FGFR1 is critical for the anti-endothelial mesenchymal transition effect of N-acetyl-seryl-aspartyl-lysyl-proline via induction of the MAP4K4 pathway (original) (raw)
Main
Endothelial-to-mesenchymal transition (EndMT) contributes to the appearance of fibroblasts and has a critical role in organ fibrosis.1, 2 In fact, EndMT is involved in various pathological processes, including kidney fibrosis,3 cardiac fibrosis4 and cancer.5 During EndMT, endothelial cells lose their cell-to-cell adhesion properties and acquire invasive and migratory properties.6 EndMT is characterized by increased expression of mesenchymal markers, such as vimentin, smooth muscle α_-actin (α_-SMA), smooth muscle protein 22_α (SM22_α) and fibroblast specific protein 1 (FSP1, also known as S100A4),7, 8, 9, 10 and decreased expression of endothelial cell markers, such as VE-cadherin and CD31 (also known as platelet endothelial cell adhesion molecule-1).11, 12 Accumulating evidence demonstrates that transforming growth factor β (TGF_β_)/smad signaling is a major inducer of EndMT, and proteins in the non-TGF_β_ pathway, such as _β_-catenin and Notch, are also involved in EndMT induction.13, 14
N_-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP) is a tetrapeptide that is enzymatically cleaved by prolyl oligopeptidase (POP) from the N-terminal sequence of thymosin_β_4 (T_β_4) and hydrolyzed by angiotensin-converting enzyme.15, 16 Furthermore, AcSDKP has been proven to have an antifibrotic effect in various organ fibrosis models.17, 18, 19, 20 Long-term treatment with AcSDKP suppresses renal fibrosis by inhibiting the TGF_β/smad signaling pathway.21 In the kidneys of diabetic mice, the decreased fibroblast growth factor receptor (FGFR) level was restored by AcSDKP treatment,22 and fibroblast growth factors (FGFs) have a key role in the normal state of vascularity.23, 24 FGF2/FGFR1 signaling is associated with the induction of the miRNA let-7, ameliorating the vascular disease state through the suppression of TGF_β_/TGF_β_R1-induced EndMT.25 Furthermore, FGFR1 is a key inhibitor of TGF_β_/smad signaling in endothelial cells.26 However, the molecular interplay between AcSDKP and FGFR1 signaling in endothelial cells is still unclear.
Mitogen-activated protein kinase kinase kinase kinase 4 (MAP4K4) belongs to the Ste20 family of kinases, which have been shown to affect various biological processes, such as embryonic development and inflammation. MAP4K4 is associated with multiple molecular pathways, including integrin signaling and the mammalian target of rapamycin (mTOR).27 In endothelial cells, MAP4K4 inhibits integrin β_1 activity by inducing moesin.28 Furthermore, integrin β_1 induces the epithelial–mesenchymal transition (EMT) and organ fibrosis via the activation of TGF_β/smad signaling.29, 30, 31 We have shown that the interaction between dipeptidyl peptidase 4 (DPP-4) and integrin β_1 induces EndMT by activating TGF_β/smad signaling.32 TGF_β/smad signaling is also inhibited by Misshapen (msn) kinases (the mammalian orthologs of MAP4K4).33
Here, we hypothesize that the AcSDKP–FGFR1–MAP4K4 axis has a key role in the suppression of TGF_β_/smad signaling and EndMT in endothelial cells.
Results
The proximity of AcSDKP and FGFR1 is essential for AcSDKP-mediated inhibition of TGF_β_/smad signaling pathway in endothelial cells
A duolink in situ proximity ligation assay (PLA) was performed to analyze the proximity between AcSDKP and FGFR1 in cultured human dermal microvascular endothelial cell (HMVEC) (Figure 1a). AcSDKP and FGFR1 displayed close proximity in normal cultured HMVECs (Figure 1a), suggesting that endogenous AcSDKP interacts with FGFR1. Exogenous AcSDKP incubation in endothelial cells further increased the close proximity between AcSDKP and FGFR1 (Figure 1a). In addition, in the presence of a neutralizing FGFR1 antibody (N-FGFR1), which suppresses FGFR1 activity in endothelial cells, the close proximity between AcSDKP and FGFR1 was markedly diminished in HMVECs, even with AcSDKP incubation (Figure 1a). We previously reported that the anti-TGF_β_1/smad and anti-kidney fibrosis effects of AcSDKP were associated with the restoration of FGFR phosphorylation (P-FGFR) and FGFR levels.22, 34 We also confirmed that AcSDKP significantly inhibited TGF_β_2-induced smad3 phosphorylation (p-smad3), p-smad2, TGF_β_R1 and TGF_β_R2 levels, and restored TGF_β_2-suppressed FGFR1 in endothelial cells (Figures 1b and c and Supplementary Figure S1a). Interferon-γ (IFN-γ) has been confirmed to inhibit FGFR1 protein levels,25 and treatment of HMVECs with AcSDKP was associated with increased FGFR1/P-FGFR1 levels and decreased expression of IFN-γ (Supplementary Figure S1b). Furthermore, T_β_4 restored TGF_β_2-suppressed FGFR1 levels, and a POP inhibitor (KYP-2047) abolished the effect of T_β_4 (Supplementary Figure S2a). The bone morphogenetic protein (BMP) pathway has been associated with EndMT induction.35 We investigated whether AcSDKP inhibits EndMT by suppressing the BMP pathway, and AcSDKP did not inhibit the BMP-2/BMP-4-induced expression of p-smad1/5/8 in endothelial cells (Supplementary Figure S2b). To further elucidate the significance of AcSDKP on FGFR1 levels and its functional relevance, three AcSDKP mutant peptides, AcSDKA, AcDSPK and AcADKP, were constructed. Western blot analysis revealed that the AcSDKP mutants had no effect on the levels of TGF_β_2-induced p-smad3, TGF_β_R1 and TGF_β_R2 and suppressed FGFR1 in endothelial cells (Figures 1d and e).
Figure 1
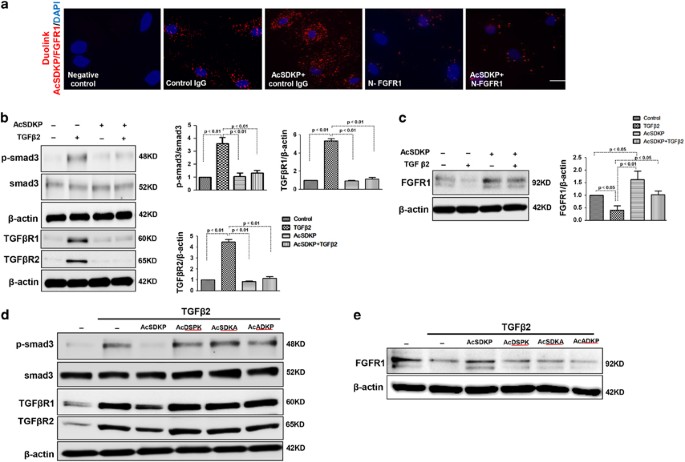
Proximity between AcSDKP and FGFR1 inhibits the TGF_β_/smad signaling pathway in HMVECs. (a) HMVECs were treated with N-FGFR1 (1.5 _μ_g/ml) for 48 h with or without preincubation with AcSDKP (100 nM) for 2 h, and the proximity between AcSDKP and FGFR1 was analyzed by the Duolink In Situ Assay. For each slide, images at a × 400 original magnification were obtained from six different areas. (b and c) HMVECs were treated with TGF_β_2 (5 ng/ml) for 15 min or 48 h with or without preincubation with AcSDKP for 2 h, and the p-smad3, TGF_β_R1, TGF_β_R2 and FGFR1 levels were analyzed by western blot. Densitometric analysis of the p-smad3/smad3, TGF_β_R1/_β_-actin, TGF_β_R2/_β_-actin and FGFR1/_β_-actin levels from each group (_n_=6) were analyzed. (d and e) HMVECs were incubated with TGF_β_2 for 15 min or 48 h with or without preincubation with AcSDKP or its mutants (AcDSPK, AcSDKA, AcADKP) (100 nM) for 2 h. The p-smad3/smad3, TGF_β_R1/_β_-actin, TGF_β_R2/_β_-actin and FGFR1/_β_-actin protein levels were analyzed by western blot
AcSDKP suppresses TGF_β_/smad signaling and EndMT through the FGFR1 pathway
N-FGFR1 suppressed FGFR1 and induced both TGF_β_R1 and TGF_β_R2 in cultured endothelial cells (Figure 2a). Treating the cultured cells with N-FGFR1 increased the p-smad3 and TGF_β_R1 protein levels, especially when N-FGFR1 and TGF_β_2 were incubated together (Figure 2b). However, in the presence of N-FGFR1, increased p-smad3 and TGF_β_R1 levels were not suppressed by AcSDKP in endothelial cells (Figure 2b), suggesting that AcSDKP-suppressed TGF_β_/smad signaling is dependent on the FGFR1 pathway. Nevertheless, AcSDKP did not bind TGF_β_R1 or TGF_β_R2 in endothelial cells (Supplementary Figure S3), likely indicating that there is no distinct interaction between AcSDKP and TGF_β_Rs. FGF2/FGFR1 has been shown to inhibit EndMT induction.25, 26 N-FGFR1 treatment in endothelial cells induced EndMT, as revealed by the decreased CD31 levels and the increased SM22_α_, FSP1 and α_-SMA, and was associated with TGF_β_R1 and TGF_β_R2 levels, especially when N-FGFR1 and TGF_β_2 were applied together (Figure 2c and Supplementary Figures S4a and b). In addition, the N-FGFR1-induced EndMT was not reversed by AcSDKP or FGF2 treatment (Figure 2c and Supplementary Figures S4a and b). Knocking down fibroblast growth factor substrate 2 (FRS2), a key adaptor of FGFR1,36 using a specific siRNA also induced p-smad3 expression and EndMT (Figure 2d). However, AcSDKP did not suppress FRS2 siRNA-induced p-smad3 and EndMT in endothelial cells (Figure 2d). In addition, a neutralizing TGF_β (1-2-3) antibody (N-TGF_β_) completely abolished the N-FGFR1-induced TGF_β_/smad signaling and EndMT (Figure 2e). These findings suggest that the anti-TGF_β_/smad and anti-EndMT effects of AcSDKP are dependent on the FGFR1/FRS2 signaling pathway.
Figure 2
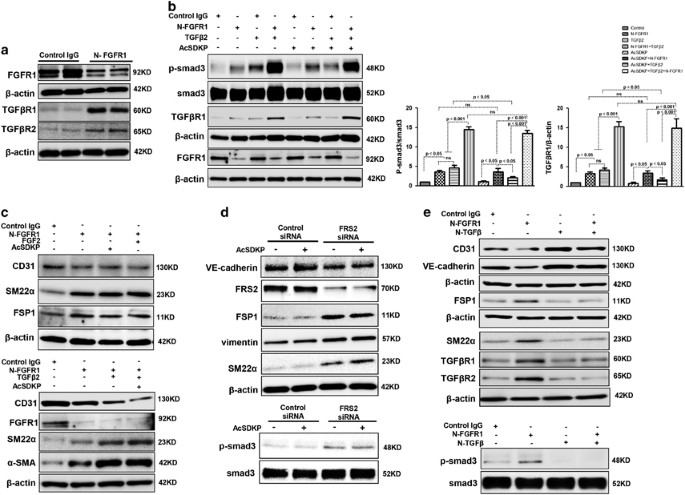
AcSDKP suppresses TGF_β_/smad signaling and EndMT through the FGFR1/FRS2 pathway. (a) HMVECs were treated with N-FGFR1 for 48 h, and the FGFR1, TGF_β_R1 and TGF_β_R2 protein levels were analyzed by western blot. (b) HMVECs were treated with TGF_β_2 in the presence or absence of N-FGFR1 for 15 min with or without AcSDKP preincubation. The p-smad3 and TGF_β_R1 protein levels were analyzed by western blot. Densitometric analysis of the p-smad3/smad3 and TGF_β_R1/β_-actin levels (n_=3) in each group was performed. (c) HMVECs were incubated with either N-FGFR1 in the presence or absence of TGF_β_2 for 48 h with or without preincubation with AcSDKP for 2 h or with N-FGFR1 in the presence or absence of TGF_β_2 for 48 h with or without 24 h of incubation with FGF2 (50 ng/ml). The CD31, SM22_α, FSP1 and α_-SMA protein levels were analyzed by western blot. (d) HMVECs were transfected with FRS2 siRNA (100 nM) for 48 h with or without AcSDKP preincubation. The VE-cadherin, FSP1, vimentin, SM22_α and p-smad3 levels were analyzed by western blot. (e) HMVECs were treated with N-FGFR1 for 48 h or 15 min in the presence or absence of N-TGF_β (1, 2, 3) (1.0 μ_g/ml). The CD31, VE-cadherin, SM22_α, FSP1, TGF_β_R1, TGF_β_R2 and p-smad3 levels were analyzed by western blot
FGF2/FGFR1 regulates MAP4K4 signaling in endothelial cells
We next investigated the mechanism responsible for decreasing the expression of phosphorylated MAP4K4 (P-MAP4K4) after the FGFR1 pathway was shut down. Immunofluorescence staining revealed that the siRNA-mediated knockdown of FRS2 in endothelial cells significantly reduced the expression of P-MAP4K4 compared with that of the control group (Figure 3a), and TGF_β_2-treated cells also showed a similar result (Figure 3a). Western blot analysis was performed to confirm these findings; the expression of P-MAP4K4 in endothelial cells was suppressed by N-FGFR1 and activated by FGF2 (Figures 3b and c). The close proximity between FGFR1 and P-MAP4K4 was observed in normal cultured endothelial cells (Figure 4a), and this close proximity was significantly decreased in the N-FGFR1-treated cells (Figure 4a); TGF_β_2 treatment also produced comparable results (Figure 4a). Furthermore, PF-06260933, a potent MAP4K4 inhibitor, significantly suppressed the proximity between FGFR1 and MAP4K4 in endothelial cells (Supplementary Figure S5), suggesting that MAP4K4 phosphorylation is essential for the proximity between FGFR1 and MAP4K4. Immunoprecipitation assays revealed a physical interaction between FGFR1 and P-MAP4K4, and N-FGFR1 decreased this interaction in endothelial cells (Figure 4b).
Figure 3
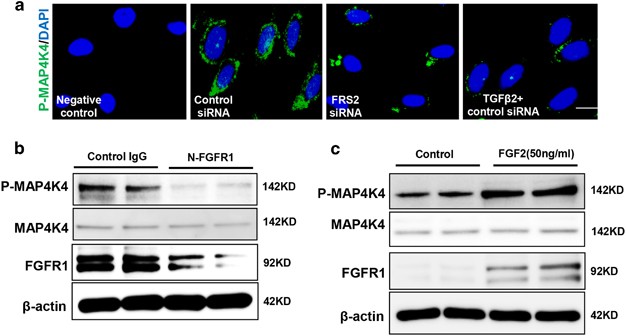
FGF2/FGFR1 mediates MAP4K4 signaling in endothelial cells. (a) Immunofluorescence microscopy analysis of P-MAP4K4 expression following FRS2 siRNA or TGF_β_2 treatment. For each slide, images of six different fields of view at × 400 magnification were evaluated. The scale bar is 60 _μ_m in each panel. (b and c) HMVECs were treated with N-FGFR1 for 48 h or FGF2 for 24 h. The P-MAP4K4 levels were analyzed by western blot
Figure 4
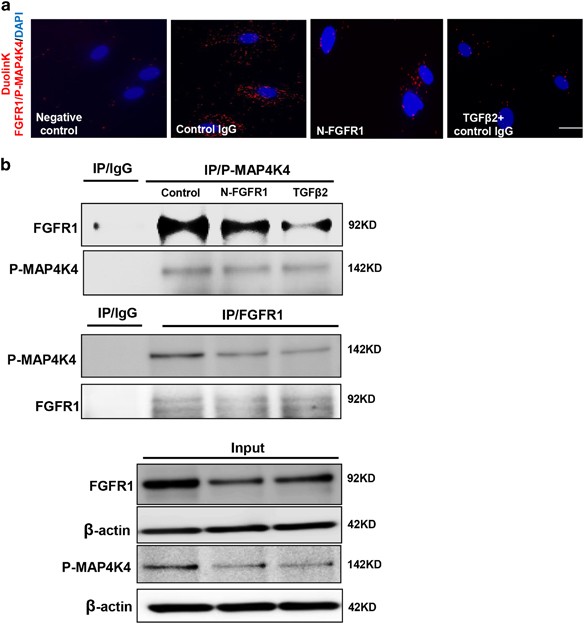
The proximity between FGFR1 and P-MAP4K4 decreases in FGFR1-deficient cells. (a) HMVECs were treated with N-FGFR1 or TGF_β_2 for 48 h. The proximity between FGFR1 and P-MAP4K4 was analyzed using the Duolink In Situ Assay. For each slide, images at × 400 original magnification were obtained from six different areas. (b) immunoprecipitation analysis with either a P-MAP4K4 or a FGFR1 antibody was performed and analyzed by western blot. Then, the FGFR1 and P-MAP4K4 levels with N-FGFR1 or TGF_β_2 treatment were analyzed by western blot in endothelial cells
MAP4K4 signaling is activated by AcSDKP in a FGFR1/FRS2-dependent manner
We next examined whether AcSDKP-activated MAP4K4 signaling in endothelial cells via the FGFR1/FRS2 pathway. Endothelial cells were treated with either N-FGFR1 or FRS2 siRNA in the presence or absence of FGF2 or AcSDKP. In the presence of N-FGFR1, the P-MAP4K4 levels were significantly decreased in endothelial cells and could not be restored by FGF2 (Figure 5a). Similarly, AcSDKP also failed to restore the N-FGFR1-suppressed P-MAP4K4 levels (Figure 5b). Furthermore, in FRS2 siRNA-treated endothelial cells, neither AcSDKP nor FGF2 restored FRS2 siRNA-suppressed P-MAP4K4 levels (Figures 5c and d). These findings suggest that FGFR1 is a key upstream molecule of MAP4K4 signaling in endothelial cells.
Figure 5
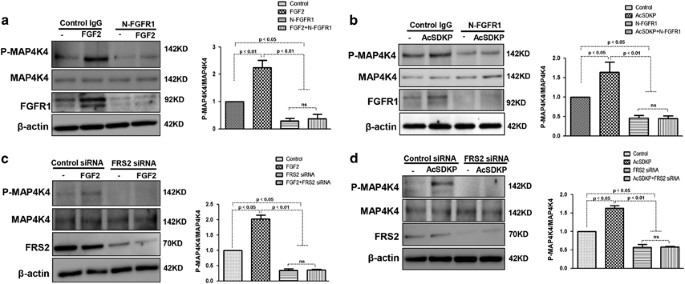
MAP4K4 signaling is mediated by AcSDKP in a FGFR1/FRS2-dependent manner. (a and b) HMVECs were treated with N-FGFR1 for 48 h in the presence or absence of FGF2 or AcSDKP. P-MAP4K4 levels were analyzed by western blot. Densitometric analysis of P-MAP4K4 levels normalized to MAP4K4. For each group, _n_=3 were analyzed. (c and d) HMVECs were transfected with FRS2 siRNA for 48 h with or without FGF2 or AcSDKP treatment. P-MAP4K4 levels were analyzed by western blot. Densitometric analysis of P-MAP4K4 levels, normalized to MAP4K4. A total of _n_=3 from each group were analyzed
MAP4K4 is a key inhibitor of TGF_β_/smad signaling and EndMT via suppression of integrin _β_1 signaling
In endothelial cells, the increased p-smad3 level induced by MAP4K4 siRNA was not suppressed by AcSDKP treatment (Figure 6a). In addition, MAP4K4 siRNA treatment led to decreased expression of the endothelial cell markers VE-cadherin and CD31 and increased expression of the mesenchymal markers FSP1, SM22_α_ and vimentin (Figure 6b), suggesting that MAP4K4 deficiency induces EndMT. However, AcSDKP did not inhibit the MAP4K4 siRNA-induced EndMT (Figure 6b). MAP4K4 has been shown to inhibit the expression of integrin _β_1 in endothelial cells,28 and we have previously reported that the interaction between DPP-4 and integrin _β_1 can induce EndMT.32 In this study, both TGF_β_2 and MAP4K4 siRNA increased the expression of integrin _β_1 in endothelial cells, especially when TGF_β_2 and MAP4K4 siRNA were applied together (Figure 6c). However, in the cells in which MAP4K4 was knocked down, AcSDKP failed to restore the increased levels of integrin _β_1 (Figure 6c).
Figure 6
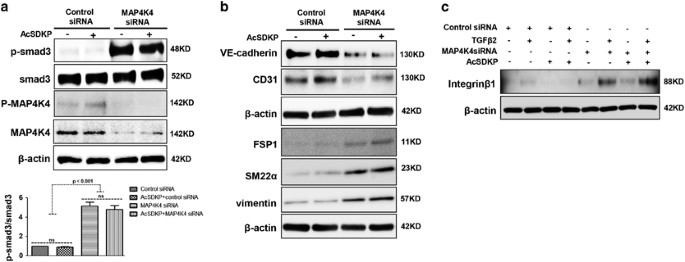
MAP4K4 deficiency induces TGF_β_/smad signaling and EndMT via activation of integrin _β_1. (a) HMVECs were transfected with MAP4K4 siRNA (100 nM) for 48 h. Next, the cells were treated with or without AcSDKP for 2 h. The p-smad3/smad3 pathway was analyzed by western blot. Densitometric analysis of the p-smad3/smad3 levels was performed, with n_=3 for each group. (b) HMVECs were treated with MAP4K4 siRNA for 48 h with or without AcSDKP treatment. The VE-cadherin, CD31, FSP1, SM22_α and vimentin protein levels were analyzed by western blot. (c) HMVECs were transfected with MAP4K4 siRNA for 48 h in the presence or absence of TGF_β_2 with or without AcSDKP. The integrin _β_1 level was analyzed by western blot
AcSDKP inhibits diabetes-induced EndMT and restores diabetes-suppressed FGFR1 and P-MAP4K4 expression in mice
AcSDKP restored the high glucose (HG)-induced EndMT in cultured endothelial cells (Supplementary Figure S7). Therefore, to investigate the role of the AcSDKP–FGFR1–MAP4K4 signaling pathway in EndMT induction in vivo, we developed AcSDKP-treated streptozotocin (STZ)-induced diabetic mice. FGFR1 and P-MAP4K4 expression in their cardiac tissue endothelial cells was analyzed by immunofluorescence, and FGFR1 and P-MAP4K4 expression was markedly suppressed in the diabetic hearts compared with the control group (Figure 7a). In addition, AcSDKP treatment restored the diabetes-suppressed endothelial FGFR1 and P-MAP4K4 expression (Figure 7a). We next analyzed endothelial cells in hearts undergoing EndMT, which were recognized by double labeling with CD31/α_-SMA or VE-cadherin/SM22_α antibodies. The diabetic hearts exhibited more endothelial cells undergoing EndMT compared with the control hearts (Figure 7b), and AcSDKP treatment restored the diabetes-induced EndMT (Figure 7b). When compared with the control mice, the diabetic mice exhibited increased endothelial p-smad3 levels in the hearts, which was inhibited by AcSDKP (Figure 7c). In the hearts, AcSDKP reversed the diabetes-suppressed FGFR1 and P-MAP4K4 levels and the diabetes-induced TGF_β_2 levels (Figure 7d). However, the TGF_β_1 and TGF_β_3 protein levels were not significantly altered (Figure 7d). In kidneys, AcSDKP also restored the diabetes-suppressed FGFR1 and P-MAP4K4 levels and the diabetes-induced TGF_β_1 and TGF_β_2 expression (Supplementary Figure S6). The TGF_β_3 protein level was not significantly altered (Supplementary Figure S6).
Figure 7
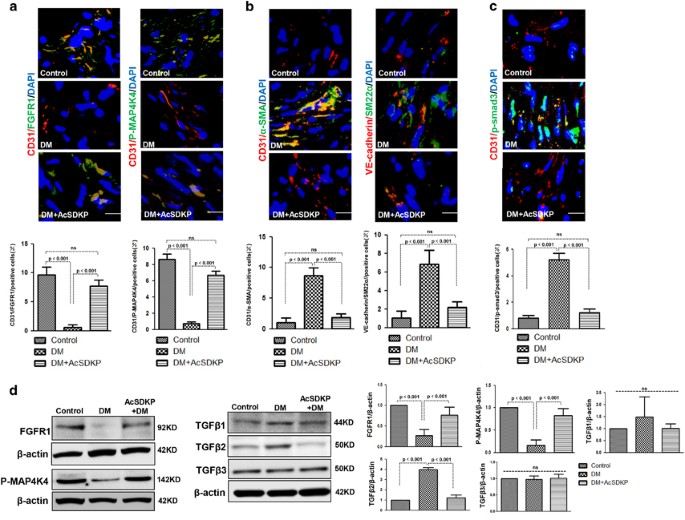
AcSDKP inhibits TGF_β_/smad signaling and EndMT and restores the FGFR1 and P-MAP4K4 levels in diabetic hearts. (a) Immunofluorescence microscopy analysis of CD31/FGFR1 and CD31/P-MAP4K4 in the heart tissues from each group of mice. The scale bar is 60 _μ_m in each panel. The CD31 and FGFR1 double-labeled cells and the CD31 and P-MAP4K4 double-labeled cells in each visual field were assessed by fluorescence microscopy and quantified. For each section, images from six different fields of view at × 400 magnification were evaluated. (b and c) Immunofluorescence microscopy analysis of CD31/α_-SMA, VE-cadherin /SM22_α and CD31/p-smad3 expression levels in the heart tissues from each group of mice. The scale bar is 60 _μ_m in each panel. The CD31 and α_-SMA double-labeled cells, the VE-cadherin and SM22_α double-labeled cells and the CD31 and p-smad3 double-labeled cells in each visual field were analyzed by fluorescence microscopy and quantified. For each section, images from six different fields of view at × 400 magnification were evaluated. Four mice from each group were analyzed. (d) Western blot analysis of the FGFR1, P-MAP4K4, TGF_β_1, TGF_β_2 and TGF_β_3 levels in cardiac tissues. A representative blot from four independent experiments was shown. The densitometric analysis of western blot data was presented (_n_=4). The diabetic mice are abbreviated as DM in the figure
Discussion
In our study, we found that AcSDKP inhibited TGF_β_/smad signaling and EndMT in endothelial cells via the FGFR1/MAP4K4 pathway in vitro and in vivo. In vitro, TGF_β_/smad signaling and the associated EndMT were induced by FGFR1 deficiency in a TGF_β_2-dependent manner. AcSDKP-activated MAP4K4 signaling through the FGFR1/FRS2 pathway. AcSDKP could not suppress the TGF_β_/smad signaling and EndMT induced by MAP4K4 deficiency. In vivo, we also confirmed that AcSDKP restored the expression of FGFR1 and P-MAP4K4 and was associated with TGF_β_/smad3 expression and EndMT in diabetic hearts and kidneys.
AcSDKP exhibits anti-EndMT and antifibrotic effects in several organ fibrosis models,22, 37 and EndMT has shown to be induced by TGF_β_2 in both a smad-dependent and a smad-independent manner.11, 13, 38 AcSDKP has also been shown to inhibit TGF_β_1-induced p-smad2/3.34 However, the detailed molecular mechanisms by which AcSDKP inhibits TGF_β_/smad signaling and EndMT in endothelial cells are not entirely clear. A recent study confirmed that FGF/FGFR suppressed TGF_β_-induced EndMT via the induction of miR-let-7s.25 Our previous study also demonstrated that AcSDKP restored diabetes-suppressed FGFR and FGFR phosphorylation levels.22 FGFR1 has been confirmed as a key inhibitor of TGF_β_-induced EndMT.26 Here, we established the FGFR1 signaling-dependent inhibitory effects of AcSDKP on TGF_β_/smad signaling and EndMT in HMVECs (Figure 8). AcSDKP exhibited anti-TGF_β_/smad3 and anti-EndMT effects and restored the TGF_β_2-suppressed FGFR1 levels. AcSDKP also suppressed IFN-γ, the potent molecule that inhibited FGFR1.The anti-TGF_β_/smad and anti-EndMT effects of AcSDKP were lost when FGFR1 signaling was disrupted.
Figure 8
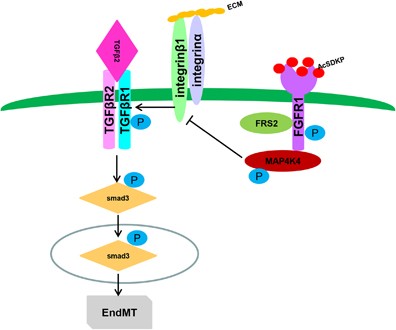
Schematic of the AcSDKP/FGFR1/MAP4K4 pathway suppression of TGF_β_/smad signaling and EndMT. In endothelial cells, the close proximity between AcSDKP and FGFR1 increased FGFR1 and induced its phosphorylation levels. Interacting with co-factor FRS2, FGFR1 recruited MAP4K4 and induced its phosphorylation. Subsequently, p-MAP4K4 suppressed integrin_β_1 (integrin_β_1 should be localized on the cell surface interacted with some of α integrins). Integrin_β_1 was a potent activator of TGF-β signaling and also EndMT. Therefore, AcSDKP could inhibit EndMT through FGFR1-MAP4K4-dependent manner
FGFR1 has a key role in the regulation of cell migration,39, 40, 41 and the interaction between FGFR1 and integrin _β_1 is essential for endothelial cell migration.42 In addition, MAP4K4 promotes endothelial cell migration via inactivation of integrin _β_1 signaling.28 Here, we confirmed that FGFR1 was essential for regulation of the MAP4K4 pathway in endothelial cells, as suppression of FGFR1 clearly decreased P-MAP4K4 levels. Conversely, FGF2-induced FGFR1 activation was associated with the induction of P-MAP4K4 (Figure 8).
Kaneko et al.33 reported that misshapen, the mammalian ortholog of MAP4K4, was a direct suppressor of the TGF_β_/smad pathway. Recent studies showed that MAP4K4 deficiency in T cells led to insulin resistance and that MAP4K4 expression was significantly suppressed in type 2 diabetic mice.43, 44 Despite these important biological roles, little information is known regarding the regulation of MAP4K4 in TGF_β_/smad signaling and EndMT induction. Our data demonstrated that AcSDKP-activated P-MAP4K4 expression was dependent on the FGFR1 signaling pathway. In addition, the suppression of MAP4K4 in endothelial cells significantly induced TGF_β_/smad signaling and EndMT, even in the presence of AcSDKP (Figure 8). In vivo, we demonstrated that AcSDKP reversed the diabetes-suppressed FGFR1 and P-MAP4K4 levels associated with the induction of TGF_β_/smad signaling and EndMT in endothelial cells in hearts and kidneys.
MAP4K4 has been shown to suppress integrin _β_1 signaling in endothelial cells28 and integrin β_1 is associated with TGF_β signaling and EndMT induction in endothelial cells.32, 45 In our study, suppression of MAP4K4 increased the integrin _β_1 levels in endothelial cells, and AcSDKP had no effect on its levels in MAP4K4-deficient cells (Figure 8). These data confirmed that MAP4K4 is a key downstream molecule for the anti-EndMT effect of AcSDKP.
Our results confirmed the functional interactions among AcSDKP, FGFR1 and MAP4K4 in endothelial cells. The close proximity between AcSDKP and FGFR1 inhibited the EndMT associated with TGF_β_/smad signaling by activating the MAP4K4 pathway. In addition, AcSDKP restored both diabetes-suppressed FGFR1 and P-MAP4K4 levels and induced TGF_β_/smad signaling and EndMT in heart and kidney. These findings reveal an AcSDKP–FGFR1–MAP4K4 signaling axis, offering new insights into endothelial cell biology and a potential target for future studies of EndMT-associated organ fibrosis.
Materials and methods
Reagents
The AcSDKP peptide was a gift from Asabio Bio Technology (Osaka, Japan). The AcSDKA (100 nmol, SQ14640), AcDSPK (100 nmol, SQ14641) and AcADKP (100 nmol, SQ14639) peptides were purchased from Scrum Inc. (Tokyo, Japan). The following antibodies were purchased from Abcam (Cambridge, UK): mouse monoclonal anti-FGFR1 (1 : 500, ab823), rabbit polyclonal anti-integrin _β_1(1 : 500, ab115146), mouse monoclonal anti-vimentin (1 : 2000, ab8978), rabbit polyclonal anti-_α_-SMA (1 : 1000, ab5694), rabbit monoclonal anti-IFN-γ (1 : 1000, ab133566), rabbit polyclonal anti-TGF_β_RII (1 : 1000, ab61213) and rabbit polyclonal anti-TGF_β_3 (1 : 500, ab 15537). The human BMP-4 peptide (1 _μ_g/ml, ab40140) and the human T_β_4 peptide (1 μ_g/ml, ab42293) were also purchased from Abcam. The mouse monoclonal anti-human CD31 (1 : 1000, AF3628), human neutralizing FGFR1 (1 : 500, MAB765) and neutralizing TGF_β (1-2-3) (1 : 500, MAB1835) antibodies were purchased from R&D Systems (Minneapolis, MN, USA). A rabbit polyclonal anti-phospho-smad3 (s423 and s425) (1 : 1000, 600-401-919) antibody was purchased from Rockland Immunochemicals (Gilbertsville, PA, USA). The mouse monoclonal anti-_β_-actin (1:10 000, A2228), rabbit anti-TGF_β_1 (1:500, SAB4502954) and rabbit anti-TGF_β_RI (1:500, SAB4502958) antibodies, as well as FGF2 human recombinant (50 _μ_g/ml, SRP4037), KYP-2047 (1 _μ_g/ml, SML0208) and PF-06260933 (1 μ_mol, PZ-0272) were obtained from Sigma (St. Louis, MO, USA). The rabbit polyclonal anti-phospho-HGK/MAP4K4 PSer801 antibody (1:500, PA5-12874) and the rabbit anti-SMAD2 (1 : 500, #51-1300) antibody were purchased from Thermo Fisher Scientific (Waltham, MA, USA). The rabbit polyclonal anti-HGK/MAP4K4 (1 : 500, NBP1-58853), rabbit polyclonal anti-SM22_α (1:1000, NBP1-33003) and rabbit monoclonal anti-VE-cadherin (1:1000, NBP1-43347) antibodies, along with the recombinant human BMP-2 protein (1 _μ_g/ml, NBP1-98923) were obtained from Novus Biologicals (Littleton, CO, USA). The rabbit polyclonal anti-S100A4 (also known as FSP1) (1:200, PRB-497P), rabbit polyclonal anti-FRS2 (1:1000, sc-8318) and goat polyclonal anti-p-smad1/5/8 (1 : 1000, sc-12353) antibodies were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). The rabbit polyclonal anti-AcSDKP (1 : 1000, A03881) antibody was purchased from Biocompare (South, San Francisco, CA, USA). The rabbit polyclonal anti-smad3 (1:1000, #9513), rabbit polyclonal anti-p-smad2 (1:400, #3108S) and rabbit anti-phospho-FGFR1 (1:500, #3471) antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). The TGF_β_2 antibody (1:1000, GTX15539) was purchased from Gene Tex (Alton Pkwy, Irvine, CA, USA).
Cell culture and treatment
The HMVECs were cultured in EBM-2 medium supplemented with EGM-2 (5.5 mmol/l glucose, fetal bovine serum, hydrocortisone, hFGF-β, VEGF, R-IGF-1, ascorbic acid, hEGF, GA-1000 and heparin) (Lonza, Alpharetta, GA, USA). When the cells reached 70–80% confluence, TGF_β_2 (5 ng/ml), N-FGFR1 (1.5 μ_g/ml) and TGF_β_2 with either the N-FGFR1 or the neutralizing TGF_β (1, 2, 3) antibody (1.0 _μ_g/ml) was added to the experimental medium (a mixture of HuMedia-MVG in serum-free RPMI 1640 medium at a 1 : 3 ratio) with or without preincubation with 100 nM AcSDKP for 2 h.
Transfection experiments
HMVECs were transfected with siRNA (100 nmol/l) targeting FRS2 (Invitrogen, Carlsbad, CA, USA) or MAP4K4 (Dharmacon, Lafayette, CO, USA) (FRS2: 5′-GCGGAAAAACCGCAAGUTT-3′ and MAP4K4: 5′-GACCAACUCUGGCUUGUUA-3′). The transfection reagent lipofectamine 2000 (Invitrogen) was used according to the manufacturer’s instructions. Cells were incubated with lipofectamine 2000 and target RNA in serum-free medium for 6 h. After this, the medium was replaced by experimental medium (a mixture of serum-free Humedia-EB2 in serum-free RPMI at a 1:3 ratio), the cells were treated as follows: for FRS2 siRNA transfection, the cells were treated with or without FGF2 (50 ng/ml) for 24 h or AcSDKP for 48 h; for the MAP4K4 siRNA transfection, the cells were treated with TGF_β_2 for 48 h in the presence or absence of AcSDKP.
Duolink in situ assay
The duolink in situ PLA was performed according to the manufacturer’s protocol. Briefly, HMVECs were treated with N-FGFR1 (1.5 ug/ml) or MAP4K4 siRNA (100 nmol) for 48 h with or without preincubation with AcSDKP. Control IgG was added in the negative control group, and TGF_β_2 and control IgG were added in the positive control group. Next, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. After blocking, the cells were incubated overnight with rabbit anti-AcSDKP (1 : 1200) and mouse anti-FGFR1 (1 : 50) or mouse anti-FGFR1 (1 : 50) and rabbit anti-P-MAP4K4 (1 : 1200). The cells were incubated with PLA probe solution for 1 h at 37 °C before being treated with a ligase solution for 30 min at 37 °C and a polymerase amplification solution for 100 min at 37 °C. The samples were immediately mounted with Duolink In Situ Mounting Medium with DAPI for 20 min and analyzed by fluorescence microscopy. For each slide, the images were analyzed from six different view fields at a × 400 magnification.
Western blot analysis
Proteins were harvested using RIPA lysis buffer (lysis buffer, PMSF, protease inhibitor cocktail and sodium orthovanadate, which was purchased from Santa Cruz Biotechnology). The protein lysates were boiled in SDS sample buffer at 100 °C for 6 min, separated on SDS-polyacrylamide gels, and then transferred onto PVDF membranes (Pall Corporation, Pensacola, FL, USA) using the semi-dry method. After blocking, the membranes were incubated overnight with primary antibodies at 4 °C, followed by the corresponding peroxidase-conjugated secondary antibody for 1 h at room temperature. The blots were developed with an enhanced chemiluminescence detection system (Pierce Biotechnology, Rockford, IL, USA) and visualized using an ImageQuant LAS 400 camera system (GE Healthcare Life Sciences, Uppsala, Sweden).
Immunoprecipitation
HMVECs were treated with either N-FGFR1 or TGF_β_2. After 48 h, the cells were harvested using the RIPA lysis buffer system and centrifuged at 14 000 × g for 15 min at 4 °C. The supernatant was then transferred to a new tube, washed twice with protein A solution (Cell Signaling Technology), and incubated with primary antibody overnight at 4 °C. Protein A was added again, and incubated for 2 h at 4 °C. Next, the solution was centrifuged, and the supernatant was discarded. The pellets were washed three times with RIPA lysis buffer. After the supernatant was discarded, 2 × SDS sample buffer was added to the pellets, and the mixture was boiled for 5 min to elute the captured proteins for western blotting.
Immunofluorescence for cell culture
The treated HMVECs were cultured on eight-well culture slides for 48 h. The cells were then fixed with 100% methanol for 10 min at −20 °C and acetone for 1 min at −20 °C. After blocking with 2% BSA/PBS for 30 min at room temperature, the cells were incubated with primary antibody for 1 h. After being washed with PBS, the slides were incubated with the corresponding secondary antibodies for 30 min. The cells were then extensively washed three times with PBS and mounted with mounting medium containing DAPI (Vector Laboratories, Burlingame, CA, USA). The images were analyzed by fluorescence microscopy (Axio Vert.A1, Carl Zeiss Microscopy GmbH, Jena, Germany).
Immunostaining analysis of mouse tissue
Frozen heart sections were fixed with acetone for 10 min at −20 °C. The sections were blocked with 2% BSA/PBS for 30 min at room temperature and incubated with primary antibodies against CD31/FGFR1, CD31/P-MAP4K4, CD31/α_-SMA, VE-cadherin/SM22_α and CD31/p-smad3 for 1 h. The sections were then incubated with the corresponding secondary antibodies for 30 min at room temperature and mounted in mounting medium containing DAPI. The images were analyzed by fluorescence microscopy (Axio Vert.A1, Carl Zeiss Microscopy GmbH).
Animal experiments
Control, STZ-treated CD-1 and AcSDKP-treated STZ mice were prepared based on our previous report.22 Briefly, 8-week-old CD-1 mice were injected with STZ (200 mg/kg). Sixteen weeks after the induction of diabetes, the diabetic mice were divided into two groups, a nontreatment group and an AcSDKP treatment group (500 _μ_g/kg BW/day using an osmotic mini-pump for 8 weeks). The experiments described in the methods were carried out in accordance with the animal protocols of Kanazawa Medical University (protocol numbers 2014-89, 2013-114 and 2014-101).
Statistical analysis
The data are expressed as the mean±S.E.M. One-way ANOVA followed by the Tukey’s multiple comparison test (statistical significance was defined as P<0.05) was used for statistical analysis using GraphPad Prism software (Ver 5.0f, La Jolla, Canada).
References
- Zeisberg EM, Potenta SE, Sugimoto H, Zeisberg M, Kalluri R . Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol 2008; 19: 2282–2287.
Article Google Scholar - He J, Xu Y, Koya D, Kanasaki K . Role of the endothelial-to-mesenchymal transition in renal fibrosis of chronic kidney disease. Clin Exp Nephrol 2013; 17: 488–497.
Article CAS Google Scholar - Srivastava SP, Koya D, Kanasaki K . MicroRNAs in kidney fibrosis and diabetic nephropathy: roles on EMT and EndMT. Biomed Res Int 2013; 2013: 125469.
Article Google Scholar - Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 2007; 13: 952–961.
Article CAS Google Scholar - Potenta S, Zeisberg E, Kalluri R . The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer 2008; 99: 1375–1379.
Article CAS Google Scholar - Lin F, Wang N, Zhang TC . The role of endothelial-mesenchymal transition in development and pathological process. IUBMB Life 2012; 64: 717–723.
Article CAS Google Scholar - Kalluri R, Zeisberg M . Fibroblasts in cancer. Nat Rev Cancer 2006; 6: 392–401.
Article CAS Google Scholar - Nakajima Y, Yamagishi T, Hokari S, Nakamura H . Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP). Anat Rec 2000; 258: 119–127.
Article CAS Google Scholar - Armstrong EJ, Bischoff J . Heart valve development: endothelial cell signaling and differentiation. Circ Res 2004; 95: 459–470.
Article CAS Google Scholar - Maleszewska M, Gjaltema RA, Krenning G, Harmsen MC . Enhancer of zeste homolog-2 (EZH2) methyltransferase regulates transgelin/smooth muscle-22α expression in endothelial cells in response to interleukin-1β and transforming growth factor-β2. Cell Signal 2015; 27: 1589–1596.
Article CAS Google Scholar - Medici D, Potenta S, Kalluri R . Transforming growth factor-β2 promotes Snail-mediated endothelial-mesenchymal transition through convergence of Smad-dependent and Smad-independent signalling. Biochem J 2011; 437: 515–520.
Article CAS Google Scholar - Moonen JR, Krenning G, Brinker MG, Koerts JA, van Luyn MJ, Harmsen MC . Endothelial progenitor cells give rise to pro-angiogenic smooth muscle-like progeny. Cardiovasc Res 2010; 86: 506–515.
Article CAS Google Scholar - Kitao A, Sato Y, Sawada-Kitamura S, Harada K, Sasaki M, Morikawa H et al. Endothelial to mesenchymal transition via transforming growth factor-beta1/Smad activation is associated with portal venous stenosis in idiopathic portal hypertension. Am J Pathol 2009; 175: 616–626.
Article CAS Google Scholar - Dejana E, Hirschi KK, Simons M . The molecular basis of endothelial cell plasticity. Nat Commun 2017; 8: 14361.
Article CAS Google Scholar - Hrenak J, Paulis L, Simko F . N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP): potential target molecule in research of heart, kidney and brain. Curr Pharm Des 2015; 21: 5135–5143.
Article CAS Google Scholar - Kanasaki K, Nagai T, Nitta K, Kitada M, Koya D . N-acetyl-seryl-aspartyl-lysyl-proline: a valuable endogenous anti-fibrotic peptide for combating kidney fibrosis in diabetes. Front Pharmacol 2014; 5: 70.
Article Google Scholar - Liu YH, D'Ambrosio M, Liao TD, Peng H, Rhaleb NE, Sharma U et al. _N-_acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling and dysfunction induced by galectin-3, a mammalian adhesion/growth-regulatory lectin. Am J Physiol Heart Circ Physiol 2009; 296: H404–H412.
Article CAS Google Scholar - Zuo Y, Chun B, Potthoff SA, Kazi N, Brolin TJ, Orhan D et al. Thymosin β4 and its degradation product, Ac-SDKP, are novel reparative factors in renal fibrosis. Kidney Int 2013; 84: 1166–1175.
Article CAS Google Scholar - Chen YW, Liu BW, Zhang YJ, Dong GF, Ding XD, Xu LM et al. Preservation of basal AcSDKP attenuates carbon tetrachloride-induced fibrosis in the rat liver. J Hepatol 2010; 53: 528–536.
Article CAS Google Scholar - Xu H, Yang F, Sun Y, Yuan Y, Cheng H, Wei Z et al. A new antifibrotic target of Ac-SDKP: inhibition of myofibroblast differentiation in rat lung with silicosis. PLoS ONE 2012; 7: e40301.
Article CAS Google Scholar - Shibuya K, Kanasaki K, Isono M, Sato H, Omata M, Sugimoto T et al. N-acetyl-seryl-aspartyl-lysyl-proline prevents renal insufficiency and mesangial matrix expansion in diabetic db/db mice. Diabetes 2005; 54: 838–845.
Article CAS Google Scholar - Nagai T, Kanasaki M, Srivastava SP, Nakamura Y, Ishigaki Y, Kitada M et al. _N-_acetyl-seryl-aspartyl-lysyl-proline inhibits diabetes-associated kidney fibrosis and endothelial-mesenchymal transition. Biomed Res Int 2014; 2014: 696475.
PubMed PubMed Central Google Scholar - Hatanaka K, Simons M, Murakami M . Phosphorylation of VE-cadherin controls endothelial phenotypes via p120-catenin coupling and Rac1 activation. Am J Physiol Heart Circ Physiol 2011; 300: H162–H172.
Article CAS Google Scholar - Murakami M, Nguyen LT, Zhuang ZW, Zhang ZW, Moodie KL, Carmeliet P et al. The FGF system has a key role in regulating vascular integrity. J Clin Invest 2008; 118: 3355–3366.
Article CAS Google Scholar - Chen PY, Qin L, Barnes C, Charisse K, Yi T, Zhang X et al. FGF regulates TGF-β signaling and endothelial-to-mesenchymal transition via control of let-7 miRNA expression. Cell Rep 2012; 2: 1684–1696.
Article CAS Google Scholar - Chen PY, Qin L, Tellides G, Simons M . Fibroblast growth factor receptor 1 is a key inhibitor of TGFβ signaling in the endothelium. Sci Signal 2014; 7: ra90.
Article Google Scholar - Lewellyn L, Cetera M, Horne-Badovinac S . Misshapen decreases integrin levels to promote epithelial motility and planar polarity in Drosophila. J Cell Biol 2013; 200: 721–729.
Article CAS Google Scholar - Vitorino P, Yeung S, Crow A, Bakke J, Smyczek T, West K et al. MAP4K4 regulates integrin-FERM binding to control endothelial cell motility. Nature 2015; 519: 425–430.
Article CAS Google Scholar - Liu S, Kapoor M, Denton CP, Abraham DJ, Leask A . Loss of beta1 integrin in mouse fibroblasts results in resistance to skin scleroderma in a mouse model. Arthritis Rheum 2009; 60: 2817–2821.
Article CAS Google Scholar - Yeh YC, Wei WC, Wang YK, Lin SC, Sung JM, Tang MJ . Transforming growth factor-{beta}1 induces Smad3-dependent {beta}1 integrin gene expression in epithelial-to-mesenchymal transition during chronic tubulointerstitial fibrosis. Am J Pathol 2010; 177: 1743–1754.
Article CAS Google Scholar - Chen X, Wang H, Liao HJ, Hu W, Gewin L, Mernaugh G et al. Integrin-mediated type II TGF-β receptor tyrosine dephosphorylation controls SMAD-dependent profibrotic signaling. J Clin Invest 2014; 124: 3295–3310.
Article CAS Google Scholar - Shi S, Srivastava SP, Kanasaki M, He J, Kitada M, Nagai T et al. Interactions of DPP-4 and integrin β1 influences endothelial-to-mesenchymal transition. Kidney Int 2015; 88: 479–489.
Article CAS Google Scholar - Kaneko S, Chen X, Lu P, Yao X, Wright TG, Rajurkar M et al. Smad inhibition by the Ste20 kinase Misshapen. Proc Natl Acad Sci USA 2011; 108: 11127–11132.
Article CAS Google Scholar - Kanasaki K, Koya D, Sugimoto T, Isono M, Kashiwagi A, Haneda M . _N_-acetyl-seryl-aspartyl-lysyl-proline inhibits TGF-beta-mediated plasminogen activator inhibitor-1 expression via inhibition of Smad pathway in human mesangial cells. J Am Soc Nephrol 2003; 14: 863–872.
Article CAS Google Scholar - Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M et al. EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 2013; 498: 492–496.
Article CAS Google Scholar - Hoch RV, Soriano P . Context-specific requirements for Fgfr1 signaling through Frs2 and Frs3 during mouse development. Development 2006; 133: 663–673.
Article CAS Google Scholar - Srivastava SP, Shi S, Kanasaki M, Nagai T, Kitada M, He J et al. Effect of antifibrotic microRNAs crosstalk on the action of _N_-acetyl-seryl-aspartyl-lysyl-proline in diabetes-related kidney fibrosis. Sci Rep 2016; 6: 29884.
Article CAS Google Scholar - Ghosh AK, Nagpal V, Covington JW, Michaels MA, Vaughan DE . Molecular basis of cardiac endothelial-to-mesenchymal transition (EndMT): differential expression of microRNAs during EndMT. Cell Signal 2012; 24: 1031–1036.
Article CAS Google Scholar - Trokovic N, Trokovic R, Mai P, Partanen J . Fgfr1 regulates patterning of the pharyngealregion. Genes Dev 2003; 17: 141–153.
Article CAS Google Scholar - Chioni AM, Grose R . FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J Cell Biol 2012; 197: 801–817.
Article CAS Google Scholar - Hardy KM, Yatskievych TA, Konieczka J, Bobbs AS, Antin PB . FGF signalling through RAS/MAPK and PI3K pathways regulates cell movement and gene expression in the chicken primitive streak without affecting E-cadherin expression. BMC Dev Biol 2011; 11: 20.
Article CAS Google Scholar - Zou L, Cao S, Kang N, Huebert RC, Shah VH . Fibronectin induces endothelial cell migration through β1 integrin and Src-dependent phosphorylation of fibroblast growth factor receptor-1 at tyrosines 653/654 and 766. J Biol Chem 2012; 287: 7190–7202.
Article CAS Google Scholar - Chuang HC, Sheu WH, Lin YT, Tsai CY, Yang CY, Cheng YJ et al. HGK/MAP4K4 deficiency induces TRAF2 stabilization and Th17 differentiation leading to insulin resistance. Nat Commun 2014; 5: 4602.
Article CAS Google Scholar - Chuang HC, Wang JS, Lee IT, Sheu WH, Tan TH . Epigenetic regulation of HGK/MAP4K4 in T cells of type 2 diabetes patients. Oncotarget 2016; 7: 10976–10989.
PubMed PubMed Central Google Scholar - Bhowmick NA, Zent R, Ghiassi M, McDonnell M, Moses HL . Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J Biol Chem 2001; 276: 46707–46713.
Article CAS Google Scholar
Acknowledgements
We thank Asabio Bio Technology for providing AcSDKP. This work was partially supported by grants from the Japan Society for the Promotion of Science awarded to TN with KK (24659264), KK (23790381, 26460403) and DK (25282028, 25670414 and 16K15472) and research grants for KK (the Takeda Science Foundation and the Naito Foundation). This work was partially supported by a grant for Collaborative Research awarded to DK (C2011-4 and C2012-1) and a grant for Promoted Research awarded to KK (S2013-13, S2014-4, S2015-3 and S2016-3). SP Srivastava is supported by the Japanese Government MEXT (Ministry of Education, Culture, Sports, Science, and Technology) Fellowship Program. This work was also supported by a Medical Research Encouragement Prize from The Japan Medical Association awarded to KK. Boehringer Ingelheim (Japan), MitsubishiTanabe Pharma and Ono Pharmaceutical contributed to establishing the Division of Anticipatory Molecular Food Science and Technology. KK is under a consultancy agreement with Boehringer Ingelheim.
Author contributions
JL performed all the in vitro experiments, as well as some animal experiments, and participated in writing the manuscript. SS helped perform the animal experiments and supervised the in vitro experiments. SPS supervised the in vitro experiments. M Kitada, TN and KN participated in the data analysis and discussions. M Kohno participated in the discussion. KK proposed the experimental concept, designed and supervised the experiments, provided intellectual input, and wrote the manuscript. DK provided intellectual input.
Author information
Authors and Affiliations
- Department of Diabetology and Endocrinology, Kanazawa Medical University, Ishikawa, Japan
Jinpeng Li, Sen Shi, Swayam Prakash Srivastava, Munehiro Kitada, Takako Nagai, Kyoko Nitta, Keizo Kanasaki & Daisuke Koya - Department of Pediatric Surgery, Kanazawa Medical University, Ishikawa, Japan,
Jinpeng Li & Miyuki Kohno - Division of Anticipatory Molecular Food Science and Technology, Medical Research Institute, Kanazawa Medical University, Ishikawa, Japan
Sen Shi, Munehiro Kitada, Keizo Kanasaki & Daisuke Koya
Authors
- Jinpeng Li
- Sen Shi
- Swayam Prakash Srivastava
- Munehiro Kitada
- Takako Nagai
- Kyoko Nitta
- Miyuki Kohno
- Keizo Kanasaki
- Daisuke Koya
Corresponding authors
Correspondence toKeizo Kanasaki or Daisuke Koya.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by J Chipuk
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information accompanies this paper on Cell Death and Disease website
Supplementary information
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article
Cite this article
Li, J., Shi, S., Srivastava, S. et al. FGFR1 is critical for the anti-endothelial mesenchymal transition effect of _N_-acetyl-seryl-aspartyl-lysyl-proline via induction of the MAP4K4 pathway.Cell Death Dis 8, e2965 (2017). https://doi.org/10.1038/cddis.2017.353
- Received: 10 March 2017
- Revised: 27 June 2017
- Accepted: 02 July 2017
- Published: 03 August 2017
- Issue date: August 2017
- DOI: https://doi.org/10.1038/cddis.2017.353