Modifications and intracellular trafficking of FADD/MORT1 and caspase-8 after stimulation of T lymphocytes (original) (raw)
Introduction
‘Death receptors’, a subgroup of the tumour necrosis factor (TNF) receptor family, such as Fas (CD95/APO-1), and their corresponding ligands act as critical regulators of apoptosis, as well as other processes in the immune system.1 The ‘death domain’ (DD) of a death receptor is essential for apoptosis induction, providing the docking site for homotypic interaction with the DD in the adaptor proteins FADD/MORT1 and TRADD.2, 3 Upon recruitment to activated death receptors at the plasma membrane, the ‘death effector domain’ (DED) of FADD binds to one of two DEDs within caspase-8 (and in humans also caspase-10), inducing their autocatalytic activation.2, 3, 4 Caspase-8 launches the caspase cascade by proteolytic cleavage and activation of downstream effector caspases (caspase-3, -6, -7), which degrade vital cellular constituents. Active caspase-8 can also trigger an amplification process through proteolytic activation of the proapoptotic Bcl-2 family member Bid.5, 6, 7 Experiments with knockout mice and transgenic mice expressing inhibitors of FADD and caspase-8 have demonstrated that FADD8, 9, 10 and caspase-811, 12, 13, 14 are essential for death receptor-induced apoptosis.
Surprisingly, it was found that FADD and caspase-8 are also critical for mitogen- and antigen receptor-induced activation and proliferation of T, B and NK cells.8, 10, 12, 15, 16 Loss of FADD function diminished both the proliferative expansion of immature T cells stimulated through the pre-TCR complex17 and the response of mature T cells to mitogens8, 10 or antigen.18 Recently, two siblings exhibiting features of autoimmune lymphoproliferative syndrome (ALPS), a disease that is usually due to mutation in the fas gene,1 were found instead to have a homozygous mutation in the _caspase_-8 gene.15 The resulting instability of caspase-8 rendered lymphocytes resistant to Fas-induced apoptosis and surprisingly, B, T and NK cells were also refractory to mitogenic and antigenic stimulation.15 An essential role for caspase-8 in T-cell activation was proven by the analysis of mice devoid of caspase-8 only in T cells.12, 16
While it is known that FADD transmits the apoptotic signal by recruiting caspase-8 and promoting its activation through conformational change, it is unclear how FADD and caspase-8 promote cell proliferation. A putative role for p53 in the proliferative defect of T cells lacking FADD function19 has not been confirmed.18 Evidence that FADD, caspase-815, 20 and cFLIP21, 22, 23 promote T-cell activation by increasing stimulation of NF-_κ_B and c-jun N-terminal kinase (JNK) was not substantiated in other studies.12, 18
Rafts are specialised microdomains, located in the plasma membranes of cells, which have the ability to include or exclude signalling proteins and aggregate in response to various stimuli, thus serving as platforms for signalling complexes. Some reports have indicated that rafts play a critical role in TCR signalling.24, 25, 26 These studies should, however, be viewed with caution as the amount of TCR/CD3 and their signal transducers recovered with rafts depends on the type of detergent used.27 Recently, it was suggested that activation of Fas leads to the recruitment of FADD and caspase-8 into lipid rafts in mouse thymocytes,28 human CD4+ T cells29 and in a human lymphoblastoid cell line,30 indicating that such intracellular trafficking might also play a role in ‘death receptor’-induced apoptosis. However, not all investigators have been able to reproduce these findings.31
We generated monoclonal antibodies to mouse FADD and caspase-8 to analyse their subcellular localisation and post-translational modification. Our studies show that FADD undergoes a mitogen-stimulation-specific post-translational modification. Moreover, although stimulation with both mitogens or Fas ligand cause recruitment of FADD and caspase-8 to distinct sites at the plasma membrane, they do not colocalise with lipid rafts. In addition, in response to these stimuli caspase-8 becomes part of a larger protein complex. These results indicate that differences in post-translational modification and subcellular trafficking may determine whether FADD and caspase-8 transduce an apoptotic or a proliferative signal in T cells.
Results
Characterisation of novel monoclonal antibodies to mouse FADD and caspase-8
To obtain monoclonal antibodies recognising mouse FADD or caspase-8, hybridomas generated by fusing spleen cells of immunised rats to myeloma cells were screened by immunofluorescence staining and flow cytometric analysis of cells transfected with FLAG epitope-tagged FADD or caspase-8.32 Antibodies specific to FADD (Figure 1c–e) or caspase-8 (Figure 2c, d) were identified by a staining profile that was comparable to that seen with an anti-FLAG antibody (Figures 1b, 2b). The lower intensity immunofluorescence peak represents background staining of parental FDC-P1 cells (Figure 1a) or untransfected 293T cells (Figure 2a), and the higher intensity peak is due to specific staining of FLAG-tagged mouse FADD or caspase-8 in transfected cells.
Figure 1
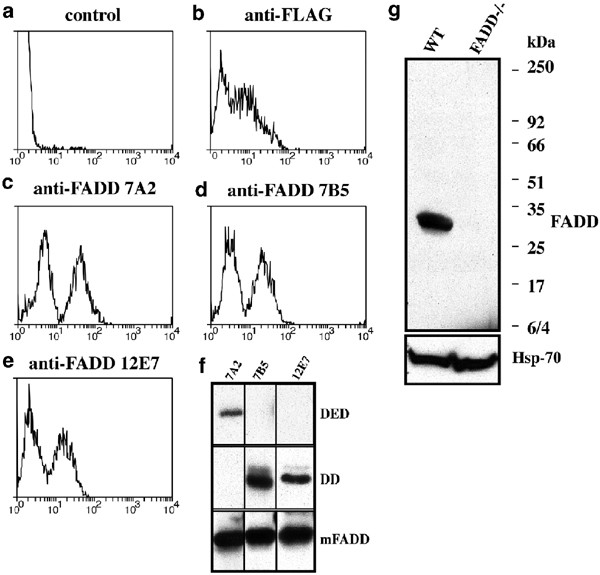
Screening for mAbs specific to mouse FADD. Hybridomas producing mAbs to mouse FADD were identified using a 1 : 1 mixture of parental FDC-P1 cells and FDC-P1 cells expressing FLAG-tagged mouse FADD. Cells were stained with medium alone (a, negative control), anti-FLAG M2 mAb (b, positive control), anti-mouse FADD mAbs 7A2 (c), 7B5 (d) or 12E7 (e). (f) Epitope mapping of anti-FADD mAbs using lysates from 293T cells transiently transfected with expression constructs for full-length mouse FADD, the DED or DD domains was performed by Western blotting. (g) Anti-FADD 7A2 mAb detected mouse FADD by Western blotting in lysates from wt but not FADD−/− ES cells. Probing with an anti-HSP70 mAb shows equal protein loading
Figure 2
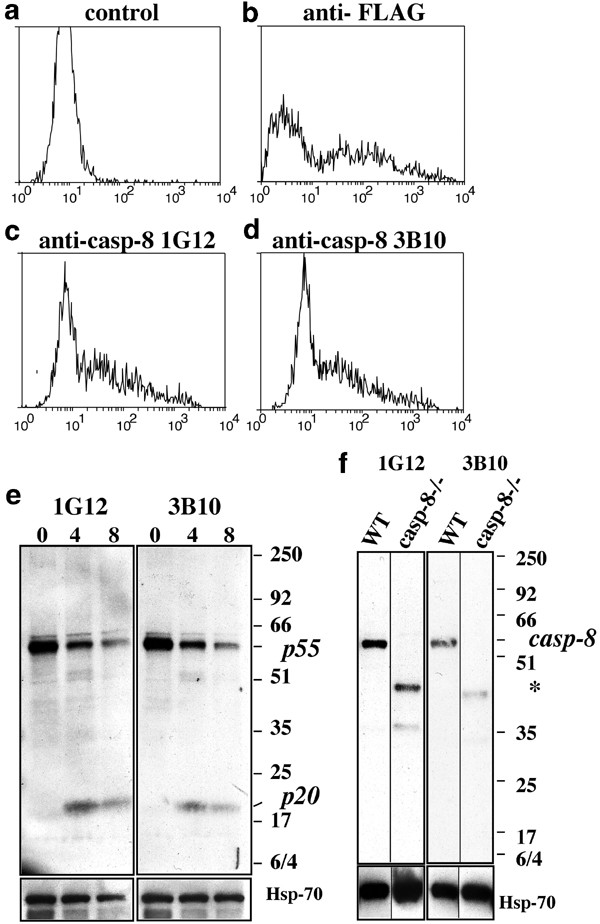
Screening for mAbs specific to mouse caspase-8. Hybridomas producing mAbs to mouse caspase-8 were identified using 293T cells transiently transfected with FLAG-tagged mutant (catalytically inactive) mouse caspase-8. Cells were stained with medium alone (a, negative control), anti-FLAG M2 mAb (b, positive control) or anti-mouse caspase-8 mAbs 1G12 (c) and 3B10 (d). (e) Anti-caspase-8 mAbs detected the products of caspase-8 processing in mouse thymocytes treated with FLAG-FasL plus anti-FLAG mAb by Western blotting. (f) An ∼55–58 kDa band corresponding to endogenous caspase-8 was detected only in wt MEFs but not in caspase-8−/− MEFs. * smaller polypeptides in lysates from caspase-8 mutant MEFs but not from wt MEFs recognised by the anti-caspase-8 mAbs, which are likely to represent truncated forms of caspase-8. Probing with an anti-HSP70 mAb shows equal protein loading
Three anti-FADD mAbs (7A2, 7B5 and 12E7) were selected and epitope mapped by Western blotting using lysates prepared from 293T cells transfected with expression constructs encoding either partial-length mouse FADD (aa 1–177), its ‘death domain’ (DD, aa 80–205) or its ‘death effector domain’ (DED, aa 1–79) (Figure 1f). The mAb 7A2 recognises an epitope within the DED of FADD (Figure 1f) and further analysis narrowed this to the region spanning aa 42–77 (data not shown). The mAbs 7B5 and 12E7 recognise epitope(s) within the DD (Figure 1f). The mAb 7A2 is specific for mouse FADD, whereas mAb 7B5 also recognises canine FADD, and mAb 12E7 recognises mouse, canine and human FADD (Figure 3d). The specificity of the anti-FADD mAbs is demonstrated by their ability to detect the 28 kDa FADD protein in lysates from wt ES cells but not binding to any proteins in lysates from FADD−/− ES cells9 (Figure 1g).
Figure 3
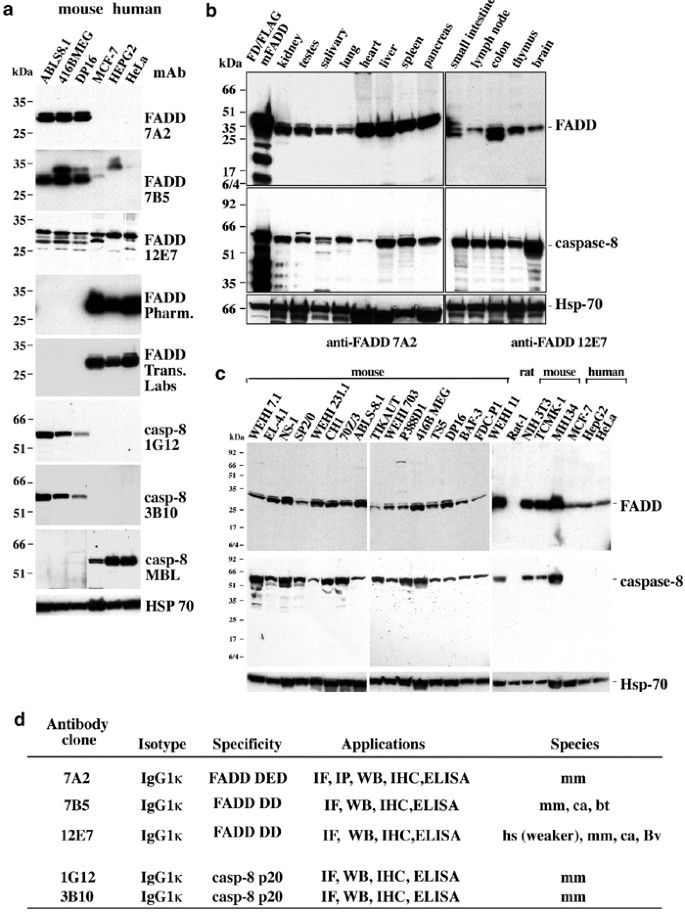
A comparison of various anti-FADD and anti-caspase-8 mAbs (a). FADD or caspase-8 protein expression was detected in lysates from 106 cultured mouse or human cell lines using our anti-FADD or anti-caspase-8 mAbs or commercial mAbs. (b) Western blot of mouse tissue lysates (25 _μ_g protein/lane) showing FADD and caspase-8 expression. (c) FADD and caspase-8 expression in cultured mouse and human cell lines. FADD and caspase-8 proteins from cell lysates of 106 cells were detected with mAbs 7A2 or 12E7 and 1G12, respectively, by Western blotting. Equal protein loading was demonstrated by immunoblotting with a mAb specific to HSP70. (d) summary of the characteristics of anti-FADD mAbs 7A2, 7B5 and 12E7 and anti-caspase-8 mAbs 1G12 and 3B10. All antibodies recognise mouse (mm) FADD protein or mm caspase-8. Monoclonal antibody 7B5 also detects canine (ca) and bovine (bv) FADD and mAb 12E7 also detects human (hs) FADD by immunofluorescence staining (IF), immunopecipitation (IP) and Western blotting (WB), albeit human more weakly than mouse FADD
Two anti-caspase-8 mAbs (1G12 and 3B10) were chosen for further experiments (Figure 2). In healthy thymocytes, these mAbs detected unprocessed caspase-8 (∼55–58 kDa). Following stimulation with FasL, processed p20 and intermediate cleavage products were also detected (Figure 2e). The anti-caspase-8 mAbs specifically recognised full-length pro-capase-8 in MEFs derived from wt but not those from caspase-8-deficient mice14 (Figure 2f). These mAbs recognised smaller proteins in lysates from caspase-8 mutant MEFs but not from wt MEFs, or any of the other cell lines tested (Figure 2, labelled with *). These proteins are likely to be truncated forms of caspase-8, since only exons 1 and 2 (encoding the two DED domains) were deleted in the caspase-8 mutant mice.14
We compared our mAbs with commercially available antibodies, which were raised against the human proteins but are nonetheless widely used to detect their mouse counterparts. Western blot analyses of lysates from several cell lines demonstrated that our mAbs detect endogenous mouse FADD or caspase-8 (Figure 3a). In contrast, the commercially available mAbs recognised only human FADD (Pharmingen), human and canine FADD (Transduction Laboratories) or human caspase-8 (MBL or Alexis; not shown), but strikingly not endogenous levels of mouse FADD or caspase-8 (Figure 3a).
Widespread expression of mouse FADD and caspase-8
We used our panel of mAbs to determine the expression of FADD and caspase-8 in cultured mouse cell lines and normal mouse tissues (Figure 3b, c). Readily detectable levels of FADD and caspase-8 were found in haematopoietic tissues, organs of the neuroendocrine system, the small intestine, liver, lung, kidney and heart (Figure 3b). Analysis of cell lines demonstrated that FADD and caspase-8 were expressed at all stages of B and T lymphopoiesis, in cells of myeloid origin and those established from kidney, breast, liver and colon (Figure 3c). These results are consistent with previous mRNA analyses (reviewed in Ashkenazi and Dixit2) and document that FADD and caspase-8 are widely expressed.
Subcellular localisation of FADD
The subcellular localisation of FADD remains controversial. Some studies have shown that FADD resides in the cytoplasm and upon ligation of ‘death receptors’, a fraction is recruited to the DISC at the plasma membrane.33 In contrast, it has been documented that overexpressed FADD causes the formation of filamentous structures in the nucleus and cytoplasm.34 More recently it has been reported35, 36 that the primary localisation of FADD is in the nucleus of adherent cells, whereas in nonadherent cells, such as Jurkat T cells, nuclear localisation of FADD was observed in 50% of cells and cytoplasmic localisation in the remainder.
Subcellular fractionation and confocal microscopy experiments were used to determine the localisation of endogenous FADD in adherent and nonadherent cells of mouse and human origin. For subcellular fractionation, we chose to Dounce homogenise cells rather than lyse them with nonionic detergents, since the latter may cause artefactual changes in protein localisation.37 When subcellular fractions of Jurkat (nonadherent) cells were analysed by immunoblotting with commercial antibodies, FADD was found exclusively in the cytosolic fraction (Figure 4a). The purity of the fractions was verified by probing blots with antibodies to cytoplasmic proteins, such as the Golgi complex resident protein _β_-COP (cytoplasmic fraction Figure 4a) and Apaf-138 (cytoplasmic fraction Figure 4b), the nuclear protein PARP (nuclear containing fraction Figure 4b) and caspase-2, found in both compartments (Figure 4a, b) as reported previously.39 Further fractionation of the cytoplasmic fraction from the adherent cell line HeLa to generate light membrane (LM) and soluble fractions (S) showed that FADD was present in the soluble fraction similar to Apaf-1 and a small amount of FADD in the light membrane fraction but none in the HM fraction where the nuclei are found (Figure 4b). The presence of _β_-COP in the HM fraction of HeLa cells (Figure 4b) is probably due to the Golgi apparatus becoming trapped by the extensive cytoskeleton of these adherent cells, compared to the nonadherent Jurkat cells (Figure 4a).
Figure 4
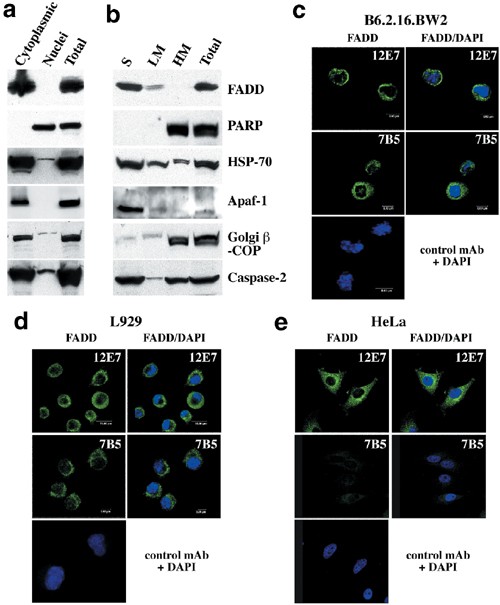
Analysis of FADD localisation by subcellular fractionation and Western blotting or confocal microscopy. (a) Lysates from Jurkat cells were separated into nuclear and cytoplasmic fractions. (b) Lysates from HeLa cells were separated into heavy membrane (HM-nuclei), light membrane (LM) and soluble fractions (S). Protein localisation in each fraction was analysed by Western blotting with mAbs of the indicated specificities. Data shown are representative of three independent experiments. (c–e) FADD is localised in the cytolasmic compartment in B6.2.16.BW2 mouse T hybridoma cells (c), L929 mouse fibroblastoid cells (d) and HeLa human cervical carcinoma cells (e). Cells were stained with anti-FADD mAb (green) plus DAPI (blue) and examined by confocal microscopy. Bars represent 8 _μ_m
The subcellular localisation of FADD was also studied by immunofluorescence staining, and confocal microscopy in nonadherent B6.2.16.BW2 mouse T-lymphoma cells as well as in adherent mouse L929 fibroblasts and human HeLa cells using anti-FADD mAb 12E7. In all cell lines tested, FADD had a cytoplasmic, non-nuclear localisation (Figure 4c–e). As negative controls, we stained HeLa cells with anti-mouse FADD mAb 7B5, which does not recognise human FADD (Figure 4e) and with isotype-matched control antibodies (Figure 4c–e). Collectively, these results demonstrate that FADD is localised in the cytoplasm but not the nucleus in the cells tested.
FADD and caspase-8 do not localise in membrane rafts
Since FADD and caspase-8 are required for TCR/CD3-induced T lymphocyte proliferation, they may relocate to the plasma membrane rafts, where several components of this signalling pathway are constitutively localised or are recruited upon TCR stimulation. We investigated localisation of FADD and caspase-8 in spleen cells or purified T lymphocytes that were either left unstimulated or had been activated for 1, 4 or 24 h with Con A, Phorbol-12-myristate-acetate (PMA) plus ionomyocin, or mitogenic mAbs to CD3 and CD28 (in the presence of IL-2). Localisation of these proteins in purified T cells or the T-lymphoma line B6.2.16.BW2 was also determined after stimulation by FasL (FLAG FasL+anti-FLAG mAb) for 10 min or as a control with anti-FLAG mAb alone. Cells were solubilised with 0.33% Brij 98 at 37°C and sucrose density gradient fractions were analysed by SDS-PAGE and Western blotting. Fractions containing rafts were identified by staining with cholera toxin (CTx) (rafts are enriched in the CTx receptor GMI) and antibodies to lck and LAT, two T-cell signalling proteins known to partition into rafts.40 Nonraft fractions were identified by blotting with antibodies to the cytoskeletal protein paxillin.41 The distribution of FADD, caspase-8 and Fas was examined by immunoblotting with our mAbs and an anti-Fas mAb. In purified T lymphocytes (Figure 5a), thymocytes (Figure 5d) or B6.2.16.BW2T lymphoma cells (Figure 5f), neither FADD nor caspase-8 was found in rafts (boxed fractions), but both were recovered exclusively in the nonraft detergent soluble fractions. Moreover, neither mitogenic stimulation nor treatment with FasL caused recruitment of FADD or caspase-8 into rafts in purified T cells (Figure 5b, c), thymocytes (Figure 5e) or B6.2.16BW2T hybridoma cells (Figure 5g). We also found that the T-cell receptor complex, specifically TCR_α_, resided in non-raft fractions, both before and during mitogenic activation and also after stimulation with FasL (Figure 5a–c, f, g). Consistent with our results, others42 have also shown that most components of the TCR complex are excluded from lipid rafts before and after T-cell activation, with only 10% of the CD3_ζ_ chain localised to the raft fraction. Interestingly, a portion of Fas partitioned to rafts (similar to the levels of LAT and lck in rafts) in resting, mitogen-stimulated and FasL-treated T cells (Figure 5).
Figure 5
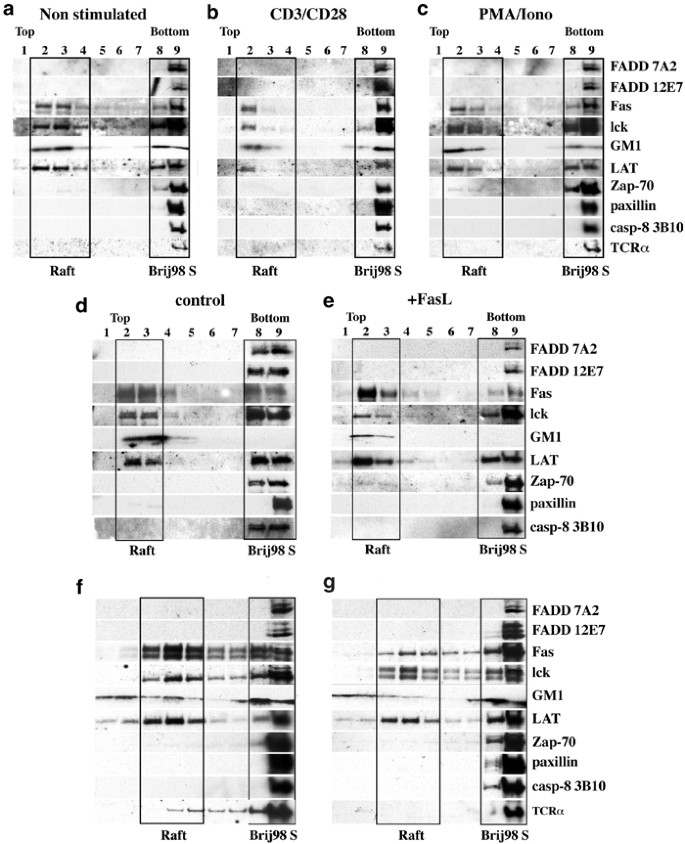
Association of FADD and caspase-8 with the soluble fractions but no association with membrane rafts in resting or stimulated T cells. Purified T cells (108) were either left untreated (a) or stimulated for 1 h with anti-CD3/CD28 mAbs plus IL-2 (b) or with PMA/ionomycin plus IL-2 (c). In total, 2 × 108 thymocytes (d, e) or 2 × 108 B6.2.16.BW2T hybridoma cells (f, g) were either stimulated by FLAG-tagged FasL crosslinked with anti-FLAG mAb (e, g) or were treated with anti-FLAG mAb alone (control; d, f) for 10 min. at 37°C. Protein distribution was assessed by immunoblotting with the Abs indicated. GM1 was detected with CTx directly conjugated to HRP. Gradient fractions 2–3 or 3–4 correspond to membrane rafts and fractions 8–9 to Brij98 soluble membrane fractions, respectively
Membrane rafts can be visualised in intact cells by staining with FITC-conjugated CTx and analysis by confocal microscopy. Staining of quiescent T cells with FITC-CTx produced a diffuse homogeneous ring-like plasma membrane staining (Figures 6b, 7b) with patches rarely visible. Staining for FADD (Figure 6a) and caspase-8 (Figure 7b) showed a diffuse cytoplasmic localisation in all cells, distinct from CTx staining. We examined whether treatment of T cells with mitogens led to redistribution of FADD or caspase-8 to rafts. Redistribution of rafts, as shown by the presence of polarised caps, was first visible by CTx-FITC staining 1 h after mitogenic stimulation and was clearly evident by 24 h (Figure 6d). Staining of mitogenically stimulated cells with anti-FADD mAbs also produced a cap, which was visible from 1 h poststimulation (78% of T cells, Table 1, Figure 6e, f). However, the FADD staining did not overlap with that of GM1 at 1 h (Table 1, Figure 6i, l) or 24 h (Table 1, Figure 6m, n) of mitogenic stimulation. Similarly, staining of T cells stimulated for 1 h with mitogens with anti-caspase-8 mAbs 1 h showed concentrated clusters at or very close to the plasma membrane in 38–47% of cells (Table 1 Figure 7d, g). A substantial proportion of T cells did not show substantial relocalisation of caspase-8 (Table 1, Figure 7j–l). In those T cells that showed caspase-8 clustering, these clusters were distinct from GM1 localisation (Table 1, Figure 7f, i). Moreover, in most cells, FADD and caspase-8 capping after mitogenic stimulation was polarised to single spots on the plasma membrane, unlike the multiple spots observed with GM1 staining. The staining specificity was demonstrated by labelling with an isotype-matched control mAb (Figures 6o, 7j, l).
Figure 6
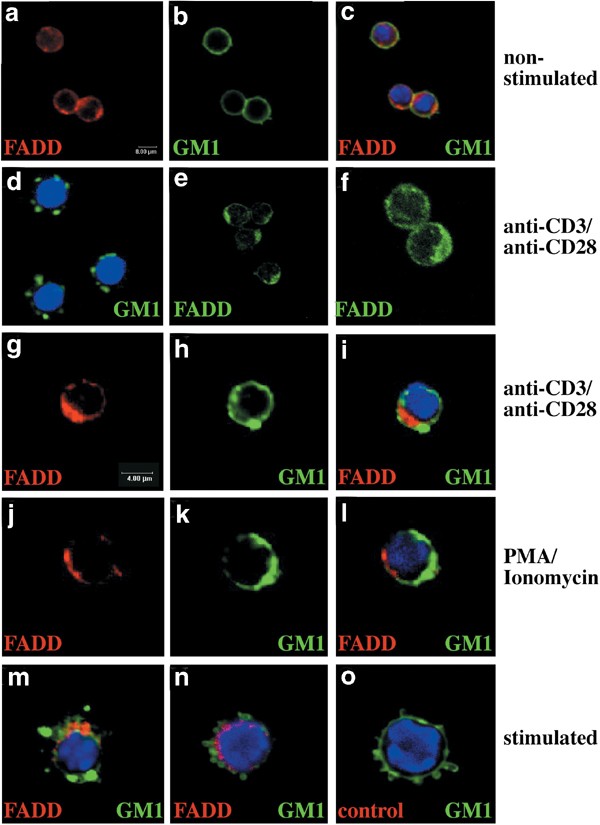
Confocal microscopic analysis shows no association of FADD with rafts after T-cell activation. T cells were stained with FITC-conjugated CTx to detect membrane rafts (green), anti-FADD mAbs (12E7) (avidin Cy-5 (red) or avidin Texas Red (n) and DAPI (blue). (a–c), Staining of quiescent T cells with FITC-CTx (b, c) or anti-FADD mAb (a, c). After stimulation, T cells were stained with FITC-CTx and DAPI (d, 24 h after stimulation, with PMA/ionomycin) or anti-FADD mAb alone (e, f, 1 h after stimulation with anti-CD3/anti-CD28 mAbs). (g–o) Staining overlays of T cells stimulated with anti-CD3/CD28 mAbs plus IL-2 (g–i-1 h, m–24 h) or with PMA/ionomycin plus IL-2 (j–l-1 h, n–24 h) stained with anti-FADD, CTx and DAPI. (o) Staining with an isotype-matched control mAb (red) of T cells after 24 h stimulation with anti-CD3/anti-CD28 mAbs. Data shown are representative of five independent experiments and represent the appearance of the majority of cells under a stated condition (see Table 1)
Figure 7
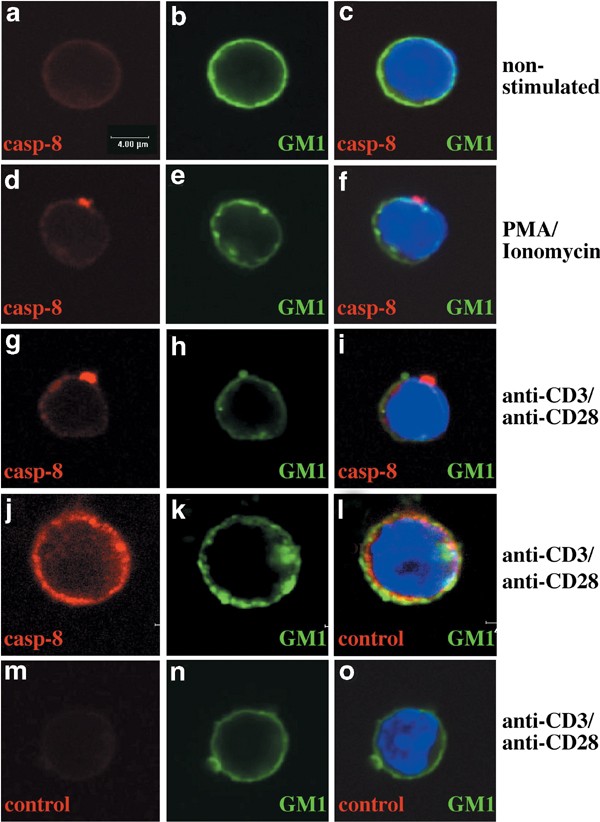
Confocal microscopic analysis shows no association of caspase-8 with rafts in mitogenically stimulated T cells. T cells were stained with FITC-conjugated CTx to detect membrane rafts (green), anti-caspase-8 mAb (1G12) or isotype control mAb (avidin Cy-5 (red)) and DAPI (blue). Staining of quiescent T cells with FITC-CTx (b, c) or anti-caspase-8 mAb (a). Cells were stimulated with PMA/ionomycin plus IL-2 (d–f) or anti-CD3/anti-CD28 mAbs plus IL-2 (g–l) for 1 or 24 h (j–l). (m, o) staining with an isotype-matched control mAb (red). Data shown are representative of two independent experiments and represent the appearance of the majority of cells under a stated condition (see Table 1)
Table 1 Distribution of FADD/caspase-8 after mitogenic stimulation or Fas ligation
FADD and caspase-8 localisation was also studied after stimulation of thymocytes or purified T cells with FasL. Confocal imaging of thymocytes (Figure 8a–o) or T cells (Figure 8p–r) showed that the homogeneous FADD, caspase-8 and GM1 patterns of staining observed in unstimulated cells (Figure 8a–f) changed after Fas ligation to concentrated clusters at or very close to the plasma membrane in all cells examined (Figure 8g–r, Table 1). Consistent with the data from the lipid raft isolation experiments (Figure 5), neither FADD nor caspase-8 staining colocalised with GM1 staining (Table 1, Figure 8g–r).
Figure 8
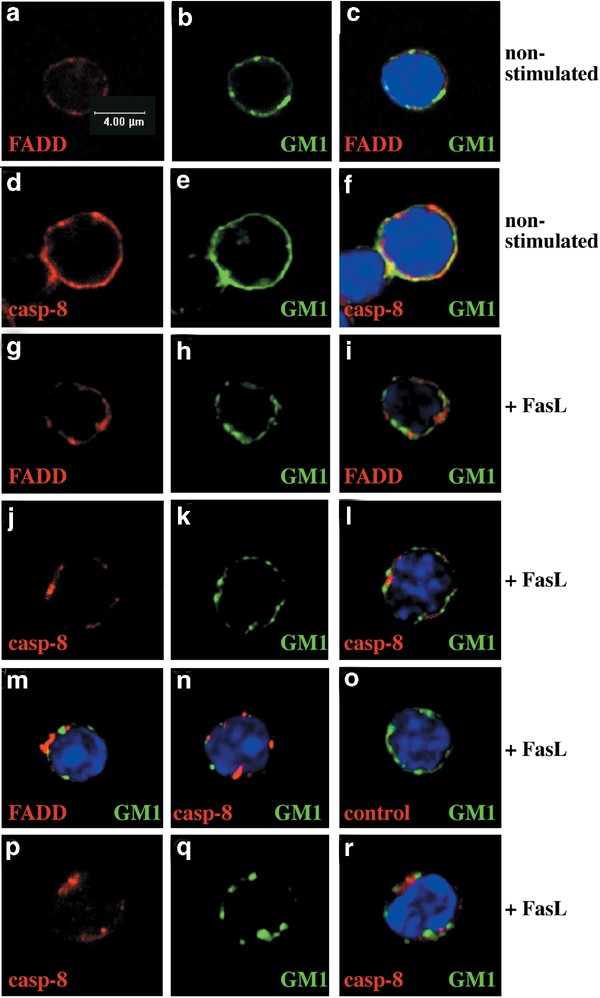
Confocal microscopic analysis shows no association of FADD or caspase-8 with membrane rafts after Fas ligation. Thymocytes (a–o) or purified T cells (p–r) were stained with FITC-CTx to detect membrane rafts (green), anti-FADD mAb (12E7, red) or anti-caspase-8 mAbs (1G12, 3B10, red) and DAPI (blue). Staining of unstimulated thymocytes for FADD (a), caspase-8 (d) or GM1 (b, e) and overlays (c, f). After stimulation with FLAG-FasL plus anti-FLAG mAb for 10 min, cells were stained for, FADD (g, m), caspase-8 (j, n, p) and GM1 (h, k, m, n, q). Overlays (i, l–n, r) or (o) staining with an isotype-matched control mAb (red) of thymocytes treated for 10 min with FasL. Data shown are representative of three independent experiments and represent the appearance of the majority of cells under a stated condition (see Table 1)
Mitogenic stimulation and FasL treatment cause different post-translational modifications in FADD and caspase-8
We also used our panel of mAbs to determine if FADD or caspase-8 underwent post-translational modifications in T cells after mitogenic stimulation or treatment with FasL (Figure 9). Western blotting on lysates from T cells that had been stimulated for 1 or 24 h with anti-CD3/CD28 mAbs or PMA/ionomycin was performed with the anti-FADD mAbs. In all cases, at 0 and 1 h post-stimulation, FADD was detected as a ∼28 kDa doublet. This finding is consistent with previous observations,43, 44, 45 which demonstrated that the doublet represents two differentially phosphorylated forms of FADD. p0 probably corresponds to nonphophorylated FADD and p1 to FADD phosphorylated on S191.43 After 24 h of mitogenic stimulation, an additional slower migrating form of FADD (apparent MW ∼30 kDa, p2) was detected by all three anti-FADD mAbs (Figure 9a). This band is only observed in response to mitogenic stimulation of T cells but not after treatment with FasL (Figure 9a). Incubation of lysates from mitogen-activated T cells with lambda phosphatase (_λ_-PPase), which dephosporylates modified serine, threonine and tyrosine residues, did not affect p2, but the p1 FADD band disappeared (Figure 9d). Therefore, the FADD p2 band probably reflects some post-translational modification other than phosphorylation. Interestingly, in cells that are continuously cycling in culture, such as B6.2.16.BW2T lymphoma cells, all three forms of FADD, including p2, are constitutively present (data not shown).
Figure 9
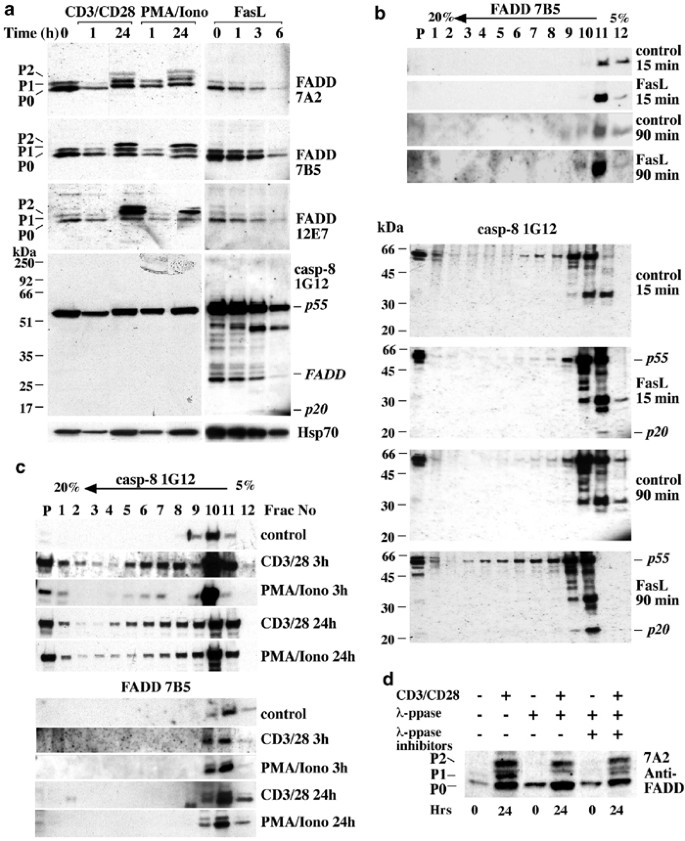
Mitogenic stimulation and FasL treatment cause post-translational modifications of FADD and caspase-8. (a) Western blot analysis of lysates from purified T cells (106 cells/lane) after mitogenic stimulation (anti-CD3/CD28 mAb or PMA/ionomycin with IL-2) or after treatment with FasL, probed with anti-FADD or anti-caspase-8 mAbs. p0: nonphosphorylated FADD, p1: phosphorylated FADD, p2: additional higher molecular weight form of FADD. Equal protein loading was demonstrated by probing with an anti-HSP70 mAb. (b, c) Western blot analysis of continuous sucrose gradient fractions from purified T cells (3 × 107) treated with FLAG-FasL plus anti-FLAG mAb, with anti-FLAG mAb (control) for 15 or 90 min (b) or stimulated with mitogens (c) as in (a) for 3 or 24 h and probed with anti-FADD or anti-caspase-8 mAbs. Cleaved caspase-8 indicated by p20. (d) Western blot analysis of lysates from purified T cells treated with _λ_-PPase, probed with anti-FADD mAb (7B5)
Since both FADD and caspase-8 are recruited to the larger DISC complex after Fas-ligation, we examined whether they might also become part of large complexes in T cells after mitogenic stimulation. Larger complexes and proteins associated with cytoskeletal components can be separated from soluble and intracellular membrane-associated proteins on continuous sucrose gradients (5–20%).46 Purified T cells were treated with FLAG-FasL plus anti-FLAG mAb for 15 or 90 min (Figure 9b), or were mitogenically stimulated for 2, 3 or 24 h (Figure 9c). In unstimulated T cells, both FADD and caspase-8 were found in lighter fractions 10–12 (Figure 9b, c), as would be expected for soluble, nonaggregated proteins. However, after both, mitogenic stimulation or Fas ligation, a shift in caspase-8 to denser fractions was observed (Figure 9b, c). Upon treatment with FasL, only a minor portion of caspase-8 appeared to be proteolysed, perhaps indicating that a small amount of caspase-8 is sufficient to launch the apoptotic caspase cascade. Most cleaved caspase-8 was found in the lighter fractions, as expected because removal of the two DEDs liberates active caspase-8 from the DISC. In FasL-treated cells, there were increased amounts of FADD and caspase-8 in the pellet fraction (Figure 9b). This is the fraction in which we would expect to find the DISC components because we generated cell extracts without using detergents. Analysis by sucrose gradient fractionation showed that most FADD proteins from mitogenically activated T cells migrated in the lighter fractions (Figure 9c).
These results indicate that mitogenic stimulation induces a substantial portion of caspase-8, but at most only a small fraction of FADD, to either associate with other proteins in a larger complex or to become part of cytoskeletal structures. It is possible that FADD is only transiently recruited with caspase-8 to this complex and that at later time points other proteins, such as FLIP, take over its position.
Discussion
Caspase-8 and FADD are not only essential for ‘death receptor’-induced apoptosis but are also required for antigen- or mitogen-induced T-cell activation. How signalling for T-cell activation and proliferation is transmitted through FADD and caspase-8 remains unresolved. Experiments with T cells lacking FADD or caspase-8 function have found no defects in activation of NF-_κ_B, JNK, MAP kinases or calcium flux (reviewed in Newton and Strasser16), indicating that FADD and caspase-8 must play critical roles in a different pathway. At the present time, it is also unclear whether FADD and caspase-8 interact with each other in the same way in both cell death and mitogenic activation.
We investigated whether mitogenic stimulation or death receptor signalling resulted in modifications in FADD and/or caspase-8 that might affect their subcellular localisaton or interaction with other proteins. To investigate these issues, we generated mAbs that could detect mouse FADD and caspase-8 at endogenous levels in multiple assay systems (Figures 1, 2 and 3). These reagents were highly specific since they detected endogenous levels of FADD or caspase-8 only in cells from wt mice but did not detect any proteins in cells from the corresponding knockout mice. Many commercially available antibodies recognise only the human proteins at endogenous levels (Figure 3), whereas the novel ones we have generated readily detect the mouse proteins.
Expression analyses demonstrated that FADD and caspase-8 are very widely expressed (Figure 3), consistent with previous analyses of FADD and caspase-8 mRNA expression, indicating that these proteins play critical roles in many cell types. Subcellular fractionation, immunofluorescence staining and confocal microscopy showed that FADD has a cytoplasmic localisation in both adherent and nonadherent cells (Figure 4). This observation is consistent with a previous report,33 but contrasts with the observation that overexpressed FADD causes the formation of filamentous structures in the nucleus.34 This discrepancy may be due to the use of overexpression systems, which can cause artefacts in subcellular protein localisation. Moreover, recent studies with adherent cells35, 36 indicated that the primary localisation of endogenous FADD was inside the nucleus. The authors used a commercial anti-human FADD mAb. We have also used this mAb and indeed on staining fixed HeLa cells it produced a nuclear signal, but when using this mAb in Western blotting of subcellular fractions, FADD was found exclusively in the non-nuclear fraction (Figure 4b). Consistent with our observation, the previous study36 also found that upon fractionation of Hela cells, most FADD proteins were found in the cytoplasmic fraction. Although, we do not have a complete explanation for the discrepancies, it may be possible that the differences are caused by differences in the specificities of the antibodies used or in the cell types analysed.
Membrane domains (lipid rafts) aggregate in response to many different stimuli and remain intact after detergent solubilisation and float in sucrose density gradients. The finding that several components of the TCR/CD3-signalling pathway are either resident in, or recruited to rafts after stimulation have led to the idea that these specialised membrane regions are essential for lymphocyte activation.40 Moreover, one study has indicated that Fas-induced apoptosis signalling occurs via ceramide-rich lipid rafts.47 These findings could, however, not be reproduced in another study on Fas signalling.31 To resolve this discrepancy, we investigated whether stimulation with FasL or mitogens resulted in the recruitment of Fas, FADD or caspase-8 to lipid rafts. Our raft isolation and confocal microscopy studies indicated that FADD and caspase-8 were neither constitutively present in lipid rafts nor recruited to rafts after mitogenic or death receptor stimulation (Figures 5, 6, 7 and 8). Similar results were obtained in studies with thymocytes, purified mature T cells and the T hybridoma line B6.2.16.BW2, using two different raft isolation methods (Brij98 at 37°C and Triton-X-100 at 4°C), indicating that the conclusions might be generalised. Interestingly, a portion of Fas was found to reside constitutively in lipid rafts of these cells. In agreement with this result, recent reports also documented the constitutive presence of a portion of Fas in lipid rafts in thymocytes and the L12.10-Fas T-cell line.28, 29, 30 In contrast to our results (Figures 5, 6, 7 and 8) and those of Algeciras-Schimnich et al,31 it has been reported that caspase-830 or both FADD and caspase-8 are recruited to lipid rafts upon Fas stimulation.28, 29 We believe that our mAbs are specific and sensitive enough to detect endogenous FADD and caspase-8 (Figures 1, 2). Moreover, since multiple known raft constituents were localised in the raft fractions, our raft isolation appears adequate (Figure 5). Finally, lack of localisation of FADD or caspase-8 to lipid rafts was confirmed by confocal microscopy (Figures 6, 7 and 8). Using the novel antibodies we have generated, in multiple techniques we do not observe recruitment of FADD nor caspase-8 to lipid rafts after stimulation with FasL, and hence the DISCs formed in response to death receptor ligation do not form in lipid rafts. Sequestration of a portion of Fas into rafts may serve to limit death receptor signalling or may play a role in receptor internalisation. Although neither FADD nor caspase-8 were recruited to lipid rafts after stimulation, the distribution of both proteins changed from a mostly diffuse cytoplasmic appearance in untreated cells to an uneven capping at or near the plasma membrane (Figures 6, 7 and 8). Such a change in localisation is consistent with recruitment of these proteins into the DISC.
The anti-FADD mAbs were sensitive enough to detect both the known nonphosphorylated and phosphorylated forms of FADD. Interestingly, an additional slower migrating form of FADD was detected in T cells after mitogenic stimulation but not after death receptor ligation (Figure 9a). This observation indicates that FADD might undergo a specific post-translational modification in response to mitogenic stimulation. Treatment of lysates with _λ_-PPase did not result in the disappearance of the highest molecular weight FADD band (p2) (Figure 9d), although its intensity was slightly reduced. Therefore, mitogenic stimulation induces a post-translational modification of FADD, the nature of which remains unknown, but is probably not due to additional phosphorylation.
In addition to FADD, caspase-8 also behaves differently in response to mitogenic stimulation versus Fas ligation. This can be seen as a shift to denser fractions in a continuous sucrose density gradient in lysates from mitogenically activated T cells (Figure 9b, c). This shift may indicate the formation of a larger caspase-8 complex. Both caspase-8 and FLIPL are recruited to the DISC after Fas-ligation.2 It is possible that the larger caspase-8 complex evident after mitogenic stimulation is also due to the binding of capase-8 to FLIPL, which has been proposed to act as a molecular switch between death and growth signals in T cells.48
In conclusion, we have shown that during both ‘death receptor’-induced apoptosis signalling and mitogenic activation, FADD and caspase-8 aggregate (less so for mitogenic stimulation) and form caps at the plasma membrane, but do not colocalise with lipid rafts. Further studies indicated that mitogenic stimulation through FADD and caspase-8 may not necessarily proceed through the same signalling pathway as cell death, since mitogenic stimulation, but not Fas ligation, induced a unique post-translational modification of FADD. Indeed, it has been shown recently that a specific mutation in FADD impedes cell proliferation but has no effect on its ability to transduce a death signal from Fas.49 The mobility shift that we found provides the first physical evidence that FADD is specifically modified after mitogenic stimulation. Our finding that caspase-8 is recruited to larger complexes after mitogenic stimulation, and not only after ‘death receptor’ ligation, strengthens the evidence that caspase-8 plays an essential role in T-cell-mediated immune responses. These differences in modifications may determine whether FADD and caspase-8 induce cell death or proliferation and may do so by determining which other signalling proteins are activated. Since endogenous FADD, caspase-8 and the DISC can be immunoprecipitated using our mAbs, we intend to use these reagents as tools to isolate FADD and/or caspase-8 containing macromolecular complexes that may play critical roles in mitogenic stimulation.
Materials and Methods
Experimental animals
All experiments with animals were performed according to the guidelines of the Melbourne Health Research Directorate Animal Ethics Committee
Production of recombinant mouse FADD and caspase-8 proteins
Mouse FADD cDNA was amplified by PCR from a C57BL/6 mouse spleen cell cDNA library and cloned into pGEX-6P-3 (Amersham Biosciences). The recombinant FADD protein fused to GST was expressed in BL21(DE3)pLysS (GOLD-Amersham Biosciences) bacteria. GST-FADD protein in the soluble fraction was purified using a glutathione–Sepharose 4B column (Amersham Pharmacia Biotech). The coding region for mouse caspase-8 large catalytic subunit (p20) was amplified by PCR and cloned into pET-32c(+) (Novagen Inc., Madison, WI, USA) containing an N-terminal His6-tag. The recombinant protein was purified from lysates prepared from Escherichia coli AD494 (DE3) bacteria using a HiTrap chelating column (Amersham Biosciences), and eluting bound protein with imidazole (Sigma).
Immunisation and hybridoma fusion
Wistar rats were immunised with recombinant GST mouse FADD or His6caspase-8 (p20) protein. Hybridomas were generated and those producing mAbs to FADD or caspase-8 identified using the previously described screening strategy.50 Hybridomas producing mAbs to FADD were identified using parental FDC-P1 cells and a subclone stably expressing N-terminally FLAG-tagged mouse FADD mixed at a 1 : 1 ratio. Hybridomas producing mAbs to caspase-8 were identified using 293T cells transiently transfected with a FLAG-tagged mutant mouse caspase-8 in which the active site cysteine had been changed to alanine. The cells were fixed in 1% paraformaldehyde, permeabilised with 0.3% saponin (Sigma) and stained with hybridoma supernatants. Bound antibodies were revealed by staining with fluorescein-isothyiocyanate (FITC)-conjugated goat anti-rat Ig antibodies (Southern Biotechnology) and analysed in a FACScan (Becton Dickinson). Hybridomas producing antibodies to FADD or caspase-8 were cloned and purified as previously described.32
Cell lines and tissue culture
The cell lines used for analysis of FADD and caspase-8 expression are listed in our previous study.39 Cell lines and lymphocytes were cultured in the high glucose version of Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal calf serum (FCS, Trace, Australia), 50 _μ_M 2-mercaptoethanol (2-ME), 13 _μ_M folic acid and 100 _μ_M L-asparagine, or were grown in DMEM or RPMI medium supplemented only with 10% FCS. Liposome (Lipofectamine, Invitrogen)-mediated transfection of 293T cells with expression constructs for FADD (aa 1–177), the DED of FADD (aa 1–79), the DD of FADD (aa 80–205) or catalytically inactive (C>A mutant) caspase-8 was performed according to the manufacturer's instructions. Spleen cells or purified T lymphocytes were stimulated with mitogens as described previously.51
Spleen cells or purified T lymphocytes were stimulated for various periods of time with: (i) 2 ng/ml PMA, 2 _μ_g/ml ionomycin (both from Sigma), and IL-2 (from X63/0 cells stably transfected with a mouse IL-2 expression vector), or (ii) plate-bound monoclonal anti-CD3 (KT3) and anti-CD28 (37N51) antibodies (10 _μ_g/ml of each antibody in the coating solution) plus IL-2. Thymocytes or purified resting T cells were incubated for 10, 15 or 20 min at 37°C with recombinant soluble human FasL/CD95L (100, 200 or 1000 ng/ml) carrying a FLAG epitope tag (Apotech Inc.). Anti-FLAG monoclonal antibodies (1 _μ_g/ml, Sigma) were used to enhance crosslinking of receptor–ligand complexes. Cells were then washed twice in cold PBS and lysates were prepared.
T cell purification and cell sorting
T lymphocytes were purified from lymph nodes of mice by negative sorting after staining for all unwanted cell types – B cells, macrophages, granulocytes, nucleated erythroid cells – with FITC-conjugated surface marker-specific mAbs (RB6-8C5 anti-Gr-1, M1/70 anti-Mac-1, F4/80, Ter119, 5.1. anti-IgM, HB58 anti-Ig_κ_, RA3-6B2 anti-CD45R/B220). Viable cells that were not stained with FITC-labelled mAbs or the vital dye propidium iodide (PI, Sigma at 2 _μ_g/ml) (FITC−PI−) were purified on a DIVA (Becton Dickinson) or MoFlo (Cytomation) cell sorter. Alternatively, unwanted cells were stained with unconjugated surface marker-specific mAbs plus goat anti-rat Ig antibody-coated magnetic beads (Paesel and Lorei) and purified by magnetic panning as previously described.52 The purity of sorted T cells was verified by staining cells with R-PE- or Tricolor-labelled monoclonal antibodies specific to CD4 and CD8 (Caltag) or with FITC-conjugated anti-Thy-1 antibodies; the purity ranged from 90 to 99%.
Western Blotting and _λ_-PPase treatment
Cell and tissue lysates were prepared as described previously.50 Proteins were eluted by boiling in SDS : PAGE gel loading buffer, size-fractionated on polyacrylamide gels (Novex) and transferred to nitrocellulose membranes (Amersham Pharmacia) by electroblotting. Nonspecific binding of antibodies to membranes was blocked by overnight incubation in 5% skimmed milk, 1% casein, 0.05% Tween-20. Membranes were then probed with the following antibodies: anti mouse FADD (clones 7A2, 7B5, 12E7), anti-human FADD (clone A66-2 Pharmingen or clone 1, Transduction Laboratories), anti-mouse caspase-8 mAbs (clones 1G12, 3B10), anti-human caspase-8 (clone 5D3, MBL or clone 12F5, Alexis) or rat anti-mouse Fas antibody (Upstate Biotechnology). The following reagents and antibodies were used for probing Western blots of fractions from sucrose density gradients: HRP-conjugated GM1 (Ctx) (Sigma), mouse anti-ZAP-70, lck and paxillin antibodies (BD Transduction Laboratories), rat anti-Fas and rabbit anti-LAT antibodies (Upstate Biotechnology) or hamster anti-TCR_α_ chain antibody (H28-710). To control for the concentration and integrity of proteins in the tissue lysates, blots were probed with mouse anti-HSP70 mAb N6 (gift from Dr R Anderson, Peter MacCallum Cancer Research Institute, Melbourne, Australia and Dr W Welch, UCSF, USA). Bound antibodies were visualised by labelling with goat anti-rat IgG (Southern Biotechnology) or sheep anti-mouse IgG antibodies (Chemicon) conjugated to HRP or with biotin-conjugated goat anti-Armenian hamster IgG antibodies (Jackson ImmunoResearch) plus HRP-conjugated streptavidin (Jackson ImmunoResearch), followed by enhanced chemiluminescence (ECL; Amersham Pharmacia).
T-cell lysates (prepared as above without NaF or Na3V04) were incubated with _λ_-PPase, (NEB) (106/200 U), at 30°C for 1 h according to the manufacturer's recommendations. _λ_-PPase activity was inhibited by the inclusion of NaF (5 mM) and Na3Vo4 (2 mM).
Subcellular fractionation
Cells were resuspended in hypotonic buffer (10 mM NaCl, 1.5 mM MgCl2, 10 mM Tris-HCL, pH 7.4) and allowed to swell on ice for 10 min at 4°C and were then lysed in a Dounce homogenizer (12–36 strokes with a type ‘B’ pestle; Konte Glassware Corporation). After 3 min centrifugation at 1000 × g at 4°C, followed by two washes in hypotonic buffer, the pelleted nuclei or heavy membranes (HM-nuclei) were resuspended in the lysis buffer used for Western blotting (see above). The resulting supernatant was fractionated further by centrifugation at 50 000 r.p.m for 30 min in a TLA 100.3 Beckman benchtop rotor to generate the soluble (S) and light membrane (LM) fractions. The latter was then resuspended in lysis buffer containing protease inhibitors.
Purification of raft fractions
Detergent-insoluble and soluble fractions were separated using two different methods. In one method,53 spleen cells, thymocytes or purified lymph node-derived T lymphocytes (5 × 107 or 108) were washed twice in cold PBS, resuspended in 2.4 ml or 1 ml 1% Triton X-100 in MNE buffer (25 mM MES pH 6.5, 150 mM NaCl, 5 mM EDTA) containing 0.5 _μ_g/ml Pefabloc, 1 _μ_g/ml leupeptin, aprotinin, soybean trypsin inhibitor and pepstatin, 5 mM NaF, 2 mM Na3VO4 and 30 mM sodium pyrophosphate and allowed to swell on ice for 20 min. Cells were lysed in a Dounce homogeniser (15 strokes with a type ‘B’ pestle). Efficient lysis (routinely ∼99%) was checked microscopically by staining an aliquot of cells with Trypan blue. Nuclei and cell debris were removed by centrifugation at 4°C at 900 × g for 3 min. The supernatants were resuspended in 1.0 ml 85% sucrose made with MNE buffer and transferred to Beckman ultracentrifuge tubes that were overlaid with 6 ml of 30% sucrose followed by 3 ml of 5% sucrose in MNE buffer. Samples were ultracentrifuged in a Beckmann SW40T rotor (33 500 r.p.m. for 16–18 h at 4°C). Fractions (12) were collected from the top of the gradient. Proteins in each fraction were precipitated with three volumes of acetone, resuspended in SDS-PAGE running buffer, size-fractionated (12% SDS-PAGE Novex) under reducing conditions and then transferred to nitrocellulose membranes by electroblotting.
Alternatively, lipid rafts were isolated with Brij98 using an adaptation of the method described by Drevot et al.54 Cells were gently sonicated (five 5 s bursts, 5W) in 1 ml of ice-cold buffer A (25 mM HEPES, 150 mM NaCl, 1 mM EGTA, containing protease inhibitors, 5 mM NaF, 2 mM Na3VO4, 2 _μ_g/ml chymostatin and 5 _μ_g/ml _α_2 macroglobulin). The postnuclear supernatant (PNS) was obtained by centrifugation at 2000 r.p.m. in a bench top centrifuge at 4°C for 3 min. PNS from 2 × 108 mouse thymocytes, 108 purified mouse T cells or 108 B6.2.16.BW2T hybridoma cells were incubated for 4 min at 37°C before Brij 98 (Sigma) was added to a final concentration of 1%. After 5 min of solubilisation at 37°C, the PNS (1 ml) was diluted with 2 ml prewarmed (37°C) buffer A containing 2 M sucrose (giving final concentrations of 1.33 M sucrose and 0.33% Brij98) and then chilled on ice (55 min) before being placed at the bottom of a step sucrose gradient (0.9–0.8–0.75–0.7–0.6–0.5–0.4–0.2 M sucrose, 1 ml each) in buffer A. Gradients were centrifuged at 38 000 r.p.m. for 16 h at 4°C in a SW40 rotor (Beckman Instruments). Fractions (1 ml) were harvested from the top, except for the last one (pooled fractions 9–11), which contained 3 ml. Aliquots (50 _μ_l) of each fraction of the gradient were resolved on SDS-PAGE under reducing conditions as indicated above.
Velocity sedimentation on sucrose gradients
For velocity sedimentation, T lymphocytes were purified as described above (3 × 107) and treated with: (i) FLAG-FasL (100 ng/ml) plus anti-FLAG mAb (1 _μ_g/ml) for 15 or 90 min, (ii) plate-bound anti-CD3 plus anti-CD28 mAbs (10 _μ_g/ml of each antibody in the coating solution) with IL-2 for 2, 3 or 24 h, or (iii) PMA (2 ng/ml), ionomycin (2_μ_g/ml) and IL-2 for 2, 3 or 24 h. Cells were harvested, washed twice in cold PBS, resuspended in 500 _μ_l 0.05 M PIPES. NaOH, 2 mM MgCl2, 0.05 M HEPES (pH 7.0), 1 mM EDTA containing protease inhibitors, 5 mM NaF, 2 mM Na3VO4 and 30 mM sodium pyrophosphate. Cells were lysed using a 1 ml Dounce homgeniser and centrifuged at 3000 r.p.m. for 5 min to remove cellular debris. Supernatants were loaded on a 5–20% sucrose gradient55 made in the lysis buffer described above, containing protease inhibitor cocktail tablets (Roche). Samples were centrifuged in a SW40 Ti rotor (Beckman) at 30 000 r.p.m. for 18 h at 15°C. A total of twelve 1 ml fractions were collected manually from the bottom. Aliquots (200 _μ_l) of each fraction of the gradient were acetone precipitated and proteins were resolved on SDS-PAGE under reducing conditions as indicated above.
Immunofluorescence staining and confocal microscopy
For immunofluorescence staining, HeLa and L929 cells were grown and stained in chamber slides using Cell Tak (Becton Dickinson) for attachment. Jurkat cells and normal T cells were stained in suspension and then cytospun onto slides. Cells were fixed for 10 min at RT with PBS containing 1% paraformaldehyde (BDH), washed and then permeabilised for 30 min at RT in 0.3% saponin (Sigma) containing 10% normal goat serum to prevent nonspecific binding of antibodies. Cells were then stained with anti-FADD mAbs 7B5 or 12E7, anti-caspase-8 mAbs 1G12 or 3B10, or isotype-matched control mAbs in PBS containing 2% FCS/0.3% saponin. Cells were then washed with PBS containing 0.03% saponin/2% FCS and incubated with FITC-conjugated goat anti-rat Ig antibodies (Southern Biotechnology) containing DAPI (2 _μ_g/ml; Molecular Probes) to stain nuclei.
Purified resting T lymphocytes and activated T-cell blasts (5 × 105) were stained for 30 min at 4°C with FITC-conjugated CTx (GM1, 1 _μ_g/ml, Sigma) to label lipid rafts. Cells were then washed twice in PBS and fixed for 15 min at RT in PBS/1% paraformaldehyde. After washing, the fixed cells were stained for 1 h at 4°C with 5–10 _μ_g/ml rat anti-mouse FADD mAb (12E7) or rat anti-mouse caspase-8 mAb (1G12 or 3B10) in balanced salt solution containing 2% FCS, 0.3% saponin and 10% normal mouse serum (NMS). After two further washes, cells were incubated with biotinylated mouse anti-rat IgG1 antibody (RG11/39.4), which was detected by streptavidin-Texas Red (Gibco BRL) or streptavidin-Cy-5 (Amersham Pharmacia) containing 2 _μ_g/ml DAPI. All washes were in balanced salt solution containing 2% FCS and 0.3% saponin. Slides were mounted in fluorescent mounting medium (Dako) containing 100 _μ_g/ml DIABCO (Sigma). Controls included staining with an isotype-matched control rat IgG1 mAb (Pharmingen) or with primary or secondary antibodies alone. Samples were analysed with a Leica confocal scanning microscope using SCANware software (Leica Lasertechnik).
Abbreviations
PMA:
phorbol-12-myristate-acetate
DD:
death domain
ALPS:
autoimmune lymphoproliferative syndrome
JNK:
c-jun N-terminal kinase
References
- Nagata S (1999) Fas ligand-induced apoptosis. Annu. Rev. Genet. 33: 29–55
Article CAS Google Scholar - Ashkenazi A and Dixit VM (1998) Death receptors: signaling and modulation. Science 281: 1305–1308
Article CAS Google Scholar - Strasser A, O'Connor L and Dixit VM (2000) Apoptosis signaling. Annu. Rev. Biochem. 69: 217–245
Article CAS Google Scholar - Muzio M, Chinnaiyan AM, Kischkel FC, O'Rourke K, Shevchenko A, Ni J, Scaffidi C, Bretz JD, Zhang M, Gentz R, Mann M, Krammer PH, Peter ME and Dixit VM (1996) FLICE, a novel FADD homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/Apo-1) death-inducing signaling complex. Cell 85: 817–827
Article CAS Google Scholar - Green DR and Reed JC (1998) Mitochondria and apoptosis. Science 281: 1309–1311
Article CAS Google Scholar - Gross A, McDonnell JM and Korsmeyer SJ (1999) Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 13: 1899–1911
Article CAS Google Scholar - Wang X (2001) The expanding role of mitochondria in apoptosis. Genes Dev. 15: 2922–2933
CAS Google Scholar - Newton K, Harris AW, Bath ML, Smith KGC and Strasser A (1998) A dominant interfering mutant of FADD/Mort1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J. 17: 706–718
Article CAS Google Scholar - Yeh WC, Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, El-Deiry WS, Lowe SW, Goeddel DV and Mak TW (1998) FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279: 1954–1958
Article CAS Google Scholar - Zhang J, Cado D, Chen A, Kabra NH and Winoto A (1998) Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature 392: 296–300
Article CAS Google Scholar - Sakamaki K, Inoue T, Asano M, Sudo K, Kazama H, Sakagami J, Sakata S, Ozaki M, Nakamura S, Toyokuni S, Osumi N, Iwakura Y and Yonehara S (2002) Ex vivo whole-embryo culture of caspase-8-deficient embryos normalize their aberrant phenotypes in the developing neural tube and heart. Cell Death Differ. 9: 1196–1206
Article CAS Google Scholar - Salmena L, Lemmers B, Hakem A, Matysiak-Zablocki E, Murakami K, Au B, Berry DM, Tamblyn L, Shehabeldin EM, Migon E, Wakeham A, Bouchard D, Chen WY, McGlade JC, Ohashi PS and Hakem R (2003) Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 17: 883–895
Article CAS Google Scholar - Smith KGC, Strasser A and Vaux DL (1996) CrmA expression in T lymphocytes of transgenic mice inhibits CD95 (Fas/APO-1)-transduced apoptosis, but does not cause lymphadenopathy or autoimmune disease. EMBO J. 15: 5167–5176
Article CAS Google Scholar - Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P and Wallach D (1998) Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9: 267–276
Article CAS Google Scholar - Chun HJ, Zheng L, Ahmad M, Wang J, Speirs CK, Siegel RM, Dale JK, Puck J, Davis J, Hall CG, Skoda-Smith S, Atkinson TP, Straus SE and Lenardo MJ (2002) Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 419: 395–399
Article CAS Google Scholar - Newton K and Strasser A (2003) Caspases signal not only apoptosis but also antigen-induced activation in cells of the immune system. Genes Dev. 17: 819–825
Article CAS Google Scholar - Newton K, Harris AW and Strasser A (2000) FADD/MORT1 regulates the pre-TCR checkpoint and can function as a tumour suppressor. EMBO J. 19: 931–941
Article CAS Google Scholar - Newton K, Kurts C, Harris AW and Strasser A (2001) Effects of a dominant interfering mutant of FADD on signal transduction in activated T cells. Curr. Biol. 11: 273–276
Article CAS Google Scholar - Zörnig M, Hueber A-O and Evan G (1998) p53-dependent impairment of T-cell proliferation in FADD dominant-negative transgenic mice. Curr. Biol. 8: 467–470
Article Google Scholar - Kennedy NJ, Kataoka T, Tschopp J and Budd RC (1999) Caspase activation is required for T cell proliferation. J. Exp. Med. 190: 1891–1896
Article CAS Google Scholar - Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, Peter ME and Yang X (2002) c-FLIPL is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 21: 3704–3714
Article CAS Google Scholar - Kataoka T, Budd RC, Holler N, Thome M, Martinon F, Irmler M, Burns K, Hahne M, Kennedy N, Kovacsovics M and Tschopp J (2000) The caspase-8 inhibitor FLIP promotes activation of NF-_κ_B and Erk signaling pathways. Curr. Biol. 10: 640–648
Article CAS Google Scholar - Wajant H, Haas E, Schwenzer R, Mühlenbeck F, Kreuz S, Schubert G, Grell M, Smith C and Scheurich P (2000) Inhibition of death receptor-mediated gene induction by a cycloheximide-sensitive factor occurs at the level of or upstream of Fas-associated death domain protein (FADD). J. Biol. Chem. 275: 24357–24366
Article CAS Google Scholar - Montixi C, Langlet C, Bernard A-M, Thimonier J, Dubois C, Wurbel M-A, Chauvin J-P, Pierres M and He H-T (1998) Engagement of T cell receptor triggers its recruitment to low-density detergent-insoluble membrane domains. EMBO J. 17: 5334–5348
Article CAS Google Scholar - Xavier R, Brennan T, Li Q, McCormack C and Seed B (1998) Membrane compartmentation is required for efficient T cell activation. Immunity 8: 723–732
Article CAS Google Scholar - Zhang W, Trible RP and Samelson LE (1998) LAT palmitoylation: its essential role in membrane microdomain targeting and tyrosine phosphorylation during T cell activation. Immunity 9: 239–246
Article CAS Google Scholar - Giurisato E, McIntosh DP, Tassi M, Gamberucci A and Benedetti A (2002) T cell receptor can be recruited to a subset of plasma membrane rafts, independently of cell signalling and attendantly to raft clustering. J. Biol. Chem. 278: 6771–6778
Article Google Scholar - Hueber AO, Bernard AM, Herincs Z, Couzinet A and He HT (2002) An essential role for membrane rafts in the initiation of Fas/CD95-triggered cell death in mouse thymocytes. EMBO Rep. 3: 190–196
Article CAS Google Scholar - Scheel-Toellner D, Wang K, Singh R, Majeed S, Raza K, Curnow SJ, Salmon M and Lord JM (2002) The death-inducing signalling complex is recruited to lipid rafts in Fas-induced apoptosis. Biochem. Biophys. Res. Commun. 297: 876–879
Article CAS Google Scholar - Garofalo T, Misasi R, Mattei V, Giammarioli AM, Malorni W, Pontieri GM, Pavan A and Sorice M (2003) Association of the death-inducing signaling complex with microdomains after triggering through CD95/Fas. Evidence for caspase-8-ganglioside interaction in T cells. J. Biol. Chem. 278: 8309–8315
Article CAS Google Scholar - Algeciras-Schimnich A, Shen L, Barnhart BC, Murmann AE, Burkhardt JK and Peter ME (2002) Molecular ordering of the initial signaling events of CD95. Mol. Cell. Biol. 22: 207–220
Article CAS Google Scholar - O'Reilly LA, Cullen L, Moriishi K, O'Connor L, Huang DCS and Strasser A (1998) Rapid hybridoma screening method for the identification of monoclonal antibodies to low abundance cytoplasmic proteins. BioTechniques 25: 824–830
Article CAS Google Scholar - Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH and Peter ME (1997) FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J. 16: 2794–2804
Article CAS Google Scholar - Perez D and White E (1998) E1B 19K inhibits Fas-mediated apoptosis through FADD-dependent sequestration of FLICE. J. Cell Biol. 141: 1255–1266
Article CAS Google Scholar - Gómez-Angelats M and Cidlowski JA (2003) Molecular evidence for the nuclear localization of FADD. Cell Death Differ. 10: 791–797
Article Google Scholar - Screaton RA, Kiessling S, Sansom OJ, Millar CB, Maddison K, Bird A, Clarke AR and Frisch SM (2003) Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: a potential link between genome surveillance and apoptosis. Proc. Natl. Acad. Sci. USA 100: 5211–5216
Article CAS Google Scholar - Hsu YT and Youle RJ (1997) Nonionic detergents induce dimerization among members of the Bcl-2 family. J. Biol. Chem. 272: 13829–13834
Article CAS Google Scholar - Hausmann G, O'Reilly LA, van Driel R, Beaumont JG, Strasser A, Adams JM and Huang DCS (2000) Pro-apoptotic apoptosis protease-activating factor 1 (Apaf-1) has a cytoplasmic localization distinct from Bcl-2 or Bcl-xL . J. Cell Biol. 149: 623–634
Article CAS Google Scholar - O'Reilly LA, Ekert P, Harvey N, Marsden V, Cullen L, Vaux DL, Hacker G, Magnusson C, Pakusch M, Cecconi F, Kuida K, Strasser A, Huang DCS and Kumar S (2002) Caspase-2 is not required for thymocyte or neuronal apoptosis even though cleavage of caspase-2 is dependent on both Apaf-1 and caspase-9. Cell Death Differ. 9: 832–841
Article CAS Google Scholar - Cherukuri A, Dykstra M and Pierce SK (2001) Floating the raft hypothesis: lipid rafts play a role in immune cell activation. Immunity 14: 657–660
Article CAS Google Scholar - Arudchandran R, Brown MJ, Peirce MJ, Song JS, Zhang J, Siraganian RP, Blank U and Rivera J (2000) The Src homology 2 domain of Vav is required for its compartmentation to the plasma membrane and activation of c-Jun NH(2)-terminal kinase 1. J. Exp. Med. 191: 47–60
Article CAS Google Scholar - Kosugi A, Saitoh S, Noda S, Yasuda K, Hayashi F, Ogata M and Hamaoka T (1999) Translocation of tyrosine-phosphorylated TCR_ζ_ chain to glycolipid-enriched membrane domains upon T cell activation. Int. Immunol. 11: 1395–1401
Article CAS Google Scholar - Scaffidi C, Volkland J, Blomberg I, Hoffmann I, Krammer PH and Peter ME (2000) Phosphorylation of FADD/MORT1 at serine 194 and association with a 70-kDa cell cycle-regulated protein kinase. J. Immunol. 164: 1236–1242
Article CAS Google Scholar - Winoto A (1997) Genes involved in T-cell receptor-mediated apoptosis of thymocytes and T-cell hybridomas. Semin. Immunol. 9: 51–58
Article CAS Google Scholar - Zhang J and Winoto A (1996) A mouse Fas-associated protein with homology to the human Mort1/FADD protein is essential for Fas-induced apoptosis. Mol. Cell. Biol. 16: 2756–2763
Article CAS Google Scholar - Puthalakath H, Villunger A, O'Reilly LA, Beaumont JG, Coultas L, Cheney RE, Huang DCS and Strasser A (2001) Bmf: a pro-apoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 293: 1829–1832
Article CAS Google Scholar - Cremesti A, Paris F, Grassme H, Holler N, Tschopp J, Fuks Z, Gulbins E and Kolesnick R (2001) Ceramide enables fas to cap and kill. J. Biol. Chem. 276: 23954–23961
Article CAS Google Scholar - Lens SM, Kataoka T, Fortner KA, Tinel A, Ferrero I, MacDonald RH, Hahne M, Beermann F, Attinger A, Orbea HA, Budd RC and Tschopp J (2002) The caspase 8 inhibitor c-FLIPL modulates T-cell receptor-induced proliferation but not activation-induced cell death of lymphocytes. Mol. Cell. Biol. 22: 5419–5433
Article CAS Google Scholar - Hua ZC, Sohn SJ, Kang C, Cado D and Winoto A (2003) A function of Fas-associated death domain protein in cell cycle progression localized to a single amino acid at its C-terminal region. Immunity 18: 513–521
Article CAS Google Scholar - O'Reilly LA, Cullen L, Visvader J, Lindeman G, Print C, Bath ML, Huang DCS and Strasser A (2000) The pro-apoptotic BH3-only protein Bim is expressed in hemopoietic, epithelial, neuronal and germ cells. Am. J. Pathol. 157: 449–461
Article CAS Google Scholar - O'Reilly LA, Huang DCS and Strasser A (1996) The cell death inhibitor Bcl-2 and its homologues influence control of cell cycle entry. EMBO J. 15: 6979–6990
Article CAS Google Scholar - O'Reilly LA, Harris AW, Tarlinton DM, Corcoran LM and Strasser A (1997) Expression of a bcl-2 transgene reduces proliferation and slows turnover of developing B lymphocytes in vivo. J. Immunol. 159: 2301–2311
CAS PubMed Google Scholar - Bi K, Tanaka Y, Coudronniere N, Sugie K, Hong S, van Stipdonk MJ and Altman A (2001) Antigen-induced translocation of PKC-θ to membrane rafts is required for T cell activation. Nat. Immunol. 2: 556–563
Article CAS Google Scholar - Drevot P, Langlet C, Guo XJ, Bernard AM, Colard O, Chauvin JP, Lasserre R and He HT (2002) TCR signal initiation machinery is pre-assembled and activated in a subset of membrane rafts. EMBO J. 21: 1899–1908
Article CAS Google Scholar - Puthalakath H, Huang DCS, O'Reilly LA, King SM and Strasser A (1999) The pro-apoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell 3: 287–296
Article CAS Google Scholar
Acknowledgements
We thank Drs W Yeh for FADD−/− ES cells, D Wallach for caspase-8−/− MEFs, Leonie Cullen for technical assistance and A Milligan, J Merryfull, A Naughton and C Tilbrook for animal care. We are grateful to Drs S Cory, J Adams, A Harris and D Vaux for helpful discussions and critical reading of this manuscript. This work was supported by grants and fellowships from the Leukemia and Lymphoma Society of America (New York), the Dr Josef Steiner Cancer Research Foundation (Bern, Switzerland), the National Health and Medical Research Council (Canberra, Australia; Reg. Key 973002), R D Wright Biomedical Career Development award (NHMRC), Sylvia and Charles Viertel Charitable Foundation, the Cancer Research Institute (New York) (CA80188) and the JDFR/NHMRC.
Author information
Author notes
- K Newton
Present address: Molecular Oncology, Genentech Inc., South San Francisco, CA, USA - K Scalzo
Present address: Austin Research Institute, Austin RMC, Melbourne, Australia
Authors and Affiliations
- The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia
L A O'Reilly, U Divisekera, K Newton, K Scalzo, H Puthalakath, D C S Huang & A Strasser - Research Center for Experimental Biology, Tokyo Institute of Technology, Yokohama, Japan
T Kataoka, M Ito & A Strasser
Authors
- L A O'Reilly
- U Divisekera
- K Newton
- K Scalzo
- T Kataoka
- H Puthalakath
- M Ito
- D C S Huang
- A Strasser
Corresponding author
Correspondence toA Strasser.
Additional information
Edited by ME Peter
Rights and permissions
About this article
Cite this article
O'Reilly, L., Divisekera, U., Newton, K. et al. Modifications and intracellular trafficking of FADD/MORT1 and caspase-8 after stimulation of T lymphocytes.Cell Death Differ 11, 724–736 (2004). https://doi.org/10.1038/sj.cdd.4401408
- Received: 25 September 2003
- Revised: 19 November 2003
- Accepted: 23 December 2003
- Published: 12 March 2004
- Issue Date: 01 July 2004
- DOI: https://doi.org/10.1038/sj.cdd.4401408