A global map for dissecting phenotypic variants in human lincRNAs (original) (raw)
Introduction
Recently, the rapid explosion of knowledge on noncoding RNAs (ncRNAs), particularly large intergenic ncRNAs (lincRNAs), has brought these previously neglected and undervalued functional molecules to the forefront.1, 2 The lincRNAs are a class of extensively transcribed ncRNA molecules in the mammalian genome, which are usually greater than 200 nt in length, and do not overlap with protein-coding regions.3 Recent studies have shown that lincRNAs are involved in diverse biological functions, such as epigenetic regulation,4 cell-cycle control and apoptosis,5 reprogramming and pluripotency6 and gene expression regulation.7 Moreover, emerging studies have revealed that significant numbers of lincRNAs are associated with human diseases, including breast cancer,8 prostate cancer,9 leukemias and carcinomas10 and nervous system diseases.11
Although the importance of lincRNAs in human diseases has been recognized, the specific elements with functional significance in the lincRNA sequences remain largely unidentified. Their discovery may depend on the identification of key genetic variants, which occur within lincRNAs and have conclusive relationships to diseases.12 Currently, several lincRNA polymorphisms have been found to be associated with human diseases. For example, single-nucleotide polymorphisms (SNPs) in ANRIL have been associated with an increased risk of atherosclerosis, type 2 diabetes and coronary heart disease.13 Thus, lincRNA polymorphisms are possible candidates for causal variants of human disease, and have important implications for biology and biomedical studies. Moreover, genome-wide association studies (GWASs) have identified a large number of phenotype-associated SNPs in intergenic regions of the human genome; it is difficult to explain the pathogenesis of these SNPs, because they cannot be directly linked to changes in protein content or function. Recent studies are beginning to link these risk SNPs to lincRNAs. For example, a meta-analysis of two existing GWAS found prostate cancer-associated SNPs in lincRNA regions.14 Therefore, linking phenotype-associated SNPs to lincRNAs will provide independent support for functional implications and lead to a greater understanding of disease pathogenesis.
Here we systematically analyzed naturally occurring variants in human lincRNAs and found a relatively low level of lincRNA polymorphisms. We also conducted a genome-wide evaluation of lincRNA polymorphisms affecting predicted RNA secondary structures, and found that some variants had large effects and might have potential significance in disease development. Using phenotype-associated SNPs from integrated GWAS data, we found that some lincRNAs were involved in specific diseases; we also identified candidate-causal SNPs and corresponding lincRNAs. These findings will help us to elucidate underlying molecular mechanisms of lincRNAs in human diseases.
Materials and methods
Human lincRNA data
Human lincRNA data were downloaded from the human body map (HBM),2 which used an integrative approach to define a reference catalog of human lincRNAs. We obtained the genomic coordinate data of 4662 stringent lincRNAs from HBM, which defined each lincRNA region as the isoform with maximal exons (including exon and intron regions).
Protein-coding gene and pre-miRNA data
Genomic locations of 38 605 human RefSeq protein-coding genes were retrieved from the UCSC genome browser,15 and the genomic coordinates of 1523 human pre-miRNAs (precursors) were downloaded from miRBase.16
SNP data
Human SNP data were downloaded from the UCSC genome browser (dbSNP build 132).15 We discarded the SNPs with only one allele or more than two alleles, insertion and deletion mutations, and SNPs having different positions in the human genome (hg19/GRCh37). Then, we identified SNPs in each lincRNA (pre-miRNA or protein-coding gene) and calculated the SNP density based on the number of SNPs divided by the total length of this lincRNA (pre-miRNA or protein-coding gene). For comparison, we also calculated the SNP density of upstream and downstream nearest neighboring protein-coding genes (and flanking regions with the same length) of each lincRNA.
Evolutionarily conserved regions
Genome-wide MultiZ multiple alignments of 46 species to the human genome (hg19/GRCh37) were downloaded from the UCSC genome browser.15 We chose evolutionarily conserved regions (ECRs) ≥100 bp long with ≥70% identity and mapped these ECRs to lincRNAs (including exon and intron regions), which resulted in identification of 6663 ECRs in human lincRNAs. These threshold criteria are informative for discriminating important functional elements.17, 18
Phenotype-associated SNP data
Phenotype-associated SNPs reported by GWAS were downloaded from NHGRI GWAS Catalog.19 Considering that additional SNPs in linkage disequilibrium (LD) with phenotype-associated SNPs might also be mapped to lincRNAs, we extracted the LD SNPs with phenotype-associated SNPs using the the CEU HapMap data (_r_2>0.5). We classified phenotypes into 22 different disease classes using a classification scheme based on the physiological system affected by the disease.20
Functional enrichment analysis
Based on the idea that some lincRNAs would most likely be involved in the regulation of their nearest neighboring protein-coding genes,1 we associated the lincRNAs with their nearest RefSeq protein-coding genes, and used these genes to evaluate the potentially functional coverage of lincRNAs. The similar strategy has been applied in several previous studies.21, 22 The enrichment analysis was done with DAVID23 by analyzing enrichment of the Gene Ontology (GO) Biological Process terms. For comparison, we used the same gene lists as those used with DAVID for functional enrichment analysis with GOstat24 and Ontologizer.25
RNA secondary structure prediction and significance calculation
To evaluate the RNA structural changes caused by each lincRNA polymorphism’s mutant and wild-type alleles quantitatively, we extracted the 200-bp flanking transcript sequences on both sides of the SNP, which took into account both accuracy and computational efficiency.26 Then, we predicted the minimum free energy (MFE, Δ_G_) based on the 401-bp transcript sequences using the RNAFold program.27 RNA structural changes (ΔΔ_G_) were calculated by the MFE differences using ΔΔ_G_=|ΔGmut−ΔGwt|, where ΔGmut was the MFE of the mutant-type allele, and ΔGwt was the MFE of the wild-type allele. Furthermore, we used a significance calculation to search for the large structural changes caused by SNPs.28 We computed a _P_-value for each lincRNA polymorphism, which was based on the total number of SNPs in a lincRNA divided by the rank of the SNP’s MFE change in this lincRNA.
Results
Human lincRNA has a relatively low level of polymorphism
We mapped 712 394 SNPs to 4662 human lincRNA sequences. The average SNP density for lincRNA regions was 8.602 SNPs/kb, which was significantly lower than pre-miRNA regions (9.268 SNPs/kb; _P_-value <0.05, independent samples _t_-test) and protein-coding gene regions (9.818 SNPs/kb; _P_-value <2.2e−14, independent samples _t_-test). SNP density in lincRNA regions was also significantly lower than in the flanking regions and in neighboring protein-coding gene regions (Figure 1a). Furthermore, we compared the SNP densities of exon and intron regions of all lincRNAs. We found that the exon regions had a significantly lower SNP density than the intron regions (7.265 SNPs/kb vs 9.728 SNPs/kb; _P_-value <2.3e−41, independent samples _t_-test), and the number of annotated exons per lincRNA did not affect the SNP density (Supplementary Figure S1a). In addition, we identified 6663 ECRs from 1554 lincRNAs, and 10 440 SNPs were mapped to ECRs (Supplementary Figure S1b). Although ECRs had different length distributions, the average SNP density in ECRs (6.605 SNPs/kb) was significantly lower than that in the lincRNA regions (Figure 1b; _P_-value <1.9e–39, independent samples _t_-test).
Figure 1
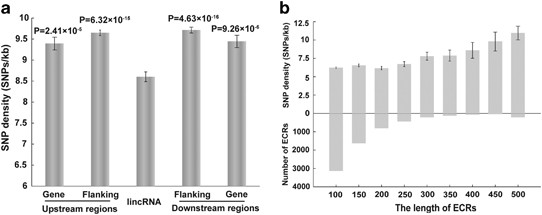
SNPs in human lincRNAs. (a) The average SNP density in lincRNA, gene and flanking regions. Flanking regions represent adjacent regions with the same length of lincRNAs, and gene regions represent nearest neighboring protein-coding genes of lincRNAs. Error bars are mean±SEM. The _t_-test _P_-values comparing lincRNA regions are shown. (b) The SNP density distribution of ECRs with different lengths. Error bars are mean±SEM.
Because the SNP density evaluation had potential functional significance, the above results indicated that lincRNAs had a low level of polymorphisms, which was likely due to their stringent functional constraint. Thus, some variants in low SNP density lincRNAs might have important functional and disease implications. For example, we found 17 SNPs in a 2943-bp lincRNA on chromosome 4, thus presenting a SNP density of 5.78 SNPs/kb, which was lower than its flanking regions and neighboring protein-coding gene regions (Supplementary Figure S2). We found a SNP of these 17 lincRNA polymorphisms, rs6843082 (located in exon 2), that was associated with atrial fibrillation.29 This SNP also had a LD(_r_2=0.763) relationship with another disease-risk SNP for atrial fibrillation, rs2634073.30 Moreover, we found a novel candidate disease-risk lincRNA polymorphism, rs12644625 (located in an intron), which had a very high LD relationship with several known atrial fibrillation risk SNPs, including rs2634073 (_r_2=0.572), rs2200733 (_r_2=0.952), rs2220427 (_r_2=0.952)30 and rs17042171 (_r_2=0.952).31
In addition, ECRs were used to identify functional noncoding regions in previous studies.32 Because the functions of the vast majority of lincRNA sequences were unknown, we believed that ECRs in human lincRNAs might be more likely to harbor variants with functional consequences. For example, we found that a attention-deficit/hyperactivity disorder (ADHD)-associated SNP (rs2823819, located in an intron) identified previously was located within a 293-bp ECR with 77% identity.33 Furthermore, we found that two other SNPs were located in the same ECR, rs17241719 (located in an intron) and rs76313674 (located in an intron), suggesting that they were novel candidate risk SNPs for ADHD.
Functional prediction of lincRNAs with different SNP densities
To provide further insight into the SNP density analysis supporting functional significance, we undertook functional enrichment analysis of the top 5% low and high SNP density lincRNAs using DAVID23 (Supplementary Figure S3a). It is worth noting that there are some important differences in significantly enriched GO terms. For example, low SNP density lincRNAs were enriched in some more general terms or biological processes, such as development, structure, transcription and metabolic regulation, indicating that these lincRNAs might cover a broad range of functions. In contrast, the high SNP density lincRNAs were enriched in more specific terms (Supplementary Figure S3b).
In addition, we also observed a slight enrichment of ECRs in the low SNP density lincRNAs (_P_-value=0.063, _χ_2-test). There were 11 614 out of a total of 17 839 ECRs (65.1%) located in lincRNAs with below average level of SNP density. We found that some lincRNAs contained multiple ECRs and computed the percent of ECR length of the total sequence length for each lincRNA. Then, we compared the significantly enriched GO terms of the top 5% highly and poorly conserved lincRNAs (Supplementary Figure S3c). Compared with poorly conserved lincRNAs, these highly conserved lincRNAs were enriched in more general terms, such as regulation of transcription, biosynthesis, gene expression and metabolic processes (Supplementary Figure S3d). The DAVID results were consistent with our previous observations and those obtained with GOStat24 and Ontologizer25 (Supplementary Table S1). We also computed function enrichment for the nearest genes of the top 100 high/low SNP density and conserved lincRNAs, which showed similar results (Supplementary Figure S4).
Many lincRNA polymorphisms have significant effects on predicted RNA secondary structures
In some cases, SNPs may cause disease by modifying RNA secondary structures.28 To investigate the effects of lincRNA polymorphisms on RNA secondary structures, we predicted the MFE (Δ_G_) changes caused by SNPs in human lincRNAs. The MFE change (ΔΔ_G_) distribution of all lincRNA polymorphisms is shown in Figure 2a. We observed that most lincRNA polymorphisms had only small effects on predicted RNA secondary structures. A small number of SNPs, however, had a large effect, suggesting that these SNPs might have important roles on lincRNA function and disease. Therefore, we next identified significant structural changes through quantitative calculations (Materials and methods). Based on the P_-value of 0.1, we identified the top 10% of lincRNA polymorphisms that had large effects on predicted RNA secondary structures. A notable increase of ΔΔ_G values was observed in these SNPs (Figure 2b), in particular in the ΔΔ_G_ values >3 kcal/mol. We further investigated whether these significant SNPs had an enrichment trend in ECRs. For all SNPs in lincRNAs, there were 1.99% of SNPs located in ECRs, but for significant SNPs, this fraction rose to 2.08% (_P_-value=0.154, χ_2-test); especially, in these SNPs that had particularly large effects on predicted RNA secondary structures (6≤ΔΔ_G ≤7), the fraction rose to 2.53%, and this enrichment was significant (Figure 2c; _P_-value=0.029, _χ_2-test).
Figure 2
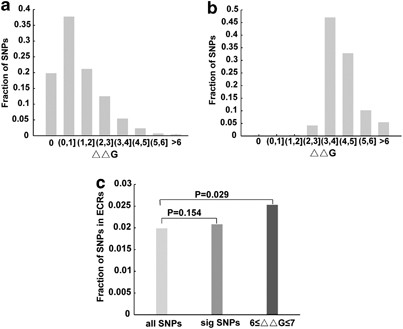
Structural effects of lincRNA polymorphisms. (a) The predicted MFE change (ΔΔ_G_) distribution for all lincRNA polymorphisms. (b) The predicted MFE change (ΔΔ_G_) distribution for significant lincRNA polymorphisms. (c) The fraction of SNPs in ECRs to all lincRNA polymorphisms, significant lincRNA polymorphisms and lincRNA polymorphisms with larger effects (6≤ΔΔ_G_ ≤7). _P_-values were from _χ_2-tests.
The consequence of RNA structural changes of lincRNA might exhibit their functional effects, even leading to disease. For example, we found a SNP, rs9858061 (located in an intron), located in an ECR of a low SNP density lincRNA region (6.87 SNPs/kb) on chromosome 3. We predicted that the MFE change (ΔΔ_G_) between G and U allele was 6 kcal/mol (Supplementary Figure S5). This SNP had a very high LD relationship with a disease-risk SNP for type 2 diabetes (rs9290240, _r_2=0.969) in the same lincRNA,34 suggesting that rs9858061 might be candidate disease-risk SNP for type 2 diabetes.
A global map of phenotype-associated variants in human lincRNAs
We investigated whether previously published phenotype-associated SNPs could take part in lincRNA-mediated regulation. We found 217 phenotype-associated SNPs located in 162 lincRNAs, which were associated with 116 phenotypes. These phenotypes were classified into 17 different disease classes based on the physiological system affected by the disease. Notably, 28 and 16 phenotype-associated lincRNA polymorphisms were assigned to ‘Psychiatric’ class and ‘Neurological’ class, respectively. This finding was consistent with previous studies, which revealed that lincRNAs had important roles in brain35 and neuropsychiatric disorders.11 We also observed a relatively large number of phenotype-associated lincRNA polymorphisms assigned to ‘Endocrine’ class (27 SNPs), ‘Cardiovascular’ class (26 SNPs) and ‘Cancer’ class (18 SNPs). These results were also consistent with previous studies.12
We constructed a circular map to gain a global view of the phenotype-associated SNPs in each human lincRNA (Figure 3), and other informations, including MFE change, SNP density and the percent of ECR length, were also shown in the map. We found that some lincRNAs contained multiple SNPs that were associated with particular diseases, suggesting that these lincRNAs might have key poles in these diseases (Supplementary Table S2). For example, we found a lincRNA on chromosome 1 with four bipolar disorder risk SNPs. Both the SNP density and ECR length of this lincRNA supported its functional importance. In particular, one of the disease-associated SNPs, rs472913,36 had a large effect on the lincRNA's secondary structure. We found four type 2 diabetes risk SNPs in a lincRNA on chromosome 5, and the SNP density of this lincRNA was below the average level. Although the lincRNA had a relatively low degree of conservation, two SNPs in this lincRNA, rs17590866 and rs980229,37 had large effects on RNA secondary structures. In addition, we found that three of the five type 1 diabetes risk SNPs in a lincRNA on chromosome 12, rs2106406, rs2106407 and rs12425190,38 had large effects on RNA secondary structures. We also found that several SNPs in human lincRNAs were associated with breast and prostate cancer. These SNPs were located in low SNP density and evolutionarily conserved lincRNAs, and several of them could significantly affect RNA secondary structures. Therefore, we believed that more SNPs located in lincRNAs would be potential functional and etiological variants.
Figure 3
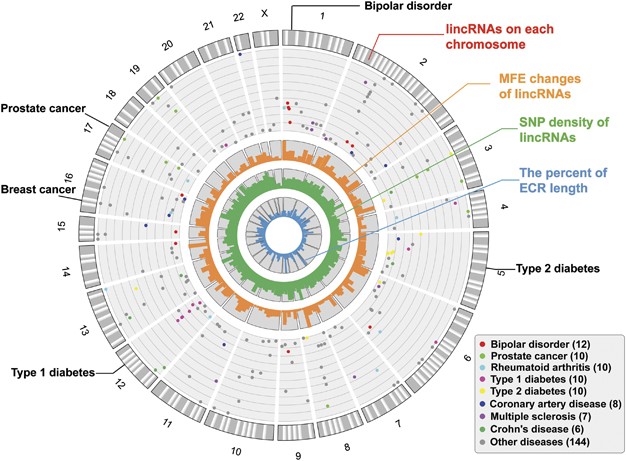
The global map of the phenotype-associated SNPs in human lincRNAs. The gray bands in the outer circle of the map represent the phenotype-associated lincRNAs on each chromosome. The dots in the map represent the phenotype-associated SNPs, with the positions of −log10 (_P_-values), and different colors of dots represent different diseases. Only diseases with at least six SNPs are colored, whereas other diseases are shown in gray. The name of the diseases and the number of SNPs are shown on the bottom right. The bar plots in the inner circles of the map represent the distribution of MFE change (dark orange), SNP density (dark green) and the percent of ECR length (dark blue). We indicated the name of diseases with at least three phenotype-associated SNPs in the lincRNA, as well as those mentioned in the text.
Furthermore, we found that a large number of lincRNA polymorphisms had high LD relationships with known phenotype-associated SNPs, which might be candidate-causal SNPs for diseases. In total, we found 1191 candidate-causal SNPs located in 318 lincRNAs, which were associated with 18 different disease classes and more than 150 phenotypes.
Discussion
In this study, using the huge wealth of SNP data in current publicly databases, especially a large number of phenotype-associated SNPs identified by GWAS, we provided a systematic analysis of SNPs in human lincRNAs. Our results indicated that the SNP density in lincRNA regions was relatively low. Because SNP density analysis providing functional implications was supported by the findings from both the protein-coding genes and the miRNAs,39, 40 we believed that some low SNP density lincRNAs might have important roles in key cellular processes. Furthermore, because the lincRNAs had differences in length distribution, we examined the relationship between lincRNA length and SNP density. We found that the lincRNA length was not correlated with the SNP density (_P_-value=0.054, Pearson’s correlation); the SNP distribution of lincRNA had a length-independent manner. In addition, we found that ECRs had lower SNP density and were thus more likely to harbor functional variants. ECR length was also not correlated with SNP density (_P_-value=0.224, Pearson’s correlation). Previous studies have revealed that small ECRs in ncRNAs could be functional domains such as binding sites for miRNAs, proteins or DNA,41 ECRs were overall small variants and contained functional variants.42 However, it is very difficult to determine the window size for ECR; small windows make it very hard to discriminate between conserved and non-conserved elements, whereas large windows could miss some important elements (Supplementary Figure S1b). For example, we found a SNP in a short ECR (32 bp) with 74% identity, rs6941421 (located in an intron), that was associated with multiple sclerosis.43 This disease-risk SNP also had a very high LD relationship with two other SNPs located in the same lincRNA, rs7755465 (_r_2=0.910, located in an intron) and rs4712249 (_r_2=0.977, located in an intron), suggesting that they might be novel candidate risk SNPs.
In addition, we evaluated the effects of lincRNA polymorphisms on predicted RNA secondary structures. Because the precise functions of the majority of lincRNA sequences remain unknown, it is difficult to identify the molecular cause of disease-associated SNPs located in lincRNAs. Previous studies have shown that functional noncoding regions in the human genome had conserved RNA secondary structures,44 and certain human diseases could be caused by variants inducing structural changes,28 suggesting RNA structural change as a potential molecular cause of the disease. Our results found that some known disease-associated SNPs located in lincRNAs had significant effects on predicted RNA secondary structures, which might have causal effects on disease risk.
Notably, lincRNA polymorphisms were associated with a broad range of human diseases, suggesting their causal roles in these diseases. Currently, despite aberrant expression of many lincRNAs observed in human diseases, there is still little understanding of their contribution to etiology. Recent studies have begun to link variants in human lincRNAs with disease risk. For example, SNPs identified as susceptibility loci for myocardial infarction have been mapped to a long ncRNA, MIAT.45 Although the precise pathogenic mechanisms remain unclear, our results suggested that there were important links between lincRNA polymorphisms and human diseases. In the future, we will continue to deeply interpret the effects of variants on lincRNA function and their direct connection to diseases.
In summary, this study dissected the properties of lincRNA polymorphisms from the perspective of human disease. All data in our study can be queried and downloaded from a user-friendly web server, called lincPoly (freely accessed at http://bioinfo.hrbmu.edu.cn/lincPoly). We hope that this study will continue to improve our knowledge and understanding of lincRNAs and their potential implications in disease.
References
- Guttman M, Amit I, Garber M et al: Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009; 458: 223–227.
Article CAS Google Scholar - Cabili MN, Trapnell C, Goff L et al: Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 2011; 25: 1915–1927.
Article CAS Google Scholar - Ponting CP, Oliver PL, Reik W : Evolution and functions of long noncoding RNAs. Cell 2009; 136: 629–641.
Article CAS Google Scholar - Nagano T, Mitchell JA, Sanz LA et al: The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 2008; 322: 1717–1720.
Article CAS Google Scholar - Hung T, Wang Y, Lin MF et al: Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet 2011; 43: 621–629.
Article CAS Google Scholar - Loewer S, Cabili MN, Guttman M et al: Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet 2010; 42: 1113–1117.
Article CAS Google Scholar - Khalil AM, Guttman M, Huarte M et al: Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci USA 2009; 106: 11667–11672.
Article CAS Google Scholar - Gupta RA, Shah N, Wang KC et al: Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010; 464: 1071–1076.
Article CAS Google Scholar - Prensner JR, Iyer MK, Balbin OA et al: Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat Biotechnol 2011; 29: 742–749.
Article CAS Google Scholar - Calin GA, Liu CG, Ferracin M et al: Ultraconserved regions encoding ncRNAs are altered in human leukemias and carcinomas. Cancer Cell 2007; 12: 215–229.
Article CAS Google Scholar - Qureshi IA, Mattick JS, Mehler MF : Long non-coding RNAs in nervous system function and disease. Brain Res 2010; 1338: 20–35.
Article CAS Google Scholar - Wapinski O, Chang HY : Long noncoding RNAs and human disease. Trends Cell Biol 2011; 21: 354–361.
Article CAS Google Scholar - Pasmant E, Sabbagh A, Vidaud M, Bieche I : ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J 2010; 25: 444–448.
Article Google Scholar - Jin G, Sun J, Isaacs SD et al: Human polymorphisms at long non-coding RNAs (lncRNAs) and association with prostate cancer risk. Carcinogenesis 2011; 32: 1655–1659.
Article CAS Google Scholar - Karolchik D, Baertsch R, Diekhans M et al: The UCSC genome browser database. Nucleic Acids Res 2003; 31: 51–54.
Article CAS Google Scholar - Kozomara A, Griffiths-Jones S : miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 2011; 39: D152–D157.
Article CAS Google Scholar - Dermitzakis ET, Reymond A, Lyle R et al: Numerous potentially functional but non-genic conserved sequences on human chromosome 21. Nature 2002; 420: 578–582.
Article CAS Google Scholar - Nobrega MA, Ovcharenko I, Afzal V, Rubin EM : Scanning human gene deserts for long-range enhancers. Science 2003; 302: 413.
Article CAS Google Scholar - Hindorff LA, Sethupathy P, Junkins HA et al: Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA 2009; 106: 9362–9367.
Article CAS Google Scholar - Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL : The human disease network. Proc Natl Acad Sci USA 2007; 104: 8685–8690.
Article CAS Google Scholar - Garmire LX, Garmire DG, Huang W, Yao J, Glass CK, Subramaniam S : A global clustering algorithm to identify long intergenic non-coding RNA—with applications in mouse macrophages. PLoS One 2011; 6: e24051.
Article CAS Google Scholar - Liao Q, Liu C, Yuan X et al: Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res 2011; 39: 3864–3878.
Article CAS Google Scholar - Huang da W, Sherman BT, Lempicki RA : Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44–57.
Article Google Scholar - Beissbarth T, Speed TP : GOstat: find statistically overrepresented gene ontologies within a group of genes. Bioinformatics 2004; 20: 1464–1465.
Article CAS Google Scholar - Bauer S, Grossmann S, Vingron M, Robinson PN : Ontologizer 2.0—a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics 2008; 24: 1650–1651.
Article CAS Google Scholar - Hariharan M, Scaria V, Brahmachari SK : dbSMR: a novel resource of genome-wide SNPs affecting microRNA mediated regulation. BMC Bioinformatics 2009; 10: 108.
Article Google Scholar - Hofacker IL : Vienna RNA secondary structure server. Nucleic Acids Res 2003; 31: 3429–3431.
Article CAS Google Scholar - Halvorsen M, Martin JS, Broadaway S, Laederach A : Disease-associated mutations that alter the RNA structural ensemble. PLoS Genet 2010; 6: e1001074.
Article Google Scholar - Ellinor PT, Lunetta KL, Glazer NL et al: Common variants in KCNN3 are associated with lone atrial fibrillation. Nat Genet 2010; 42: 240–244.
Article CAS Google Scholar - Gudbjartsson DF, Arnar DO, Helgadottir A et al: Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 2007; 448: 353–357.
Article CAS Google Scholar - Benjamin EJ, Rice KM, Arking DE et al: Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat Genet 2009; 41: 879–881.
Article CAS Google Scholar - Hardison RC : Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet 2000; 16: 369–372.
Article CAS Google Scholar - Mick E, Todorov A, Smalley S et al: Family-based genome-wide association scan of attention-deficit/hyperactivity disorder. J Am Acad Child Adolesc Psychiatry 2010; 49: 898–905 e893.
Article Google Scholar - Sladek R, Rocheleau G, Rung J et al: A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 2007; 445: 881–885.
Article CAS Google Scholar - Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS : Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci USA 2008; 105: 716–721.
Article CAS Google Scholar - Scott LJ, Muglia P, Kong XQ et al: Genome-wide association and meta-analysis of bipolar disorder in individuals of European ancestry. Proc Natl Acad Sci USA 2009; 106: 7501–7506.
Article CAS Google Scholar - Saxena R, Voight BF, Lyssenko V et al: Genome-wide association analysis identifiesloci for type 2 diabetes and triglyceride levels. Science 2007; 316: 1331–1336.
Article CAS Google Scholar - Consortium TWTCC: Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature 2007; 447: 661–678.
Article Google Scholar - Zhao Z, Fu YX, Hewett-Emmett D, Boerwinkle E : Investigating single nucleotide polymorphism (SNP) density in the human genome and its implications for molecular evolution. Gene 2003; 312: 207–213.
Article CAS Google Scholar - Saunders MA, Liang H, Li WH : Human polymorphism at microRNAs and microRNA target sites. Proc Natl Acad Sci USA 2007; 104: 3300–3305.
Article CAS Google Scholar - Prensner JR, Chinnaiyan AM : The emergence of lncRNAs in cancer biology. Cancer Discov 2011; 1: 391–407.
Article CAS Google Scholar - Drake JA, Bird C, Nemesh J et al: Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat Genet 2006; 38: 223–227.
Article CAS Google Scholar - Baranzini SE, Wang J, Gibson RA et al: Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum Mol Genet 2009; 18: 767–778.
Article CAS Google Scholar - Washietl S, Hofacker IL, Lukasser M, Huttenhofer A, Stadler PF : Mapping of conserved RNA secondary structures predicts thousands of functional noncoding RNAs in the human genome. Nat Biotechnol 2005; 23: 1383–1390.
Article CAS Google Scholar - Ishii N, Ozaki K, Sato H et al: Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J Hum Genet 2006; 51: 1087–1099.
Article CAS Google Scholar
Acknowledgements
This research was supported by the National Natural Science Foundation of China (Grant Nos. 91129710, 61073136 and 61170154), the Specialized Research Fund for the Doctoral Program of Higher Education of China (Grant Nos. 20102307110022) and the Science Foundation of Heilongjiang Province (Grant No. D200834).
Author information
Author notes
- Shangwei Ning, Peng Wang, Jingrun Ye and Xiang Li: These authors contributed equally to this work.
Authors and Affiliations
- College of Bioinformatics Science and Technology, Harbin Medical University, Harbin, China
Shangwei Ning, Peng Wang, Jingrun Ye, Xiang Li, Ronghong Li, Zuxianglan Zhao, Xiao Huo, Li Wang, Feng Li & Xia Li
Authors
- Shangwei Ning
You can also search for this author inPubMed Google Scholar - Peng Wang
You can also search for this author inPubMed Google Scholar - Jingrun Ye
You can also search for this author inPubMed Google Scholar - Xiang Li
You can also search for this author inPubMed Google Scholar - Ronghong Li
You can also search for this author inPubMed Google Scholar - Zuxianglan Zhao
You can also search for this author inPubMed Google Scholar - Xiao Huo
You can also search for this author inPubMed Google Scholar - Li Wang
You can also search for this author inPubMed Google Scholar - Feng Li
You can also search for this author inPubMed Google Scholar - Xia Li
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toXia Li.
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Ning, S., Wang, P., Ye, J. et al. A global map for dissecting phenotypic variants in human lincRNAs.Eur J Hum Genet 21, 1128–1133 (2013). https://doi.org/10.1038/ejhg.2013.7
- Received: 16 August 2012
- Revised: 18 December 2012
- Accepted: 08 January 2013
- Published: 06 March 2013
- Issue Date: October 2013
- DOI: https://doi.org/10.1038/ejhg.2013.7