Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies (original) (raw)
- Letter
- Published: 15 January 2006
Nature Genetics volume 38, pages 209–213 (2006)Cite this article
- 5972 Accesses
- 1066 Citations
- 9 Altmetric
- Metrics details
A Corrigendum to this article was published on 01 March 2006
Abstract
Genome-wide association is a promising approach to identify common genetic variants that predispose to human disease1,2,3,4. Because of the high cost of genotyping hundreds of thousands of markers on thousands of subjects, genome-wide association studies often follow a staged design in which a proportion (πsamples) of the available samples are genotyped on a large number of markers in stage 1, and a proportion (πsamples) of these markers are later followed up by genotyping them on the remaining samples in stage 2. The standard strategy for analyzing such two-stage data is to view stage 2 as a replication study and focus on findings that reach statistical significance when stage 2 data are considered alone2. We demonstrate that the alternative strategy of jointly analyzing the data from both stages almost always results in increased power to detect genetic association, despite the need to use more stringent significance levels, even when effect sizes differ between the two stages. We recommend joint analysis for all two-stage genome-wide association studies, especially when a relatively large proportion of the samples are genotyped in stage 1 (πsamples ≥ 0.30), and a relatively large proportion of markers are selected for follow-up in stage 2 (πmarkers ≥ 0.01).
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Additional access options:
Figure 1: Power of a two-stage design for joint and replication-based analysis with 1,000 cases and 1,000 controls genotyped on 300,000 independent markers with αgenome = 0.05.
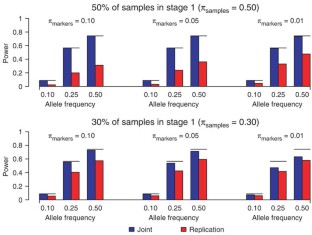
Figure 2: Power of a two-stage design for joint and replication-based analysis with 1,000 cases and 1,000 controls genotyped on 300,000 independent markers with αgenome = 0.05, using a GRR of 1.40 and prevalence of 0.10.

Figure 3: Power of a two-stage design for joint and replication-based analysis with 1,000 cases and 1,000 controls genotyped on 300,000 independent markers with αgenome = 0.05, using a GRR of 1.40 and prevalence of 0.10.

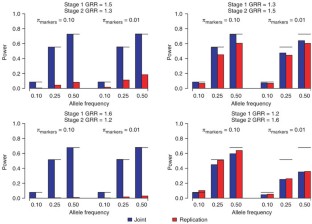
Figure 4: Power of a two-stage design for joint and replication-based analyses in the presence of between-stage heterogeneity with 1,000 cases and 1,000 controls genotyped on 300,000 independent markers with αgenome = 0.05.
Similar content being viewed by others
References
- Risch, N. & Merikangas, K. The future of genetic studies of complex human diseases. Science 273, 1516–1517 (1996).
Article CAS Google Scholar - Hirschhorn, J.N. & Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 6, 95–108 (2005).
Article CAS Google Scholar - Kruglyak, L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat. Genet. 22, 139–144 (1999).
Article CAS Google Scholar - Cardon, L.R. & Bell, J.I. Association study designs for complex diseases. Nat. Rev. Genet. 2, 91–99 (2001).
Article CAS Google Scholar - Klein, R.J. et al. Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 (2005).
Article CAS Google Scholar - Sachidanandam, R. et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 409, 928–933 (2001).
Article CAS Google Scholar - The International HapMap Consortium. The International HapMap Project. Nature 426, 789–796 (2003).
- Hinds, D.A. et al. Whole-genome patterns of common DNA variation in three human populations. Science 307, 1072–1079 (2005).
Article CAS Google Scholar - Johnson, G.C.L. et al. Haplotype tagging for the identification of common disease genes. Nat. Genet. 29, 233–237 (2001).
Article CAS Google Scholar - Ke, X. & Cardon, L.R. Efficient selective screening of haplotype tag SNPs. Bioinformatics 19, 287–288 (2003).
Article CAS Google Scholar - Stram, D.O. et al. Choosing haplotype-tagging SNPS based on unphased genotype data using a preliminary sample of unrelated subjects with an example from the multiethnic cohort study. Hum. Hered. 55, 27–36 (2003).
Article Google Scholar - Satagopan, J.M., Venkatraman, E.S. & Begg, C.B. Two-stage designs for gene-disease association studies with sample size constraints. Biometrics 60, 589–597 (2004).
Article Google Scholar - Satagopan, J.M., Verbel, D.A., Venkatraman, E.S., Offit, K.E. & Begg, C.B. Two-stage designs for gene-disease association studies. Biometrics 58, 163–170 (2002).
Article Google Scholar - Thomas, D., Xie, R.R. & Gebregziabher, M. Two-stage sampling designs for gene association studies. Genet. Epidemiol. 27, 401–414 (2004).
Article Google Scholar - Devlin, B., Roeder, K. & Wasserman, L. Genomic control, a new approach to genetic-based association studies. Theor. Popul. Biol. 60, 155–166 (2001).
Article CAS Google Scholar - Hinds, D.A. et al. Matching strategies for genetic association studies in structured populations. Am. J. Hum. Genet. 74, 317–325 (2004).
Article CAS Google Scholar - Pritchard, J.K. & Donnelly, P. Case-control studies of association in structured or admixed populations. Theor. Popul. Biol. 60, 227–237 (2001).
Article CAS Google Scholar - Ripatti, S., Pitkaniemi, J. & Sillanpaa, M.J. Joint modeling of genetic association and population stratification using latent class models. Genet. Epidemiol. 21, S409–S414 (2001).
Article Google Scholar - Satten, G.A., Flanders, W.D. & Yang, Q.H. Accounting for unmeasured population substructure in case-control studies of genetic association using a novel latent-class model. Am. J. Hum. Genet. 68, 466–477 (2001).
Article CAS Google Scholar - Shmulewitz, D., Zhang, J.Y. & Greenberg, D.A. Case-control association studies in mixed populations: Correcting using genomic control. Hum. Hered. 58, 145–153 (2004).
Article CAS Google Scholar - Yang, B.Z., Zhao, H.Y., Kranzler, H.R. & Gelernter, J. Practical population group assignment with selected informative markers: characteristics and properties of Bayesian clustering via STRUCTURE. Genet. Epidemiol. 28, 302–312 (2005).
Article Google Scholar
Acknowledgements
This research was supported by the US National Institutes of Health.
Author information
Authors and Affiliations
- Department of Biostatistics and Center for Statistical Genetics, University of Michigan, 1420 Washington Heights, Ann Arbor, 48109-2029, Michigan, USA
Andrew D Skol, Laura J Scott, Gonçalo R Abecasis & Michael Boehnke
Authors
- Andrew D Skol
You can also search for this author inPubMed Google Scholar - Laura J Scott
You can also search for this author inPubMed Google Scholar - Gonçalo R Abecasis
You can also search for this author inPubMed Google Scholar - Michael Boehnke
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toMichael Boehnke.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Table 1
Significance thresholds and power of joint and replication-based analysis for a number of two-stage genome-wide association designs. (PDF 219 kb)
Supplementary Table 2
Power of joint and replication-based analysis for a number of two-stage genome-wide association designs. (PDF 190 kb)
Rights and permissions
About this article
Cite this article
Skol, A., Scott, L., Abecasis, G. et al. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies.Nat Genet 38, 209–213 (2006). https://doi.org/10.1038/ng1706
- Received: 31 May 2005
- Accepted: 05 November 2005
- Published: 15 January 2006
- Issue Date: 01 February 2006
- DOI: https://doi.org/10.1038/ng1706