The inflammasome adaptor ASC regulates the function of adaptive immune cells by controlling Dock2-mediated Rac activation and actin polymerization (original) (raw)
Main
Inflammasomes are intracellular multiprotein complexes that are emerging as key regulators of the innate immune response. Deregulated inflammasome activity has been linked to autoimmune diseases, including inflammatory bowel diseases1,2,3,4,5, vitiligo6, gouty arthritis7 and type I and type II diabetes8,9 and less common autoinflammatory disorders collectively referred to as 'cryopyrinopathies'10,11. Distinct inflammasome complexes are assembled around members of the Nod-like receptor (NLR) family or DNA-binding HIN-200 family in a pathogen-specific manner12,13,14. NLRC4 assembles an inflammasome in macrophages infected with intracellular pathogens such as Salmonella typhimurium, Legionella pneumophila, Pseudomonas aeruginosa and Shigella flexneri15,16,17,18,19,20,21,22. In contrast, lethal toxin from Bacillus anthracis triggers activation of the NLRP1B inflammasome in mouse macrophages, and Nlrp1b has been identified as the key susceptibility locus for anthrax lethal toxin–induced macrophage death23. NLRP3 mediates activation of the cysteine protease caspase-1 in lipopolysaccharide (LPS)-primed macrophages that are exposed to microbial components with diverse molecular structures such as viral RNA and DNA or microbial toxins such as the ionophore nigericin17,24,25,26,27. In addition, the endogenous danger-associated molecules monosodium urate and calcium pyrophosphate dehydrate crystals also activate the NLRP3 inflammasome, which suggests a role for this inflammasome in the etiology of gouty arthritis and pseudogout7. The NLRP3 inflammasome contributes to host defense against Salmonella infection in vivo28. Moreover, published reports have characterized the critical role of the NLRP3 inflammasome in protection against colitis and colitis-associated tumorigenesis2,3,5. Finally, DNA viruses such as vaccinia virus and cytomegalovirus and the bacterial pathogens Francisella tularensis and Listeria monocytogenes induce activation of caspase-1 through the HIN-200 family member AIM2 (refs. 29,30,31,32,33,34). Once activated, caspase-1 cleaves and allows secretion of bioactive interleukin 1β (IL-1β) and IL-18. In addition, caspase-1 mediates a specialized form of cell death in infected macrophages and dendritic cells (DCs), a process that contributes substantially to the pathophysiology of several infectious diseases12,35.
The adaptor ASC (Pycard) was initially believed to exert its effects on immune signaling mainly by bridging the interaction between NLRs or HIN-200 proteins and caspase-1 in inflammasome complexes36,37. However, emerging evidence has indicated important inflammasome-independent roles for ASC in controlling immune responses. For instance, the adjuvant quality of the oil-in-water emulsion MF59 has been shown to require ASC, whereas the inflammasome components NLRP3 and caspase-1 are dispensable38. Consequently, the induction of antigen-specific γ-immunoglobulin (IgG) antibodies to influenza vaccines in MF59 is impaired in mice that lack ASC (_Pycard_−/−; called '_Asc_−/−' here) but not in NLRP3-deficient (_Nlrp3_−/−) mice or caspase-1-deficient (_Casp1_−/−) mice38. Moreover, granuloma formation and host defense in chronic Mycobacterium tuberculosis infection depends on ASC but not on NLRP3 or caspase-1 (ref. 39). Finally, _Asc_−/− mice, but not _Nlrp3_−/− mice or _Casp1_−/− mice, are substantially protected against disease progression in experimental models of rheumatoid arthritis and experimental autoimmune encephalomyelitis (EAE), respectively40,41,42. Those last studies have indicated the existence of cell-intrinsic roles for ASC in DC and lymphocyte populations. However, the molecular effector mechanism by which ASC regulates adaptive immune responses independently of inflammasomes has remained unclear.
Here we show that ASC has a critical cell-intrinsic role in regulating immune-cell functions in lymphocytes and DCs in an inflammasome-independent manner. The chemotaxis of _Asc_−/− lymphocytes, but not of _Nlrp3_−/− or _Casp1_−/− lymphocytes, was considerably impaired despite normal expression of chemokine receptors and downstream mitogen-activated protein kinase (MAPK) signaling. Instead, _Asc_−/− lymphocytes failed to migrate because of defective actin polymerization induced by the small GTPase Rac at the leading edge of leukocytes. Consequently, total lymphocyte and DC populations in secondary lymphoid organs were much smaller in _Asc_−/− mice but not in _Nlrp3_−/− or _Casp1_−/− mice. Rac activation and actin polymerization have a critical role in other cell type functions as well, including the uptake of antigens by DCs43. Accordingly, ASC-deficient DCs were impaired in their ability to take up antigens and to drive T cell proliferation. Again, inflammasome signaling was dispensable for ASC-mediated antigen presentation and T cell activation. Instead, transcriptome analysis showed that ASC specifically regulated the transcript abundance of Dock2, a guanine nucleotide–exchange factor specific for cells of the immune response that mediates Rac-dependent actin polymerization and migration of T cells and B cells44. As in _Asc_−/− cells, we identified a critical role for Dock2 in antigen uptake by DCs. Moreover, ectopic expression of Dock2 in _Asc_−/− lymphocytes and DCs restored ASC-dependent immune functions, which confirmed that Dock2 is the main effector that drives the inflammasome-independent functions of ASC in cells of the immune response. Thus, ASC regulates adaptive immune responses by controlling Dock2 expression in DCs and lymphocytes. Our results identify a previously unknown mechanism that regulates the function of cells of the adaptive immune response and provide a mechanistic explanation for the critical inflammasome-independent role of ASC in driving T cell and humoral responses after vaccination and in autoimmune disease.
Results
Antigen uptake and presentation require ASC
The presentation of antigenic peptides on receptors for major histocompatibility complex class II of 'professional' phagocytes represents a critical step in the activation of adaptive immune responses that is dependent on actin polymerization43. We confirmed the critical role of ASC in antigen presentation by receptors for major histocompatibility complex class II with the observation that _Asc_−/− DCs were severely impaired in inducing the proliferation of bovine serum albumin (BSA)-specific T cells (Fig. 1a). Moreover, wild-type T cells incubated with _Asc_−/− DCs in the presence of BSA produced significantly lower amounts cytokines of the T helper type 1 (TH1) subset (interferon-γ; Fig. 1b), the TH2 subset (IL-6 and IL-10; Fig. 1c and Supplementary Fig. 1) and the TH17 subset of helper T cells (IL-17; Fig. 1d), which further established the critical role for ASC in priming T cells. Unlike _Asc_−/− DCs, _Nlrp3_−/− DCs and _Casp1_−/− DCs elicited a T cell–proliferative response similar to that induced by wild-type DCs (data not shown), which emphasizes the role of ASC in antigen presentation by DCs that is independent of its role in inflammasome activation.
Figure 1: ASC controls antigen uptake and presentation independently of inflammasomes.
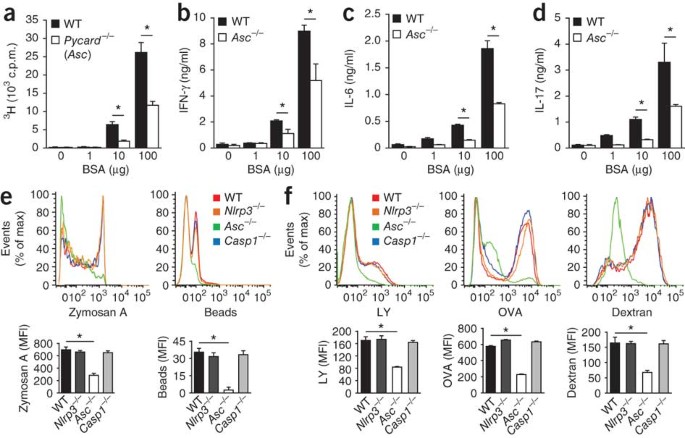
(a–d) Dose-dependent antigen-specific proliferation of lymphocytes among DCs obtained from naive wild-type (WT) and _Asc_−/− mice (n = 4–6 per group) and cultured for 72 h with wild-type CD4+ T cells in the presence of 0–100 μg BSA, assessed as uptake of [3H]thymidine (a) and as the concentration of interferon-γ (IFN-γ; b), IL-6 (c) and IL-17 (d). *P < 0.01 (two-tailed Student's _t_-test). (e) Flow cytometry analysis of the phagocytosis of fluorescein-labeled zymosan A or polystyrene beads by wild-type, Nlrp3_−/−, Asc_−/− and _Casp1_−/− BMDCs after incubation together for 3 h at 37 °C. MFI, mean fluorescence intensity. *P < 0.0005 (two-tailed Student's _t_-test). (f) Flow cytometry analysis of the macropinocytosis of fluorescein-labeled luciferase yellow (LY), OVA or dextran by wild-type, Nlrp3_−/−, Asc_−/− and _Casp1_−/− BMDCs after incubation as in e. *P < 0.005 (two-tailed Student's _t_-test). Data are representative of three independent experiments (a–d; mean and s.d.) or at least three independent experiments (e,f; mean and s.e.m. of triplicates).
The recognition and uptake of antigen by professional antigen-presenting cells represents one of the first steps in presenting peptides to receptors for major histocompatibility class II (ref. 43). To explore the possibility that ASC is required for antigen uptake by professional phagocytes, we incubated wild-type, _Nlrp3_−/−, _Asc_−/− and _Casp1_−/− bone marrow–derived DCs (BMDCs) for 3 h with fluorescein isothiocyanate (FITC)-labeled zymosan A or polystyrene beads and measured phagocytosis by flow cytometry. Internalization of zymosan A or polystyrene beads by _Asc_−/− BMDCs was impaired, whereas phagocytosis by _Nlrp3_−/− and _Casp1_−/− BMDCs was similar to that of wild-type cells (Fig. 1e). This result confirmed that _Asc_−/− BMDCs were defective in the phagocytosis of particulate antigens. To determine whether the uptake of small soluble antigens, which proceeds via fluid endocytosis (macropinosis), was also affected by ASC deficiency, we incubated wild-type, _Nlrp3_−/−, _Asc_−/− and _Casp1_−/− BMDCs for 3 h with FITC-labeled dextran, FITC-labeled ovalbumin (OVA) or lucifer yellow, then washed the cells and assessed macropinosis of these substances by flow cytometry. _Asc_−/− DCs were defective in internalizing all three; in contrast, macropinosis of these by _Nlrp3_−/− and _Casp1_−/− BMDCs was not affected (Fig. 1f). Together these results indicate that ASC controls the uptake and presentation of antigens in professional antigen-presenting cells independently of inflammasomes.
ASC is critical for lymphocyte chemotaxis and migration
In addition to its role in regulating antigen uptake by professional phagocytes, ASC has been proposed to exert cell-intrinsic functions in lymphocytes41,42. In agreement with that, the total number of spleen and lymph node cells in _Asc_−/− mice was about half of that in wild-type mice, with counts of CD4+ T cells, CD8+ T cells, B cells and CD11c+ cells all much lower in _Asc_−/− spleens and lymph nodes (Fig. 2a). _Nlrp3_−/− and _Casp1_−/− mice had numbers similar to those of wild-type mice in both the spleen and lymph nodes for each cell population (Supplementary Fig. 2). To determine whether ASC has an important role during lymphocyte migration and T cell development, we analyzed the migration of congenically marked wild-type and _Asc_−/− lymphocytes to peripheral lymph nodes and spleens of wild-type mice. The intrinsic migratory ability of _Asc_−/− T cells and B cells was impaired, as indicated by the significantly lower number of both cell types retrieved from the spleen and lymph nodes of wild-type hosts (Fig. 2b). To confirm the intrinsic migratory defect of _Asc_−/− lymphocytes, we created mixed chimeras by injecting an equal ratio of wild-type and _Asc_−/− bone marrow into lethally irradiated wild-type mice. At 6 weeks after reconstitution, ∼98% of the CD4+ T cells found in blood and secondary lymphoid organs were derived from wild-type bone marrow (Fig. 2c). Similarly, most circulating B cells were wild-type cells (Fig. 2c). In addition to being the result of intrinsic defects in migration, the very high ratio of wild-type lymphocytes to _Asc_−/− lymphocytes in mixed chimeras could also have been due to differences in lymphocyte development and cell survival. Although leukocyte populations in _Asc_−/− mice (Supplementary Fig. 3) and chimera mice with _Asc_−/− bone marrow (Supplementary Fig. 4) indeed contained slightly fewer single-positive (CD4+ or CD8+) thymocytes and modestly more double-negative (CD4−CD8−) T cells, these differences in thymocyte development were only minor. Nevertheless, to further confirm the migratory phenotype of _Asc_−/− T cells and B cells, we studied the in vitro migration of these cells toward chemokines. In agreement with a critical role for ASC in lymphocyte chemotaxis, splenic CD4+ T cells from _Asc_−/− mice were nearly completely unable to migrate toward the chemokines SDF-1 and SLC (Fig. 2d). Similarly, B cells from _Asc_−/− mice were defective in their migration toward both SDF-1 and the lymphocyte chemokine BLC (Fig. 2e). In contrast, splenic T lymphocytes and B lymphocytes of _Nlrp3_−/− and _Casp1_−/− mice migrated toward these chemokines to a degree similar to that of wild-type lymphocytes (Supplementary Fig. 5). Thus, ASC is required for lymphocyte migration in vitro and in vivo.
Figure 2: ASC is required for lymphocyte migration in vitro and in vivo.
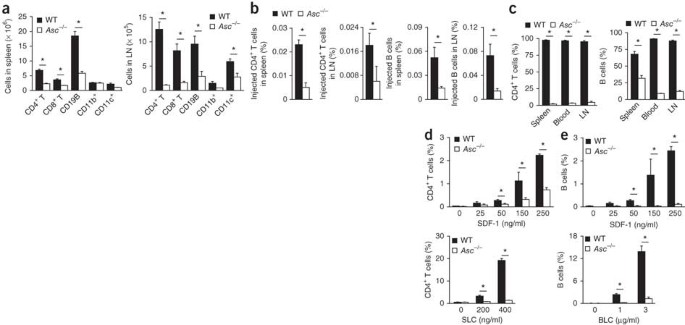
(a) Total number of various cell populations (horizontal axis) among wild-type and _Asc_−/− spleen cells (left) and lymph node cells (LN; right) stained for CD4, CD8, CD19, CD11b and CD11c. (b) Migration of congenically marked wild-type and _Asc_−/− CD4+ T cells and B cells into the spleen and axillary lymph nodes of naive wild-type mice 48 h after injection of an equal ratio of congenically marked wild-type and _Asc_−/− CD4+ T cells or B cells, presented as a percentage of the cells injected. (c) Frequency of congenically marked CD4+ T cells and B cells in the spleen, blood and axillary lymph nodes (LN) of lethally irradiated mice 6 weeks after injection of an equal ratio of congenically marked wild-type and _Asc_−/− bone marrow cells, presented as a percentage of total bone marrow–derived lymphocytes. (d,e) Transwell chemotaxis assay of the migration of wild-type and _Asc_−/− splenocytes in vitro (n = 3–4 mice per group) toward SDF-1, SLC or BLC, presented as frequency among total migratory CD4+ T lymphocytes (d) or B cells (e). *P < 0.005 (two-tailed Student's _t_-test). Data are representative of three independent experiments (mean and s.d. of triplicates).
ASC controls Rac activation and actin polymerization
Both efficient endocytosis of antigens and the migration of lymphocytes toward a chemokine gradient require the activation of small GTPases such as Cdc42 and Rac to induce F-actin polymerization and cytoskeletal reorganization43. However, the expression and activation of Cdc42 were not altered in _Asc_−/− BMDCs and lymphocytes, respectively (Supplementary Fig. 6). In contrast, _Asc_−/− BMDCs were impaired in Rac activation (Fig. 3a) and F-actin polymerization (Fig. 3b) when incubated with OVA. Because lymphocyte migration relies on Rac activation and actin polymerization induced by chemokine receptors, we assessed the extent of Rac activation after chemokine stimulation. Although wild-type CD4+ T cells and B cells activated Rac (Fig. 3c) and induced F-actin polymerization (Fig. 3d) after incubation with SDF-1 (500 ng/ml), these responses were much lower in _Asc_−/− T cells and B cells (Fig. 3c,d). The role of ASC in Rac activation and F-actin polymerization induced by chemokines in lymphocytes was specific, because SDF-1-induced activation of the MAPK Erk was not affected in _Asc_−/− T lymphocytes and B lymphocytes (Supplementary Fig. 7). Thus, defective antigen uptake by _Asc_−/− DCs and chemotaxis of _Asc_−/− lymphocytes is linked to impaired Rac activation and F-actin polymerization in these cells.
Figure 3: ASC is essential for Rac activation and actin polymerization, induced by antigens or chemokines in DCs or lymphocytes, respectively.
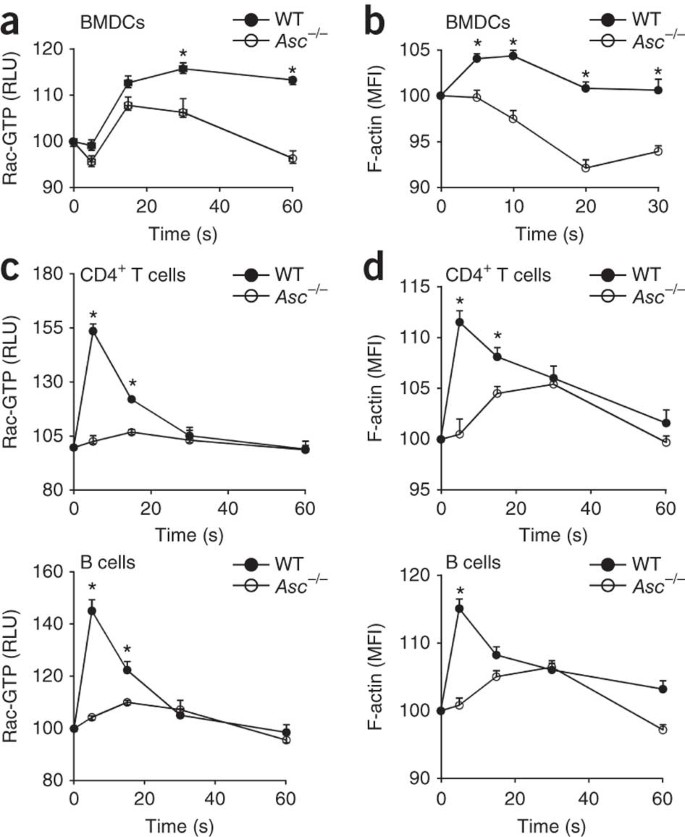
(a,b) Flow cytometry analysis of Rac GTPase activity (a) and F-actin polymerization (b) in wild-type and _Asc_−/− BMDCs treated for 0–60 s (a) or 0–30 s (b) with OVA, presented as relative light units (RLU; a) or mean fluorescence intensity (b) relative to baseline, set as 100. (c) Small G protein–activation assay of Rac activation in lysates of CD4+ T cells and B cells isolated from spleens of wild-type and _Asc_−/− mice and treated for 0–60 s in vitro with SDF-1 (500 ng/ml). (d) Flow cytometry analysis of F-actin polymerization in CD4+ T cells and B cells isolated from wild-type and _Asc_−/− mice (n = 1–3 per group) and treated for 0–60 s in vitro with SDF-1 (500 ng/ml). *P <0.05 (two-tailed Student's _t_-test). Data are representative of at least three independent experiments (mean and s.d. of triplicates).
ASC regulates Dock2 expression independently of inflammasomes
Our results showed that ASC regulates antigen uptake in DCs and the migration of lymphocytes independently of inflammasomes through the modulation of Rac activation and F-actin polymerization. To characterize the molecular mechanism involved, we did microarray experiments to identify genes dysregulated in _Asc_−/− BMDCs despite their normal expression in _Nlrp3_−/− or _Casp1_−/− cells. Notably, among the more than 39,000 transcripts present on the microarray, the transcripts of only 5 genes were downregulated at least 66% in _Asc_−/− BMDCs relative to their expression in wild-type BMDCs (Fig. 4a). In addition to Asc, these included _Dock2 (_80%) and the genes encoding follistatin-like 1 protein (Fstl1; 80%), galanin (Gal; 73%) and fatty acid–binding protein 4 (Fabp4; 70%). Quantitative PCR analysis confirmed the lower expression of Dock2 mRNA in _Asc_−/− BMDCs (Fig. 4b), but the expression of Fstl1, Gal and Fabp4 was unaltered in _Asc_−/− BMDCs (data not shown). Unlike _Asc_−/− BMDCs, _Nlrp3_−/− and _Casp1_−/− BMDCs had normal abundance of Dock2 mRNA (Fig. 4b), which confirmed that Dock2 transcript abundance is regulated by ASC in an inflammasome-independent manner. We observed similarly lower Dock2 transcript abundance in the absence of ASC in CD4+ T cells and isolated B cells (Fig. 4c). In contrast, Dock2 mRNA expression in _Nlrp3_−/− and _Casp1_−/− CD4+ T cells and B cells was similar to that of wild-type cells (data not shown).
Figure 4: ASC regulates Dock2 expression independently of inflammasomes and TLRs.
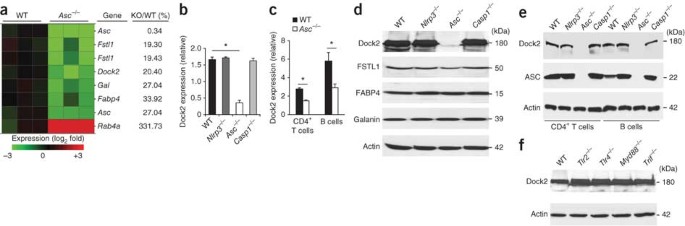
(a) Microarray analysis gene expression among RNA from naive wild-type and _Asc_−/− BMDCs; right margin, genes with a transcript difference of threefold or more in _Asc_−/− cells relative to the expression in wild-type cells. (b) Quantitative PCR analysis of Dock2 mRNA expression in naive wild-type, _Nlrp3_−/−, _Asc_−/− and _Casp1_−/− BMDCs, presented relative to the expression of Gapdh (encoding glyceraldehyde phosphate dehydrogenase). (c) Quantitative PCR analysis of Dock2 mRNA expression in purified wild-type and _Asc_−/− CD4+ T cells and B cells, presented as in b. (d) Immunoblot analysis of Dock2, FSTL1, FABP4 and galanin in naive wild-type, _Nlrp3_−/−, _Asc_−/− and _Casp1_−/− BMDCs. kDa, kilodaltons. (e) Immunoblot analysis of Dock2 and ASC in purified wild-type, Nlrp3_−/−, Asc_−/− and _Casp1_−/− CD4+ T cells and B cells. (f) Immunoblot analysis of Dock2 in lysates of naive wild-type, _Tlr2_−/−, _Tlr4_−/−, _Myd88_−/− and _Trif_−/− BMDCs. Actin serves as a loading control throughout. *P < 0.005 (two-tailed Student's _t_-test). Data are representative of at least three independent experiments (mean and s.d. in b,c).
We next prepared lysates of wild-type, _Nlrp3_−/−, _Asc_−/− and _Casp1_−/− BMDCs to assess the expression of Dock2, FSTL1, FABP4 and galanin by immunoblot analysis. Notably, Dock2 expression was nearly abolished in _Asc_−/− BMDCs but was not altered in _Nlrp3_−/− and _Casp1_−/− BMDCs (Fig. 4d). The expression of FSTL1, galanin and FABP4 was not altered in _Asc_−/− BMDCs (Fig. 4d), which confirmed the results of quantitative PCR. Additionally, activation of the NLRP3 inflammasome by stimulation with LPS and ATP did not upregulate Dock2 expression in _Asc_−/− BMDCs or macrophages (Supplementary Fig. 8). Moreover, Dock2 expression remained stable in _Nlrp3_−/− and _Casp1_−/− BMDCs treated with those stimuli (Supplementary Fig 8). We confirmed ASC-dependent Dock2 expression in the BMDMs used above and BMDCs from an independently generated line of ASC-deficient mice45 (Supplementary Fig. 9). We also observed the nearly complete absence of Dock2 protein in _Asc_−/− T cells and B cells (Fig. 4e). These results indicate that ASC specifically regulates Dock2 expression in an inflammasome-independent manner in myeloid cells and lymphocytes. We also explored whether ASC regulates Dock2 expression in a Toll-like receptor (TLR)-dependent way, but Dock2 expression was normal in BMDCs isolated from mice deficient in TLR2 or TLR4, as well as in cells lacking the common TLR adaptors MyD88 or TRIF (Fig. 4f). Thus, ASC specifically regulates Dock2 expression in myeloid cells and lymphocytes in a TLR- and inflammasome-independent way.
ASC regulates Dock2 mRNA stability
To characterize the mechanism by which ASC regulates Dock2 mRNA abundance, we first analyzed the subcellular localization of ASC in BMDCs by subcellular fractionation. Notably, we detected ASC in both the cytosolic and nuclear fractions of naive BMDCs (Fig. 5a). The subcellular localization of ASC to these compartments remained largely stable after stimulation with LPS and ATP (Fig. 5a). As expected, the antibody to ASC did not detect immunoreactive bands in lysates of _Asc_−/− BMDCs, which confirmed its specificity. Unlike ASC, caspase-1 was located exclusively in the cytosol of naive and BMDCs stimulated with LPS and ATP (Fig. 5a), which suggested that the nuclear pool of ASC is not recruited to inflammasome complexes. In agreement with that, analysis of confocal micrographs indicated that ASC and caspase-1 localized together in the cytosol of BMDCs stimulated with LPS and ATP but not in untreated BMDCs (Fig. 5b). In contrast, ASC located in the nuclear compartment failed to localize together with caspase-1 under these conditions (Fig. 5b), which suggested that nuclear ASC may be responsible for regulating Dock2 mRNA abundance in an inflammasome-independent manner.
Figure 5: ASC localizes to the nucleus and controls the stability of Dock2 mRNA.
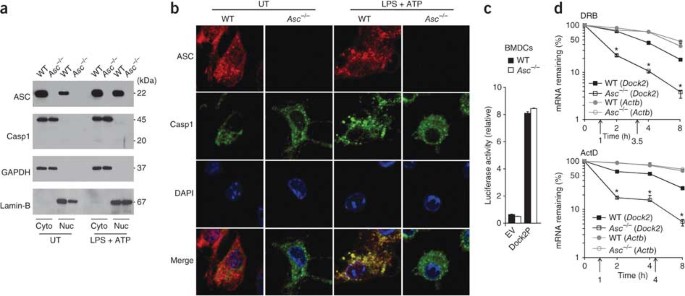
(a) Immunoblot analysis of ASC and caspase-1 (Casp1) in the cytosolic (Cyto) and nuclear (Nuc) compartments of wild-type and _Asc_−/− BMDCs left untreated (UT) or primed for 4 h with LPS (1 μg/ml), in the presence of ATP (5 mM) for the final 15 min (LPS + ATP). GAPDH and lamin-B serve as compartment-specific markers for the cytosolic and nuclear compartments, respectively. (b) Microscopy of wild-type and _Asc_−/− BMDCs left untreated or primed and treated as in a, then washed in PBS, fixed, made permeable and stained for ASC and caspase-1. The DNA-intercalating dye DAPI stains nuclei. Original magnification, ×63. (c) Luciferase activity of wild-type and _Asc_−/− BMDCs transfected by nucleofection with empty reporter vector (EV) or luciferase vector containing the Dock2 promoter (Dock2P); results are presented relative to renilla luciferase activity. (d) Quantitative RT-PCR analysis of Dock2 and Actb mRNA among total RNA from wild-type and _Asc_−/− BMDCs treated with DRB (50 μM) or actinomycin D (ActD; 5 μg/ml), normalized to the expression of Gapdh mRNA and presented relative to baseline expression, set as 100%; the half-life of mRNA (upward arrows) was calculated as the time required for decay to 50% of baseline. *P < 0.005 (Student's _t_-test). Data are representative of at least three independent experiments (mean and s.d. in c,d).
We next assessed whether nuclear ASC regulated the transcriptional activity of the Dock2 promoter. For this, we cloned the Dock2 promoter into a luciferase reporter vector and used this to analyze Dock2 promoter–driven luciferase production in wild-type and _Asc_−/− BMDCs. Luciferase expression was induced similarly in wild-type and _Asc_−/− BMDCs (Fig. 5c), which suggested that ASC may regulate Dock2 mRNA abundance via mRNA stability instead. To investigate this possibility, we assessed Dock2 mRNA stability in wild-type and _Asc_−/− BMDCs after treatment with the reversible transcription inhibitor DRB or actinomycin D to block new transcription. The half-life of Dock2 transcripts was much lower in _Asc_−/− BMDCs (30 min) than in wild-type cells (∼4 h; Fig. 5d). Unlike the half-life of Dock2 mRNA, the half-life of Actb mRNA (encoding β-actin) was almost completely unaffected in _Asc_−/− BMDCs (Fig. 5d), which confirmed the specificity of these results. Thus, regulation of Dock2 mRNA stability represents a major mechanism by which ASC controls Dock2 expression. To examine the potential contribution of post-translational events to the regulation of Dock2 expression, we pretreated wild-type and _Asc_−/− cells for 1, 4 or 6 h with the proteasome inhibitor MG132 before probing lysates for Dock2 expression. Proteasome inhibition resulted in slightly higher Dock2 expression in wild-type cells, but this was not sufficient to restore expression in _Asc_−/− cells (Supplementary Fig. 10).
Dock2 is critical for antigen uptake by DCs
Dock2 is member of a conserved family of guanine nucleotide–exchange factors that is specific to cells of the immune response and has been identified as a central regulator of the migration of lymphocytes and plasmacytoid DCs that controls Rac-dependent actin polymerization and cytoskeletal reorganization in these cells44,46,47. However, its role in DC function has not been characterized. We hypothesized that similar to _Asc_−/− BMDCs (Fig. 1), professional phagocytes lacking Dock2 might be impaired in antigen uptake and endocytosis. We first examined the macropinosis of FITC-labeled dextran, FITC-OVA and lucifer yellow by wild-type and _Dock2_−/− BMDCs. The uptake of dextran (Fig. 6a), OVA (Fig. 6b) and lucifer yellow (Fig. 6c) by _Dock2_−/− BMDCs was significantly impaired relative to their uptake by wild-type BMDCs, which indicated the importance of Dock2 in the endocytosis of soluble antigens by professional phagocytes. The uptake of larger particles and insoluble antigens proceeds through phagocytosis rather than macropinosis48. To determine the role of Dock2 in phagocytosis, we compared the uptake of FITC-labeled zymosan A and FITC-labeled polystyrene beads by wild-type and _Dock2_−/− BMDCs. In agreement with an important role for Dock2 in phagocytosis, _Dock2_−/− DCs were impaired in the internalization of zymosan A (Fig. 6d) and polystyrene beads (Fig. 6e) relative to the internalization of these by wild-type control cells. Notably, ASC expression and inflammasome activation were normal in _Dock2_−/− BMDCs (Supplementary Fig. 11), which further confirmed that ASC- and Dock2-mediated endocytosis is uncoupled from inflammasome signaling.
Figure 6: Dock2 is critical for antigen uptake by DCs and restores immune-cell functions in the absence of ASC.
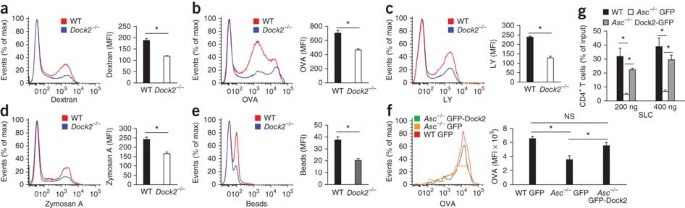
(a–c) Macropinocytosis of FITC-labeled dextran (a), OVA (b) or lucifer yellow (c) by wild-type and _Dock2_−/− BMDCs after incubation together for 3 h at 37 °C. (d–e) Phagocytosis of fluorescein-labeled zymosan A (d) or polystyrene beads (e) by wild-type and _Dock2_−/− BMDCs after incubation as in a. (f) Macropinocytosis of fluorescein-labeled OVA by wild-type and _Asc_−/− BMDCs transfected by nucleofection with plasmid expressing GFP or GFP-Dock2, assessed by flow cytometry 24 h after transfection. (g) Transwell chemotaxis assay of the in vitro migration of wild-type and _Asc_−/− CD4+ T lymphocytes expressing either GFP or GFP-Dock2 toward SLC; results are presented as the frequency among the total migrating T cell population. NS, not significant; *P < 0.05 (two-tailed Student's _t_-test). Data are representative of at least three independent experiments (a–g; mean and s.e.m. of triplicates).
Ectopic expression of Dock2 restores ASC-mediated functions
The observation that Dock2 was critical for the internalization of soluble and insoluble antigens via both macropinosis and phagocytosis by professional antigen-presenting cells, together with our results showing that ASC controlled Dock2 expression in BMDCs and lymphocytes (Figs. 4 and 5), suggested that ASC may act upstream of Dock2 in controlling Dock2-dependent immune functions. Therefore, we assessed whether the defective Rac activation, actin polymerization and antigen uptake by _Asc_−/− BMDCs and impaired migration of _Asc_−/− lymphocytes was due to the specific downregulation of Dock2 in these cells. For this, we transfected _Asc_−/− BMDCs by nucleofection with plasmid encoding green fluorescent protein (GFP) alone or a GFP-Dock2 fusion protein. Endocytosis of OVA by _Asc_−/− BMDCs expressing GFP-Dock2 was significantly higher than its uptake by _Asc_−/− BMDCs expressing GFP alone (Fig. 6f), which confirmed the requirement for Dock2 downstream of ASC for antigen uptake by professional phagocytes. Similarly, ectopic expression of Dock2 in _Asc_−/− T cells restored their migration toward the chemokine SLC (Fig. 6g). Collectively, these data show that the functional defects of _Asc_−/− BMDCs and lymphocytes were due to a specific decrease in Dock2 mRNA stability that led to impaired Dock2 expression in the absence of ASC and that ectopic expression of Dock2 in the cells was sufficient to restore ASC-dependent immune-cell functions.
Discussion
The adaptor ASC is well known to contribute to innate immune responses by enabling activation of the cysteine protease caspase-1 in inflammasomes45,49. Studies have provided evidence indicating that _Asc_−/− mice are protected from disease progression in animal models of arthritis and multiple sclerosis, whereas mice lacking the inflammasome components NLRP3 or caspase-1 are not40,41,42. ASC has been suggested to affect the immune-cell functions of lymphocytes41,42 and DCs40. Although such observations suggest that ASC controls immune-cell functions through inflammasome-independent mechanisms, the molecular pathways involved have remained unclear. ASC has been reported to regulate the activation of MAPKs in macrophages by interacting with the phosphatase DUSP10 in response to TLR ligands and infection with bacterial pathogens50. However, this function may be stimulus dependent, as earlier reports did not observe differences in MAPK signaling in activated and infected _Asc_−/− macrophages45. Furthermore, we failed to detect changes in chemokine-induced phosphorylation of Erk in _Asc_−/− cells of the immune response.
Instead, we found that antigen presentation by DCs and the migration of T lymphocytes and B lymphocytes were considerably diminished in _Asc_−/− mice as a result of impaired Rac-mediated actin polymerization. Microarray and immunoblot analysis of naive and stimulated _Asc_−/− DCs and lymphocytes showed that ASC specifically regulated Dock2 expression. The role of ASC in regulating the abundance of Dock2 mRNA was highly specific, as only 5 transcripts of the 39,000 transcripts analyzed were up- or downregulated by at least 66% in _Asc_−/− BMDCs relative to their expression in wild-type BMDCs. Moreover, Dock2 was the only ASC-regulated gene whose expression was altered at the protein level as well. Instead of directly affecting Dock2 promoter activity, ASC had a critical role in stabilizing Dock2 mRNA, as the half-life of Dock2 transcripts was much lower (87.5%) in _Asc_−/− DCs. Further analysis is needed to determine the specific contribution of the nuclear and cytosolic pools of ASC to this process. Given that ASC lacks intrinsic enzymatic activity, ASC most probably affects Dock2 abundance by assembling one or more protein complex(es) in the cytosol and/or nucleus, distinct from the inflammasomes. Moreover, in addition to regulating the decay rate of Dock2 transcripts, ASC may contribute to as-yet-unknown processes that regulate Dock2 mRNA and Dock2 protein at the post-transcriptional and/or post-translational level. These may include microRNAs that target Dock2, splicing of Dock2 mRNA linked to nonsense-mediated mRNA decay, trafficking of Dock2 mRNA, the rate of Dock2 translation and the degree of post-translational modification that may influence the trafficking and degradation of Dock2 in lysosomes. However, the notable effect of ASC deficiency on the decay rate of Dock2 mRNA may hamper detailed analysis of the potential role of ASC in regulating Dock2 expression through these additional mechanisms. Regardless of what proves to be true, we confirmed downregulated Dock2 expression as the mechanism that drove the defective phagocytosis and antigen presentation by DCs and dysfunctional chemotaxis of lymphocytes in the absence of ASC. For example, ectopic expression of Dock2 in ASC-deficient cells restored lymphocyte migration and antigen uptake by DCs. Thus, the observation that ASC controlled Dock2 expression and Dock2-mediated regulation of Rac activation and actin polymerization provides a molecular basis for the defective antigen presentation by DCs and the lymphocyte-intrinsic defects in cell migration observed in the studies cited above.
In conclusion, we have provided genetic evidence of a critical and unexpected role for ASC in regulating the motility of T lymphocytes and B lymphocytes and antigen uptake by professional antigen-presenting cells independently of inflammasomes. Instead, ASC controlled the stability of Dock2 transcripts and expression of Dock2. Our observations identify a previously unknown role for ASC in regulating adaptive immune responses that is intrinsic to lymphocytes and DCs. The vital role of ASC in regulating both adaptive and innate immune responses suggests that modulating ASC expression and function may represent a powerful therapeutic strategy for the treatment of inflammatory and autoimmune disorders.
Methods
Mice.
Both independent _Asc_−/− mouse lines used in this study have been characterized45,51. _Nlrp3_−/−, _Casp1_−/−, _Tlr2_−/−, _Tlr4_−/−, _MyD88_−/−, _Trif_−/− and _Dock2_−/− mice have been described2,44. All mice were backcrossed at least ten generations to the C57BL/6J genetic background. Mice were housed in a pathogen-free facility and animal studies were conducted under protocols approved by St. Jude Children's Research Hospital Committee on Use and Care of Animals or by the Ghent University Hospital ethical committee.
Microarray analysis and quantitative PCR.
The quality of isolated RNA was confirmed with an Agilent 2100 Bioanalyzer before microarray analysis. RNA samples were processed on an HT MG-430 PM array plate with the Affymetrix GeneTitan system. Three biological replicates of naive wild-type and _Asc_−/− BMDCs were analyzed by microarray, and transcript expression values were summarized by the robust multi-array average method52. Differences between wild-type and _Asc_−/− BMDCs in gene expression were analyzed by an empirical Bayesian method (Cyber-T statistics program)53, and the false-discovery rate was estimated as described54. For quantitative PCR analysis of Dock2, samples were amplified with specific primers (forward, 5′-TTGCTCAGCCAGCTACTGTATG-3′; reverse, 5′-TTGGTGATGACAGGAAGCAGAAT-3′). Quantification was normalized relative to expression of the reference gene Gapdh.
Subcellular fractionation.
Cytoplasmic and nuclear fraction extractions were prepared with NE-PER Nuclear and Cytoplasmic Extraction Reagents according to the manufacturer's instructions (Pierce).
Immunoblot analysis.
Standardized protein concentrations of cellular lysates were separated by SDS-PAGE. Antibody to Dock2 (anti-Dock2; 09-454), anti-ASC (AL177; Enzo Life Sciences), anti-caspase-1 (a gift from P. Vandenabeele), anti-FSTL1 (AF1738; R&D Systems), anti-FABP4 (2120; Cell Signaling Technology), anti-galanin (ab99452; Abcam) and anti-β-actin (4970; Cell Signaling Technology) were used at a final dilution of 1:1,000. Proteins were detected by horseradish peroxidase–based enhanced chemiluminescence (34095; Thermo Scientific).
Transfection.
Immature BMDCs were transfected with a Nucleofector kit (VPA-1009; Lonza) and were incubated at 37 °C for 24 h before antigen-uptake assays.
Retroviral transduction.
GFP-Dock2 and GFP were cloned into a mouse stem cell virus retroviral vector. Phoenix-Eco packaging cells were transfected with Lipofectamine 2000 (11668-027; Invitrogen), and recombinant viruses were collected 48 h and 72 h after transfection. Total spleen cells were isolated and cultured for 24 h with anti-CD3 (2.5 μg/ml; 145-2C11; eBioscience), anti-CD28 (5 μg/ml; 37.51; eBioscience) and IL-2 (100 U/ml) and then transduced with retrovirus by 'spin inoculation'. GFP+ cells were sorted by flow cytometry and then used for chemotaxis assays.
T cell proliferation.
Popliteal lymph nodes were collected 10 d after immunization with 10 μg BSA (Sigma, 85040) in complete Freund's adjuvant emulsion. CD4+ T cells were isolated by negative selection by a mouse CD4+ T cell–enrichment strategy (AutoMACS; 130-095-248; Milteny). CD4+CD11c+ DCs (130-091-262) were isolated from spleens of naive wild-type and _Asc_−/− mice treated with collagenase-D (11088874103) according to the manufacturer's instructions (Roche). Cocultures were started with 5 × 105 CD4+ T cells per well and 2.5 × 105 DCs per well and the appropriate concentration of antigen. Cultures were maintained for 72 h at 37 °C and 5% CO2 in U-bottomed plates in 300 μl HL-1 medium (7720; Lonza) supplemented with penicillin G (50 U/ml) and streptomycin (50 μg/ml), 2-mercaptoethanol (50 μM), L-glutamine (292 μg/ml) and 0.1% (wt/vol) BSA. Supernatants were collected for evaluation of cytokine production and cultures were pulsed with [3H]thymidine (1 μCi/well in 10 μl), then were incubated for an additional 18 h and collected onto UniFilter GF/C plates, then radioactivity was measured as counts per minute on a TopCount NXT microplate scintillation counter (PerkinElmer).
Cytokine measurement.
Cytokines were measured with a Milliplex ELISA kit (Millipore).
Flow cytometry.
For flow cytometry, cells were stained with anti-CD4 (L3T4), anti-CD8 (53-6.7), anti-CD11b (M1/70) or anti-B220 (RA3-6B2; all from eBioscience); or anti-CD8α (53-6.7), anti-TCRβ (H57-597), anti-CD44 (IM7), anti-CD69 (H1.2F3), anti-CD11c (N418), anti-CD45.1 (A20) or anti-CD45.2 (104; all from Biolegend); after staining, cells were analyzed on an LSR II (Becton-Dickinson). For analysis of antigen uptake and phagocytosis, DCs were incubated for 3 h with fluorochrome-conjugated OVA, dextran, luciferase yellow, zymosan A or beads. Cells were washed several times and analyzed by flow cytometry. For analysis of actin polymerization, isolated T cells and B cells were fixed in 4% (vol/vol) paraformaldehyde, made permeable with Permeabilization Wash Buffer (421002; Biolegend) and stained with Alexa Fluor 488–labeled phalloidin (BD Biosciences) for flow cytometry.
Confocal microscopy.
BMDCs (1 × 105) were plated on glass cover slips (BD Biosciences) and, 24 h later, cells were stimulated, washed in PBS and fixed for 10 min in 4% (vol/vol) paraformaldehyde. Cells were made permeable for 5 min with 0.1% (vol/vol) Triton X-100. Blockade was achieved by incubation for 60 min with blocking buffer (1% (vol/vol) BSA in PBS and 0.1% (vol/vol) Triton X-100). Cells were stained for 1 h with mouse monoclonal anti-ASC (1 μg/ml; 04-147; Millipore) and rabbit polyclonal antibody to caspase-1 (ref. 51; a gift from P. Vandenabeele) in blocking buffer. After cells were washed, bound antibodies were detected for 1 h at 25 °C with Alexa Fluor 488–conjugated goat anti-rabbit (for caspase-1 (green); A-11034; Molecular probes) and Alexa Fluor 647–conjugated chicken anti-mouse (for ASC (red); A-21463; Molecular probes). Slides were mounted with ProLong Gold-DAPI mounting media (P-36931; Invitrogen) and were analyzed with an inverted spinning-disk confocal microscope (Zeiss) with a 63× objective lens with Slidebook software. The specificity of anti-ASC and anti-caspase-1 was confirmed through the use of _Asc_−/− and _Casp1_−/− BMDCs, respectively, as a negative control.
Luciferase (promoter activity) assay.
The Dock2 promoter (positions −3931 to +132) was amplified from mouse genomic DNA (forward primer, 5′-GATCGTCGACGCAAGGTCAGAAATTTTGTTAGAAAAGATTTTAAA-3′; reverse primer, 5′-CATGGGATCCCCCACACTTGCCCACACCTAC-3′) and was cloned in to the pGL3-Enhancer vector upstream to the firefly luciferase reporter gene. BMDCs were transfected with combinations of the pGL3-Enhancer–Dock2 promoter reporter plasmid or pGL3-Enhancer empty vector and control renilla luciferase plasmid (pTK-RL) through the use of an Amaxa DC Nucleofector kit (Lonza). Luciferase activity was quantified 24 h after transfection with the Dual-Luciferase Reporter Assay System according to the manufacturer's instructions (Promega). Dock2 promoter activity (firefly luciferase) was normalized to the internal control (renilla luciferase).
Competitive-reconstitution mixed chimeras.
Wild-type CD45.1+ congenic mice were lethally irradiated with a split dose of 1,200 rads. Bone marrow from CD45.1+CD45.2+ wild-type mice and CD45.2+_Asc_−/− mice was injected intravenously at a ratio of 1:1. After 6 weeks of reconstitution, the frequency of wild-type and _Asc_−/− CD4+ T cells and B cells was calculated with anti-CD45.1 (A20; Biolegend) and anti-CD45.2 (104; Biolegend).
Chemotaxis assay.
Splenocytes (1.5 × 106) in 100 μl RPMI medium were placed in Transwell permeable supports (pore size, 5 μm), which were placed onto 24-well plates containing 500 μl RPMI medium supplemented with chemokines (R&D Systems) at various concentrations. Cells were incubated for 3 h at 37 °C. Cells placed in the Transwell supports and those that migrated to the lower chamber were collected and stained with anti-TCRβ (H57-597; eBioscience) and anti-B220 (RA3-6B2; eBioscience).
GTPase activity assay.
The GTPase activity of Cdc42 and Rac1 in cell extracts was measured by G-LISA activation assay (BK127 (for Cdc42) and BK127 (for Rac1); Cytoskeleton).
Assay of mRNA stability.
New transcription was inhibited by culture of BMDCs in the presence of DRB (5,6-dichloro-1-β-ribofuranosyl benzamidazole; 50 μM; D191; Sigma-Aldrich) or actinomycin D (5 μg/ml; A1410; Sigma-Aldrich). Total RNA was then isolated for measurement of the abundance of Dock2 and Actb transcripts by real-time quantitative RT-PCR. Transcript abundance was normalized to that of Gapdh.
Statistical analysis.
P values were calculated with Student's _t_-test. P values of less than 0.05 were considered significant.
Change history
01 June 2012
In the version of this article initially published, the authors identified a defect in Dock2 expression in ASC-deficient (Pycard–/–) mice that was not related to the inflammasome. With this addendum statement and figure, the authors now report that not all ACS-deficient strains have this defect, to alert the community to this finding.
References
- Villani, A.C. et al. Common variants in the NLRP3 region contribute to Crohn's disease susceptibility. Nat. Genet. 41, 71–76 (2009).
Article CAS Google Scholar - Zaki, M.H. et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391 (2010).
Article CAS Google Scholar - Allen, I.C. et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056 (2010).
Article CAS Google Scholar - Bauer, C. et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199 (2010).
Article CAS Google Scholar - Dupaul-Chicoine, J. et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32, 367–378 (2010).
Article CAS Google Scholar - Jin, Y. et al. NALP1 in vitiligo-associated multiple autoimmune disease. N. Engl. J. Med. 356, 1216–1225 (2007).
Article CAS Google Scholar - Martinon, F., Petrilli, V., Mayor, A., Tardivel, A. & Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241 (2006).
Article CAS Google Scholar - Magitta, N.F. et al. A coding polymorphism in NALP1 confers risk for autoimmune Addison's disease and type 1 diabetes. Genes Immun. 10, 120–124 (2009).
Article CAS Google Scholar - Larsen, C.M. et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 356, 1517–1526 (2007).
Article CAS Google Scholar - Agostini, L. et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325 (2004).
Article CAS Google Scholar - Hoffman, H.M., Mueller, J.L., Broide, D.H., Wanderer, A.A. & Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 29, 301–305 (2001).
Article CAS Google Scholar - Lamkanfi, M. Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 11, 213–220 (2011).
Article CAS Google Scholar - Lamkanfi, M. & Dixit, V.M. The inflammasomes. PLoS Pathog. 5, e1000510 (2009).
Article Google Scholar - Lamkanfi, M. & Dixit, V.M. Inflammasomes: guardians of cytosolic sanctity. Immunol. Rev. 227, 95–105 (2009).
Article CAS Google Scholar - Amer, A. et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281, 35217–35223 (2006).
Article CAS Google Scholar - Franchi, L. et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat. Immunol. 7, 576–582 (2006).
Article CAS Google Scholar - Mariathasan, S. et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232 (2006).
Article CAS Google Scholar - Miao, E.A. et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat. Immunol. 7, 569–575 (2006).
Article CAS Google Scholar - Suzuki, T. et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 3, e111 (2007).
Article Google Scholar - Sutterwala, F.S. et al. Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245 (2007).
Article CAS Google Scholar - Miao, E.A., Ernst, R.K., Dors, M., Mao, D.P. & Aderem, A. Pseudomonas aeruginosa activates caspase 1 through Ipaf. Proc. Natl. Acad. Sci. USA 105, 2562–2567 (2008).
Article CAS Google Scholar - Franchi, L. et al. Critical role for Ipaf in _Pseudomonas aeruginosa_-induced caspase-1 activation. Eur. J. Immunol. 37, 3030–3039 (2007).
Article CAS Google Scholar - Boyden, E.D. & Dietrich, W.F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 38, 240–244 (2006).
Article CAS Google Scholar - Kanneganti, T.D. et al. Critical role for cryopyrin/Nalp3 in activation of caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 281, 36560–36568 (2006).
Article CAS Google Scholar - Sutterwala, F.S. et al. Critical role for NALP3/CIAS1/cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24, 317–327 (2006).
Article CAS Google Scholar - Kanneganti, T.D. et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 26, 433–443 (2007).
Article CAS Google Scholar - Muruve, D.A. et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452, 103–107 (2008).
Article CAS Google Scholar - Broz, P. et al. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 207, 1745–1755 (2010).
Article CAS Google Scholar - Wu, J., Fernandes-Alnemri, T. & Alnemri, E.S. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 30, 693–702 (2010).
Article CAS Google Scholar - Warren, S.E. et al. Cutting edge: cytosolic bacterial DNA activates the inflammasome via Aim2. J. Immunol. 185, 818–821 (2010).
Article CAS Google Scholar - Tsuchiya, K. et al. Involvement of absent in melanoma 2 in inflammasome activation in macrophages infected with Listeria monocytogenes. J. Immunol. 185, 1186–1195 (2010).
Article CAS Google Scholar - Sauer, J.D. et al. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7, 412–419 (2010).
Article CAS Google Scholar - Rathinam, V.A. et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402 (2010).
Article CAS Google Scholar - Fernandes-Alnemri, T. et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393 (2010).
Article CAS Google Scholar - Miao, E.A. et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142 (2010).
Article CAS Google Scholar - Kanneganti, T.D., Lamkanfi, M. & Nunez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559 (2007).
Article CAS Google Scholar - Srinivasula, S.M. et al. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 277, 21119–21122 (2002).
Article CAS Google Scholar - Ellebedy, A.H. et al. Inflammasome-independent role of the apoptosis-associated speck-like protein containing CARD (ASC) in the adjuvant effect of MF59. Proc. Natl. Acad. Sci. USA 108, 2927–2932 (2011).
Article CAS Google Scholar - McElvania Tekippe, E. et al. Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS ONE 5, e12320 (2010).
Article Google Scholar - Ippagunta, S.K. et al. Inflammasome-independent role of apoptosis-associated speck-like protein containing a CARD (ASC) in T cell priming is critical for collagen-induced arthritis. J. Biol. Chem. 285, 12454–12462 (2010).
Article CAS Google Scholar - Kolly, L. et al. Inflammatory role of ASC in antigen-induced arthritis is independent of caspase-1, NALP-3, and IPAF. J. Immunol. 183, 4003–4012 (2009).
Article CAS Google Scholar - Shaw, P.J. et al. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J. Immunol. 184, 4610–4614 (2010).
Article CAS Google Scholar - Trombetta, E.S. & Mellman, I. Cell biology of antigen processing in vitro and in vivo. Annu. Rev. Immunol. 23, 975–1028 (2005).
Article CAS Google Scholar - Fukui, Y. et al. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature 412, 826–831 (2001).
Article CAS Google Scholar - Mariathasan, S. et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218 (2004).
Article CAS Google Scholar - Sanui, T. et al. DOCK2 regulates Rac activation and cytoskeletal reorganization through interaction with ELMO1. Blood 102, 2948–2950 (2003).
Article CAS Google Scholar - Gotoh, K. et al. Differential requirement for DOCK2 in migration of plasmacytoid dendritic cells versus myeloid dendritic cells. Blood 111, 2973–2976 (2008).
Article CAS Google Scholar - Doherty, G.J. & McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 78, 857–902 (2009).
Article CAS Google Scholar - Kanneganti, T.D. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 10, 688–698 (2010).
Article CAS Google Scholar - Taxman, D.J. et al. The NLR adaptor ASC/PYCARD regulates DUSP10, mitogen-activated protein kinase (MAPK), and chemokine induction independent of the inflammasome. J. Biol. Chem. 286, 19605–19616 (2011).
Article CAS Google Scholar - Kanneganti, T.D. et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236 (2006).
Article CAS Google Scholar - Irizarry, R.A. et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 (2003).
Article Google Scholar - Baldi, P. & Long, A.D. A Bayesian framework for the analysis of microarray expression data: regularized t-test and statistical inferences of gene changes. Bioinformatics 17, 509–519 (2001).
Article CAS Google Scholar - Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J.R. Stat. Soc. B 57, 289–300 (1995).
Google Scholar
Acknowledgements
We thank R. Flavell (Yale University School of Medicine), G. Nunez (University of Michigan) and S. Akira (Osaka University) for mutant mice; P. Vandenabeele (Ghent University) for anti-caspase-1; and H. Chi (St. Jude Children's Research Hospital) for retroviral plasmids and mutant mice. Supported by the US National Institutes of Health (R01AR056296 and R21AI088177 to T.-D.K.), the American Lebanese Syrian Associated Charities (T.-D.K.), the European Union Framework Program 7 (Marie-Curie grant 256432 to M.L.) and the Fund for Scientific Research-Flanders (M.L. and L.V.W.).
Author information
Author notes
- Sirish K Ippagunta, R K Subbarao Malireddi and Patrick J Shaw: These authors contributed equally to this work.
Authors and Affiliations
- Department of Immunology, St. Jude Children's Research Hospital, Memphis, Tennessee, USA
Sirish K Ippagunta, R K Subbarao Malireddi, Patrick J Shaw, Douglas R Green & Thirumala-Devi Kanneganti - Department of Hartwell Center for Bioinformatics & Biotechnology, St. Jude Children's Research Hospital, Memphis, Tennessee, USA
Geoffrey A Neale - Department of Biochemistry, Ghent University, Ghent, Belgium
Lieselotte Vande Walle & Mohamed Lamkanfi - Department of Medical Protein Research, VIB, Ghent, Belgium
Lieselotte Vande Walle & Mohamed Lamkanfi - Division of Immunogenetics, Kyushu University, Kyushu, Japan
Yoshinori Fukui - Department of Immunobiology and Neuroscience Medical Institute of Bioregulation, Kyushu University, Kyushu, Japan
Yoshinori Fukui
Authors
- Sirish K Ippagunta
You can also search for this author inPubMed Google Scholar - R K Subbarao Malireddi
You can also search for this author inPubMed Google Scholar - Patrick J Shaw
You can also search for this author inPubMed Google Scholar - Geoffrey A Neale
You can also search for this author inPubMed Google Scholar - Lieselotte Vande Walle
You can also search for this author inPubMed Google Scholar - Douglas R Green
You can also search for this author inPubMed Google Scholar - Yoshinori Fukui
You can also search for this author inPubMed Google Scholar - Mohamed Lamkanfi
You can also search for this author inPubMed Google Scholar - Thirumala-Devi Kanneganti
You can also search for this author inPubMed Google Scholar
Contributions
T.-D.K., M.L., S.K.I., P.J.S. and R.K.S.M. designed research; S.K.I., P.J.S., R.K.S.M., did research; G.A.N. did bioinformatic analyses; L.V.W. confirmed ASC-dependent Dock2 expression in an independently generated line of ASC-deficient mice; D.R.G. contributed to the writing of the manuscript and conceptual insights; Y.F. provided reagents; T.-D.K., M.L., S.K.I., P.J.S., R.K.S.M., G.A.N. and Y.F. analyzed data; P.J.S., M.L. and T.-D.K. wrote the paper; and T.-D.K. conceived of the study, designed the experiments and provided overall direction.
Corresponding authors
Correspondence toMohamed Lamkanfi or Thirumala-Devi Kanneganti.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
About this article
Cite this article
Ippagunta, S., Malireddi, R., Shaw, P. et al. The inflammasome adaptor ASC regulates the function of adaptive immune cells by controlling Dock2-mediated Rac activation and actin polymerization.Nat Immunol 12, 1010–1016 (2011). https://doi.org/10.1038/ni.2095
- Received: 13 June 2011
- Accepted: 27 July 2011
- Published: 04 September 2011
- Issue Date: October 2011
- DOI: https://doi.org/10.1038/ni.2095