Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques (original) (raw)
Journal Article
CENARGEN-EMBRAPA, SAIN-Parque Rural
,
W5 Norte. C.P. 02372, CEP 70849-970 Brasilia, DF, Brazil
* To whom correspondence should be addressed. Tel: +55 61 340 3589; Fax: +55 61 340 3624; Email: [email protected]
Search for other works by this author on:
1
Norwegian Institute of Fisheries and Aquaculture
,
PO Box 2511, Tromso, Norway
Search for other works by this author on:
Accepted:
05 September 1997
Published:
01 November 1997
 PDF
PDF Cite
Salah M. Aljanabi, Iciar Martinez, Universal and rapid salt-extraction of high quality genomic DNA for PCR-based techniques, Nucleic Acids Research, Volume 25, Issue 22, 1 November 1997, Pages 4692–4693, https://doi.org/10.1093/nar/25.22.4692
Close
Navbar Search Filter Mobile Enter search term Search
Abstract
A very simple, fast, universally applicable and reproducible method to extract high quality megabase genomic DNA from different organisms is decribed. We applied the same method to extract high quality complex genomic DNA from different tissues (wheat, barley, potato, beans, pear and almond leaves as well as fungi, insects and shrimps' fresh tissue) without any modification. The method does not require expensive and environmentally hazardous reagents and equipment. It can be performed even in low technology laboratories. The amount of tissue required by this method is ∼50–100 mg. The quantity and the quality of the DNA extracted by this method is high enough to perform hundreds of PCR-based reactions and also to be used in other DNA manipulation techniques such as restriction digestion, Southern blot and cloning.
To study the molecular systematics of any organism, high quality DNA is required. So far there is no one common and simple procedure for genomic DNA extraction that can be used on a large scale for different eukaryotic organisms. Usually different (tricky) tissues required different protocols and different tissue preparation steps. The need for a universal procedure is urgent especially when hundreds of samples need to be analyzed. We have applied PCR-based techniques for phylogenetic and mapping studies such as AP-PCR ( 1 , 2 ) and cycle sequencing ( 3 ) on collections of wheat ( Triticum aestivum ), barley ( Hordium vulgaris ), potato ( Solanum tuberosum ), beans ( Vicia faba ), pear ( Pyrus syrica ), wild almond ( Prunus amygdalus ), fungi ( Nomuraea relieye, Alternaria spp. ), grasshoppers ( Schistocerca pallens ), and shrimps ( Pandalus borealis ), lettuce ( Lactuca sativa ) and eucalyptus ( Eucaliptus grandis ). Most if not all genomic DNA extraction protocols such as the plant DNA extraction protocol of Murray and Thompson ( 4 ) and its many derivatives so far, human genomic DNA extraction methods ( 5 , 6 ) and animal genomic DNA extraction ( 7 ), require the use of liquid nitrogen and/or freeze-drying (lypholization) of the tissue for the initial grinding which are difficult to obtain in regions of the world where most germplasm collections of many organisms have evolved or can be sampled. We present here a protocol for DNA extraction from fresh tissue that is universally applicable on a variety of organisms regardless of the complexity of their genomes.
About 50–100 mg (1 cm 2 ) of young field or greenhouse-grown plant leaves, filtered and dried mycelium, the muscle of one back leg of a grasshopper and shrimp muscle were used for DNA extraction. The fresh tissue was homogenized in 400 µl of sterile salt homogenizing buffer (0.4 M NaCl 10 mM Tris-HCl pH 8.0 and 2 mM EDTA pH 8.0), using a Polytron Tissue Homogenizer, for 10–15 s. Then 40 µl of 20% SDS (2% final concentration) and 8 µl of 20 mg/ml protenase K (400 µg/ml final concentration) were added and mixed well. The samples were incubated at 55–65°C for at least 1 h or overnight, after which 300 µl of 6 M NaCl (NaCl saturated H 2 O) was added to each sample. Samples were vortexed for 30 s at maximum speed, and tubes spun down for 30 min at 10 000 g . The supernatant was transferred to fresh tubes. An equal volume of isopropanol was added to each sample, mixed well, and samples were incubated at −20°C for 1 h. Samples were then centrifuged for 20 min, 4°C, at 10 000 g . The pellet was washed with 70% ethanol, dried and finally resuspended in 300–500 µl sterile dH 2 O. Genomic DNA (5–15 ng) in 10 µl of ddH 2 O was used for RAPD amplification of genomic DNA ( 1 ). Ten micrograms of each genomic DNA sample was incubated overnight with 5 U Bam HI, Eco RI, Hin dIII and Cla I, and analyzed on 1.5% agarose gels. The samples were incubated for 16 h to ensure complete digestion and also to allow the detection of nuclease activity.
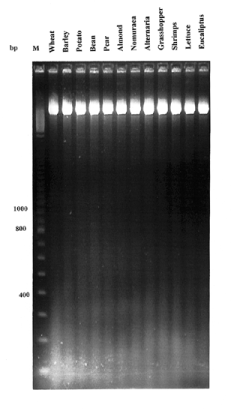
Figure 1
Example of an agarose gel electrophoresis of undigested genomic DNA of all organisms where 3 µg of genomic DNA was loaded from each sample.
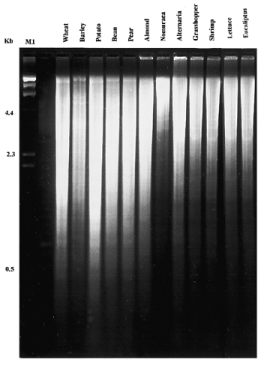
Figure 2
Example of agarose gel electrophoresis of Bam HI restriction digest of genomic DNA extracted by our protocol from all organisms under study. M1, λ/ Hin dIII molecular weight marker.
The purity of the DNA, determined from the A260/A280 ratio averaged >1.77 for all organisms. There was no RNA contamination in all samples nor any sign of degraded DNA during preparation ( Fig. 1 ). The yield of DNA ranged from 500 to 800 ng/mg fresh weight for all individuals sampled. The amount of tissue required for this method is minimal, but we scaled up the amount of tissue 10-fold without any reduction in DNA quality and quantity. The average number of PCR reactions that can be performed using DNA extracted from 50 mg tissue was >3000.
Figure 2 shows one example of an agarose gel of a Bam HI restriction digestion reaction of genomic DNA extracted by our method (we used also Eco RI, Hin dIII and Cla I, data not shown). DNA was completely digested with the restriction enzymes and there was no evidence of the presence of nucleases in any sample. All DNA samples were subjected to PCR amplification using 10mer random primers (Operon Technologies, Alameda, CA) ( Fig. 3 ). All genomic DNAs produced a clear, sharp and reproducible PCR product pattern. We repeated the PCR experiment over a period of 3 months and obtained the same banding pattern which indicates the reproducibility of the results and the integrity of the DNA ( 8 ).
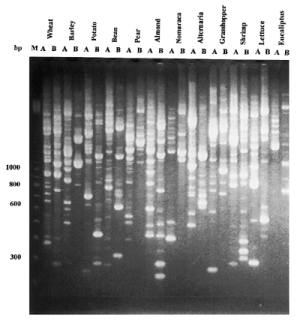
Figure 3
RAPD fingerprints of all DNAs with primers OPB-18 (5′-CCA CAG CAG T-3′) and OPJ-10 (5′-AAG CCC GAG G-3′) and 2 U Stoffel fragment Taq DNA polymerase. The amplification program is as previously described ( 1 ). The amplification products were resolved on 2% agarose gel. The organism is indicated at the top of the figure. A, primer OPB-18; B, primer OPJ-10. M1, 100 bp molecular weight DNA ladder (BRL/Gibco).
The efficiency, speed, universality and requirement of no expensive facilities or toxic chemicals makes the present method an attractive alternative to the existing methods of genomic DNA extraction; none of which is ideal or universal ( 4–7 , 9 ). These results show that the DNA produced by our universal, simple, low cost, fast and safe protocol is of high quality and can be used reliably in DNA manipulation and PCR-based techniques on a wide range of organisms even in low technology laboratories.
Acknowledgements
The authors would like to express their gratitude to the Norwegian Institute of Fisheries and Aquaculture and Cenargen-Embrapa, Brazil, for their support in performing this work. Also we thank Dr Peter Inglis for valuable and helpful comments.
References
1
, . ,
Nucleic Acids Res.
,
1990
, vol.
18
(pg.
7213
-
7218
)
2
, , , , . ,
Nucleic Acids Res.
,
1990
, vol.
18
(pg.
6531
-
6535
)
3
, . ,
Proc. Natl Acad. Sci. USA
,
1991
, vol.
88
(pg.
2815
-
2819
)
4
, . ,
Nucleic Acids Res.
,
1980
, vol.
8
(pg.
4321
-
4325
)
5
, , . ,
Nucleic Acids Res.
,
1988
, vol.
16
pg.
1215
6
, , . ,
J. Med. Genet.
,
1990
, vol.
27
(pg.
569
-
570
)
7
, . ,
Rep. Int. Whal. Comm.
,
1991
, vol.
13
(pg.
99
-
103
)
(special issue)
8
, , . ,
BioTechniques
,
1993
, vol.
14
(pg.
214
-
218
)
9
. ,
Nucleic Acids Res.
,
1994
, vol.
22
(pg.
1772
-
1773
)
© 1997 Oxford University Press
I agree to the terms and conditions. You must accept the terms and conditions.
Submit a comment
Name
Affiliations
Comment title
Comment
You have entered an invalid code
Thank you for submitting a comment on this article. Your comment will be reviewed and published at the journal's discretion. Please check for further notifications by email.