Comprehensive molecular portraits of human breast tumours (original) (raw)
Main
Breast cancer is one of the most common cancers with greater than 1,300,000 cases and 450,000 deaths each year worldwide. Clinically, this heterogeneous disease is categorized into three basic therapeutic groups. The oestrogen receptor (ER) positive group is the most numerous and diverse, with several genomic tests to assist in predicting outcomes for ER+ patients receiving endocrine therapy1,2. The HER2 (also called ERBB2) amplified group3 is a great clinical success because of effective therapeutic targeting of HER2, which has led to intense efforts to characterize other DNA copy number aberrations4,5. Triple-negative breast cancers (TNBCs, lacking expression of ER, progesterone receptor (PR) and HER2), also known as basal-like breast cancers6, are a group with only chemotherapy options, and have an increased incidence in patients with germline BRCA1 mutations7,8 or of African ancestry9.
Most molecular studies of breast cancer have focused on just one or two high information content platforms, most frequently mRNA expression profiling or DNA copy number analysis, and more recently massively parallel sequencing10,11,12. Supervised clustering of mRNA expression data has reproducibly established that breast cancers encompass several distinct disease entities, often referred to as the intrinsic subtypes of breast cancer13,14. The recent development of additional high information content assays focused on abnormalities in DNA methylation, microRNA (miRNA) expression and protein expression, provide further opportunities to characterize more completely the molecular architecture of breast cancer. In this study, a diverse set of breast tumours were assayed using six different technology platforms. Individual platform and integrated pathway analyses identified many subtype-specific mutations and copy number changes that identify therapeutically tractable genomic aberrations and other events driving tumour biology.
Samples and clinical data
Tumour and germline DNA samples were obtained from 825 patients. Different subsets of patients were assayed on each platform: 466 tumours from 463 patients had data available on five platforms including Agilent mRNA expression microarrays (n = 547), Illumina Infinium DNA methylation chips (n = 802), Affymetrix 6.0 single nucleotide polymorphism (SNP) arrays (n = 773), miRNA sequencing (n = 697), and whole-exome sequencing (n = 507); in addition, 348 of the 466 samples also had reverse-phase protein array (RPPA) data (n = 403). Owing to the short median overall follow up (17 months) and the small number of overall survival events (93 out of 818), survival analyses will be presented in a later publication. Demographic and clinical characteristics are presented in Supplementary Table 1.
Significantly mutated genes in breast cancer
Overall, 510 tumours from 507 patients were subjected to whole-exome sequencing, identifying 30,626 somatic mutations comprised of 28,319 point mutations, 4 dinucleotide mutations, and 2,302 insertions/deletions (indels) (ranging from 1 to 53 nucleotides). The point mutations included 6,486 silent, 19,045 missense, 1,437 nonsense, 26 read-through, 506 splice-site mutations, and 819 mutations in RNA genes. Comparison to COSMIC and OMIM databases identified 619 mutations across 177 previously reported cancer genes. Of 19,045 missense mutations, 9,484 were predicted to have a high probability of being deleterious by Condel15. The MuSiC package16, which determines the significance of the observed mutation rate of each gene based on the background mutation rate, identified 35 significantly mutated genes (excluding LOC or Ensembl gene IDs) by at least two tests (convolution and likelihood ratio tests) with false discovery rate (FDR) <5% (Supplementary Table 2).
In addition to identifying nearly all genes previously implicated in breast cancer (PIK3CA, PTEN, AKT1, TP53, GATA3, CDH1, RB1, MLL3, MAP3K1 and CDKN1B), a number of novel significantly mutated genes were identified including TBX3, RUNX1, CBFB, AFF2, PIK3R1, PTPN22, PTPRD, NF1, SF3B1 and CCND3. TBX3, which is mutated in ulnar-mammary syndrome and involved in mammary gland development17, harboured 13 mutations (8 frame-shift indels, 1 in-frame deletion, 1 nonsense, and 3 missense), suggesting a loss of function. Additionally, 2 mutations were found in TBX4 and 1 mutation in TBX5, which are genes involved in Holt–Oram syndrome18. Two other transcription factors, CTCF and FOXA1, were at or near significance harbouring 13 and 8 mutations, respectively. RUNX1 and CBFB, both rearranged in acute myeloid leukaemia and interfering with haematopoietic differentiation, harboured 19 and 9 mutations, respectively. PIK3R1 contained 14 mutations, most of which clustered in the PIK3CA interaction domain similar to previously identified mutations in glioma19 and endometrial cancer20. We also observed a statistically significant exclusion pattern among PIK3R1, PIK3CA, PTEN and AKT1 mutations (P = 0.025). Mutation of splicing factor SF3B1, previously described in myelodysplastic syndromes21 and chronic lymphocytic leukaemia22, was significant with 15 non-silent mutations, of which 4 were a recurrent K700E substitution. Two protein tyrosine phosphatases (PTPN22 and PTPRD) were also significantly mutated; frequent deletion/mutation of PTPRD is observed in lung adenocarcinoma23.
Mutations and mRNA-expression subtype associations
We analysed the somatic mutation spectrum within the context of the four mRNA-expression subtypes, excluding the normal-like group owing to small numbers (n = 8) (Fig. 1). Several significantly mutated genes showed mRNA-subtype-specific (Supplementary Figs 1–3) and clinical-subtype-specific patterns of mutation (Supplementary Table 2). Significantly mutated genes were considerably more diverse and recurrent within luminal A and luminal B tumours than within basal-like and HER2-enriched (HER2E) subtypes; however, the overall mutation rate was lowest in luminal A subtype and highest in the basal-like and HER2E subtypes. The luminal A subtype harboured the most significantly mutated genes, with the most frequent being PIK3CA (45%), followed by MAP3K1, GATA3, TP53, CDH1 and MAP2K4. Twelve per cent of luminal A tumours contained likely inactivating mutations in MAP3K1 and MAP2K4, which represent two contiguous steps in the p38–JNK1 stress kinase pathway24. Luminal B cancers exhibited a diversity of significantly mutated genes, with TP53 and PIK3CA (29% each) being the most frequent. The luminal tumour subtypes markedly contrasted with basal-like cancers where TP53 mutations occurred in 80% of cases and the majority of the luminal significantly mutated gene repertoire, except PIK3CA (9%), were absent or near absent. The HER2E subtype, which has frequent HER2 amplification (80%), had a hybrid pattern with a high frequency of TP53 (72%) and PIK3CA (39%) mutations and a much lower frequency of other significantly mutated genes including PIK3R1 (4%).
Figure 1: Significantly mutated genes and correlations with genomic and clinical features.
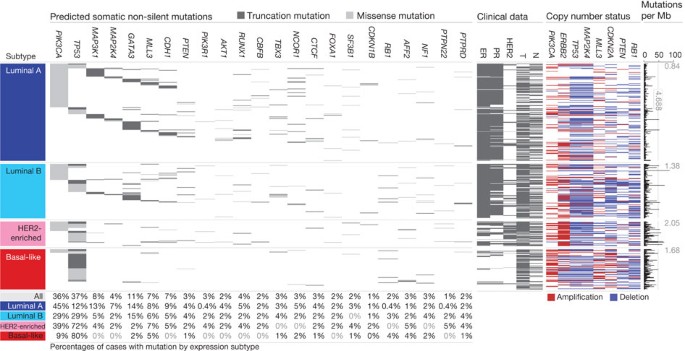
Tumour samples are grouped by mRNA subtype: luminal A (n = 225), luminal B (n = 126), HER2E (n = 57) and basal-like (n = 93). The left panel shows non-silent somatic mutation patterns and frequencies for significantly mutated genes. The middle panel shows clinical features: dark grey, positive or T2–4; white, negative or T1; light grey, N/A or equivocal. N, node status; T, tumour size. The right panel shows significantly mutated genes with frequent copy number amplifications (red) or deletions (blue). The far-right panel shows non-silent mutation rate per tumour (mutations per megabase, adjusted for coverage). The average mutation rate for each expression subtype is indicated. Hypermutated: mutation rates >3 s.d. above the mean (>4.688, indicated by grey line).
Intrinsic mRNA subtypes differed not only by mutation frequencies but also by mutation type. Most notably, TP53 mutations in basal-like tumours were mostly nonsense and frame shift, whereas missense mutations predominated in luminal A and B tumours (Supplementary Fig. 1). Fifty-eight somatic GATA3 mutations, some of which were previously described25, were detected including a hotspot 2-base-pair deletion within intron 4 only in the luminal A subtype (13 out of 13 mutants) (Supplementary Fig. 2). In contrast, 7 out of 9 frame-shift mutations in exon 5 (DNA binding domain) occurred in luminal B cancers. PIK3CA mutation frequency and spectrum also varied by mRNA subtype (Supplementary Fig. 3); the recurrent PIK3CA E545K mutation was present almost exclusively within luminal A (25 out of 27) tumours. CDH1 mutations were common (30 out of 36) within the lobular histological subtype and corresponded with lower CDH1 mRNA (Supplementary Fig. 4) and protein expression. Finally, we identified 4 out of 8 somatic variants in HER2 within lobular cancers, three of which were within the tyrosine kinase domain.
We performed analyses on a selected set of genes26 using the normal tissue DNA data and detected a number of germline predisposing variants. These analyses identified 47 out of 507 patients with deleterious germline variants, representing nine different genes (ATM, BRCA1, BRCA2, BRIP1, CHEK2, NBN, PTEN, RAD51C and TP53; Supplementary Table 3), supporting the hypothesis that ∼10% of sporadic breast cancers may have a strong germline contribution. These data confirmed the association between the presence of germline BRCA1 mutations and basal-like breast cancers7,8.
Gene expression analyses (mRNA and miRNA)
Several approaches were used to look for structure in the mRNA expression data. We performed an unsupervised hierarchical clustering analysis of 525 tumours and 22 tumour-adjacent normal tissues using the top 3,662 variably expressed genes (Supplementary Fig. 5); SigClust analysis identified 12 classes (5 classes with >9 samples per class). We performed a semi-supervised hierarchical cluster analysis using a previously published ‘intrinsic gene list’14, which identified 13 classes (9 classes with >9 samples per class) (Supplementary Fig. 6). We also classified each sample using the 50-gene PAM50 model14 (Supplementary Fig. 5). High concordance was observed between all three analyses; therefore, we used the PAM50-defined subtype predictor as a common classification metric. There were only eight normal-like and eight claudin-low tumours27, thus we did not perform focussed analyses on these two subtypes.
MicroRNA expression levels were assayed via Illumina sequencing, using 1,222 miRBase28 v16 mature and star strands as the reference database of miRNA transcripts/genes. Seven subtypes were identified by consensus non-negative matrix factorization (NMF) clustering using an abundance matrix containing the 25% most variable miRNAs (306 transcripts/genes or MIMATs (miRNA IDs)). These subtypes correlated with mRNA subtypes, ER, PR and HER2 clinical status (Supplementary Fig. 7). Of note, miRNA groups 4 and 5 showed high overlap with the basal-like mRNA subtype and contained many TP53 mutations. The remaining miRNA groups (1–3, 6 and 7) were composed of a mixture of luminal A, luminal B and HER2E with little correlation with the PAM50 defined subtypes. With the exception of _TP53_—which showed a strong positive correlation—and PIK3CA and _GATA3_—which showed negative associations with groups 4 and 5, respectively—there was little correlation with mutation status and miRNA subtype.
DNA methylation
Illumina Infinium DNA methylation arrays were used to assay 802 breast tumours. Data from HumanMethylation27 (HM27) and HumanMethylation450 (HM450) arrays were combined and filtered to yield a common set of 574 probes used in an unsupervised clustering analysis, which identified five distinct DNA methylation groups (Supplementary Fig. 8). Group 3 showed a hypermethylated phenotype and was significantly enriched for luminal B mRNA subtype and under-represented for PIK3CA, MAP3K1 and MAP2K4 mutations. Group 5 showed the lowest levels of DNA methylation, overlapped with the basal-like mRNA subtype, and showed a high frequency of TP53 mutations. HER2-positive (HER2+) clinical status, or the HER2E mRNA subtype, had only a modest association with the methylation subtypes.
A supervised analysis of the DNA methylation and mRNA expression data was performed to compare DNA methylation group 3 (N = 49) versus all tumours in groups 1, 2 and 4 (excluding group 5, which consisted predominantly of basal-like tumours). This analysis identified 4,283 genes differentially methylated (3,735 higher in group 3 tumours) and 1,899 genes differentially expressed (1,232 downregulated); 490 genes were both methylated and showed lower expression in group 3 tumours (Supplementary Table 4). A DAVID (database for annotation, visualization and integrated discovery) functional annotation analysis identified ‘extracellular region part’ and ‘Wnt signalling pathway’ to be associated with this 490-gene set; the group 3 hypermethylated samples showed fewer PIK3CA and MAP3K1 mutations, and lower expression of Wnt-pathway genes.
DNA copy number
A total of 773 breast tumours were assayed using Affymetrix 6.0 SNP arrays. Segmentation analysis and GISTIC were used to identify focal amplifications/deletions and arm-level gains and losses (Supplementary Table 5). These analyses confirmed all previously reported copy number variations and highlighted a number of significantly mutated genes including focal amplification of regions containing PIK3CA, EGFR, FOXA1 and HER2, as well as focal deletions of regions containing MLL3, PTEN, RB1 and MAP2K4 (Supplementary Fig. 9); in all cases, multiple genes were included within each altered region. Importantly, many of these copy number changes correlated with mRNA subtype including characteristic loss of 5q and gain of 10p in basal-like cancers5,29 and gain of 1q and/or 16q loss in luminal tumours4. NMF clustering of GISTIC segments identified five copy number clusters/groups that correlated with mRNA subtypes, ER, PR and HER2 clinical status, and TP53 mutation status (Supplementary Fig. 10). In addition, this aCGH subtype classification was highly correlated with the aCGH subtypes recently defined by ref. 30 (Supplementary Fig. 11).
Reverse phase protein arrays
Quantified expression of 171 cancer-related proteins and phospho-proteins by RPPA was performed on 403 breast tumours31. Unsupervised hierarchical clustering analyses identified seven subtypes; one class contained too few cases for further analysis (Supplementary Fig. 12). These protein subtypes were highly concordant with the mRNA subtypes, particularly with basal-like and HER2E mRNA subtypes. Closer examination of the HER2-containing RPPA-defined subgroup showed coordinated overexpression of HER2 and EGFR with a strong concordance with phosphorylated HER2 (pY1248) and EGFR (pY992), probably from heterodimerization and cross-phosphorylation. Although there is a potential for modest cross reactivity of antibodies against these related total and phospho-proteins, the concordance of phosphorylation of HER2 and EGFR was confirmed using multiple independent antibodies.
In RPPA-defined luminal tumours, there was high protein expression of ER, PR, AR, BCL2, GATA3 and INPP4B, defining mostly luminal A cancers and a second more heterogeneous protein subgroup composed of both luminal A and luminal B cancers. Two potentially novel protein-defined subgroups were identified: reactive I consisted primarily of a subset of luminal A tumours, whereas reactive II consisted of a mixture of mRNA subtypes. These groups are termed ‘reactive’ because many of the characteristic proteins are probably produced by the microenvironment and/or cancer-activated fibroblasts including fibronectin, caveolin 1 and collagen VI. These two RPPA groups did not have a marked difference in the percentage tumour cell content when compared to each other, or the other protein subtypes, as assessed by SNP array analysis or pathological examination. In addition, supervised analyses of reactive I versus II groups using miRNA expression, DNA methylation, mutation, or DNA copy number data identified no significant differences between these groups, whereas similar supervised analyses using protein and mRNA expression identified many differences.
Multiplatform subtype discovery
To reveal higher-order structure in breast tumours based on multiple data types, significant clusters/subtypes from each of five platforms were analysed using a multiplatform data matrix subjected to unsupervised consensus clustering (Fig. 2). This ‘cluster of clusters’ (C-of-C) approach illustrated that basal-like cancers had the most distinct multiplatform signature as all the different platforms for the basal-like groups clustered together. To a great extent, the four major C-of-C subdivisions correlated well with the previously published mRNA subtypes (driven, in part, by the fact that the four intrinsic subtypes were one of the inputs). Therefore, we also performed C-of-C analysis with no mRNA data present (Supplementary Fig. 13) or with the 12 unsupervised mRNA subtypes (Supplementary Fig. 14), and in each case 4–6 groups were identified. Recent work identified ten copy-number-based subgroups in a 997 breast cancer set30. We evaluated this classification in a C-of-C analysis instead of our five-class copy number subtypes, with either the PAM50 (Supplementary Fig. 15) or 12 unsupervised mRNA subtypes (Supplementary Fig. 16); each of these C-of-C classifications was highly correlated with PAM50 mRNA subtypes and with the other C-of-C analyses (Fig. 2). The transcriptional profiling and RPPA platforms demonstrated a high correlation with the consensus structure, indicating that the information content from copy number aberrations, miRNAs and methylation is captured at the level of gene expression and protein function.
Figure 2: Coordinated analysis of breast cancer subtypes defined from five different genomic/proteomic platforms.
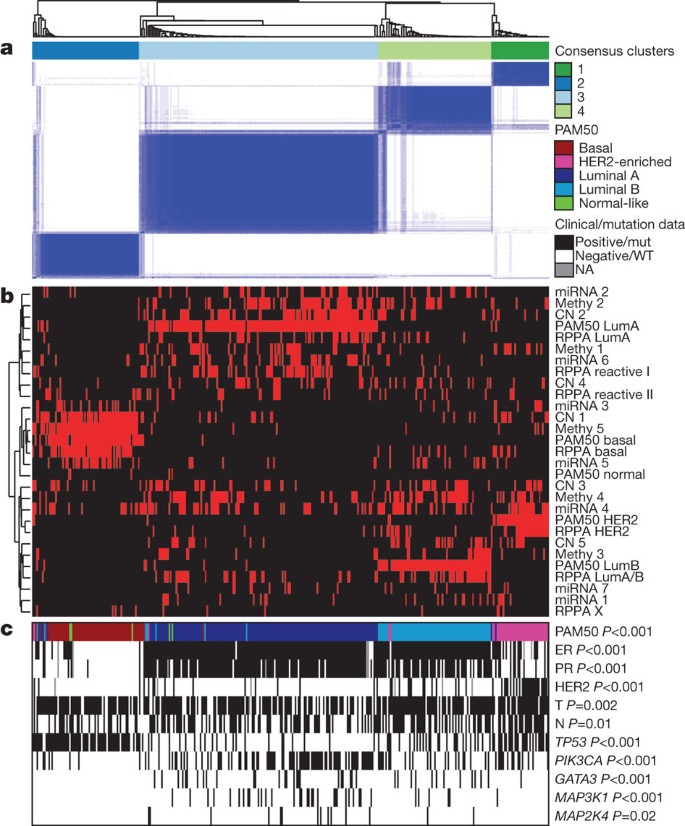
a, Consensus clustering analysis of the subtypes identifies four major groups (samples, n = 348). The blue and white heat map displays sample consensus. b, Heat-map display of the subtypes defined independently by miRNAs, DNA methylation, copy number (CN), PAM50 mRNA expression, and RPPA expression. The red bar indicates membership of a cluster type. c, Associations with molecular and clinical features. P values were calculated using a chi-squared test.
Luminal/ER+ summary analysis
Luminal/ER+ breast cancers are the most heterogeneous in terms of gene expression (Supplementary Fig. 5), mutation spectrum (Fig. 1), copy number changes (Supplementary Fig. 9) and patient outcomes1,14. One of the most dominant features is high mRNA and protein expression of the luminal expression signature (Supplementary Fig. 5), which contains ESR1, GATA3, FOXA1, XBP1 and MYB; the luminal/ER+ cluster also contained the largest number of significantly mutated genes. Most notably, GATA3 and FOXA1 were mutated in a mutually exclusive fashion, whereas ESR1 and XBP1 were typically highly expressed but infrequently mutated. Mutations in RUNX1 and its dimerization partner CBFB may also have a role in aberrant ER signalling in luminal tumours, as RUNX1 functions as an ER ‘DNA tethering factor’32. PARADIGM33 analysis comparing luminal versus basal-like cancers further emphasized the presence of a hyperactivated FOXA1–ER complex as a critical network hub differentiating these two tumour subtypes (Supplementary Fig. 17).
A confirmatory finding here was the high mutation frequency of PIK3CA in luminal/ER+ breast cancers34,35. Through multiple technology platforms, we examined possible relationships between PIK3CA mutation, PTEN loss, INPP4B loss and multiple gene and protein expression signatures of pathway activity. RPPA data demonstrated that pAKT, pS6 and p4EBP1, typical markers of phosphatidylinositol-3-OH kinase (PI(3)K) pathway activation, were not elevated in _PIK3CA_-mutated luminal A cancers; instead, they were highly expressed in basal-like and HER2E mRNA subtypes (the latter having frequent PIK3CA mutations) and correlated strongly with INPP4B and PTEN loss, and to a degree with PIK3CA amplification. Similarly, protein36 and three mRNA signatures37,38,39 of PI(3)K pathway activation were enriched in basal-like over luminal A cancers (Fig. 3a). This apparent disconnect between the presence of PIK3CA mutations and biomarkers of pathway activation has been previously noted36.
Figure 3: Integrated analysis of the PI(3)K, TP53 and RB1 pathways.
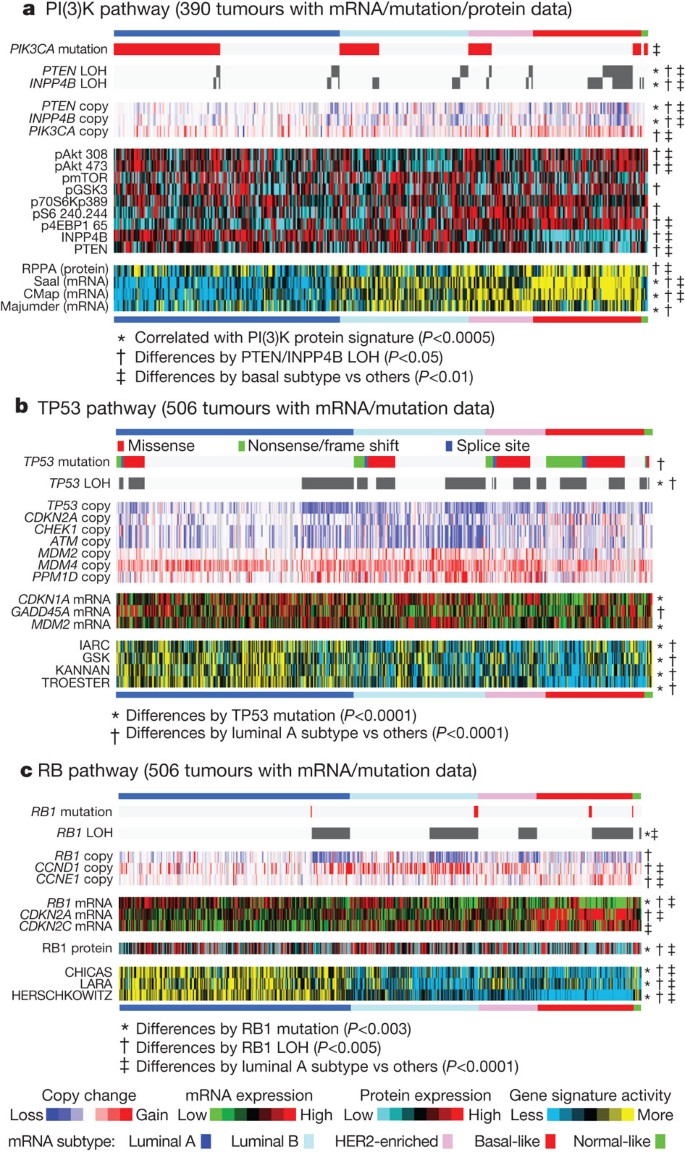
Breast cancer subtypes differ by genetic and genomic targeting events, with corresponding effects on pathway activity. a–c, For PI(3)K (a), TP53 (b) and RB1 (c) pathways, key genes were selected using prior biological knowledge. Multiple mRNA expression signatures for a given pathway were defined (details in Supplementary Methods; PI(3)K:Saal, PTEN loss in human breast tumours; CMap, PI(3)K/mTOR inhibitor treatment in vitro; Majumder, Akt overexpression in mouse model; TP53: IARC, expert-curated p53 targets; GSK, TP53 mutant versus wild-type cell lines; KANNAN, TP53 overexpression in vitro; TROESTER, TP53 knockdown in vitro; RB: CHICAS, RB1 mouse knockout versus wild type; LARA, RB1 knockdown in vitro; HERSCHKOWITZ, RB1 loss of heterozygosity (LOH) in human breast tumours) and applied to the gene expression data, in order to score each tumour for relative signature activity (yellow, more active). The PI(3)K panel includes a protein-based (RPPA) proteomic signature. Tumours were ordered first by mRNA subtype, although specific ordering differs between the panels. P values were calculated by a Pearson’s correlation or a Chi-squared test.
Another striking luminal/ER+ subtype finding was the frequent mutation of MAP3K1 and MAP2K4, which represent two contiguous steps within the p38–JNK1 pathway24,40. These mutations are predicted to be inactivating, with MAP2K4 also a target of focal DNA loss in luminal tumours (Supplementary Fig. 9). To explore the possible interplay between PIK3CA, MAP3K and MAP2K4 signalling, MEMo analysis41 was performed to identify mutually exclusive alterations targeting frequently altered genes likely to belong to the same pathway (Fig. 4). Across all breast cancers, MEMo identified a set of modules that highlight the differential activation events within the receptor tyrosine kinase (RTK)–PI(3)K pathway (Fig. 4a); mutations of PIK3CA were very common in luminal/ER+ cancers whereas PTEN loss was more common in basal-like tumours. Almost all _MAP3K1_and MAP2K4 mutations were in luminal tumours, yet MAP3K1 and MAP2K4 appeared almost mutually exclusive relative to one another.
Figure 4: Mutual exclusivity modules in cancer (MEMo) analysis.
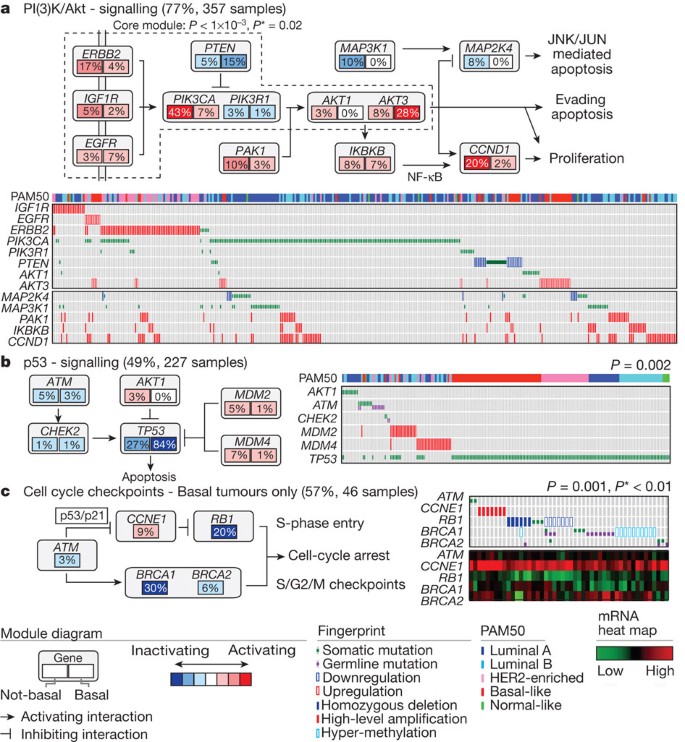
Mutual exclusivity modules are represented by their gene components and connected to reflect their activity in distinct pathways. For each gene, the frequency of alteration in basal-like (right box) and non-basal (left box) is reported. Next to each module is a fingerprint indicating what specific alteration is observed for each gene (row) in each sample (column). a, MEMo identified several overlapping modules that recapitulate the RTK–PI(3)K and p38–JNK1 signalling pathways and whose core was the top-scoring module. b, MEMo identified alterations to TP53 signalling as occurring within a statistically significant mutually exclusive trend. c, A basal-like only MEMo analysis identified one module that included ATM mutations, defects at BRCA1 and BRCA2, and deregulation of the RB1 pathway. A gene expression heat map is below the fingerprint to show expression levels.
The TP53 pathway was differentially inactivated in luminal/ER+ breast cancers, with a low TP53 mutation frequency in luminal A (12%) and a higher frequency in luminal B (29%) cancers (Fig. 1). In addition to TP53 itself, a number of other pathway-inactivating events occurred including ATM loss and MDM2 amplification (Figs 3b and 4b), both of which occurred more frequently within luminal B cancers. Gene expression analysis demonstrated that individual markers of functional TP53 (GADD45A and CDKN1A), and TP53 activity42,43 signatures, were highest in luminal A cancers (Fig. 3b). These data indicate that the TP53 pathway remains largely intact in luminal A cancers but is often inactivated in the more aggressive luminal B cancers44. Other PARADIGM-based pathway differences driving luminal B versus luminal A included hyperactivation of transcriptional activity associated with MYC and FOXM1 proliferation.
The critical retinoblastoma/RB1 pathway also showed mRNA-subtype-specific alterations (Fig. 3c). RB1 itself, by mRNA and protein expression, was detectable in most luminal cancers, with highest levels within luminal A. A common oncogenic event was cyclin D1 amplification and high expression, which preferentially occurred within luminal tumours, and more specifically within luminal B. In contrast, the presumed tumour suppressor CDKN2C (also called p18) was at its lowest levels in luminal A cancers, consistent with observations in mouse models45. Finally, RB1 activity signatures were also high in luminal cancers46,47,48. Luminal A tumours, which have the best prognosis, are the most likely to retain activity of the major tumour suppressors RB1 and TP53.
These genomic characterizations also provided clues for druggable targets. We compiled a drug target table in which we defined a target as a gene/protein for which there is an approved or investigational drug in human clinical trials targeting the molecule or canonical pathway (Supplementary Table 6). In luminal/ER+ cancers, the high frequency of PIK3CA mutations suggests that inhibitors of this activated kinase or its signalling pathway may be beneficial. Other potential significantly mutated gene drug candidates include AKT1 inhibitors (11 out of 12 AKT1 variants were luminal) and PARP inhibitors for BRCA1/BRCA2 mutations. Although still unapproved as biomarkers, many potential copy-number-based drug targets were identified including amplifications of fibroblast growth factor receptors (FGFRs) and IGFR1, as well as cyclin D1, CDK4 and CDK6. A summary of the general findings in luminal tumours and the other subtypes is presented in Table 1.
Table 1 Highlights of genomic, clinical and proteomic features of subtypes
HER2-based classifications and summary analysis
DNA amplification of HER2 was readily evident in this study (Supplementary Fig. 9) together with overexpression of multiple HER2-amplicon-associated genes that in part define the HER2E mRNA subtype (Supplementary Fig. 5). However, not all clinically HER2+ tumours are of the HER2E mRNA subtype, and not all tumours in the HER2E mRNA subtype are clinically HER2+. Integrated analysis of the RPPA and mRNA data clearly identified a HER2+ group (Supplementary Fig. 12). When the HER2+ protein and HER2E mRNA subtypes overlapped, a strong signal of EGFR, pEGFR, HER2 and pHER2 was observed. However, only ∼50% of clinically HER2+ tumours fall into this HER2E-mRNA-subtype/HER2-protein group, the rest of the clinically HER2+ tumours were observed predominantly in the luminal mRNA subtypes.
These data indicate that there exist at least two types of clinically defined HER2+ tumours. To identify differences between these groups, a supervised gene expression analysis comparing 36 HER2E-mRNA-subtype/HER2+ versus 31 luminal-mRNA-subtype/HER2+ tumours was performed and identified 302 differentially expressed genes (_q_-value = 0%) (Supplementary Fig. 18 and Supplementary Table 7). These genes largely track with ER status but also indicated that HER2E-mRNA-subtype/HER2+ tumours showed significantly higher expression of a number of RTKs including FGFR4, EGFR, HER2 itself, as well as genes within the HER2 amplicon (including GRB7). Conversely, the luminal-mRNA-subtype/HER2+ tumours showed higher expression of the luminal cluster of genes including GATA3, BCL2 and ESR1. Further support for two types of clinically defined HER2+ disease was evident in the somatic mutation data supervised by either mRNA subtype or ER status; TP53 mutations were significantly enriched in HER2E or ER-negative tumours whereas GATA3 mutations were only observed in luminal subtypes or ER+ tumours.
Analysis of the RPPA data according to mRNA subtype identified 36 differentially expressed proteins (_q_-value <5%) (Supplementary Fig. 18G and Supplementary Table 8). The EGFR/pEGFR/HER2/pHER2 signal was again observed and present within the HER2E-mRNA-subtype/HER2+ tumours, as was high pSRC and pS6; conversely, many protein markers of luminal cancers again distinguished the luminal-mRNA-subtype/HER2+ tumours. Given the importance of clinical HER2 status, a more focused analysis was performed based on the RPPA-defined protein expression of HER2 (Supplementary Fig. 19)—the results strongly recapitulated findings from the RPPA and mRNA subtypes including a high correlation between HER2 clinical status, HER2 protein by RPPA, pHER2, EGFR and pEGFR. These multiple signatures, namely HER2E mRNA subtype, HER2 amplicon genes by mRNA expression, and RPPA EGFR/pEGFR/HER2/pHER2 signature, ultimately identify at least two groups/subtypes within clinically HER2+ tumours (Table 1). These signatures represent breast cancer biomarker(s) that could potentially predict response to anti-HER2 targeted therapies.
Many therapeutic advances have been made for clinically HER2+ disease. This study has identified additional somatic mutations that represent potential therapeutic targets within this group, including a high frequency of PIK3CA mutations (39%), a lower frequency of PTEN and PIK3R1 mutations (Supplementary Table 6), and genomic losses of PTEN and INPP4B. Other possible druggable mutations included variants within HER family members including two somatic mutations in HER2, two within EGFR, and five within HER3. Pertuzumab, in combination with trastuzumab, targets the HER2–HER3 heterodimer49; however, these data suggest that targeting EGFR with HER2 could also be beneficial. Finally, the HER2E mRNA subtype typically showed high aneuploidy, the highest somatic mutation rate (Table 1), and DNA amplification of other potential therapeutic targets including FGFRs, EGFR, CDK4 and cyclin D1.
Basal-like summary analysis
The basal-like subtype was discovered more than a decade ago by first-generation cDNA microarrays13. These tumours are often referred to as triple-negative breast cancers (TNBCs) because most basal-like tumours are typically negative for ER, PR and HER2. However, ∼75% of TNBCs are basal-like with the other 25% comprised of all other mRNA subtypes6. In this data set, there was a high degree of overlap between these two distinctions with 76 TNBCs, 81 basal-like, and 65 that were both TNBCs and basal-like. Given the known heterogeneity of TNBCs, and that the basal-like subtype proved to be distinct on every platform, we chose to use the basal-like distinction for comparative analyses.
Basal-like tumours showed a high frequency of TP53 mutations (80%)9, which when combined with inferred TP53 pathway activity suggests that loss of TP53 function occurs within most, if not all, basal-like cancers (Fig. 3b). In addition to loss of TP53, a MEMo analysis reconfirmed that loss of RB1 and BRCA1 are basal-like features (Fig. 4c)47,50. PIK3CA was the next most commonly mutated gene (∼9%); however, inferred PI(3)K pathway activity, whether from gene37,38,39, protein36, or high PI(3)K/AKT pathway activities, was highest in basal-like cancers (Fig. 3a). Alternative means of activating the PI(3)K pathway in basal-like cancers probably includes loss of PTEN and INPP4B and/or amplification of PIK3CA. A recent paper12 performed exome sequencing of 102 TNBCs. Five of the top six most frequent TNBC mutations in ref. 12 were also observed at a similar frequency in our TNBC subset (Myo3A not present here); of those five, three passed our test as a significantly mutated gene in TNBCs (Supplementary Table 2).
Expression features of basal-like tumours include a characteristic signature containing keratins 5, 6 and 17 and high expression of genes associated with cell proliferation (Supplementary Fig. 5). A PARADIGM33 analysis of basal-like versus luminal tumours emphasized the importance of hyperactivated FOXM1 as a transcriptional driver of this enhanced proliferation signature (Supplementary Fig. 17). PARADIGM also identified hyperactivated MYC and HIF1-α/ARNT network hubs as key regulatory features of basal-like cancers. Even though chromosome 8q24 is amplified across all subtypes (Supplementary Fig. 9), high MYC activation seems to be a basal-like characteristic51.
Given the striking contrasts between basal-like and luminal/HER2E subtypes, we performed a MEMo analysis on basal-like tumours alone. The top-scoring module included ATM mutations, BRCA1 and BRCA2 inactivation, RB1 loss and cyclin E1 amplification (Fig. 4c). Notably, these same modules were identified previously for serous ovarian cancers41. Furthermore, the basal-like (and TNBC) mutation spectrum was reminiscent of the spectrum seen in serous ovarian cancers52 with only one gene (that is, TP53) at >10% mutation frequency. To explore possible similarities between serous ovarian and the breast basal-like cancers, we performed a number of analyses comparing ovarian versus breast luminal, ovarian versus breast basal-like, and breast basal-like versus breast luminal cancers (Fig. 5). Comparing copy number landscapes, we observed several common features between ovarian and basal-like tumours including widespread genomic instability and common gains of 1q, 3q, 8q and 12p, and loss of 4q, 5q and 8p (Supplementary Fig. 20A). Using a more global copy number comparison, we examined the overall fraction of the genome altered and the overall copy number correlation of ovarian cancers versus each breast cancer mRNA subtype (Supplementary Fig. 20A, B); in both cases, basal-like tumours were the most similar to the serous ovarian carcinomas.
Figure 5: Comparison of breast and serous ovarian carcinomas.
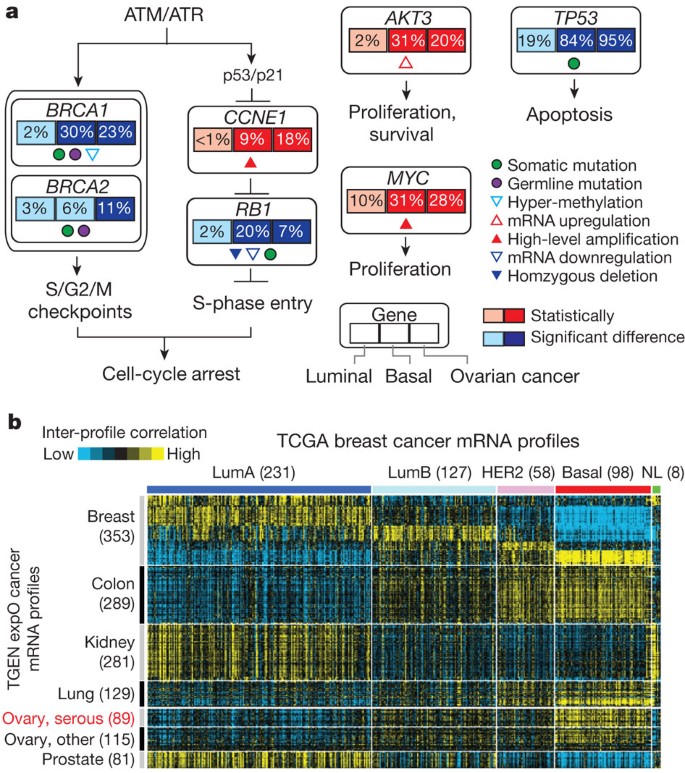
a, Significantly enriched genomic alterations identified by comparing basal-like or serous ovarian tumours to luminal cancers. b, Inter-sample correlations (yellow, positive) between gene transcription profiles of breast tumours (columns; TCGA data, arranged by subtype) and profiles of cancers from various tissues of origin (rows; external ‘TGEN expO’ data set, GSE2109) including ovarian cancers.
We systematically looked for other common features between serous ovarian and basal-like tumours when each was compared to luminal. We identified: (1) BRCA1 inactivation; (2) RB1 loss and cyclin E1 amplification; (3) high expression of AKT3; (4) MYC amplification and high expression; and (5) a high frequency of TP53 mutations (Fig. 5a). An additional supervised analysis of a large, external multitumour type transcriptomic data set (Gene Expression Omnibus accession GSE2109) was performed where each TCGA (The Cancer Genome Atlas) breast tumour expression profile was compared via a correlation analysis to that of each tumour in the multitumour set. Basal-like breast cancers clearly showed high mRNA expression correlations with serous ovarian cancers, as well as with lung squamous carcinomas (Fig. 5b). A PARADIGM analysis that calculates whether a gene or pathway feature is both differentially activated in basal-like versus luminal cancers and has higher overall activity across the TCGA ovarian samples was performed; this identified comparably high pathway activity of the HIF1-α/ARNT, MYC and FOXM1 regulatory hubs in both ovarian and basal-like cancers (Supplementary Fig. 20C). The common findings of TP53, RB1 and BRCA1 loss, with MYC amplification, strongly suggest that these are shared driving events for basal-like and serous ovarian carcinogenesis. This suggests that common therapeutic approaches should be considered, which is supported by the activity of platinum analogues and taxanes in breast basal-like and serous ovarian cancers.
Given that most basal-like cancers are TNBCs, finding new drug targets for this group is critical. Unfortunately, the somatic mutation repertoire for basal-like breast cancers has not provided a common target aside from BRCA1 and BRCA2. Here we note that ∼20% of basal-like tumours had a germline (n = 12) and/or somatic (n = 8) BRCA1 or BRCA2 variant, which suggests that one in five basal-like patients might benefit from PARP inhibitors and/or platinum compounds53,54. The copy number landscape of basal-like cancers showed multiple amplifications and deletions, some of which may provide therapeutic targets (Supplementary Table 6). Potential targets include losses of PTEN and INPP4B, both of which have been shown to sensitize cell lines to PI(3)K pathway inhibitors55,56. Interestingly, many of the components of the PI(3)K and RAS–RAF–MEK pathway were amplified (but not typically mutated) in basal-like cancers including PIK3CA (49%), KRAS (32%), BRAF (30%) and EGFR (23%). Other RTKs that are plausible drug targets and amplified in some basal-like cancers include FGFR1, FGFR2, IGFR1, KIT, MET and PDGFRA. Finally, the PARADIGM identification of high HIF1-α/ARNT pathway activity suggests that these malignancies might be susceptible to angiogenesis inhibitors and/or bioreductive drugs that become activated under hypoxic conditions.
Concluding remarks
The integrated molecular analyses of breast carcinomas that we report here significantly extends our knowledge base to produce a comprehensive catalogue of likely genomic drivers of the most common breast cancer subtypes (Table 1). Our novel observation that diverse genetic and epigenetic alterations converge phenotypically into four main breast cancer classes is not only consistent with convergent evolution of gene circuits, as seen across multiple organisms, but also with models of breast cancer clonal expansion and in vivo cell selection proposed to explain the phenotypic heterogeneity observed within defined breast cancer subtypes.
Methods Summary
Specimens were obtained from patients with appropriate consent from institutional review boards. Using a co-isolation protocol, DNA and RNA were purified. In total, 800 patients were assayed on at least one platform. Different numbers of patients were used for each platform using the largest number of patients available at the time of data freeze; 466 samples (463 patients) were in common across 5 out of 6 platforms (excluding RPPA) and 348 patients were in common on 6 out of 6 platforms. Technology platforms used include: (1) gene expression DNA microarrays52; (2) DNA methylation arrays; (3) miRNA sequencing; (4) Affymetrix SNP arrays; (5) exome sequencing; and (6) reverse phase protein arrays. Each platform, except for the exome sequencing, was used in a de novo subtype discovery analysis (Supplementary Methods) and then included in a single analysis to define an overall subtype architecture. Additional integrated across-platform computational analyses were preformed including PARADIGM33 and MEMo41.
Change history
03 October 2012
The spelling of an author name (J.B.) was corrected.
References
- Paik, S. et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 351, 2817–2826 (2004)
Article CAS Google Scholar - van 't Veer, L. J. et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536 (2002)
Article CAS Google Scholar - Slamon, D. J. et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182 (1987)
Article ADS CAS Google Scholar - Chin, K. et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 10, 529–541 (2006)
Article CAS Google Scholar - Bergamaschi, A. et al. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosom. Cancer 45, 1033–1040 (2006)
Article CAS Google Scholar - Perou, C. M. Molecular stratification of triple-negative breast cancers. Oncologist 16 (suppl. 1). 61–70 (2011)
Article Google Scholar - Sorlie, T. et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl Acad. Sci. USA 100, 8418–8423 (2003)
Article ADS CAS Google Scholar - Foulkes, W. D. et al. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl Cancer Inst. 95, 1482–1485 (2003)
Article CAS Google Scholar - Carey, L. A. et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. J. Am. Med. Assoc. 295, 2492–2502 (2006)
Article CAS Google Scholar - Ding, L. et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005 (2010)
Article ADS CAS Google Scholar - Shah, S. P. et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461, 809–813 (2009)
Article ADS CAS Google Scholar - Shah, S. P. et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486, 395–399 (2012)
Article ADS CAS Google Scholar - Perou, C. M. et al. Molecular portraits of human breast tumours. Nature 406, 747–752 (2000)
Article ADS CAS Google Scholar - Parker, J. S. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27, 1160–1167 (2009)
Article Google Scholar - González-Pérez, A. & Lopez-Bigas, N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 88, 440–449 (2011)
Article Google Scholar - Dees, N. D. et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 22, 1589–1598 (2012)
Article CAS Google Scholar - Bamshad, M. et al. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nature Genet. 16, 311–315 (1997)
Article CAS Google Scholar - Li, Q. Y. et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nature Genet. 15, 21–29 (1997)
Article Google Scholar - The Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008)
- Cheung, L. W. et al. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of PTEN protein stability. Cancer Discov. 1, 170–185 (2011)
Article CAS Google Scholar - Malcovati, L. et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 118, 6239–6246 (2011)
Article CAS Google Scholar - Wang, L. et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 365, 2497–2506 (2011)
Article CAS Google Scholar - Ding, L. et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075 (2008)
Article ADS CAS Google Scholar - Johnson, G. L. & Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298, 1911–1912 (2002)
Article ADS CAS Google Scholar - Usary, J. et al. Mutation of GATA3 in human breast tumors. Oncogene 23, 7669–7678 (2004)
Article CAS Google Scholar - Walsh, T. et al. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc. Natl Acad. Sci. USA 107, 12629–12633 (2010)
Article ADS CAS Google Scholar - Prat, A. et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 12, R68 (2010)
Article Google Scholar - Kozomara, A. & Griffiths-Jones, S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 39, D152–D157 (2011)
Article CAS Google Scholar - Weigman, V. J. et al. Basal-like breast cancer DNA copy number losses identify genes involved in genomic instability, response to therapy, and patient survival. Breast Cancer Res. Treat. 133, 865–880 (2011)
Article Google Scholar - Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012)
Article CAS Google Scholar - Hennessy, B. T. et al. A technical assessment of the utility of reverse phase protein arrays for the study of the functional proteome in non-microdissected human breast cancers. Clin. Proteomics 6, 129–151 (2010)
Article CAS Google Scholar - Daub, H. et al. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol. Cell 31, 438–448 (2008)
Article CAS Google Scholar - Vaske, C. J. et al. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 26, 237–245 (2010)
Article Google Scholar - Campbell, I. G. et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 64, 7678–7681 (2004)
Article CAS Google Scholar - Bachman, K. E. et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol. Ther. 3, 772–775 (2004)
Article CAS Google Scholar - Stemke-Hale, K. et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 68, 6084–6091 (2008)
Article CAS Google Scholar - Creighton, C. J. et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 12, R40 (2010)
Article Google Scholar - Majumder, P. K. et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nature Med. 10, 594–601 (2004)
Article CAS Google Scholar - Saal, L. H. et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nature Genet. 40, 102–107 (2008)
Article CAS Google Scholar - Wagner, E. F. & Nebreda, A. R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nature Rev. Cancer 9, 537–549 (2009)
Article CAS Google Scholar - Ciriello, G., Cerami, E., Sander, C. & Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 22, 398–406 (2012)
Article CAS Google Scholar - Kannan, K. et al. DNA microarrays identification of primary and secondary target genes regulated by p53. Oncogene 20, 2225–2234 (2001)
Article CAS Google Scholar - Troester, M. A. et al. Gene expression patterns associated with p53 status in breast cancer. BMC Cancer 6, 276 (2006)
Article Google Scholar - Deisenroth, C., Thorner, A. R., Enomoto, T., Perou, C. M. & Zhang, Y. Mitochondrial Hep27 is a c-Myb target gene that inhibits Mdm2 and stabilizes p53. Mol. Cell. Biol. 30, 3981–3993 (2010)
Article CAS Google Scholar - Pei, X. H. et al. CDK inhibitor p18_INK4c_ is a downstream target of GATA3 and restrains mammary luminal progenitor cell proliferation and tumorigenesis. Cancer Cell 15, 389–401 (2009)
Article CAS Google Scholar - Chicas, A. et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 17, 376–387 (2010)
Article CAS Google Scholar - Herschkowitz, J. I., He, X., Fan, C. & Perou, C. M. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 10, R75 (2008)
Article Google Scholar - Lara, M. F. et al. Gene profiling approaches help to define the specific functions of retinoblastoma family in epidermis. Mol. Carcinog. 47, 209–221 (2008)
Article CAS Google Scholar - Baselga, J. et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N. Engl. J. Med. 366, 109–119 (2012)
Article CAS Google Scholar - Jiang, Z. et al. Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J. Clin. Invest. 120, 3296–3309 (2010)
Article CAS Google Scholar - Chandriani, S. et al. A core MYC gene expression signature is prominent in basal-like breast cancer but only partially overlaps the core serum response. PLoS ONE 4, e6693 (2009)
Article ADS Google Scholar - The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 (2011)
- Audeh, M. W. et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376, 245–251 (2010)
Article CAS Google Scholar - Fong, P. C. et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 361, 123–134 (2009)
Article CAS Google Scholar - Fedele, C. G. et al. Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc. Natl Acad. Sci. USA 107, 22231–22236 (2010)
Article ADS CAS Google Scholar - Gewinner, C. et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell 16, 115–125 (2009)
Article CAS Google Scholar
Acknowledgements
We thank M. Sheth and S. Lucas for administrative coordination of TCGA activities, and C. Gunter for critical reading of the manuscript. This work was supported by the following grants from the USA National Institutes of Health: U24CA143883, U24CA143858, U24CA143840, U24CA143799, U24CA143835, U24CA143845, U24CA143882, U24CA143867, U24CA143866, U24CA143848, U24CA144025, U54HG003079, P50CA116201 and P50CA58223. Additional support was provided by the Susan G. Komen for the Cure, the US Department of Defense through the Henry M. Jackson Foundation for the Advancement of Military Medicine, and the Breast Cancer Research Foundation. The views expressed in this paper are those of the authors and do not reflect the official policy of the Department of Defense, or US Government.
Author information
Authors and Affiliations
- The Genome Institute, Washington University, St Louis, Missouri 63108, USA.,
Daniel C. Koboldt, Robert S. Fulton, Michael D. McLellan, Heather Schmidt, Joelle Kalicki-Veizer, Joshua F. McMichael, Lucinda L. Fulton, David J. Dooling, Li Ding, Elaine R. Mardis & Richard K. Wilson - Department of Genetics, Washington University, St Louis, Missouri 63110, USA.,
Li Ding, Elaine R. Mardis & Richard K. Wilson - Siteman Cancer Center, Washington University, St Louis, Missouri 63110, USA.,
Elaine R. Mardis, Richard K. Wilson, Matthew J. Ellis & Ron Bose - Canada’s Michael Smith Genome Sciences Centre, BC Cancer Agency, Vancouver, British Columbia V5Z, Canada.,
Adrian Ally, Miruna Balasundaram, Yaron S. N. Butterfield, Rebecca Carlsen, Candace Carter, Andy Chu, Eric Chuah, Hye-Jung E. Chun, Robin J. N. Coope, Noreen Dhalla, Ranabir Guin, Carrie Hirst, Martin Hirst, Robert A. Holt, Darlene Lee, Haiyan I. Li, Michael Mayo, Richard A. Moore, Andrew J. Mungall, Erin Pleasance, A. Gordon Robertson, Jacqueline E. Schein, Arash Shafiei, Payal Sipahimalani, Jared R. Slobodan, Dominik Stoll, Angela Tam, Nina Thiessen, Richard J. Varhol, Natasja Wye, Thomas Zeng, Yongjun Zhao, Inanc Birol, Steven J. M. Jones & Marco A. Marra - The Broad Institute of MIT and Harvard, Cambridge, 02142, Massachusetts, USA
Andrew D. Cherniack, Gordon Saksena, Robert C. Onofrio, Nam H. Pho, Scott L. Carter, Steven E. Schumacher, Barbara Tabak, Bryan Hernandez, Jeff Gentry, Huy Nguyen, Andrew Crenshaw, Kristin Ardlie, Rameen Beroukhim, Wendy Winckler, Gad Getz, Stacey B. Gabriel & Matthew Meyerson - Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, Massachusetts 02115, USA.,
Steven E. Schumacher & Barbara Tabak - Department of Medicine, Harvard Medical School, Boston, 02215, Massachusetts, USA
Rameen Beroukhim - Departments of Cancer Biology and Medical Oncology, and the Center for Cancer Genome Discovery, Dana-Farber Cancer Institute, Boston, Massachusetts 02115, USA.,
Rameen Beroukhim - Department of Medical Oncology and the Center for Cancer Genome Discovery, Dana-Farber Cancer Institute, Boston, Massachusetts 02115, USA.,
Matthew Meyerson & Lynda Chin - Department of Pathology, Harvard Medical School, Boston, 02215, Massachusetts, USA
Matthew Meyerson & Andrew H. Beck - Belfer Institute for Applied Cancer Science, Dana-Farber Cancer Institute, Boston, Massachusetts 02115, USA.,
Lynda Chin - The Center for Biomedical Informatics, Harvard Medical School, Boston, 02115, Massachusetts, USA
Peter J. Park, Nils Gehlenborg & Peter J. Park - Department of Genetics, Harvard Medical School and Division of Genetics, Brigham and Women’s Hospital, Boston, 02115, Massachusetts, USA
Raju Kucherlapati - Department of Genetics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA.,
Katherine A. Hoadley, Xiaping He, Hann-Hsiang Chao, Aleix Prat, Grace O. Silva, Michael D. Iglesia, Wei Zhao, Jonathan S. Berg, Michael Adams & Charles M. Perou - Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, 27599, North Carolina, USA
Katherine A. Hoadley, Cheng Fan, Yidi J. Turman, Yan Shi, Ling Li, Michael D. Topal, Xiaping He, Hann-Hsiang Chao, Aleix Prat, Grace O. Silva, Michael D. Iglesia, Wei Zhao, Jerry Usary, Jonathan S. Berg, Junyuan Wu, Anisha Gulabani, Tom Bodenheimer, Alan P. Hoyle, Janae V. Simons, Matthew G. Soloway, Lisle E. Mose, Stuart R. Jefferys, Saianand Balu, Joel S. Parker, D. Neil Hayes, Charles M. Perou, W. Kimryn Rathmell, Leigh Thorne, Mei Huang, Lori Boice & Ashley Hill - Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA.,
J. Todd Auman - Institute for Pharmacogenetics and Individualized Therapy, University of North Carolina at Chapel Hill, Chapel Hill, 27599, North Carolina, USA
J. Todd Auman - Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, Chapel Hill, Chapel Hill, 27599, North Carolina, USA
Michael D. Topal, Jessica Booker & Charles M. Perou - Department of Internal Medicine, Division of Medical Oncology, University of North Carolina at Chapel Hill, Chapel Hill, 27599, North Carolina, USA
D. Neil Hayes - USC Epigenome Center, University of Southern California, Los Angeles, California 90033, USA.,
Simeen Malik, Swapna Mahurkar, Hui Shen, Daniel J. Weisenberger, Timothy Triche Jr, Phillip H. Lai, Moiz S. Bootwalla, Dennis T. Maglinte, Benjamin P. Berman, David J. Van Den Berg & Peter W. Laird - Cancer Biology Division, The Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins University, Baltimore, 21231, Maryland, USA
Stephen B. Baylin - Dan L Duncan Cancer Center, Baylor College of Medicine, Houston, 77030, Texas, USA
Chad J. Creighton & Lawrence A. Donehower - Human Genome Sequencing Center, Baylor College of Medicine, Houston, 77030, Texas, USA
Chad J. Creighton & Lawrence A. Donehower - Department of Molecular and Cellular Biology, Baylor College of Medicine, Houston, 77030, Texas, USA
Lawrence A. Donehower - Department of Molecular Virology and Microbiology, Baylor College of Medicine, Houston, Texas 77030, USA.,
Lawrence A. Donehower - The Eli and Edythe L. Broad Institute of Massachusetts Institute Of Technology and Harvard University, Cambridge, 02142, Massachusetts, USA
Gad Getz, Michael Noble, Doug Voet, Gordon Saksena, Nils Gehlenborg, Daniel DiCara, Hailei Zhang, Spring Yingchun Liu, Michael S. Lawrence, Lihua Zou, Andrey Sivachenko, Pei Lin, Petar Stojanov, Rui Jing, Juok Cho, Raktim Sinha, Richard W. Park, Marc-Danie Nazaire, Jim Robinson, Helga Thorvaldsdottir, Jill Mesirov & Lynda Chin - Department of Genomic Medicine, Institute for Applied Cancer Science, University of Texas MD Anderson Cancer Center, Houston, 77054, Texas, USA
Juinhua Zhang & Lynda Chin - Department of Genomic Medicine, University of Texas MD Anderson Cancer Center, Houston, 77054, Texas, USA
Chang-Jiun Wu - Division of Genetics, Brigham and Women’s Hospital, Boston, 02115, Massachusetts, USA
Peter J. Park - Informatics Program, Children’s Hospital, Boston, 02115, Massachusetts, USA
Peter J. Park - Institute for Systems Biology, Seattle, 98109, Washington, USA
Sheila Reynolds, Richard B. Kreisberg, Brady Bernard, Ryan Bressler, Jake Lin, Vesteinn Thorsson & Ilya Shmulevich - Tampere University of Technology, Tampere, Finland
Timo Erkkila - Cancer Genomics Core Laboratory, MD Anderson Cancer Center, Houston, 77030, Texas, USA
Wei Zhang - Computational Biology Center, Memorial Sloan-Kettering Cancer Center, New York, 10065, New York, USA
Giovanni Ciriello, Nils Weinhold, Nikolaus Schultz, Jianjiong Gao, Ethan Cerami, Benjamin Gross, Anders Jacobsen, Rileen Sinha, B. Arman Aksoy, Yevgeniy Antipin, Boris Reva, Barry S. Taylor & Chris Sander - Department of Epidemiology and Biostatistics, Memorial Sloan-Kettering Cancer Center, New York, 10065, New York, USA
Ronglai Shen - Human Oncology and Pathogenesis Program, Memorial Sloan-Kettering Cancer Center, New York, 10065, New York, USA
Marc Ladanyi - Oregon Health and Science University, 3181 Southwest Sam Jackson Park Road, Portland, Oregon 97239, USA.,
Pavana Anur & Paul T. Spellman - Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston, 77030, Texas, USA
Yiling Lu, Gordon B. Mills, Ana Maria Gonzalez-Angulo & Ana Maria Gonzalez-Angulo - Kleberg Center for Molecular Markers, The University of Texas MD Anderson Cancer Center, Houston, 77030, Texas, USA
Yiling Lu & Gordon B. Mills - Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, 77030, Texas, USA
Wenbin Liu, Roel R. G. Verhaak, Rehan Akbani, Nianxiang Zhang, Bradley M. Broom, Tod D. Casasent, Chris Wakefield, Anna K. Unruh, Keith Baggerly, Kevin Coombes & John N. Weinstein - Department of Biomolecular Engineering and Center for Biomolecular Science and Engineering, University of California Santa Cruz, Santa Cruz, 95064, California, USA
David Haussler, Joshua M. Stuart, Stephen C. Benz, Jingchun Zhu, Christopher C. Szeto, Evan O. Paull, Daniel Carlin, Christopher Wong, Artem Sokolov, Sam Ng, Theodore C. Goldstein, Kyle Ellrott, Mia Grifford, Christopher Wilks, Singer Ma & Brian Craft - Howard Hughes Medical Institute, University of California Santa Cruz, Santa Cruz, 95064, California, USA
David Haussler - Buck Institute for Research on Aging, Novato, 94945, California, USA
Christopher C. Benz, Gary K. Scott, Christina Yau, Janita Thusberg & Sean Mooney - Center for Bioinformatics and Information Technology, National Cancer Institute, Rockville, 20852, Maryland, USA
Chunhua Yan, Ying Hu, Daoud Meerzaman & Kenneth Buetow - Department of Pathology, The Ohio State University College of Medicine, Columbus, 43205, Ohio, USA
Julie M. Gastier-Foster, Nilsa C. Ramirez & Robert E. Pyatt - Department Pediatrics, The Ohio State University College of Medicine, Columbus, 43205, Ohio, USA
Julie M. Gastier-Foster & Peter White - The Research Institute at Nationwide Children’s Hospital, Columbus, 43205, Ohio, USA
Julie M. Gastier-Foster, Jay Bowen, Nilsa C. Ramirez, Aaron D. Black, Robert E. Pyatt, Peter White, Erik J. Zmuda, Jessica Frick, Tara M. Lichtenberg, Robin Brookens, Myra M. George, Mark A. Gerken, Hollie A. Harper, Kristen M. Leraas, Lisa J. Wise, Teresa R. Tabler, Cynthia McAllister, Thomas Barr & Melissa Hart-Kothari - ABS Inc., Indianapolis, 46204, Indiana, USA
Katie Tarvin - ABS Inc., Wilmington, 19801, Delaware, USA
Charles Saller - Indiana University School of Medicine, Indianapolis, 46202, Indiana, USA
George Sandusky & Colleen Mitchell - Helen F. Graham Cancer Center, Christiana Care, Newark, 19713, Delaware, USA
Mary V. Iacocca, Jennifer Brown, Brenda Rabeno, Christine Czerwinski & Nicholas Petrelli - Moscow City Clinical Oncology Dispensary 1 and the Central IHC Laboratory of the Moscow Health Department, Moscow 105005, Russia.,
Oleg Dolzhansky - Russian Cancer Research Center, Moscow 115478, Russia.,
Mikhail Abramov - Cureline, Inc., South San Francisco, 94080, California, USA
Olga Voronina & Olga Potapova - Department of Surgery, Duke University Medical Center, Durham, 27710, North Carolina, USA
Jeffrey R. Marks & Jeffrey R. Marks - The Greater Poland Cancer Centre, Poznań 61-866, Poland.,
Wiktoria M. Suchorska, Dawid Murawa, Witold Kycler, Matthew Ibbs, Konstanty Korski, Arkadiusz Spychała, Paweł Murawa, Jacek J. Brzeziński, Hanna Perz, Radosław Łaźniak, Marek Teresiak, Honorata Tatka, Ewa Leporowska, Marta Bogusz-Czerniewicz, Julian Malicki, Andrzej Mackiewicz, Maciej Wiznerowicz & Maciej Wiznerowicz - Poznan University of Medical Sciences, Poznań 61-701, Poland.,
Marta Bogusz-Czerniewicz, Julian Malicki, Andrzej Mackiewicz, Maciej Wiznerowicz & Maciej Wiznerowicz - ILSBio, LLC, Chestertown, 21620, Maryland, USA
Xuan Van Le & Bernard Kohl - Ministry of Health, Hanoi, Vietnam
Nguyen Viet Tien - ILSBio LLC, Karachi, Pakistan
Richard Thorp & Khurram Zaki Khan - Hue Central Hospital, Hue City, Vietnam.,
Nguyen Van Bang & Bui Duc Phu - Stanford University Medical Center, Stanford, 94305, California, USA
Howard Sussman - Center for Minority Health Research, University of Texas, MD Anderson Cancer Center, Houston, 07703, Texas, USA
Richard Hajek - National Cancer Institute, Hanoi, Vietnam
Nguyen Phi Hung - Ho Chi Minh City Cancer Center, Vietnam
Tran Viet The Phuong - Can Tho Cancer Center, Can Tho, Vietnam.,
Huynh Quyet Thang - International Genomics Consortium, Phoenix, 85004, Arizona, USA
Robert Penny, David Mallery, Erin Curley, Candace Shelton & Peggy Yena - Mayo Clinic, Rochester, 55905, Minnesota, USA
James N. Ingle, Fergus J. Couch, Wilma L. Lingle, Fergus J. Couch & James N. Ingle - Department of Surgery, Breast Service, Memorial Sloan-Kettering Cancer Center, New York, 10065, New York, USA
Tari A. King & Tari A. King - Department of Breast Medical Oncology, The University of Texas, MD Anderson Cancer Center, Houston, 77030, Texas, USA
Ana Maria Gonzalez-Angulo, Gordon B. Mills, Mary D. Dyer, Shuying Liu, Xiaolong Meng, Modesto Patangan & Ana Maria Gonzalez-Angulo - University of California at San Francisco,
Frederic Waldman & Frederic Waldman - San Francisco, 94143, California, USA
Frederic Waldman & Frederic Waldman - Cancer Diagnostics; Nichols Institute, Quest Diagnostics; San Juan Capistrano,
Frederic Waldman & Frederic Waldman - Nichols Institute, Quest Diagnostics, 92675, California, USA
Frederic Waldman & Frederic Waldman - Helen Diller Family Comprehensive Cancer Center, University of California San Francisco, San Francisco, 94115, California, USA
Hubert Stöppler - Department of Pathology, UNC Tissue Procurement Facility, UNC Lineberger Cancer Center, Chapel Hill, 27599, North Carolina, USA
Leigh Thorne, Mei Huang & Lori Boice - Department of Pathology, Roswell Park Cancer Institute, Buffalo, 14263, New York, USA
Carl Morrison, Carmelo Gaudioso & Wiam Bshara - Department of Pathology, University of Miami Miller School of Medicine, Sylvester Comprehensive Cancer Center, Miami, 33136, Florida, USA
Kelly Daily, Sophie C. Egea, Mark D. Pegram & Carmen Gomez-Fernandez - University of Pittsburgh, Pittsburgh, 15213, Pennsylvania, USA
Rajiv Dhir - Magee-Womens Hospital of University of Pittsburgh Medical Center, Pittsburgh, 15213, Pennsylvania, USA
Rohit Bhargava & Adam Brufsky - Walter Reed National Military Medical Center, Bethesda, 20899-5600, Maryland, USA
Craig D. Shriver, Jeffrey A. Hooke & Jamie Leigh Campbell - Windber Research Institute, Windber, 15963, Pennsylvania, USA
Richard J. Mural, Hai Hu, Stella Somiari, Caroline Larson, Brenda Deyarmin, Leonid Kvecher, Hai Hu, Richard J. Mural, Chunqing Luo & Yaqin Chen - MDR Global, LLC, Windber, 15963, Pennsylvania, USA
Albert J. Kovatich - Breast Cancer Program, Washington University, St Louis, Missouri 63110, USA.,
Matthew J. Ellis & Ron Bose - Department of Internal Medicine, Division of Oncology, Washington University, St Louis, Missouri 63110, USA.,
Matthew J. Ellis & Ron Bose - Institute for Genomics and Systems Biology, University of Chicago, Chicago, 60637, Illinois, USA
Thomas Stricker & Kevin White - Center for Clinical Cancer Genetics, The University of Chicago, Chicago, 60637, Illinois, USA
Olufunmilayo Olopade - Department of Pathology and Immunology, Washington University School of Medicine, St Louis, Missouri 63110, USA.,
Li-Wei Chang - SRA International, 4300 Fair Lakes Court, Fairfax, 22033, Virginia, USA
Todd Pihl, Mark Jensen, Robert Sfeir, Ari Kahn, Anna Chu, Prachi Kothiyal, Zhining Wang, Eric Snyder, Joan Pontius, Brenda Ayala, Mark Backus, Jessica Walton, Julien Baboud, Dominique Berton, Matthew Nicholls, Deepak Srinivasan, Rohini Raman, Stanley Girshik, Peter Kigonya, Shelley Alonso, Rashmi Sanbhadti, Sean Barletta & David Pot - The Cancer Genome Atlas Program Office, Center for Cancer Genomics, National Cancer Institute, Bethesda, 20852, Maryland, USA
Margi Sheth, John A. Demchok, Kenna R. Mills Shaw, Liming Yang, Roy W. Tarnuzzer, Jiashan Zhang, Laura A. L. Dillon & Peter Fielding - TCGA Consultant, Scimentis, LLC, Statham, Georgia 30666, USA.,
Greg Eley - MLF Consulting, Arlington, 02474, Massachusetts, USA
Martin L. Ferguson - National Human Genome Research Institute, National Institutes of Health, Bethesda, 20892, Maryland, USA
Bradley A. Ozenberger, Mark S. Guyer, Heidi J. Sofia & Jacqueline D. Palchik
Consortia
The Cancer Genome Atlas Network
Genome sequencing centres: Washington University in St Louis
- Daniel C. Koboldt
- , Robert S. Fulton
- , Michael D. McLellan
- , Heather Schmidt
- , Joelle Kalicki-Veizer
- , Joshua F. McMichael
- , Lucinda L. Fulton
- , David J. Dooling
- , Li Ding
- , Elaine R. Mardis
- & Richard K. Wilson
Genome characterization centres: BC Cancer Agency
- Adrian Ally
- , Miruna Balasundaram
- , Yaron S. N. Butterfield
- , Rebecca Carlsen
- , Candace Carter
- , Andy Chu
- , Eric Chuah
- , Hye-Jung E. Chun
- , Robin J. N. Coope
- , Noreen Dhalla
- , Ranabir Guin
- , Carrie Hirst
- , Martin Hirst
- , Robert A. Holt
- , Darlene Lee
- , Haiyan I. Li
- , Michael Mayo
- , Richard A. Moore
- , Andrew J. Mungall
- , Erin Pleasance
- , A. Gordon Robertson
- , Jacqueline E. Schein
- , Arash Shafiei
- , Payal Sipahimalani
- , Jared R. Slobodan
- , Dominik Stoll
- , Angela Tam
- , Nina Thiessen
- , Richard J. Varhol
- , Natasja Wye
- , Thomas Zeng
- , Yongjun Zhao
- , Inanc Birol
- , Steven J. M. Jones
- & Marco A. Marra
Broad Institute
- Andrew D. Cherniack
- , Gordon Saksena
- , Robert C. Onofrio
- , Nam H. Pho
- , Scott L. Carter
- , Steven E. Schumacher
- , Barbara Tabak
- , Bryan Hernandez
- , Jeff Gentry
- , Huy Nguyen
- , Andrew Crenshaw
- , Kristin Ardlie
- , Rameen Beroukhim
- , Wendy Winckler
- , Gad Getz
- , Stacey B. Gabriel
- & Matthew Meyerson
Brigham & Women’s Hospital & Harvard Medical School
- Lynda Chin
- , Peter J. Park
- & Raju Kucherlapati
University of North Carolina, Chapel Hill
- Katherine A. Hoadley
- , J. Todd Auman
- , Cheng Fan
- , Yidi J. Turman
- , Yan Shi
- , Ling Li
- , Michael D. Topal
- , Xiaping He
- , Hann-Hsiang Chao
- , Aleix Prat
- , Grace O. Silva
- , Michael D. Iglesia
- , Wei Zhao
- , Jerry Usary
- , Jonathan S. Berg
- , Michael Adams
- , Jessica Booker
- , Junyuan Wu
- , Anisha Gulabani
- , Tom Bodenheimer
- , Alan P. Hoyle
- , Janae V. Simons
- , Matthew G. Soloway
- , Lisle E. Mose
- , Stuart R. Jefferys
- , Saianand Balu
- , Joel S. Parker
- , D. Neil Hayes
- & Charles M. Perou
University of Southern California/Johns Hopkins
- Simeen Malik
- , Swapna Mahurkar
- , Hui Shen
- , Daniel J. Weisenberger
- , Timothy Triche Jr
- , Phillip H. Lai
- , Moiz S. Bootwalla
- , Dennis T. Maglinte
- , Benjamin P. Berman
- , David J. Van Den Berg
- , Stephen B. Baylin
- & Peter W. Laird
Genome data analysis: Baylor College of Medicine
- Chad J. Creighton
- & Lawrence A. Donehower
Broad Institute
- Gad Getz
- , Michael Noble
- , Doug Voet
- , Gordon Saksena
- , Nils Gehlenborg
- , Daniel DiCara
- , Juinhua Zhang
- , Hailei Zhang
- , Chang-Jiun Wu
- , Spring Yingchun Liu
- , Michael S. Lawrence
- , Lihua Zou
- , Andrey Sivachenko
- , Pei Lin
- , Petar Stojanov
- , Rui Jing
- , Juok Cho
- , Raktim Sinha
- , Richard W. Park
- , Marc-Danie Nazaire
- , Jim Robinson
- , Helga Thorvaldsdottir
- , Jill Mesirov
- , Peter J. Park
- & Lynda Chin
Institute for Systems Biology
- Sheila Reynolds
- , Richard B. Kreisberg
- , Brady Bernard
- , Ryan Bressler
- , Timo Erkkila
- , Jake Lin
- , Vesteinn Thorsson
- , Wei Zhang
- & Ilya Shmulevich
Memorial Sloan-Kettering Cancer Center
- Giovanni Ciriello
- , Nils Weinhold
- , Nikolaus Schultz
- , Jianjiong Gao
- , Ethan Cerami
- , Benjamin Gross
- , Anders Jacobsen
- , Rileen Sinha
- , B. Arman Aksoy
- , Yevgeniy Antipin
- , Boris Reva
- , Ronglai Shen
- , Barry S. Taylor
- , Marc Ladanyi
- & Chris Sander
Oregon Health & Science University
- Pavana Anur
- & Paul T. Spellman
The University of Texas MD Anderson Cancer Center
- Yiling Lu
- , Wenbin Liu
- , Roel R. G. Verhaak
- , Gordon B. Mills
- , Rehan Akbani
- , Nianxiang Zhang
- , Bradley M. Broom
- , Tod D. Casasent
- , Chris Wakefield
- , Anna K. Unruh
- , Keith Baggerly
- , Kevin Coombes
- & John N. Weinstein
University of California, Santa Cruz/Buck Institute
- David Haussler
- , Christopher C. Benz
- , Joshua M. Stuart
- , Stephen C. Benz
- , Jingchun Zhu
- , Christopher C. Szeto
- , Gary K. Scott
- , Christina Yau
- , Evan O. Paull
- , Daniel Carlin
- , Christopher Wong
- , Artem Sokolov
- , Janita Thusberg
- , Sean Mooney
- , Sam Ng
- , Theodore C. Goldstein
- , Kyle Ellrott
- , Mia Grifford
- , Christopher Wilks
- , Singer Ma
- & Brian Craft
NCI
- Chunhua Yan
- , Ying Hu
- & Daoud Meerzaman
Biospecimen core resource: Nationwide Children’s Hospital Biospecimen Core Resource
- Julie M. Gastier-Foster
- , Jay Bowen
- , Nilsa C. Ramirez
- , Aaron D. Black
- , Robert E. Pyatt
- , Peter White
- , Erik J. Zmuda
- , Jessica Frick
- , Tara M. Lichtenberg
- , Robin Brookens
- , Myra M. George
- , Mark A. Gerken
- , Hollie A. Harper
- , Kristen M. Leraas
- , Lisa J. Wise
- , Teresa R. Tabler
- , Cynthia McAllister
- , Thomas Barr
- & Melissa Hart-Kothari
Tissue source sites: ABS-IUPUI
- Katie Tarvin
- , Charles Saller
- , George Sandusky
- & Colleen Mitchell
Christiana
- Mary V. Iacocca
- , Jennifer Brown
- , Brenda Rabeno
- , Christine Czerwinski
- & Nicholas Petrelli
Cureline
- Oleg Dolzhansky
- , Mikhail Abramov
- , Olga Voronina
- & Olga Potapova
Duke University Medical Center
- Jeffrey R. Marks
The Greater Poland Cancer Centre
- Wiktoria M. Suchorska
- , Dawid Murawa
- , Witold Kycler
- , Matthew Ibbs
- , Konstanty Korski
- , Arkadiusz Spychała
- , Paweł Murawa
- , Jacek J. Brzeziński
- , Hanna Perz
- , Radosław Łaźniak
- , Marek Teresiak
- , Honorata Tatka
- , Ewa Leporowska
- , Marta Bogusz-Czerniewicz
- , Julian Malicki
- , Andrzej Mackiewicz
- & Maciej Wiznerowicz
ILSBio
- Xuan Van Le
- , Bernard Kohl
- , Nguyen Viet Tien
- , Richard Thorp
- , Nguyen Van Bang
- , Howard Sussman
- , Bui Duc Phu
- , Richard Hajek
- , Nguyen Phi Hung
- , Tran Viet The Phuong
- , Huynh Quyet Thang
- & Khurram Zaki Khan
International Genomics Consortium
- Robert Penny
- , David Mallery
- , Erin Curley
- , Candace Shelton
- & Peggy Yena
Mayo Clinic
- James N. Ingle
- , Fergus J. Couch
- & Wilma L. Lingle
MSKCC
- Tari A. King
MD Anderson Cancer Center
- Ana Maria Gonzalez-Angulo
- , Gordon B. Mills
- , Mary D. Dyer
- , Shuying Liu
- , Xiaolong Meng
- & Modesto Patangan
University of California San Francisco
- Frederic Waldman
- & Hubert Stöppler
University of North Carolina
- W. Kimryn Rathmell
- , Leigh Thorne
- , Mei Huang
- , Lori Boice
- & Ashley Hill
Roswell Park Cancer Institute
- Carl Morrison
- , Carmelo Gaudioso
- & Wiam Bshara
University of Miami
- Kelly Daily
- , Sophie C. Egea
- , Mark D. Pegram
- & Carmen Gomez-Fernandez
University of Pittsburgh
- Rajiv Dhir
- , Rohit Bhargava
- & Adam Brufsky
Walter Reed National Military Medical Center
- Craig D. Shriver
- , Jeffrey A. Hooke
- , Jamie Leigh Campbell
- , Richard J. Mural
- , Hai Hu
- , Stella Somiari
- , Caroline Larson
- , Brenda Deyarmin
- , Leonid Kvecher
- & Albert J. Kovatich
Disease working group
- Matthew J. Ellis
- , Tari A. King
- , Hai Hu
- , Fergus J. Couch
- , Richard J. Mural
- , Thomas Stricker
- , Kevin White
- , Olufunmilayo Olopade
- , James N. Ingle
- , Chunqing Luo
- , Yaqin Chen
- , Jeffrey R. Marks
- , Frederic Waldman
- , Maciej Wiznerowicz
- , Ron Bose
- , Li-Wei Chang
- , Andrew H. Beck
- & Ana Maria Gonzalez-Angulo
Data coordination centre
- Todd Pihl
- , Mark Jensen
- , Robert Sfeir
- , Ari Kahn
- , Anna Chu
- , Prachi Kothiyal
- , Zhining Wang
- , Eric Snyder
- , Joan Pontius
- , Brenda Ayala
- , Mark Backus
- , Jessica Walton
- , Julien Baboud
- , Dominique Berton
- , Matthew Nicholls
- , Deepak Srinivasan
- , Rohini Raman
- , Stanley Girshik
- , Peter Kigonya
- , Shelley Alonso
- , Rashmi Sanbhadti
- , Sean Barletta
- & David Pot
Project team: National Cancer Institute
- Margi Sheth
- , John A. Demchok
- , Kenna R. Mills Shaw
- , Liming Yang
- , Greg Eley
- , Martin L. Ferguson
- , Roy W. Tarnuzzer
- , Jiashan Zhang
- , Laura A. L. Dillon
- , Kenneth Buetow
- & Peter Fielding
National Human Genome Research Institute
- Bradley A. Ozenberger
- , Mark S. Guyer
- , Heidi J. Sofia
- & Jacqueline D. Palchik
Contributions
TCGA research network contributed collectively to this study. Biospecimens were provided by tissue source sites and processed by a Biospecimens Core Resource. Data generation and analyses were performed by genome sequencing centres, cancer genome characterization centres, and genome data analysis centres. RPPA analysis was performed at the MD Anderson Cancer Center in association with the genome data analysis centre. All data were released through the Data Coordinating Center. Project activities were coordinated by NCI and NHGRI project teams. We also acknowledge the following TCGA investigators of the Breast Analysis Working Group who contributed substantially to the analysis and writing of this manuscript: Project leaders, C.M.P., M.J.E.; manuscript coordinator, C.M.P., K.A.H.; data coordinator, K.A.H.; analysis coordinator, C.M.P., K.A.H.; DNA sequence analysis, D.C.K., L.D.; mRNA microarray analysis; K.A.H., C.F.; miRNA sequence analysis, A.G.R., A.C.; DNA methylation analysis, S. Malik, S. Mahurkar, P.W.L.; copy number analysis; A.D.C., M.M.; protein analysis, W.L., R.G.W.V., G.B.M.; pathway/integrated analysis, C.J.C., C.Y., J.M.S., C.C.B., G.C., C.S., S.R., I.S.; biospecimen core resource, T.L., J.B., J.M.G.; pathology and clinical expertise, T.A.K., H.H., R.J.M., J.N.I., T.S., F.W.
Corresponding author
Correspondence toCharles M. Perou.
Ethics declarations
Competing interests
Charles M. Perou and Matthew J. Ellis are inventors on patent filing for PAM50 and have equity interest in Bioclassifier LLC. Joel S. Parker is an inventor on patent filing for PAM50.
Additional information
All of the primary sequence files are deposited in CGHub (https://cghub.ucsc.edu/); all other data including mutation annotation file are deposited at the Data Coordinating Center (http://cancergenome.nih.gov/). Sample lists, data matrices and supporting data can be found at http://tcga-data.nci.nih.gov/docs/publications/brca_2012/. The data can be explored via the ISB Regulome Explorer (http://explorer.cancerregulome.org/) and the cBio Cancer Genomics Portal (http://cbioportal.org). Data descriptions can be found at https://wiki.nci.nih.gov/display/TCGA/TCGA+Data+Primer and in Supplementary Methods.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1-20, Supplementary Methods 1-15 (with additional figures and tables) and Supplementary References. (PDF 14439 kb)
Supplementary Tables
This zipped file contains Supplementary Tables 1-8. This file was replaced on 15 November 2012 to correct an error in Supplementary Table 5. (ZIP 1009 kb)
PowerPoint slides
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/).
About this article
Cite this article
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours.Nature 490, 61–70 (2012). https://doi.org/10.1038/nature11412
- Received: 22 March 2012
- Accepted: 11 July 2012
- Published: 23 September 2012
- Issue Date: 04 October 2012
- DOI: https://doi.org/10.1038/nature11412