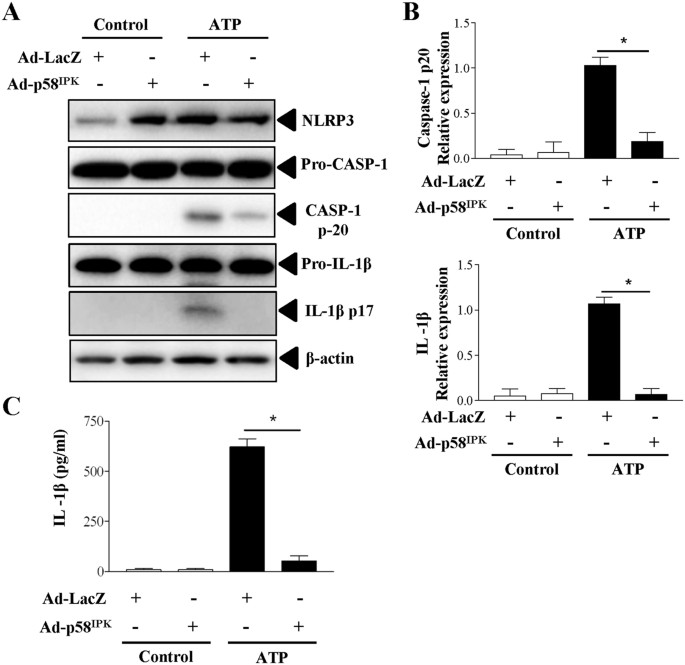
Overexpression of p58IPK reduces inflammasome activation and IL-1β secretion.
(A,B) BMDM from WT mice were transduced with Ad-p58IPK or Ad-LacZ at MOI of 50 for 24 h. Cells were then treated with LPS and ATP to induce inflammasome activation. Whole cell lysates were subjected to western blot analysis. Protein levels were quantified by densitometry and normalized by β-actin. Data represent means ± SD of 3 independent experiments. *P < 0.01. (C) Levels of IL-1β secreted into the medium of BMDM were measured by ELISA. Data represent means ± SD of 3 independent experiments. *P < 0.01.
p58IPK does not alter the adhesion capacity of LPS-activated macrophages to endothelial cells
Macrophage adhesion and interaction with endothelial cells is considered a key event in the pathogenesis of vascular injury in diabetic complications25. We determined whether deficiency of p58IPK results in overactivation of macrophages thereby enhancing macrophage adhesion to endothelial cells. BMDM from p58IPK−/− and WT mice were treated with 250 ng/ml LPS or vehicle for 6 h, labeled with fluorescence dye and resuspended for adhesion assay. As shown in Fig. 4, deficiency of p58IPK did not result in significant change in adhesion of LPS-stimulated BMDM to HUVECs. Non-treated p58IPK−/− macrophages has a tendency to increase adhesion but the difference was not significant.
Figure 4
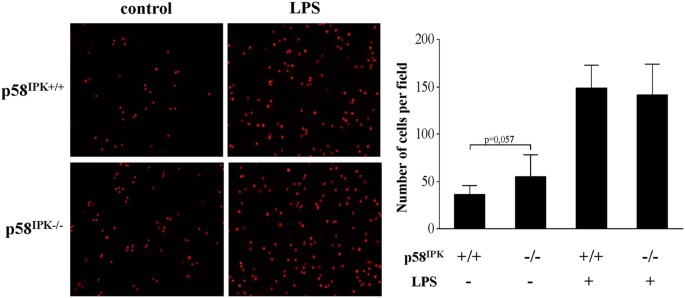
p58IPK deficiency does not alter macrophage adhesion to endothelial cells.
BMDM from WT (p58IPK+/+) or p58IPK knockout (p58IPK−/−) mice were treated with LPS (250 ng/ml) or vehicle for 6 h. Cells were labeled with fluorescent dye PKH26 and then added to HUVECs for adhesion assay. (A) Representative images of showing BMDM (red) adhering to HUVECs. LPS treatment increased BMDM adhesion to endothelial cells. However, no difference was observed in LPS-treated p58IPK−/− macrophages compared to p58IPK+/+ controls. (B) Quantification of adhered BMDM to HUVECs.
p58IPK interacts with PKR in macrophages
To determine whether p58IPK interacts with PKR and TLR4 downstream signaling molecules in macrophages, we performed immunoprecipitation to explore protein binding in BMDM treated with LPS for 15 min when significant activation of TLR4 signaling molecules was observed, or in cells with inflammasome activation. We found that p58IPK was successfully coimmunoprecipitated with p-PKR in both treatment groups (Fig. 5A). In contrast, we did not observe association of p58IPK with other signaling molecules including p-NF-κB p65, p-p38 and p-JNK (Fig. 5B). These results further confirmed that the inhibitory effect of p58IPK on TLR4 signaling and inflammasome activation is mediated by its regulation of PKR.
Figure 5
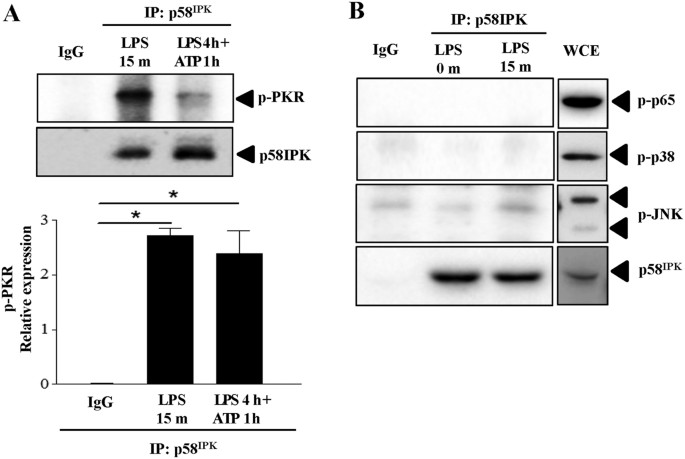
Interaction between p58IPK and PKR in BMDM.
(A) Mouse BMDM were treated as indicated and harvested. Cell lysate was subjected to immunoprecipitation using antibody against p58IPK or non-immunized serum (IgG) as a negative control, followed by western blot analysis. Relative protein levels of PKR were determined by densitometry. Data represent means ± SD of 3 independent experiments. (B) No positive binding of p58IPK with NF-κB p65, p38 or JNK was revealed in BMDM treated with LPS for 15 min, but strong activation of each molecule was observed in the whole cell extract (WCE) from cells under the same treatment. The gels were run under the same experimental conditions and full-length blots are presented in Supplementary data.
Discussion
p58IPK is a key regulator of protein translation8,9,10 and protein folding11 depending on its subcellular localization26. In addition, p58IPK suppresses the activity of PKR and regulates the host innate defense response during viral infection27. Although the function of p58IPK in protecting against cell death is well characterized, its role in macrophage-derived proinflammatory cytokine production and inflammatory response remain elusive. In the present study we investigated the potential role of p58IPK in the regulation of pro-inflammatory gene expression and inflammasome activation in macrophages. Our results provide strong evidence that p58IPK functions as a potent inhibitor of inflammasome activation and IL-1β production in macrophages. Furthermore, we demonstrate that p58IPK suppresses TLR4 downstream signaling pathways of NF-κB and JNK and these effects are mediated by inhibition of PKR (Fig. 6).
Figure 6
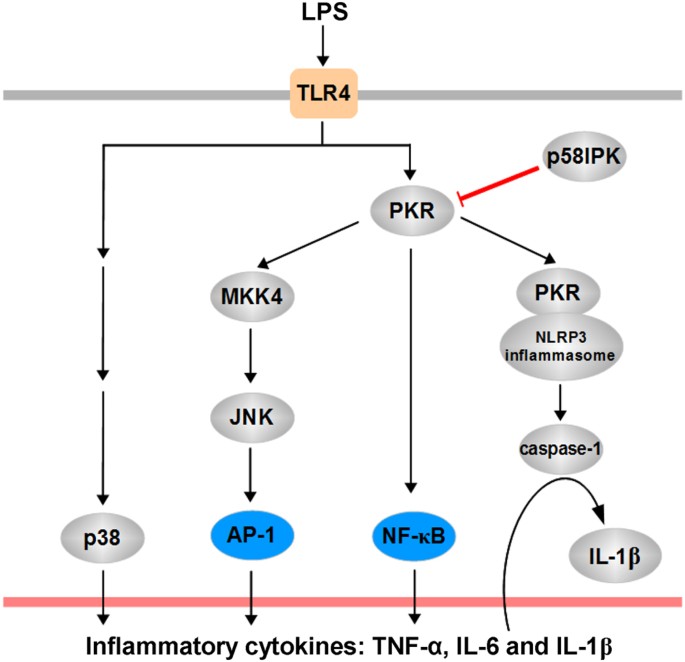
Schematic summary of the role of p58IPK in regulation PKR-dependent signaling and proinflammatory cytokine production.
PKR is a well-established regulator of innate immune response to viral infection playing an important role in host cell survival and viral amplification. Recently activation of PKR has been linked to chronic inflammation pertinent to metabolic disorders such as diabetes28,29. Interestingly, the role of PKR in inflammatory signaling pathways appears to vary in different cell types according to prior studies20,21,30. In mouse embryonic fibroblasts, PKR regulates the activity of p38 and JNK MAPKs in response to LPS and pro-inflammatory cytokines20. In contrast, a study using BMDM reported that the PKR is dispensable for activation of p38 and JNK MAPKs or activation of NF-κB30. Yet, recent work by Cabanski and colleagues shows that in alveolar macrophages PKR is critical for the activation of NF-κB and JNK, but not p38 MAPK, pathways21. Using BMDM isolated from p58IPK knockout mice and pharmacological PKR inhibitor, we demonstrated that deletion of p58IPK augments PKR, NF-κB p65 and JNK MAPK activation, but reduces p38 activation, while inhibition of PKR by specific pharmacological inhibitor largely abolished NF-κB p65 and JNK, but not p38 MAPK activation. These results support that manipulating PKR by either endogenous or exogenous inhibitors differentially regulates p38 and JNK MAPKs and NF-κB p65 activation in macrophages. Consistent with these results, we found that deficiency of p58IPK exacerbated LPS-induced expression of TNF-α and IL-1β, but did not alter the induction of IL-6 in macrophages. Future investigation is needed to evaluate the effect of p58IPK on TNF-α and IL-6 protein production and secretion from macrophages.
In addition to regulation of canonical inflammatory pathways, recent studies provide compelling evidence that supports a new and potential role of PKR in inflammasome activation31. Initial work by Lu and associates18 suggests that PKR physically interacts with inflammasome components including NLRP3, NLRP4 and absent in melanoma 2 (AIM2). Furthermore, autophosphorylation of PKR in a cell-free system with recombinant NLRP3, ASC and pro-caspase-1 is sufficient to reconstitute inflammasome activity, suggesting that PKR is a potent inducer of inflammation activation. Thus, identifying the endogenous inhibitors for PKR-mediated inflammasome activation could lead to new treatment approaches to reduce inflammation and mitigate inflammasome-related cell death. Our major findings in this study have identified p58IPK as a strong inhibitor of PKR signaling and inflammasome activation. Deletion of p58IPK significantly intensified LPS-induced inflammasome activation and IL-1β secretion, which was completely eliminated by pharmacological inhibition of PKR. Furthermore, we show that overexpression of p58IPK significantly reduced IL-1β maturation and secretion. Interestingly, we found that the adhesion capacity of LPS-activated macrophages was not affected by p58IPK deficiency, suggesting a relative specific role of p58IPK on inflammasome activation and IL-1β secretion. Given the important role of IL-1β in broad inflammatory diseases, manipulating p58IPK may serve as a new therapeutic target in controlling signaling pathways of IL-1β production and inflammation.
In summary, our study demonstrates an important role of p58IPK in regulation of PKR-mediated TLR4 downstream NF-κB and JNK MAPK pathways, inflammasome activation and IL-1β secretion from macrophages. Therefore, enhancing the function of p58IPK could lead to a new approach to prevent inflammasome and IL-1β-related tissue injury and pathological consequences such as neurodegeneration.
Methods
Materials
Ultrapure LPS was purchased from Sigma Laboratories (St. Louis, MO, USA). NLRP3 inflammasome agonist ATP was obtained from InvivoGen (San Diago, CA, USA). Fetal bovine serum, Dulbecco’s modified eagle’s medium (DMEM), 1 × MEM on-essential amino acids, 1 × MEM vitamins and sodium bicarbonate were obtained from Gibco laboratories (Grand Island, NY, USA). RIPA buffer, inhibitor mixture, PMSF and sodium orthovanadate were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Mice
p58IPK knockout (KO) mice were kindly provided by Dr. Michael G. Katze (University of Washington). All methods were carried out in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. All experimental protocols were approved by the Institutional Animal Care and Use Committees at the University at Buffalo, State University of New York.
Isolation, culture and adenoviral transduction of mouse bone marrow derived macrophages (BMDM)
Mouse BMDM were isolated from bone marrow and cultured as described previously32,33. To activate TLR4 signaling and induce inflammatory gene expression, cells were treated with LPS (250 ng/ml) for up to 24 h. To induce inflammasome activation, cells were primed with LPS (500 ng/ml) for 4 h and then stimulated with ATP (5 mM) for 1 h18. In experiments with overexpression of p58IPK, BMDM were transduced with adenoviruses expressing p58IPK (Ad-p58IPK) or β-galactosidase (Ad-LacZ) as control at a multiplicity of infection (MOI) of 50 as described previously34. Twenty-four hours after transduction, cells were treated LPS and ATP to induce inflammasome activation.
Western blot analysis
Cells were lysed and sonicated in radioimmunoprecipitation (RIPA) buffer with protease inhibitor mixture, PMSF and sodium orthovanadate. Protein concentration was measured by BCA protein assay (Thermo scientific, Rockford, IL, USA). The samples were resolved by SDS-PAGE and transferred to nitrocellulose membrane and blotted with specific antibodies: anti-p58IPK, anti-p-NF-κB, anti-NF-κB, anti-p-p38, anti-p38, anti-p-JNK, anti-JNK, (Cell Signaling Technology, Boston, MA, USA), anti-p-PKR, anti-PKR, anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-IL-1β (R&D Systems, Inc., Minneapolis, MN, USA), anti-caspase-1 (p-20) and anti-NLRP3 (AdipoGen, Inc, San Diego, CA, USA). The immunoblots were developed using chemiluminescence (SuperSignal West Dura Extended Duration Substrate, Thermo Scientific, USA) and visualized under Chemidoc MP Imaging System (Bio-Rad, Hercules, CA, USA).
Quantitative Real-time RT–PCR
Total RNA was extracted using an E.Z.N.A. total RNA kit I (Omega bio-tek, Georgia, GA) following the manufacturer’s instructions. The quantity of total RNA was determined by spectrophotometry using the Synergy HT BioTek (Winooski, Vermont, USA). A Maxima First Strand cDNA synthesis kit (Thermo Scientific, Grand Island, NY) was used for cDNA synthesis. Real-time RT-PCR was performed using SYBR Green PCR Master Mix (Bio-Rad Laboratories, Hercules, CA). 18S ribosomal RNA was chosen as endogenous control to normalize quantification of target genes. The primer sequences are as follows: mouse TNFα (forward, CGG TGCCTATGTCTCAGCCT; reverse, TTGGGCAGATTGACCTCAGC); mouse IL-1β (forward, CAGGCAGGCAGTATCACTCA; reverse, GAGGATGGGCTCTTCTTCAA); mouse IL-6 (forward, AGTCCGGAGAGGAGACTTCA; reverse, TTGCCATTGCACAACTCTTT).
IL-1β ELISA
The protein levels of IL-1β in cell culture supernatants were measured using IL-1β ELISA kits (R&D Systems) according to the manufacturer’s instructions.
Immunoprecipitation
Antibodies against the p58IPK were used to precipitate proteins from cell lysate in the presence of 20 μl protein A/G beads (Santa Cruz) overnight at 4 °C. Protein complexes were washed five times with RIPA buffer and then incubated at 95 °C for 3–5 minutes and resolved by western blotting.
Macrophage adhesion assay
Macrophage adhesion assay was carried out using human umbilical vein endothelial cells (HUVEC) and primary mouse BMDM. Briefly, HUVECs were cultured to confluence. BMDM were isolated and cultured for 8 days. Before adhesion assay, BMDM were washed three times with serum-free RPMI medium 1640 and labeled with dye PKH26 (Sigma Aldrich). Approximately 1 ml of 20,000 BMDM were incubated with HUVEC for 20 min. Unadhered cells were then washed out three times with serum-free RPMI medium 1640. The adherent cells were counted in 6–8 randomly selected optical fields in each well as described previously25. Fluorescence microphotographs of the cells were taken under an Olympus microscope (Tokyo, Japan).
Statistical analysis
The quantitative data were expressed as mean ± SD. Statistical analyses were performed using an unpaired student’s t-test when comparing two groups and one-way analysis of variance (ANOVA) test for three groups and more. Statistical differences were considered significant at a p value of less than 0.05.
Additional Information
How to cite this article: Boriushkin, E. et al. p58IPK suppresses NLRP3 inflammasome activation and IL-1β production via inhibition of PKR in macrophages. Sci. Rep. 6, 25013; doi: 10.1038/srep25013 (2016).
References
- Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820, 10.1016/j.cell.2010.01.022 (2010).
Article CAS PubMed Google Scholar - Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832, 10.1016/j.cell.2010.01.040 (2010).
Article CAS PubMed Google Scholar - Federici, S., Martini, A. & Gattorno, M. The central role of anti-IL-1 blockade in the treatment of monogenic and multifactorial autoinflammatory diseases. Front. Immunol. 4, 10.3389/fimmu.2013.00351 (2013).
- Wen, H. et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415, 10.1038/ni.2022 (2011).
Article CAS PubMed Google Scholar - Hewett, S. J., Jackman, N. A. & Claycomb, R. J. Interleukin-1β in Central Nervous System Injury and Repair. Eur. J. Neurodegener. Dis. 1, 195–211 (2012).
PubMed Google Scholar - Larsen, C. M. et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 356, 1517–1526, 10.1056/NEJMoa065213 (2007).
Article CAS PubMed Google Scholar - Mandrup-Poulsen, T., Pickersgill, L. & Donath, M. Y. Blockade of interleukin 1 in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 6, 158–166, 10.1038/nrendo.2009.271 (2010).
Article CAS PubMed Google Scholar - Melville, M. W., Hansen, W. J., Freeman, B. C., Welch, W. J. & Katze, M. G. The molecular chaperone hsp40 regulates the activity of P58IPK, the cellular inhibitor of PKR. Proc. Natl. Acad. Sci. USA 94, 97–102 (1997).
Article CAS ADS PubMed Google Scholar - Polyak, S. J., Tang, N., Wambach, M., Barber, G. N. & Katze, M. G. The P58 cellular inhibitor complexes with the interferon-induced, double-stranded RNA-dependent protein kinase, PKR, to regulate its autophosphorylation and activity. J. Biol. Chem. 271, 1702–1707 (1996).
Article CAS PubMed Google Scholar - Tang, N. M., Ho, C. Y. & Katze, M. G. The 58-kDa cellular inhibitor of the double stranded RNA-dependent protein kinase requires the tetratricopeptide repeat 6 and DnaJ motifs to stimulate protein synthesis in vivo. J. Biol. Chem. 271, 28660–28666 (1996).
Article CAS PubMed Google Scholar - Rutkowski, D. T. et al. The role of p58IPK in protecting the stressed endoplasmic reticulum. Mol. Biol. Cell. 18, 3681–3691, 10.1091/mbc.E07-03-0272 (2007).
Article CAS PubMed Google Scholar - Samuel, C. E. The eIF-2 alpha protein kinases, regulators of translation in eukaryotes from yeasts to humans. J. Biol. Chem. 268, 7603–7606 (1993).
CAS PubMed Google Scholar - Takada, Y., Ichikawa, H., Pataer, A., Swisher, S. & Aggarwal, B. B. Genetic deletion of PKR abrogates TNF-induced activation of IkappaBalpha kinase, JNK, Akt and cell proliferation but potentiates p44/p42 MAPK and p38 MAPK activation. Oncogene 26, 1201–1212, 10.1038/sj.onc.1209906 (2007).
Article CAS PubMed Google Scholar - Bonnet, M. C., Weil, R., Dam, E., Hovanessian, A. G. & Meurs, E. F. PKR stimulates NF-kappaB irrespective of its kinase function by interacting with the IkappaB kinase complex. Mol. Cell. Biol. 20, 4532–4542 (2000).
Article CAS PubMed PubMed Central Google Scholar - Zamanian-Daryoush, M., Mogensen, T. H., DiDonato, J. A. & Williams, B. R. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol. Cell. Biol. 20, 1278–1290 (2000).
Article CAS PubMed PubMed Central Google Scholar - Zhang, P. & Samuel, C. E. Induction of protein kinase PKR-dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter IPS-1 signaling. J. Biol. Chem. 283, 34580–34587, 10.1074/jbc.M807029200 (2008).
Article CAS PubMed PubMed Central Google Scholar - McAllister, C. S. et al. Mechanisms of protein kinase PKR-mediated amplification of beta interferon induction by C protein-deficient measles virus. J. Virol. 84, 380–386, 10.1128/JVI.02630-08 (2010).
Article CAS PubMed Google Scholar - Lu, B. et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 488, 670–674, 10.1038/nature11290 (2012).
Article CAS ADS PubMed PubMed Central Google Scholar - He, Y., Franchi, L. & Núñez, G. The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur. J. Immunol. 43, 1147–1152, 10.1002/eji.201243187 (2013).
Article CAS PubMed PubMed Central Google Scholar - Goh, K. C., deVeer, M. J. & Williams, B. R. G. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J. 19, 4292–4297, 10.1093/emboj/19.16.4292 (2000).
Article CAS PubMed Google Scholar - Cabanski, M. et al. PKR Regulates TLR2/TLR4-Dependent Signaling in Murine Alveolar Macrophages. Am. J. Respir. Cell Mol. Biol. 38, 26–31, 10.1165/rcmb.2007-0010OC (2008).
Article CAS PubMed Google Scholar - Kishore, R., McMullen, M. R., Cocuzzi, E. & Nagy, L. E. Lipopolysaccharide-mediated signal transduction: Stabilization of TNF-alpha mRNA contributes to increased lipopolysaccharide-stimulated TNF-alpha production by Kupffer cells after chronic ethanol feeding. Comp. Hepatol. 3 Suppl 1, S31, 10.1186/1476-5926-2-S1-S31 (2004).
Article PubMed Google Scholar - Hiscott, J. et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol. Cell. Biol. 13, 6231–6240 (1993).
Article CAS PubMed Google Scholar - Libermann, T. A. & Baltimore, D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol. Cell. Biol. 10, 2327–2334 (1990).
Article CAS PubMed Google Scholar - Huang, H., Jing, G., Wang, J., Sheibani, N. & Zhang, S. ATF4 is a novel regulator of MCP-1 in microvascular endothelial cells. J. Inflamm. 12, 31 (2015).
Article Google Scholar - Roobol, A. et al. p58IPK is an inhibitor of the eIF2alpha kinase GCN2 and its localization and expression underpin protein synthesis and ER processing capacity. Biochem. J. 465, 213–225, 10.1042/BJ20140852 (2015).
Article CAS PubMed Google Scholar - Goodman, A. G. et al. P58IPK: A Novel “CIHD” Member of the Host Innate Defense Response against Pathogenic Virus Infection. PLoS Pathog. 5, e1000438, 10.1371/journal.ppat.1000438 (2009).
Article CAS PubMed Google Scholar - Nakamura, T. et al. Double-Stranded RNA-Dependent Protein Kinase Links Pathogen Sensing with Stress and Metabolic Homeostasis. Cell 140, 338–348, 10.1016/j.cell.2010.01.001 (2010).
Article CAS PubMed Google Scholar - Nakamura, T., Arduini, A., Baccaro, B., Furuhashi, M. & Hotamisligil, G. S. Small-Molecule Inhibitors of PKR Improve Glucose Homeostasis in Obese Diabetic Mice. Diabetes 63, 526–534, 10.2337/db13-1019 (2014).
Article CAS PubMed Google Scholar - Hsu, L.-C. et al. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature 428, 341–345 (2004).
Article CAS ADS PubMed Google Scholar - Yim, H. C. H. & Williams, B. R. G. Protein Kinase R and the Inflammasome. Interferon Cytokine Res. 34, 447–454, 10.1089/jir.2014.0008 (2014).
Article CAS Google Scholar - Li, J. et al. Macrophage Metalloelastase (MMP-12) Deficiency Mitigates Retinal Inflammation and Pathological Angiogenesis in Ischemic Retinopathy. PLoS ONE 7, e52699, 10.1371/journal.pone.0052699 (2012).
Article CAS ADS PubMed Google Scholar - Zhang, X., Goncalves, R. & Mosser, D. M. The Isolation and Characterization of Murine Macrophages. Curr. Protoc. Immunol, 10.1002/0471142735.im1401s83 (2008).
- Boriushkin, E. et al. Identification of p58IPK as a Novel Neuroprotective Factor for Retinal Neurons. Invest. Ophthalmol. Vis. Sci. 56, 1374–86, 10.1167/iovs.1114-15196 (2015).
Article CAS PubMed PubMed Central Google Scholar
Acknowledgements
We thank Dr. Michael Katze (University of Washington, Seattle, WA) for kindly providing P58IPK knockout mice. This work is supported, in part, by NIH/NEI grants EY019949 and EY025061, by a grant from American Diabetes Association and by an Unrestricted Grant to the Department of Ophthalmology, SUNY-Buffalo, from Research to Prevent Blindness.
Author information
Authors and Affiliations
- Department of Ophthalmology/Ross Eye Institute, University at Buffalo, the State University of New York, Buffalo, 14214, NY, USA
Evgenii Boriushkin, Joshua J. Wang, Junhua Li, Maulasri Bhatta & Sarah X. Zhang - SUNY Eye Institute, the State University of New York, Buffalo, 14214, NY, USA
Evgenii Boriushkin, Joshua J. Wang, Junhua Li, Maulasri Bhatta & Sarah X. Zhang - Department of Biochemistry, University at Buffalo, the State University of New York, Buffalo, 14214, NY, USA
Sarah X. Zhang
Authors
- Evgenii Boriushkin
You can also search for this author inPubMed Google Scholar - Joshua J. Wang
You can also search for this author inPubMed Google Scholar - Junhua Li
You can also search for this author inPubMed Google Scholar - Maulasri Bhatta
You can also search for this author inPubMed Google Scholar - Sarah X. Zhang
You can also search for this author inPubMed Google Scholar
Contributions
S.X.Z., J.J.W. and E.B. Conceived the experiments; E.B., J.J.W., J.L. and M.B. performed the experiments; S.X.Z., E.B., J.J.W., J.L. and M.B. analysed the data; E.B., J.J.W. and S.X.Z. wrote and revised the manuscript. All authors reviewed and approved the final version.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article
Cite this article
Boriushkin, E., Wang, J., Li, J. et al. p58IPK suppresses NLRP3 inflammasome activation and IL-1β production via inhibition of PKR in macrophages.Sci Rep 6, 25013 (2016). https://doi.org/10.1038/srep25013
- Received: 18 December 2015
- Accepted: 04 April 2016
- Published: 26 April 2016
- DOI: https://doi.org/10.1038/srep25013