Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome (original) (raw)
Abstract
In fruit fly research, chromosomal deletions are indispensable tools for mapping mutations, characterizing alleles and identifying interacting loci. Most widely used deletions were generated by irradiation or chemical mutagenesis. These methods are labor-intensive, generate random breakpoints and result in unwanted secondary mutations that can confound phenotypic analyses. Most of the existing deletions are large, have molecularly undefined endpoints and are maintained in genetically complex stocks. Furthermore, the existence of haplolethal or haplosterile loci makes the recovery of deletions of certain regions exceedingly difficult by traditional methods, resulting in gaps in coverage. Here we describe two methods that address these problems by providing for the systematic isolation of targeted deletions in the D. melanogaster genome. The first strategy used a P element–based technique to generate deletions that closely flank haploinsufficient genes and minimize undeleted regions. This deletion set has increased overall genomic coverage by 5–7%. The second strategy used FLP recombinase and the large array of FRT-bearing insertions described in the accompanying paper1 to generate 519 isogenic deletions with molecularly defined endpoints. This second deletion collection provides 56% genome coverage so far. The latter methodology enables the generation of small custom deletions with predictable endpoints throughout the genome and should make their isolation a simple and routine task.
Similar content being viewed by others
Main
Deletion screens conducted at the Bloomington Drosophila Stock Center are based on the observation that P transposase often induces chromosomal aberrations involving the sites of two P insertions2. When two insertions are present at different sites on homologous chromosomes, deletions that remove the region between the two insertions can be recovered. In appropriately marked crosses, the deletions can be recognized from the recombination of visible, flanking markers. Our results are consistent with studies of P transposition showing that faulty transposition can produce deletions when the 5′ end of one P element and the 3′ end of a different P element are inserted in a chromosome as if they were the two ends of a single element3,4 (Fig. 1a). This mistake in the normal transposition process has been called hybrid element insertion (HEI), because the paired 5′ and 3′ ends from different P elements inserted at a new chromosomal site by P transposase can be thought of as a temporary, synthetic or 'hybrid' P element3. Resulting deletions are flanked by one of the starting P constructs (Fig. 1a). This provides a convenient molecular tag for subsequent breakpoint characterization. Because a hybrid element can potentially insert anywhere in the genome, many different chromosomal rearrangements can result in addition to simple deletions. We isolated other rearrangements (data not shown) but specifically selected for deletions among recombinant progeny by complementation tests against mutations mapping between the original P insertion sites.
Figure 1: Schematics for deficiency generation.
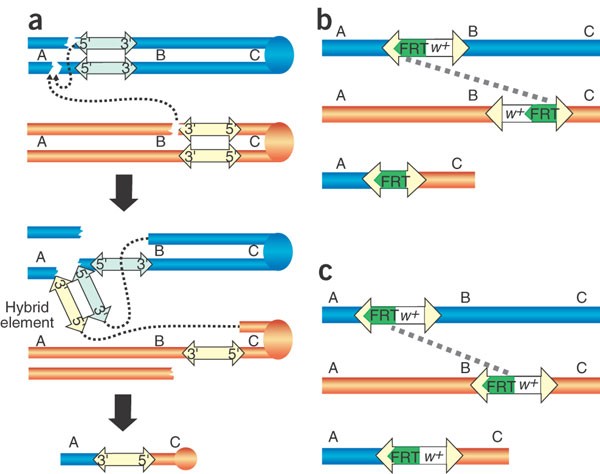
The starting pairs of chromosomes are shown in orange and blue, with the transposon insertion elements indicated in light yellow or light blue and FRT elements in green. The order of theoretical markers A, B and C along the chromosomes is indicated. (a) The HEI strategy used at Bloomington. (b) Using FLP-FRT recombination to generate _w_− deficiencies from Exelixis transposon insertions. (c) Using FLP-FRT recombination to generate _w_+ deficiencies from Exelixis transposon insertions.
The success of the approach depends on a strong inherent bias of hybrid elements to insert in or near one of the original P insertions. If HEI were random, only one deletion breakpoint would be predictable. We recovered only two deletions in all our screens in which the breakpoints differed from the sites of the original P insertions at the level of polytene cytology (Df(2R)BSC25 and one not shown). The basis of this bias is unknown, but it resembles, at least superficially, the bias of single P elements to transpose locally5. Although the deletion endpoints are not entirely predictable, the method is useful because deletions can be targeted to particular genomic regions, the screens are moderately efficient and the size of deletions seems to be limited only by aneuploidy effects.
Figure 2a shows the crosses we used in HEI deletion screens and Table 1a gives markers and stocks we found particularly useful. When flanking markers and P insertions are linked (Fig. 2a), deletions will be recovered among progeny with both recessive marker phenotypes. With different linkage relationships, deletions will be recovered among progeny with different marker combinations. We recovered deletions with the two P insertions in the same and in opposite orientations. As P element ends are the only _cis_-acting sequences required for transposition, it seems that any two P constructs can be combined to produce HEI deletions. We successfully used combinations of P{PZ}, P{lacW}, P{SUPor-P}, P{EP} and P{GT1} insertions.
Figure 2: Genetic crossing schemes used to generate deletions.
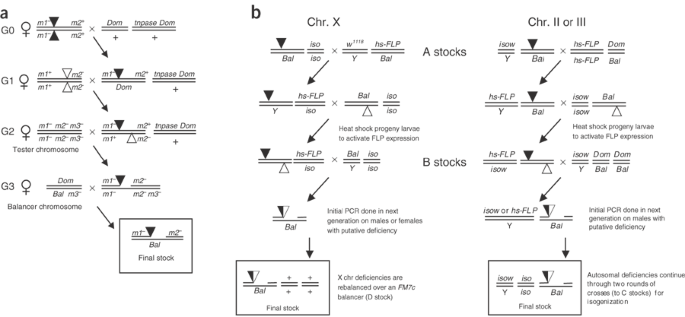
(a) Genetic scheme used for _P_-element transposase-based deletions. The crosses place P insertions (triangles) marked with flanking visible marker mutations (m1 − and m2 −) in males together with a source of P transposase (tnpase), so that hybrid element insertions can occur in premeiotic germ cells. Deletion events accompanied by recombination of flanking visible markers are identified in crosses to females carrying appropriate tester chromosomes. Deletions are established in stocks with balancer chromosomes (Bal). Dom and m3 − represent other convenient dominant and recessive markers, respectively. (b) Genetic scheme used for FLP-FRT–based deletions. Crosses place two FRT-bearing transposon insertions (triangles) in trans in the presence of heat shock–driven FLP recombinase (hs-FLP). Activation of FLP recombinase results in the generation of deletions that can be detected by PCR. Deletions are established in isogenized stocks with balancers (Bal). Genotypes for nontransposon stocks (A, B, C, and D stocks) are provided in Table 1. Dom, dominant visible marker mutation; iso, isogenized chromosome; Y, Y chromosome.
Table 1 Stock genotypes used in the generation of deletions
So far, Bloomington has recovered 45 unique deletions using the HEI approach with _trans_-heterozygous P insertions (Supplementary Table 1 online). Table 2a summarizes their contribution to genomic coverage. All the screens were designed to fill gaps in deletion coverage, and most were designed to extend from a point adjacent to a haploinsufficient locus into an existing deletion. The fact that most haploinsufficient genes in D. melanogaster encode protein components of ribosomes6 simplified the planning. By minimizing the undeleted regions to the small intervals between the two P insertions most closely linked to haploinsufficients, we could maximize coverage without the need for chromosomal duplications. For example, Df(2L)BSC32 and Df(2L)BSC36 flank the haploinsufficient locus RpL9, leaving only it and four other genes undeleted. So far, we have maximized deletion coverage around 14 known or potential haploinsufficients and maximized coverage to one side of an additional 10 haploinsufficients.
Table 2 Genome coverage provided by D. melanogaster deletion sets
This method may be useful in certain experimental situations where P insertions are favorably located, but the method described below will probably prove more useful in most situations and will be used at Bloomington for further efforts to improve the Stock Center deletion collection. As described by Thibault et al.1 in this issue of Nature Genetics, investigators at Exelixis generated a large isogenic collection of P and piggyBac insertion lines containing FRT sites. In the presence of FLP recombinase, FRT-bearing transposon insertions can be used to efficiently isolate deletions with precisely defined endpoints7. We used this method to generate a set of isogenic deletions as described below. The DrosDel Consortium is using a similar method (see URL).
The Exelixis deletion series is based on three of the four classes of transposon constructs described in the accompanying paper1. These transposon insertions contain FRT sites of 199 bp either 5′ (in XP and WH transposons) or 3′ (in RB transposons) of the white+ transgene (w+; Fig. 3a). In the presence of FLP recombinase, efficient _trans_-recombination between FRT elements results in a genomic deletion with a single residual element tagging the deletion site (Fig. 1b,c). In deletions generated using XPs, which contain tandem FRT sites, the _cis_- recombination event occurs earlier than that in trans. Depending on the pair of starting insertions, their genomic orientation and the relative position of _w_+ with respect to the FRTs, some deletions can initially be detected in the progeny by a loss of the _w_+ marker (Table 3). The crosses outlined in Figure 2b and Table 1b allow the efficient recovery of deletions in four generations. Progeny are screened for the presence of the residual element by PCR detection of a resulting hybrid element (e.g., XP:WH) using element-specific primers, or by detection of residual element ends using paired element-specific and genome-specific primers (Fig. 3b). Final confirmation of the deletion ends can be provided by PCR or sequencing with genome-specific primers. In general, PCR screening of five _w_− individual progeny from _w_− deletion generation crosses, or 50 progeny from _w_+ deletion generation crosses, resulted in four confirmed deletions. In addition to deletions, tandem duplications could also be isolated using these insertions and an appropriately designed PCR strategy.
Figure 3: FRT-bearing transposons and PCR strategies used in FLP-FRT–based deletions.
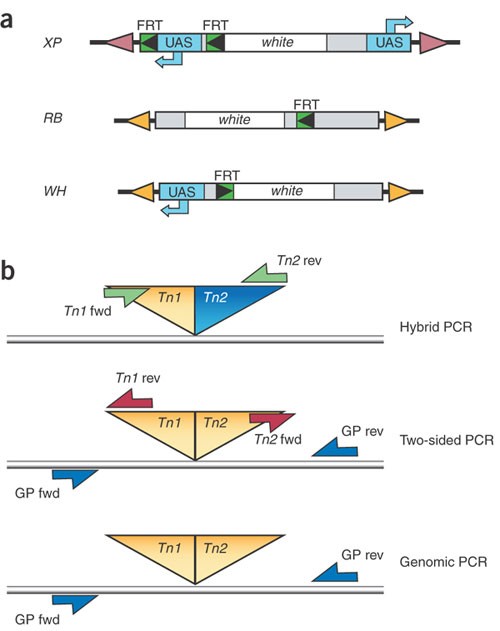
(a) Schematic of the three transposon types used in FLP-FRT–based deletions. FRT sites, UAS-containing sequences and the gene white are indicated. See Thibault et al.1 for details. FRT orientation is indicated by the direction of the arrowheads (FRT sites must be in the same orientation for deletion generation). (b) For transposon combinations that generate a recombinant hybrid element (composed of two different element types), transposon-specific primers were used to generate a fragment of known size across the newly formed hybrid (hybrid PCR). For transposon combinations that regenerate an intact element, a genomic primer in combination with a transposon-specific primer for each end of the transposon was used (two-sided PCR). We also carried out PCR using only genomic primers for additional confirmation (genomic PCR). Table 3 lists the PCR confirmation method used for initial detection of the various deletion types. Supplementary Methods online provides transposon primer sequences and expected fragment sizes.
Table 3 Allowable pairwise combinations of Exelixis transposons for deletion generation
Supplementary Table 2 online provides a list of 519 deletion stocks, covering approximately 56% of CG isoforms in the genome (see Table 2b), which have been transferred to the Bloomington Drosophila Stock Center. These deletions average 140 kb in length (ranging from 4.5 to 659 kb) and delete an average of 25 CG isoforms (ranging from 1 to 83). The density of starting insertions in the Exelixis collection and the precise nature of deletion endpoints allow the design of very small customized deletions. For example, one can generate null alleles for single genes for which a null point mutant is lacking (see examples in Supplementary Table 2 online: Df(2L)Exel9033, Df(2R)Exel6062, Df(3L)Exel9028 and Df(3R)Exel9056). This targeted strategy can be also used to exclude problematic regions, such as haploinsufficient loci, or to generate precisely overlapping regions for high resolution coverage. Additionally, as deletions generated by this method are carried in the same isogenic background, meaningful comparisons between deletion-based modification phenotypes are relatively straightforward. We used this approach to refine regions of phenotypic interest by sequential deletion mapping (K.A.E., H.L.F.-L., M.B. Mahoney & A.L.P., unpublished data) and to carry out genome-wide screens for specific phenotypes (S. Thibault, personal communication). Previously available deletions made such analyses more difficult, owing to the complex differences in the genetic backgrounds of stocks, large individual deletion sizes and imprecisely defined endpoints.
In summary, we describe the highest resolution deletion coverage of a metazoan genome so far. Improvements to genome coverage and breakpoint distribution will be continued by the Bloomington Drosophila Stock Center using the FRT-based deletion strategy described in this manuscript, as well as by the DrosDel Consortium. Furthermore, the establishment of these methods and the widespread distribution of FRT-bearing insertion stocks1 and tools allows individual scientists to generate customized aberrations in the D. melanogaster genome in an efficient and straightforward manner.
Methods
P transposase–based deletion screens.
Visible marker mutations must first be combined in cis with each P insertion by standard meiotic crossover crosses. Once marked, the P insertion chromosomes may be used in the genetic scheme shown in Figure 2a. Many different visible markers could potentially be used, but Table 1a lists markers and stocks we found useful for various chromosomal intervals.
Recombinant chromosomes can arise either from HEI events involving two different P insertions or from events involving the two copies of a single P insertion present on sister chromatids. The latter yield small deletions flanking the original P insertion site4, which were of no interest to us. We distinguished deletions extending between different P insertion sites from other recombinant chromosomes by complementation tests. We characterized deletions in polytene chromosome preparations. In successful screens, an average of one in every four recombinant premeiotic clones was associated with a deletion extending from one P insertion site to the other, and a unique _P_-to-P deletion clone was detected in 1 of every 3,000 progeny. There was wide variation in the efficiency of screens: approximately one-third of the P insertion pairs produced no recombinant _P_-to-P deletions, despite repeated attempts. We attribute this variability to inherent differences in the ability of P insertions to participate in hybrid element insertions. P insertions may be poorly mobile and rarely give rise to recombinant deletions, or they may be extremely mobile, excise efficiently in the intermediate G1 generation (where a single P insertion is combined with a transposase source) and leave few intact insertions to participate in HEI events in the G2 generation. Consequently, we generally designed screens of ∼25,000 progeny and repeated the screens twice more before switching to a different pair of P insertions.
Most deletions generated by P transposase in the presence of _trans_-heterozygous P insertions will be tagged with one of the original P constructs (Fig. 1a). There is, however, a class of _P_-to-P deletions that will not be flanked by a P construct. When the two original P insertions are present in the same relative orientation, deletions may also form by a DNA repair process called hybrid excision and repair, the rejoining of the broken chromatid ends left behind when the 5′ and 3′ ends of two different P elements excise and participate in a HEI event3. No P construct will be present at the junction point. Hybrid excision and repair deletions have been recovered in _trans_-heterozygous P element screens, but they seem to be generated less often than HEI deletions (A. Paré and J. Ewer, personal communication).
Two variations on the HEI deletion method have been successful. First, one can eliminate the process of marking P insertions with flanking visible markers and scoring recombinants by testing for deletions among progeny that have lost the internal marker carried by one of the P constructs if one has an efficient assay for distinguishing deletions from the larger number of simple excisions occurring in the intermediate G1 generation. Such an approach was previously reported8. Second, one can recover deletions between the sites of P insertions if two insertions are first recombined in cis by meiotic crossover9.
Genetic crosses for generation of FLP-FRT deletions.
Figure 2b provides a schematic of genetic crosses we used for FLP-FRT deletion generation; genotypes used can be found in Table 1b. The following describes autosomal deletion screens (X-chromosome deletion crosses are similar; Fig. 2b). We carried out all crosses at 25 °C unless otherwise stated. We selected transposon elements flanking the region to be deleted (Table 3). Males carrying one element were mated with females carrying a FLP recombinase transgene. Progeny males carrying both the element and FLP recombinase were then mated to females carrying the second element. After 2 d, we subjected parents and progeny (progeny contain both the FRT-bearing elements in trans and FLP recombinase) to a 1-h heat shock by placing the bottles into a 37 °C water bath. We removed parents after 72 h of total egg-laying time and subjected the bottles to daily 1-h heat shocks for 4 d more. We raised progeny to adulthood, collected virgin females and crossed them to males containing marked balancer chromosomes. Individual progeny males (five _w_− males for a _w_− deletion; 50 males for a _w_+ deletion) were then crossed pairwise to virgin females to generate additional progeny for PCR confirmation analysis and to balance the stocks in an isogenic background.
As we were unable to maintain healthy balanced FM7c stocks in our isogenic background, most X chromosome deletions were stocked in a nonisogenized FM7c background (D stock, Table 1b). Because the X chromosomes are derived from the same isogenized X and the backgrounds of the X deletion stocks are similar, phenotypic comparisons between X chromosome deletions should still be valid. Alternatively, one could balance X chromosome deletions with the isogenized B stock for X deletions (a Binsinscy balancer in an isogenized background).
PCR confirmation of FLP-FRT–based deletions.
We carried out all PCR reactions using purified genomic DNA from five homogenized flies obtained from each isolate line (for genomic DNA preparation, see Supplementary Methods online). In general, it was sufficient to test 5 putative lines for each _w_− deletion and 50 putative lines for each _w_+ deletion. We used three different PCR strategies for deletion confirmation. Figure 3b provides a schematic and description of these strategies. Table 3 lists the PCR confirmation strategies used for initial detection of the various deletion types. Supplementary Methods online provide transposon primer sequences and expected fragment sizes for these combinations, as well as additional primer pair sequences for 'two-sided' PCR that may be used for secondary confirmation.
For products longer than 3 kb (see Supplementary Methods online), we carried out PCR using TaKaRa La Taq (Takara Bio). For products less than 3 kb, we used AmpliTaq Gold (Applied Biosystems). We used a standard touchdown program with both systems. Genomic primers were chosen using an in-house primer design tool and made by Biosource.
URL.
DrosDel Consortium is available at http://www.drosdel.org.uk/.
Note: Supplementary information is available on the Nature Genetics website.
References
- Thibault, S.T. et al. A complementary transposon toolkit for Drosophila melanogaster. Nat. Genet. advance online publication, 22 February 2004 (doi:10.1038/ng1314).
- Berg, R., Engels, W.R. & Kreber, R.A. Site-specific X-chromosome rearrangements from hybrid dysgenesis in Drosophila melanogaster. Science 210, 427–429 (1980).
Article CAS Google Scholar - Gray, Y.H., Tanaka, M.M. & Sved, J.A. _P_-element-induced recombination in Drosophila melanogaster: hybrid element insertion. Genetics 144, 1601–1610 (1996).
CAS PubMed PubMed Central Google Scholar - Preston, C.R., Sved, J.A. & Engels W.R. Flanking duplications and deletions associated with _P_-induced male recombination in Drosophila. Genetics 144, 1623–1638 (1996).
CAS PubMed PubMed Central Google Scholar - Tower, J., Karpen, G.H., Craig, N. & Spradling A.C. Preferential transposition of _Drosophila P_-elements to nearby chromosomal sites. Genetics 133, 347–359 (1993).
CAS PubMed PubMed Central Google Scholar - Ashburner, M. Drosophila: A Laboratory Handbook (Cold Spring Harbor Laboratory Press, 1989).
Google Scholar - Golic, K.G. & Golic, M.M. Engineering the Drosophila genome: chromosome rearrangements by design. Genetics 144, 1693–1711 (1996).
CAS PubMed PubMed Central Google Scholar - Terman, J.R., Mao, T., Pasterkamp, R.J., Yu, H.H. & Kolodkin, A.L. MICALs, a family of conserved flavoprotein oxidoreductases, function in plexin-mediated axonal repulsion. Cell 109, 887–900 (Supplemental Data)(2002).
Article CAS Google Scholar - Cooley, L., Thompson, D. & Spradling, A.C. Constructing deletions with defined endpoints in Drosophila. Proc. Natl. Acad. Sci. USA 87, 3170–3173 (1990).
Article CAS Google Scholar
Acknowledgements
We thank J. Roote, D. Gubb, D. Coulson, T. Morley and M. Ashburner for early development of the _trans_-heterozygous P element–deletion technique and for introducing it to Bloomington; K. Matthews for advice and support; N. Klitgord for additional bioinformatics analysis of Exelixis deletions; J. McLaughlin for help with figures; the Exelixis genome biochemistry team for sequencing support; and the fly genetics team and bioinformatics staff for hands-on work and sequence analysis of the Exelixis D. melanogaster knockout collection. The work at Bloomington was supported by grants from the US National Institutes of Health National Center for Research Resources and the US National Science Foundation Division of Biological Infrastructure to K.R.C. and T.C.K.
Author information
Author notes
- Kari Huppert
Present address: Department of Molecular Biology and Pharmacology, Washington University School of Medicine, Campus Box 8103, 660 S. Euclid Ave., St Louis, Missouri, 63110, USA - Marie Marcinko
Present address: Scripps Research Institute, 10550 N. Torrey Pines, SP 145, La Jolla, California, 92037, USA - Brett Milash
Present address: Huntsman Cancer Institute, 2000 Circle of Hope, University of Utah, Salt Lake City, Utah, 84132, USA - Millicent A Winner
Present address: Department of Medicine, University of Louisville, Baxter II, Room 334, 570 S. Preston St., Louisville, Kentucky, 40202, USA - Matthew A Singer
Present address: Chemicon International, 28820 Single Oak Drive, Temecula, California, 92590, USA - Casey Kopczynski
Present address: Ercole Biotech, 7030 Kit Creek Road, P.O. Box 12295, Research Triangle Park, North Carolina, 27709, USA - Daniel Curtis
Present address: Novartis Institutes for BioMedical Research, Developmental & Molecular Pathways, 100 Technology Square, Office 5653, Cambridge, Massachusetts, 02139, USA - Annette L Parks, Kevin R Cook and Marcia Belvin: These authors contributed equally to this work.
Authors and Affiliations
- Exelixis, 170 Harbor Way, South San Francisco, 94083-0511, California, USA
Annette L Parks, Marcia Belvin, Nicholas A Dompe, Robert Fawcett, Kari Huppert, Lory R Tan, Christopher G Winter, Deanna Grant, Marie Marcinko, Wesley Y Miyazaki, Stephanie Robertson, Kenneth J Shaw, Mariano Tabios, Valentina Vysotskaia, Lora Zhao, Kyle A Edgar, Elizabeth Howie, Keith Killpack, Brett Milash, Amanda Norton, Doua Thao, Kellie Whittaker, Lori Friedman, Jonathan Margolis, Matthew A Singer, Casey Kopczynski, Daniel Curtis, Gregory D Plowman, Geoffrey Duyk & Helen L Francis-Lang - Department of Biology, Bloomington Drosophila Stock Center, Indiana University, 1001 E. Third St., Bloomington, 47405-3700, Indiana, USA
Kevin R Cook, Kevin P Bogart, Jennifer E Deal, Megan E Deal-Herr, Rachel S Andrade, Millicent A Winner & Thomas C Kaufman - Department of Molecular Biology and Pharmacology, Washington University School of Medicine, Campus Box 8103, 660 S. Euclid Ave., St Louis, Missouri, 63110, USA
Marie Marcinko, Brett Milash, Millicent A Winner, Matthew A Singer, Casey Kopczynski & Daniel Curtis
Authors
- Annette L Parks
You can also search for this author inPubMed Google Scholar - Kevin R Cook
You can also search for this author inPubMed Google Scholar - Marcia Belvin
You can also search for this author inPubMed Google Scholar - Nicholas A Dompe
You can also search for this author inPubMed Google Scholar - Robert Fawcett
You can also search for this author inPubMed Google Scholar - Kari Huppert
You can also search for this author inPubMed Google Scholar - Lory R Tan
You can also search for this author inPubMed Google Scholar - Christopher G Winter
You can also search for this author inPubMed Google Scholar - Kevin P Bogart
You can also search for this author inPubMed Google Scholar - Jennifer E Deal
You can also search for this author inPubMed Google Scholar - Megan E Deal-Herr
You can also search for this author inPubMed Google Scholar - Deanna Grant
You can also search for this author inPubMed Google Scholar - Marie Marcinko
You can also search for this author inPubMed Google Scholar - Wesley Y Miyazaki
You can also search for this author inPubMed Google Scholar - Stephanie Robertson
You can also search for this author inPubMed Google Scholar - Kenneth J Shaw
You can also search for this author inPubMed Google Scholar - Mariano Tabios
You can also search for this author inPubMed Google Scholar - Valentina Vysotskaia
You can also search for this author inPubMed Google Scholar - Lora Zhao
You can also search for this author inPubMed Google Scholar - Rachel S Andrade
You can also search for this author inPubMed Google Scholar - Kyle A Edgar
You can also search for this author inPubMed Google Scholar - Elizabeth Howie
You can also search for this author inPubMed Google Scholar - Keith Killpack
You can also search for this author inPubMed Google Scholar - Brett Milash
You can also search for this author inPubMed Google Scholar - Amanda Norton
You can also search for this author inPubMed Google Scholar - Doua Thao
You can also search for this author inPubMed Google Scholar - Kellie Whittaker
You can also search for this author inPubMed Google Scholar - Millicent A Winner
You can also search for this author inPubMed Google Scholar - Lori Friedman
You can also search for this author inPubMed Google Scholar - Jonathan Margolis
You can also search for this author inPubMed Google Scholar - Matthew A Singer
You can also search for this author inPubMed Google Scholar - Casey Kopczynski
You can also search for this author inPubMed Google Scholar - Daniel Curtis
You can also search for this author inPubMed Google Scholar - Thomas C Kaufman
You can also search for this author inPubMed Google Scholar - Gregory D Plowman
You can also search for this author inPubMed Google Scholar - Geoffrey Duyk
You can also search for this author inPubMed Google Scholar - Helen L Francis-Lang
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toKevin R Cook.
Ethics declarations
Competing interests
Some authors are past or present employees of Exelixis, and some of them have personal financial interests as shareholders in Exelixis.
Supplementary information
Rights and permissions
About this article
Cite this article
Parks, A., Cook, K., Belvin, M. et al. Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome.Nat Genet 36, 288–292 (2004). https://doi.org/10.1038/ng1312
- Received: 05 January 2004
- Accepted: 28 January 2004
- Published: 22 February 2004
- Issue Date: 01 March 2004
- DOI: https://doi.org/10.1038/ng1312