Catalytic activity of caspase-3 is required for its degradation: stabilization of the active complex by synthetic inhibitors (original) (raw)
Introduction
Apoptosis, a form of programmed cell death, is a highly regulated process essential for the tidy disposal of dying cells in many multicellular organisms.1,2 This mode of death is critical for normal development as well as the maintenance of cellular homeostasis.3 On the molecular level, members of a family of cysteine proteases, designated caspases, are responsible for programmed cell death.4,5 In response to an apoptotic stimulus a caspase cascade is activated in which initiator caspases are first activated through dimerization and they, in turn, act to cleave and activate downstream effector caspases.6 Once matured, effector caspases can cleave specific protein substrates within the cell, resulting in the apoptotic phenotype.5,7
Numerous studies have implicated caspase-3 as one of the main effector caspases in mammalian cells.5 The importance of this gene is exemplified by the phenotype of the caspase-3-null mice that are characterized by severe defects in neuronal development due to a lack of normal programmed cell death.8 Other studies performed using caspase-3-null MCF-7 breast carcinoma cells demonstrate that caspase-3 is essential for inactivation of ICAD (inhibitor of caspase-activated DNase) and is therefore the primary activator of apoptotic DNA fragmentation.9,10 Accordingly, results using caspase-3 immunodepleted cell-free extracts indicate that caspase-3 is the main effector caspase for many apoptotic processes, like chromatin margination, DNA fragmentation and nuclear collapse in this system.11
Caspase-3 is synthesized as a dormant proenzyme (p32), consisting of a 3 kDa prodomain, the large subunit (p17) and the small subunit (p12). Maturation is executed via a two-step mechanism.5 First, the large and the small subunit are generated by proteolytic cleavage mediated via an initiator caspase12 or through pH-dependent autocatalytic processing.13 In a second step, the preactivated caspase-3 self-catalyses removal of its prodomain from the large subunit to generate the p17 polypeptide. The X-ray crystal structure of active caspase-3 has been solved.14,15 The mature enzyme was shown to exist as a tetramer with two independent active sites, each containing elements from one large (p17) and one small (p12) subunit. Catalysis is mediated by a mechanism typical of cysteine proteases involving a catalytic dyad, composed of Cys163 and His121, which are harbored by the large subunit. The substrate-binding cleft recognizes a short 4 amino-acid stretch within protein substrates, directly N-terminal to the cleavage site.14
This tetrapeptide motif, which is sufficient to bind specifically to the active caspase, is the basis for the design of the synthetic inhibitors. Comparative inhibitor analysis clearly indicates that peptide-derived inhibitors harboring the optimal substrate tetrapeptide-motif for a specific caspase are highly potent and in some cases selective.16 To date, all synthetic caspase inhibitors, as well as the endogenous, macromolecular inhibitors that have been identified are competitive inhibitors which act by occupying the active site of the enzyme.
Caspase inhibitors typically contain an electrophilic group or ‘warhead’, which binds covalently to the active-site cysteine,16 and depending on the chemistry of this ‘warhead’, caspase inhibitors can be characterized as being either reversible inhibitors that include aldehydes, nitriles and ketones or irreversible inhibitors such as diazomethylketones, acyloxymethylketones and halomethylketones. Although in general peptide-based inhibitors show substantial disadvantages, like poor cell penetration and poor metabolic stability in cell-based assays, some classes have proven effective at inhibiting recombinant caspases in vitro, as well as in cell-based systems. The aspartic acid-esterified nonselective tripeptide-fluoromethylketone, Z-VAD(OMe)-CH2F, which is presumably de-esterified and activated upon entry into cells, is efficacious at inhibiting cell death in vivo;5,17 this is despite the drawbacks of the active counterpart, which include the lack of chemical stability and electrophilic promiscuity, that potentially results in activity against other biological targets.5
The mechanisms that regulate caspase-3 activation have been extensively studied, but few studies have investigated the fate of caspase-3 once activated or for that matter once inhibited. However, this phenomenon is important as cells may not always be destined to die following activation of caspase-3, particularly if they are treated with inhibitors or are required on a transient basis to serve some regulatory role. In this study, we sought to directly analyze the fate of active caspase-3 in intact cells. We report, that active caspase-3 is rapidly degraded by a mechanism that is dependent on the catalytic activity of the active enzyme. This degradation can be inhibited by synthetic caspase-3 inhibitors which concomitantly results in the stabilization of active caspase-3. The stabilizing effect mediated by synthetic inhibitors is primarily a function of the inhibitor's intrinsic potency as well as its cell permeability. The possible implications of these observations are discussed.
Results
Although several studies have elegantly elucidated the mechanism of caspase processing and activation, the fate of caspases, once activated, is poorly understood. 6,12 For instance, following the induction of apoptosis, we and others18,19 have noted the stark lack of an appropriate precursor–product relationship between the proenzyme and the mature (p17) subunit of caspase-3. As shown by immunoblot analysis this phenomenon is readily observed in HeLa cells induced by staurosporine treatment to undergo apoptosis (Figure 1). The loss of p17 signal, as determined by Western blotting, following the conversion from the proenzyme to the active form, is not due to a difference in the affinity of the caspase-3 antibody for the p17 subunit or to poor transfer efficiency of the p17 subunit to the nitrocellulose membrane, as experiments with recombinant caspase-3 have demonstrated (data not shown).
Figure 1
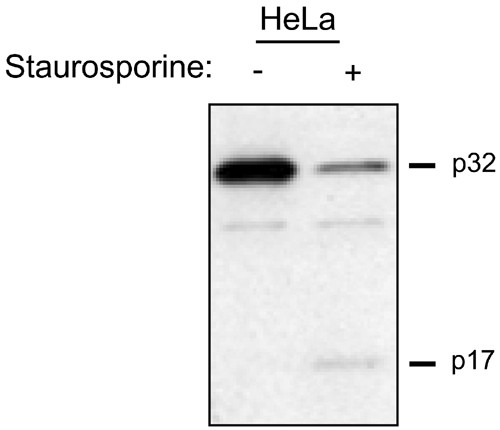
Lack of precursor product relationship between procaspase-3 and mature caspase-3 in apoptotic HeLa cells. Caspase-3 was activated in HeLa cells by treatment of the cells with 1 _μ_M staurosporine for 4 h. Cytoplasmic lysate (15 _μ_g) from untreated and treated cells was analyzed by SDS-PAGE and Western blotting using an antibody directed against the large subunit (p17) of human caspase-3. The nonspecific band migrating between the p32 and p17 polypeptides serves as a loading control
To address the fate of active caspase-3 in HeLa cells, we employed a tetracycline-responsive expression system which transiently coexpresses the large and small subunits of caspase-3 from a bidirectional promoter. This expression system can be used to directly overproduce active caspase-3 enzyme, while circumventing the need for a proapoptotic stimulus. Consistent with the rapid turnover of mature endogenous caspase-3, we failed to detect significant amounts of the large subunit by Western blot analysis at 48 h post-transfection using an antibody specific for the p17 subunit (Figure 2a, lane 3). In contrast, procaspase-3 (p32) was detected at high levels following transfection using the same expression system (Figure 2a, lane 1). A corresponding lack of detectable p12 polypeptide has been similarly noted in other immunoblot experiments (data not shown). The following observations make it unlikely that a complete lack of expression of the p17 and p12 subunits was responsible for this difference. First, similar amounts of mRNA encoded by the transfected constructs (p17/p12, p32) were detected by RT-PCR (Figure 2a, lanes 1 and 3). Second, unlike cells transfected with plasmid-DNA encoding the proenzyme (p32), cells coexpressing both subunits of the active enzyme, p17 and p12, underwent apoptosis (Figure 2b, compare top left and right panels). This clearly indicates that active enzyme was indeed being expressed. In addition, the cell death observed could be specifically linked to the expression of p17/p12, as repression of the p17/p12 construct through addition of doxycycline to the culture medium resulted in healthy cells (Figure 2b, bottom right panel). Third, detection of a cotransfected EGFP protein shows that expression of exogenous proteins was clearly not being abolished in the p17/p12 transfected cells, although the induction of apoptosis did have a mild effect on the expression efficiency (Figure 2c). As illustrated in Figure 2c, clusters of dying cells (indicated by the arrows) could be easily identified as being GFP-positive by fluorescence microscopy. The EGFP protein was reduced in its amount, but was not totally absent (Figure 2c). The same holds true for a coexpressed inactive procaspase-3 mutant, which cannot be processed (Figure 5, lane 8). Together, these results suggest that, once generated, the active enzyme is rapidly degraded.
Figure 2
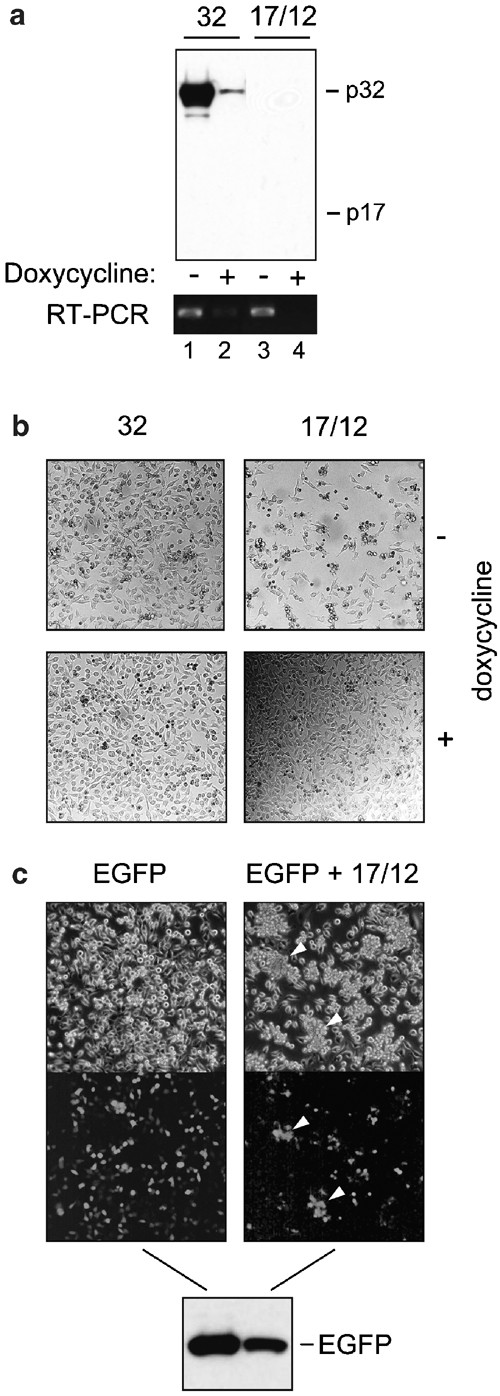
Transgene expression is not completely abolished by toxicity induced from coexpression of caspase-3 large and small. HeLa Tet-Off cells were transfected with either 2.5 _μ_g of vector encoding procaspase-3 (32), 2.5 _μ_g of vector encoding both large and small caspase-3 subunits (17/12), or cotransfected with either 1.25 _μ_g of empty tet-responsive bidirectional expression vector and 1.25 _μ_g of vector encoding EGFP (EGFP), or 1.25 _μ_g of vector encoding both large and small caspase-3 subunits and 1.25 _μ_g of vector encoding EGFP (EGFP+17/12). Expression was for 48 h either in the presence or absence (a and b) or absence (c) of the transgene-specific transcription inhibitor doxycycline. (a) SDS-PAGE and Western blot using an anticaspase-3 large subunit antibody (upper panel) on 15 _μ_g of cytoplasmic lysate and RT-PCR analysis (lower panel) on 100 ng of total RNA using caspase-3-specific primers. (b) light micrographs of HeLa Tet-Off cells post-transfection. (c), light and fluorescent micrographs and SDS-PAGE/Western blot using an anti-GFP antibody on HeLa Tet-Off cells post-transfection
Figure 5
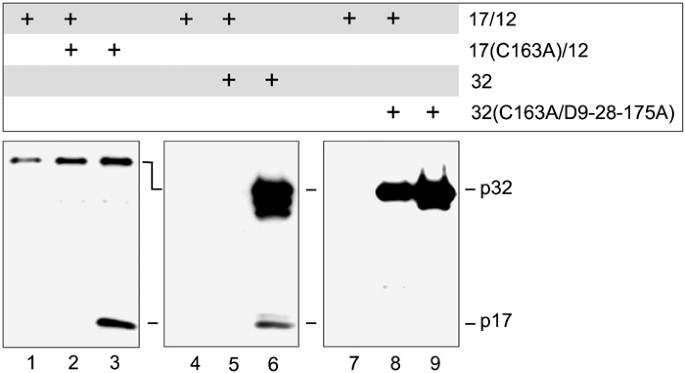
An active conformation is necessary for caspase-3 turnover. HeLa Tet-Off cells were cotransfected with either 1.25 _μ_g of empty tet-responsive bidirectional expression vector and 1.25 _μ_g of vector encoding both large and small caspase-3 subunits (17/12), 1.25 _μ_g of vector encoding both large subunit containing cysteine 163 to alanine mutation and small caspase-3 subunit and 1.25 _μ_g of vector encoding both large and small caspase-3 subunits (17/12+17(C163A)/12), 1.25 _μ_g of empty tet-responsive bidirectional expression vector and 1.25 _μ_g of vector encoding both large subunit containing cysteine 163 to alanine mutation and small caspase-3 subunit (17(C163A)/12), 1.25 _μ_g of vector encoding procaspase-3 and 1.25 _μ_g vector encoding both large and small caspase-3 subunits (32+17/12), 1.25 _μ_g of empty tet-responsive bidirectional expression vector and 1.25 _μ_g of vector encoding procaspase-3 (32), 1.25 _μ_g of vector encoding inactive and uncleavable procaspase-3 and 1.25 _μ_g vector encoding both large and small caspase-3 subunits (32(C163A/D9-28-175A)+17/12), or 1.25 _μ_g of vector encoding inactive and uncleavable procaspase-3 and 1.25 _μ_g of empty tet-responsive bidirectional expression vector (32(C163A/D9-28-175A)). Expression was maintained for 48 h followed by SDS-PAGE and Western blots using an anticaspase-3 large subunit antibody on 15 _μ_g of cytoplasmic lysates
In order to test whether the catalytic site of caspase-3 was involved in conferring instability to the active enzyme, we performed transfection experiments with either proenzyme(p32)-expressing or p17/p12-expressing vectors in the presence of an irreversible pan-caspase inhibitor (zVAD-FMK). Interestingly, the inhibitor produced a notable increase in the level of p17 such that it could easily be detected by Western blot analysis (Figure 3a, compare lanes 3 and 4). This suggested that caspase inhibitors could have either a direct stabilizing effect on the active enzyme complex or enhance the expression through a cytoprotective effect. Experiments with the overexpressed proenzyme strongly support a mechanism involving enzyme stabilization. A small fraction of the highly expressed proenzyme was found to spontaneously undergo processing to its active subunits. In the presence of inhibitor, a significant elevation of the p17 subunit could be detected, thus corroborating the inhibitor stabilizing effect on the active enzyme (Figure 3a, compare lanes 1 and 2). The increase is even more significant in light of the fact that the inhibitor is also acting to prevent procaspase-3 processing to its active subunits through an inhibition of upstream activator caspases (i.e. caspases-8, -9) or autolytic processing. Hence, in addition to inhibiting the catalytic activity of caspases, synthetic inhibitors appear to influence the rate of degradation of active caspase-3, likely through stabilization of the heteromeric complex. A similar effect on p17 turnover was observed when a more selective and reversible caspase-3 inhibitor (MF452) was administered to the cell cultures (Figure 3b). This inhibitor is intrinsically at least two orders of magnitude more potent than zVAD-FMK and can bind active caspase-3 with much greater affinity. This is consistent with the more pronounced level of complex stabilization seen with MF452. The enhanced stabilizing effect of MF452 could not be attributed to more efficient rescue from cell death (i.e. healthier cell cultures) given that more cell death was observed in the MF452-treated cultures than in the zVAD-FMK-treated cultures (Figure 3b). In these experiments, zVAD-FMK, a less potent but pan-caspase inhibitor, was likely mediating its cytoprotective effects through inhibition of multiple caspases. To further explore the relationship between the intrinsic potency of different caspase-3 inhibitors, their ability to inhibit apoptosis as measured by DNA fragmentation, and their ability to stabilize the enzyme in vivo, we compared the relative stabilizing effect of a panel of selective inhibitors (Figure 3c; Table 1). When comparing inhibitors with similar ability to inhibit apoptosis (i.e those within Group I, II, or III), we found that the magnitude of the stabilization tracked directly with intrinsic potency against caspase-3. Furthermore, the inhibitors which were the most potent at inhibiting apoptosis (Figure 3c, Group III, lanes 7–9) were not able to stabilize the enzyme any better than weaker inhibitors of apoptosis (Figure 3c, Group I, lanes 2–4). Thus, the ability of the inhibitors to bind specifically to caspase-3 primarily determines their capacity to stabilize the enzyme complex.
Figure 3
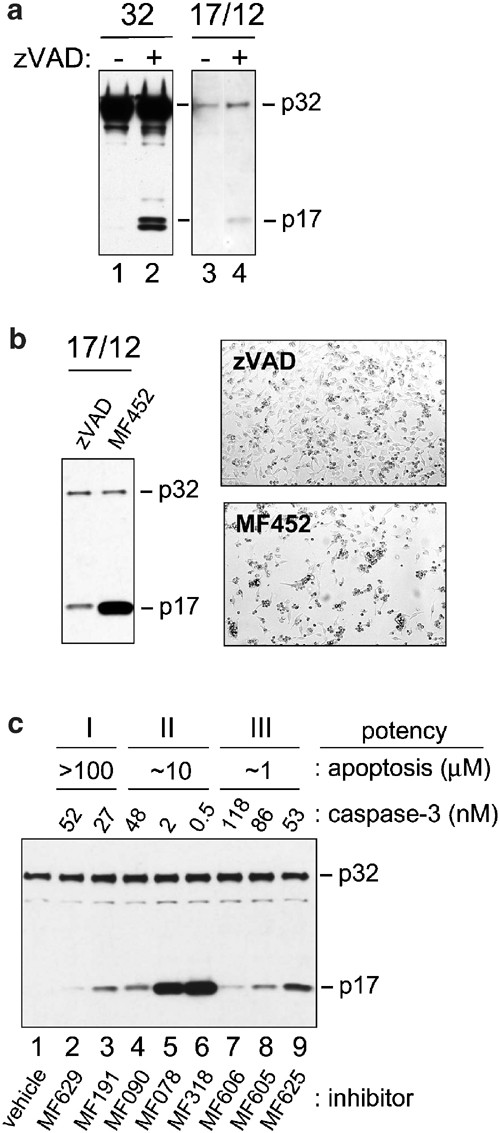
Caspase-3 inhibitors stabilize the matured enzyme. HeLa Tet-Off cells were transfected with either 2.5 _μ_g of vector encoding procaspase-3 (32) or 2.5 _μ_g of vector encoding both large and small caspase-3 subunits (17/12) in the absence or presence of the caspase-3 inhibitors zVAD, MF452, MF629, MF191, MF090, MF078, MF318, MF606, MF605, or MF625 (100 _μ_M). Expression was for 48 h. (a and c), SDS-PAGE and Western blot using an anticaspase-3 large subunit antibody on 15 _μ_g of cytoplasmic lysate from transfected cells. (b), SDS-PAGE and Western blot using an anticaspase-3 large subunit antibody on 15 _μ_g of cytoplasmic lysate and light micrograph of transfected cells 48 post-transfection
Table 1 Structure and Inhibition Profile of Caspase-3 Inhibitors Used in this Study
To further examine the requirement for stabilization of an active conformation of caspase-3, we asked whether a caspase-3 selective inhibitor can also stabilize the p17 large subunit when it is expressed in the absence of the p12 subunit. Under these conditions the p17 subunit could not be detected in significant amounts by immunoblot analysis (Figure 4, lane 3). Even in the presence of inhibitor the turnover of the large subunit was not suppressed (Figure 4, lane 4), indicating the inhibition of turnover is dependent on coexpression of the small subunit. To confirm proper expression of the large subunit from the vector encoding only the large subunit, cotransfection with a second vector encoding the small subunit was performed. Coexpression of p12 partly rescued the ability of the inhibitor to stabilize the large subunit (Figure 4, lane 6). The rapid degradation of the p17 subunit, when expressed alone, likely results from its failure to assemble into a stable heteromeric complex. We conclude from these results that the formation of the p17/p12 complex is a prerequisite for inhibitor-mediated stabilization.
Figure 4
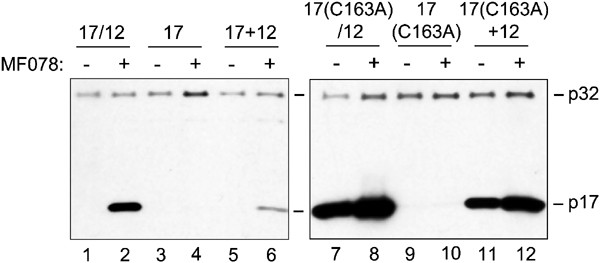
Inhibitor-mediated stabilization requires an active caspase-3 conformation. HeLa Tet-Off cells were transfected with either 2.5 _μ_g of vector encoding both large and small caspase-3 subunits (17/12), 2.5 _μ_g of vector encoding caspase-3 large subunit (17), 2.5 _μ_g vector encoding both large subunit containing cysteine 163 to alanine mutation and small caspase-3 subunit (17(C163A)/12), 2.5 _μ_g of vector encoding caspase-3 large subunit containing cysteine 163 to alanine mutation (17(C163A)), or cotransfected with either 1.25 _μ_g of vector encoding caspase-3 large subunit and 1.25 _μ_g vector encoding caspase-3 small subunit (17+12), or cotransfected with 1.25 _μ_g vector encoding caspase-3 large subunit containing cysteine 163 to alanine mutation and 1.25 _μ_g vector encoding caspase-3 small subunit (17(C163A)+12). Expression was for 48 h either in the absence or presence of the caspase-3 inhibitor MF078 (100 _μ_M). SDS-PAGE and Western blots using an anticaspase-3 antibody on 15 _μ_g of cytoplasmic lysates from transfected cells is shown
In an effort to determine whether the catalytic activity of caspase-3 was required for rapid turnover to occur, we examined the stability of a p17 polypeptide mutant harboring a point mutation which inactivates the catalytic cysteine (Cys163Ala). Upon dimerization with p12, to form the bipartite substrate binding site, the resulting caspase-3 heterodimer was no longer able to catalyze cleavage of substrate. Cells were transfected with plasmids expressing both the p17(C163A) and the p12 subunit. In contrast to the active wild-type p17 subunit, the mutant p17 subunit could be easily detected by Western blot analysis 48 h after transfection, indicating that the catalytically inactive mutant p17 polypeptide was being expressed in a stable manner (Figure 4, lane 7). Thus, it can be concluded that the rapid degradation of wild-type p17/p12 complex requires the catalytic activity of the enzyme. When caspase activity was abolished, as was the case with the mutant p17(C163A)/p12 complex, the p17 subunit was stabilized. The small increase in expression level of the mutant heterodimer in the presence of the inhibitor MF078 is believed to be a result of enhanced synthesis secondary to cytoprotection of the cells by MF078 post-transfection. This cytoprotective effect is also observed on transfection of other nonapoptotic genes (data not shown) and can be seen at the level of endogenous p32 activation.
Furthermore, we observed that stable expression of mutant p17 was dependent on the coexpression of the small p12 subunit. The stabilizing effect of p12 was visible either when it was encoded separately from or together with the mutant p17 subunit (Figure 4, lane 7 and 11). When expressed alone, the p17(C163A) protein could not be detected (Figure 4, lane 9). The same observation was made for the expression of the wild-type p17 subunit when expressed alone (Figure 4, lane 3). In summary, these results strongly reinforce the hypothesis that the large subunit is only stable when assembled as a heteromeric complex together with the small subunit. Moreover, the mechanism of degradation of the unassembled wild-type or mutant p17 subunit likely differs from the mechanism mediating turnover of the active assembled p17/p12 complex, because turnover of the unassembled p17 subunit is independent of the catalytic activity of the caspase-3, whereas the degradation of p17/12 complex is not. Stabilization of the p17(C163A)/p12 complex indicates that caspase-3 activity is required for the turnover of the p17/p12 complex.
Thus far, we have demonstrated that caspase-3 catalytic activity is required for the turnover of mature caspase-3 and that the active form of caspase-3 can be stabilized by caspase inhibitors. Given this, two possibilities for the mechanism of active caspase-3 degradation can be proposed. On the one hand, the catalytic activity may be required to catalyze a cleavage event which is necessary to initiate the turnover of caspase-3. This cleavage event may act in cis to provide the signal for autolysis, although at the moment evidence for this possibility does not exist, since even at high concentrations, experiments with the active recombinant enzyme indicate that it is quite stable (data not shown), or in trans, propagated via other proteolytic components in the cell. Alternatively, an intact active site may be required to attract a ‘destructor’ molecule which targets caspase-3 for degradation. To address these possibilities, we asked whether active caspase-3 can mediate turnover of the inactive p17/p12 complex via a transacting mechanism (Figure 5). To this end, we coexpressed active p17/p12 subunits in combination with mutant p17(C163A)/p12 subunits. Even if hybrid catalytic domains are formed, one-quarter of the tetramers would be expected to be composed purely of mutant active sites. Under these conditions, the mutated large subunit p17(C163A) could not be detected, which is in strong contrast to its prominent presence in cells when expressed alone (Figure 4, lane 7 and Figure 5, lanes 2 and 3). This result demonstrated that active caspase-3 can act in trans to mediate turnover of heteromeric p17/p12 complexes.
Although the p17(C163A)/p12 heterodimer is catalytically inactive, it can most likely adopt a conformation equivalent to the endogenous p17/p12 complex. A pseudoactive conformation might be important for the transacting mechanism of caspase-3 degradation. To test this hypothesis, we generated an uncleavable active site mutant of procaspase-3 through site-directed mutagenesis of the three primary maturation sites (Asp9, Asp28, and Asp175) to glutamic acids and conversion of the catalytic cysteine to an alanine residue. Upon coexpression with the active p17/p12 subunits the mutant form of procaspase-3 remained intact (Figure 5, lane 8), while wild-type procaspase-3 was degraded (Figure 5, lane 5), probably via processing to its mature subunits. The mutant procaspase-3 (D9,18,171A; C163A) polypeptide could not be processed and consequently, formation of a heteromeric, pseudo-active conformation was precluded. The lack of degradation of this mutant suggests that an active conformation is necessary for turnover of caspase-3 in the presence of enzymatic activity. In summary, three major conclusions can be drawn from our current work. First, caspase activity is critical to the promotion of its own turnover. Second, the active conformation of caspase-3 triggers its rapid degradation and this can occur via a _trans_-acting mechanism. Third, synthetic inhibitors can bind and stabilize the active enzyme, thereby preventing degradation of the mature enzyme in intact cells.
Discussion
Caspases are synthesized on a constitutive basis as inactive proenzymes. In response to numerous proapoptotic stimuli procaspase-3 is rapidly processed to generate a large (p17) and small (p12) polypeptide subunit, and two copies of each subunit form a catalytically active tetrameric complex.5,12 Work from our lab and that of others18,19 demonstrates that the active form of caspase-3 is highly labile and is rapidly turned over relative to its proenzyme form. In addition, we show that turnover of caspase-3 is dependent on its catalytic activity and that as a consequence of inhibition, caspase inhibitors act to efficiently stabilize the active form of the enzyme. The rapid degradation of caspase-3 likely reflects the cell's need to ensure the timely suppression of a destructive proteolytic activity. This represents an efficient strategy to eliminate induced or even spontaneously generated caspase activity and thereby helps to protect cells. The control of enzyme activity via a degradative pathway is superimposed upon existing regulatory mechanisms that act to modulate caspases, including proteolytic processing (i.e. proenzyme maturation) and protein–protein interaction (i.e. IAP-mediated inhibition).5,20,21 Temporal and/or spatial restriction of caspase-3 activity has previously been proposed to be important in cell cycle control22 and neuronal plasticity.23 Caspases have also been shown to be activated as part of the denucleation process during the terminal differentiation of lens epithelial cells.24 Similarly, active caspases are detected following T-lymphocyte stimulation in the absence of apoptosis, suggesting a regulatory role during this process.25,26 Once activated, caspases that have fulfilled their regulatory functions would need to be eliminated to avoid driving the cells into apoptosis. Other examples of proapoptotic proteins, which are regulated by degradation include SMAC, tBid and APAF-1. In the case of the tBid the protein was shown to be degraded following cleavage by caspase-8 and a degradation-resistant form of t-Bid was found to cause less apoptosis compared to wt-tBid in transfection experiments.27 Similarly, degradation of the cytochrome _c_-binding protein APAF-1 inhibits caspase activation during Ca(2+)-mediated neuronal cell death.28 In addition, two independent groups have demonstrated that IAP-mediated proteosomal degradation of SMAC involves ubiquitination.29,30
With regard to the mechanism of turnover, the stabilization of the active enzyme by caspase inhibitors, as well as the increased stability of the catalytically inactive mutant suggests that the activity of caspase-3 may trigger its own degradation either directly or indirectly. Moreover, this activity can be provided in trans, such that an inactive caspase-3 mutant(C163A) can be efficiently degraded in the presence of active caspase-3. Self-cleavage of active caspase-3 seems unlikely as a potential mechanism for degradation given that the purified active enzyme does not display autolytic properties. Nonetheless, it is conceivable that caspase-3-dependent cleavage of one or more proteins, could in turn promote caspase-3 degradation. In this case, the activation of a specific degradative pathway for caspase-3 would be initiated in a feedback loop by caspase-3 itself.
Our studies reveal that only active caspase-3 or inactive caspase-3 arranged in a pseudoactive conformation seems to be degraded, while a noncleavable procaspase-3 molecule with a mutation in the catalytic site (C163A) is stable even in the presence of the active enzyme. This implies that, in addition to catalytic activity, the active conformation influences the turnover of active caspase-3. With respect to the requirement for an active conformation, recent data indicate that XIAP, a member of the IAP family, mediates ubiquitination of caspase-3 and thereby targets caspase-3 for proteasomal degradation.31 It has been shown previously that XIAP binds to activated caspase-3 or to a catalytically inactive caspase-3 with an active conformation, but does not bind to the proenzyme.32,33 We do not believe, that the turnover of mature caspase-3 in our system involves XIAP-induced proteasomal degradation, since our studies demonstrate a requirement for an intact catalytic site. Furthermore, in our experiments, proteasome inhibitors (MG132, lactacystin) failed to stabilize active caspase-3 complexes in HeLa cells, possibly reflecting a cell-specific difference in the mode of degradation (unpublished data).
Finally, while it seems paradoxical, that on the one hand caspase inhibitors protect cells from death and on the other, help stabilize the active form of the enzyme, in effect elevating the intracellular pool of caspase-3, it should be stressed that the inhibitor-bound enzyme is catalytically inactive. Also, in spite of the rapid turnover of active caspase-3 there are generally sufficient molecules remaining to generate the apoptotic phenotype upon induction. Furthermore, through direct proteolytic inactivation of antiapoptotic proteins (e.g. IAP's) caspases can help evade their inhibition.
In summary, our studies demonstrate that active caspase-3 is less stable than procaspase-3 and that it can be stabilized with synthetic inhibitors, consistent with the observation that caspase-3 activity may trigger its own degradation. Future studies will be aimed at further elucidating the precise molecular pathway of degradation and its putative role as a mode of regulation for caspase activity.
Materials and Methods
Constructs
Procaspase-3 was amplified by polymerase chain reaction (PCR) (Clontech, Advantage HF PCR kit) as an MluI/NheI fragment using the following oligonucleotide pair: 5′-gcgcacgcgtgccgccaccatggagaacactgaaaactcagtggattc-3′ and 5′-gcgcgctagcttagtgataaaaa tagagttcttttgtgag-3′; protein limits: MENT…YFYH. The _Mlu_I/_Nhe_I fragment was subcloned into the multiple cloning site (MCS I) of the pBI vector (Clontech). Similarly, the large and small subunits of caspase-3 were amplified by PCR as _Pst_I/_Sal_I (5′-gcgcctgcaggccgccaccatgtctggaatatccctggacaacag-3′ and 5′-gcgcgtcgacttagtctgtctcaatgccacagtccag-3′; protein limits: MSGIS…IETD) and _Mlu_I/_Nhe_I (5′-gcgcacgcgtgccgccaccatgagtggtgttgatgatgacatggcg-3′ and 5′-gcgcgctagcttagtgataaaaatagagttcttttgtgag-3′; protein limits: MSGVD…YFYH) fragments and subcloned into MCS II and MCS I of the pBI vector, respectively, either together (17/12) or individually (17 or 12). A Kozak consensus motif (gccgccaccatg) was engineered into the initiator methionione residue of all coding sequences. Point mutations were created by site-directed mutagenesis using overlapping oligonucleotides combined with PCR or by employing the Quickchange™ mutagenesis kit (Stratagene). EGFP expression was encoded by the pBI-EGFP vector (Clontech).
Transfections
HeLa-Tet-Off cells (Clontech) were subcultured onto 60 mm tissue culture dishes (5 × 105 cells per dish) and transfected the following day with 2.5 _μ_g. of plasmid DNA using the Lipofectamine Plus™ reagent (Life Technologies). Cells were allowed to express the transgenes for 48 h prior to harvesting. When doxycycline (2 mg/ml in water) was included during the transfection and expression period, 2 _μ_g/ml final concentration was used. When caspase-3 inhibitors (25 mM in DMSO) were included during the transfection and expression period, 100 _μ_M final concentration was used. Cells were harvested by scraping into 75 _μ_l of CHEG buffer (50 mM HEPES/KOH, pH 7. 0.1 % CHAPS, 2 mM EDTA, 10 % glycerol) containing a protease inhibitor cocktail (Complete™, Boehringer Mannheim) in order to prepare the cytoplasmic lysates. Protein concentrations were determined using the BioRad Protein Assay (Bio Rad).
Immunoblot analysis
Immunoblot blot analysis was performed as described13 using Novex reagents. For immunoblot analysis of caspase-3, a 1 : 2000 dilution of the R280 rabbit antiserum (large subunit of caspase-3 used as immunogen; generated in-house) was used. For immunoblot of EGFP, a 1 : 1000 dilution of a monoclonal anti-GFP antibody (Cat# 1814460, Boehringer Mannheim) was used. A 1 : 3000 dilution of a horseradish peroxidase-conjugated donkey anti-rabbit or sheep anti-mouse secondary antibody (Amersham) was used.
RT-PCR
RNA from transfected cells was prepared from half of the cells cultured on a 60 mm dish after a 48 h expression period using the High Pure™ RNA isolation kit (Boehringer Mannheim). RT-PCR was performed on 100 ng of RNA and a primer pair specific for the small subunit of caspase-3 (5′-gtgttgatgatgacatggcgtgtc-3′ and 5′-ttagtgataaaaatagagttcttttg-3′) using the Titan™ One Tube RT-PCR System (Boehringer Mannheim).
Determination of inhibitor potency
For determination of caspase potency, inhibitors were titrated against recombinant purified caspases (caspase-1 (1 nM), caspase-3 (118 pM), caspase-7 (600 pM), and caspase-8 (4 nM)) and caspase activity measured immediately upon addition of fluorogenic tetrapeptide substrates at their Km (ac-DEVD-AMC (Biomol) at 10, 7.5,. and 50 _μ_M for caspases 3, 7, and 8, respectively, or ac-YVAD-AMC (Biomol) at 10 _μ_M for caspase-1) over 10 min at room temperature in CHEG buffer using a 96-well fluorometer (Cytofluor, Perseptive Biosystems). The potency assay involved testing the inhibitors against all four caspases concurrently and always included a well-studied inhibitor that served to make sure all enzymes were behaving normally. Enzyme and inhibitors were mixed just prior to substrate addition without any preincubation. Potencies were calculated and expressed as the concentration of inhibitor needed to inhibit 50% of the control caspase-3 activity (i.e. enzyme receiving vehicle alone (no inhibitor)) over the 10 min timecourse as measured by the slope of the AMC accumulation over time. Curvature in the slope for the tight binding inhibitor MF191 (ac-DEVD-cho) was negligible over this time frame.
For determination of inhibitor potency against apoptosis, human teratocarcinoma cells (NT2) plated in 96-well plates were induced to undergo apoptosis for 5 h with 5 _μ_g/ml camptothecin (Sigma) in the presence and absence of inhibitor titrations and the level of nucleosomal DNA fragmentation was measured using the Cell Death ELISA kit (Boehringer Mannheim). Potencies were calculated and expressed as the concentration of inhibitor needed to inhibit 50% of the control level of nucleosomal DNA fragmentation (i.e. cells receiving vehicle alone (no inhibitor))
Abbreviations
DEVD-CHO:
tetrapeptide aldehyde
Z-VAD(OMe)-CH2F:
tripeptide fluoromethylketone
DEVD-AMC:
tetrapeptide aminomethyl coumarin
IAP:
inhibitor of apoptosis
GFP:
green fluorescent protein
References
- Wyllie AH, Kerr JF and Currie AR (1980) Cell death: the significance of apoptosis. Int. Rev. Cytol. 68: 251–306
Article CAS Google Scholar - Vaux DL and Korsmeyer SJ (1999) Cell death in development. Cell 96: 245–254
Article CAS Google Scholar - Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267: 1456–1462
Article CAS Google Scholar - Thornberry NA and Lazebnik Y (1998) Caspases: enemies within. Science 281: 1312–1316
Article CAS Google Scholar - Nicholson DW (1999) Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 6: 1028–1042
Article CAS Google Scholar - Budihardjo I, Oliver H, Lutter M, Luo X and Wang X (1999) Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15: 269–290
Article CAS Google Scholar - Earnshaw WC, Martins LM and Kaufmann SH (1999) Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 68: 383–424
Article CAS Google Scholar - Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P and Flavell RA (1996) Decreased apoptosis in the brain and premature lethality in CPP32- deficient mice. Nature 384: 368–372
Article CAS Google Scholar - Tang D and Kidd VJ (1998) Cleavage of DFF-45/ICAD by multiple caspases is essential for its function during apoptosis. J. Biol. Chem. 273: 28549–28552
Article CAS Google Scholar - Wolf BB, Schuler M, Echeverri F and Green DR (1999) Caspase-3 is the primary activator of apoptotic DNA fragmentation via DNA fragmentation factor-45/inhibitor of caspase-activated DNase inactivation. J. Biol. Chem. 274: 30651–30656
Article CAS Google Scholar - Slee EA, Adrain C and Martin SJ (2001) Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 276: 7320–7326
Article CAS Google Scholar - Hengartner MO (2000) The biochemistry of apoptosis. Nature 407: 770–776
Article CAS Google Scholar - Roy S, Bayly CI, Gareau Y, Houtzager VM, Kargman S, Keen SL, Rowland K, Seiden IM, Thornberry NA and Nicholson DW (2001) Maintenance of caspase-3 proenzyme dormancy by an intrinsic ‘safety catch’ regulatory tripeptide. Proc. Natl. Acad. Sci. USA 98: 6132–6137
Article CAS Google Scholar - Rotonda J, Nicholson DW, Fazil KM, Gallant M, Gareau Y, Labelle M, Peterson EP, Rasper DM, Ruel R, Vaillancourt JP, Thornberry NA and Becker JW (1996) The three-dimensional structure of apopain/CPP32, a key mediator of apoptosis. Nat. Struct. Biol. 3: 619–625
Article CAS Google Scholar - Mittl PR, Di Marco S, Krebs JF, Bai X, Karanewsky DS, Priestle JP, Tomaselli KJ and Grutter MG (1997) Structure of recombinant human CPP32 in complex with the tetrapeptide acetyl-Asp-Val-Ala-Asp fluoromethyl ketone. J. Biol. Chem. 272: 6539–6547
Article CAS Google Scholar - Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW and Thornberry NA (1998) Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 273: 32608–32613
Article CAS Google Scholar - Nicholson DW (2000) From bench to clinic with apoptosis-based therapeutic agents. Nature 407: 810–816
Article CAS Google Scholar - Luo X, Budihardjo I, Zou H, Slaughter C and Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481–490
Article CAS Google Scholar - Granville DJ, Carthy CM, Jiang H, Shore GC, McManus BM and Hunt DW (1998) Rapid cytochrome c release, activation of caspases 3, 6, 7 and 8 followed by Bap31 cleavage in HeLa cells treated with photodynamic therapy [In Process Citation]. FEBS Lett 437: 5–10
Article CAS Google Scholar - Deveraux QL and Reed JC (1999) IAP family proteins – suppressors of apoptosis. Genes Dev. 13: 239–252
Article CAS Google Scholar - Shi Y (2002) Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell. 9: 459–470
Article CAS Google Scholar - Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC and Altieri DC (1998) Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 396: 580–584
Article CAS Google Scholar - Cowan CM, Thai J, Krajewski S, Reed JC, Nicholson DW, Kaufmann SH and Roskams AJ (2001) Caspases 3 and 9 send a pro-apoptotic signal from synapse to cell body in olfactory receptor neurons. J. Neurosci. 21: 7099–7109
Article CAS Google Scholar - Ishizaki Y, Jacobson MD and Raff MC (1998) A role for caspases in lens fiber differentiation. J. Cell. Biol. 140: 153–158
Article CAS Google Scholar - Miossec C, Dutilleul V, Fassy F and Diu-Hercend A (1997) Evidence for CPP32 activation in the absence of apoptosis during T lymphocyte stimulation. J. Biol. Chem. 272: 13459–13462
Article CAS Google Scholar - Alam A, Cohen LY, Aouad S and Sekaly RP (1999) Early activation of caspases during T lymphocyte stimulation results in selective substrate cleavage in nonapoptotic cells. J. Exp. Med. 190: 1879–1890
Article CAS Google Scholar - Breitschopf K, Zeiher AM and Dimmeler S (2000) Ubiquitinmediated degradation of the proapoptotic active form of bid. A functional consequence on apoptosis induction. J. Biol. Chem. 275: 21648–21652
Article CAS Google Scholar - Reimertz C, Kogel D, Lankiewicz S, Poppe M and Prehn JH (2001) Ca(2+)-induced inhibition of apoptosis in human SH-SY5Y neuroblastoma cells: degradation of apoptotic protease activating factor-1 (APAF-1). J. Neurochem. 78: 1256–1266
Article CAS Google Scholar - Hu S and Yang X (2003) Cellular inhibitor of apoptosis 1 and 2 are ubiquitin ligases for the apoptosis inducer Smac/DIABLO. J. Biol. Chem. 278: 10055–10060
Article CAS Google Scholar - MacFarlane M, Merrison W, Bratton SB and Cohen GM (2002) Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J. Biol. Chem. 277: 36611–36616
Article CAS Google Scholar - Suzuki Y, Nakabayashi Y and Takahashi R (2001) Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc. Natl. Acad. Sci. USA 98: 8662–8667
Article CAS Google Scholar - Deveraux QL, Takahashi R, Salvesen GS and Reed JC (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388: 300–304
Article CAS Google Scholar - Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC and Salvesen GS (2001) Structural basis for the inhibition of caspase-3 by XIAP. Cell 104: 791–800
Article CAS Google Scholar
Author information
Author notes
- K Hell
Present address: Institut fuer Physiologische Chemie, Ludwig Maximilians Universitaet Muenchen, Butenandtstr. 5, Gebaeude B, 81377, München, Germany - P Tawa and K Hell: Coauthors contributed equally to the work
Authors and Affiliations
- Department of Biochemistry & Molecular Biology, Merck Frosst Centre for Therapeutic Research, Kirkland, Quebec, Canada
P Tawa, K Hell, A Giroux, E Grimm, Y Han, D W Nicholson & S Xanthoudakis
Authors
- P Tawa
- K Hell
- A Giroux
- E Grimm
- Y Han
- D W Nicholson
- S Xanthoudakis
Corresponding author
Correspondence toS Xanthoudakis.
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
Tawa, P., Hell, K., Giroux, A. et al. Catalytic activity of caspase-3 is required for its degradation: stabilization of the active complex by synthetic inhibitors.Cell Death Differ 11, 439–447 (2004). https://doi.org/10.1038/sj.cdd.4401360
- Received: 12 May 2003
- Revised: 26 September 2003
- Accepted: 27 October 2003
- Published: 09 January 2004
- Issue Date: 01 April 2004
- DOI: https://doi.org/10.1038/sj.cdd.4401360