Kinetin in Familial Dysautonomia Carriers: Implications for a New Therapeutic Strategy Targeting mRNA Splicing (original) (raw)
Main
Familial dysautonomia (FD) [Riley-Day syndrome/hereditary sensory and autonomic neuropathy type III, OMIM 223900] is an autosomal recessive disease characterized by pervasive sensory and autonomic dysfunction and premature death, with a carrier frequency of 1 in 27 in the Ashkenazi Jewish population (1,2). Neuropathologic studies suggest the disease results from the incomplete embryonic development of unmyelinated and small myelinated neurons as well as progressive and relentless neurodegeneration. Although penetrance is complete, there is pronounced phenotypic variability. As there is failure of neuronal migration and maturation, some symptoms are apparent at birth such as hypotonia and poor suck with failure to thrive. Consistent with sympathetic denervation, there is a lack of autonomic homeostasis, which presents as orthostatic hypotension without compensatory tachycardia and paroxysmal hypertension. Decreased unmyelinated neurons result in sensory dysfunction that predominantly affects temperature and pain perception. Patients lack overflow tears and have gastrointestinal dysfunction with dysphagia and reflux. With advancing age, function deteriorates, however, with supportive and preventative care, morbidity and mortality has been reduced. Currently, approximately 54% of patients are over 15 y and the probability of survival to 40 y is 50% (1–3).
In 2001, mutations in IκB kinase–associated protein gene (IKBKAP) were shown to cause FD (4,5). Over 99% of patients are homozygous for the most common mutation, a T to C DNA change located at the sixth base pair of intron 20, within the exon 20 splice donor site. The major mutation leads to variable, tissue-specific skipping of exon 20 in the RNA message, resulting in reduced protein expression of IκB kinase–associated protein (IKAP) (4–6). IKAP, alternatively known as elongator protein 1 (ELP-1) is an integral component of the human elongator complex, a transcriptional regulator. A subset of elongator's target genes play a role in cell migration, and therefore decreased IKAP/ELP-1, leading to decreased functional elongator, is consistent with FD neuropathology (7).
IKBKAP is expressed ubiquitously in humans, and FD patients retain the capacity to produce wild-type mRNA and functional protein (IKAP/ELP-1 and elongator). The alternative splicing of IKBKAP in FD is also tissue specific, and the most profound aberrant splicing (that leads to overproduction of mutant IKBKAP mRNA and low levels of functional protein) occurs in neuronal tissue (5–8). Although carriers seem to be clinically asymptomatic, they demonstrate some degree of reduced splicing efficiency (9), and may have subclinical neurophysiologic manifestations that have not yet been described.
As part of a National Institute of Neurological Disorders and Stroke sponsored drug screen, it was demonstrated that kinetin (6-furfurylaminopurine; C10H9N5; molecular weight 215.21) had a dramatic effect on exon 20 inclusion (10 μM to 100 μM) in vitro (10–13). Kinetin is a natural dietary ingredient, a plant growth regulator, and an ingredient in various skin creams. However, it has never been used as a therapeutic agent for human disease (14–16). Preclinical studies in rodents demonstrated that kinetin was absorbed orally, readily distributed into the plasma and central nervous system (CNS), and was not clastogenic. Toxicity occurred only at extremely high doses (Sitek studies No. 0849-M208, No. 0849-1721.4, No. 0849-1731.C, SITEK Research Laboratories, Rockville, MD, 2005–2007).
We hypothesized that oral kinetin would also affect IKBKAP mRNA splicing in vivo. Therefore, to safely determine dose response curves in a healthy human population who also demonstrate a degree of aberrant IKBKAP splicing, we administered oral kinetin to healthy carriers of the FD common mutation for 8 d and found that kinetin increased IKBKAP mRNA in peripheral blood leukocytes. Given that approximately 15% of all known disease-causing mutations alter mRNA splicing, and that this proportion will likely increase as the regulatory effects of single nucleotide polymorphisms associated with complex, common disease are worked out, our work illustrates that treatments which target precise molecular splicing defects can be successfully developed. This mechanism offers great promise as a new, unexplored direction for pharmacologic intervention in disease.
METHODS
Study population.
Twenty-nine carriers of the common IVS20+6T>C FD mutation, [mean age 46.75 ± 6.76 y; 17 males], were recruited for enrollment from the FD registry database at the New York University (NYU) Dysautonomia Center (a convenience sample). Heterozygosity was confirmed by DNA analysis. Two subjects from cohort 1 and three subjects from cohort 2 agreed to participate for a second trial of kinetin with the understanding that it would be administered at a higher dose with food (cohorts will be defined below). The study was conducted in accordance with the requirements of the New York University School of Medicine Institutional Research Board and written informed consent was obtained from all participants.
Dose limiting toxicity.
All subjects had baseline and exit screening for blood, bone marrow, liver, and renal abnormalities. Dose limiting toxicity is defined as either transaminase and serum creatinine measurements that exceed usual laboratory reference values or a decline in white blood count, red blood cell count, and platelet count below usual reference values; or any serious adverse event as defined by the Food and Drug Administration.
Dose escalation/cohort determination.
On the basis of rodent studies performed by SITEK Research Laboratories, we accepted the no observed adverse events level (NOAEL) to be 200 mg/kg/d, as no toxicity was observed at that dose in 35-d and 90-d studies (Sitek studies No. 0849-M208, No. 0849-1721.4, No. 0849-1731.C, SITEK Research Laboratories, 2005–2007). The NOAEL in rats was converted to a human equivalent dose using 6.2 as a conversion factor. The human equivalent dose of 33.5 mg/kg/d was further reduced by a factor of 10. This starting dose, labeled “x,” (3.35 mg/kg/d) was used for the subjects in the first cohort. Doses for subsequent cohorts were determined from the modified Fibonacci dose escalation scheme: x, 2x, 3.3x, 5x, and 7x (17). There were five subjects in cohort 1, five in cohort 2, five in cohort 3, seven in cohort 4, seven in cohort 5; five of the 29 subjects agreed to participate for repeat study in cohort 6. For both cohorts 5 (fasting) and 6 (fed), the maximum dose was used (7x or 23.5 mg/kg/d). Dose escalation continued until kinetin plasma levels reached our target concentration of 10 μM (2150 ng/mL).
Kinetin administration.
Kinetin (Nature's Best Inc. Hauppauge, NY) was supplied as a clear capsule containing 250 mg of kinetin and 120 mg of maltodextrin and was administered daily for eight consecutive days. The contents of the capsules were emptied into a small amount of juice and taken in the fasting state in cohorts 1 through 5. An additional cohort (6) was studied to ascertain possible pharmacokinetic changes in the fed state. Subjects received the first dose of kinetin at the NYU GCRC (general clinical research center) and were monitored for 24 h.
Kinetin pharmacokinetics.
Blood was drawn and centrifuged at 0.5, 1, 2, 6, 12, and 24 h after the first kinetin dose. Plasma samples were divided in duplicate and labeled with the subject's numerical code and the time of draw. One sample was shipped on dry ice to ADMET Laboratories for analysis; the other was frozen at −20°C. The plasma kinetin level at each interval was plotted against time to determine when the target level of 2150 ng/mL was achieved and how long it was sustained. The target kinetin level was extrapolated from previous cell culture studies and based on the 10 μM concentration that affected splicing (12). Plots for each subject were developed and responses analyzed for each cohort at a particular dose. All subjects had a final kinetin plasma level drawn upon exiting the study on day 8, 1 h after the last dose.
Assessment of IKBKAP mRNA levels.
Each subject had two IKBKAP mRNA levels at baseline and one level at exit. The baseline measurements were obtained before administration of first kinetin dose to assure that there was consistency in an individual. Samples were collected using the PAXgene Blood RNA System. Each tube was labeled with the subject's numerical code and the time of draw, and was shipped to The Center for Human Genetic Research.
RNA extraction was performed using the PAXgene Blood RNA Kit (PreAnalytiX-Qiagen/BD, Germantown, MD) and quantification was performed using UV absorbance. Reverse transcription was performed using 500 ng total RNA, oligo(dT)/random hexamer primers (Promega, Madison, WI), and Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA). In our previous studies, we have quantified the use of exon 20 as a marker for splicing modification and have shown that kinetin acts directly on splicing and not by altering transcription or nonsense-mediated decay (NMD) of the mutant message (5,8,12,13). Quantitative polymerase chain reaction analysis of wild-type human IKBKAP was performed using the BIORAD iCycler, iQ SYBER Green supermix (BIORAD) and primers, F: 5′-TTCACGGATTGTCACTGTTGTG-3′ and R: 5′-TGTCCAACCACTTCCGAATCTGAG-3′ specific to the WT spliced isoform of IKBKAP. To ensure that the same amount of starting cDNA was present in each tube, samples were normalized to the human GAPDH gene, a reference gene with consistent expression in mRNA isolated from peripheral blood samples. Two GAPDH primers were used F: 5′-TGCACCACCAACTGCTTAGC-3′ and R: 5-GGCATGGACTGTGGTCATGAG-3′. The primer pairs were tested against a standard curve and the correlation coefficient (_r_2) and amplification efficiencies fell within accepted range. Quantitative polymerase chain reaction was performed in triplicate and the relative amount of WT IKBKAP was measured between the samples using the Livak method for relative gene expression analysis (18). This method measures the difference in each sample (Δ_C_t) between the threshold cycles of the target gene (IKBKAP) to the reference gene (GAPDH). The mean Δ_C_t for each triplicate was graphed and standard deviations for each mean were calculated.
Pharmacokinetic analysis.
Values and times of maximal (_C_max, _T_max) and postmaximal minimal (_C_min, _T_min) concentration were estimated on the basis of graphs generated in SigmaPlot 8.0 (cubic spline interpolation). Area under curve (AUC) was also estimated in SigmaPlot. Elimination rate constant (_K_el) and time of half-life (_T_1/2) were estimated according to the following formulas.
Kel=Ln(Cmax/Cmin)/(Tmax−Tmin)
T½=0.693/Kel
Statistical analysis.
The distributions of IKBKAP mRNA levels and kinetin blood levels at day 8 and the pharmacokinetic parameters associated with kinetin were summarized using descriptive summary statistics and graphical displays. IKBKAP mRNA levels were positively skewed; therefore, the levels were transformed to the logarithmic scale for statistical analyses that require the assumption of normality of data.
Statistical comparisons of all pharmacokinetic parameters among the dosing groups were performed using nonparametric statistics because of the lack of normality in the distributions of these parameters. The Jonkeheere-Terpstra test was used to test for a monotone trend in the pharmacokinetic parameters as kinetin dose increased. Dosing cohorts 1 through 5, given kinetin doses varying from 3.35 to 23.5 mg/kg/d in the fasting state, were used in this analysis. The Mann–Whitney U test was used to compare pharmacokinetic parameters between dosing cohorts 5 (fasting) and 6 (fed), both receiving the same kinetin dose, 23.5 mg/kg/d.
The intraclass correlation coefficient (ICC) was used to assess the consistency of the two replicate baseline measurements of IKBKAP mRNA levels. Linear regression analysis was used to describe and assess the association between log kinetin plasma levels and log change from baseline in IKBKAP mRNA expression. Correlation coefficients were used to describe the strength of this association.
Statistical analyses were performed using SPSS Version 14.0. The significance level for all tests was set at 0.05 (two-sided).
Five patients participated twice during the study, once in either cohort 1 or 2 in the fasting state (for the analysis of the association between kinetin level IKBKAP mRNA level) and then in cohort 6 (for the analysis of the effect of food on kinetin pharmacokinetics and IKBKAP mRNA). Group 6 was compared only with Group 5 to examine the differences at the maximum dose between the fed and fasting states.
RESULTS
Pharmacokinetics of oral kinetin.
Table 1 displays the descriptive summary statistics for the pharmacokinetic parameters obtained from the dose escalation. There were significant increases with increased dosage (based on Jonkeheere-Terpstra tests) for the following pharmacokinetic parameters when kinetin was taken in the fasting state: _C_max (ng/mL) (p < 0.001), _T_max (h) (p = 0.005), _C_min (ng/mL) (p = 0.002), _T_min (h) (p = 0.005), AUC (p < 0.001). There were no significant differences between any of the pharmacokinetic parameters for subjects on the highest dose of kinetin (23.5 mg/kg/d, seven times the starting dose), when the drug was taken in the fasting (cohort 5) or in the fed state (cohort 6).
Table 1 Pharmacokinetic parameters by dosing group for oral kinetin in human subjects
Our target plasma kinetin level was 2150 ng/mL, based on extrapolation from cell culture studies (12). In cohort 5 (fasting), this level was achieved in 86% of subjects at 2-h postdose; in cohort 6 (fed), this level was achieved in 100% of subjects at 2-h postdose (Table 2). At 6 h, kinetin levels were still above 2150 ng/mL in 43% of subjects in cohort 5 and 40% of subjects in cohort 6 (Table 2; Fig. 1). After 8 d of daily administration, kinetin levels taken 1 h after the eighth day dose were above 2150 ng/mL in 100% of subjects in cohort 5 (4 of 4 subjects) and in 50% of subjects in cohort 6 (2 of 4 subjects) (Table 2). It should be noted that none of the subjects in cohort 6 participated as subjects in cohort 5.
Table 2 Number of subjects achieving plasma kinetin levels of 2150 ng/mL or greater by cohort (percent per cohort)
Figure 1
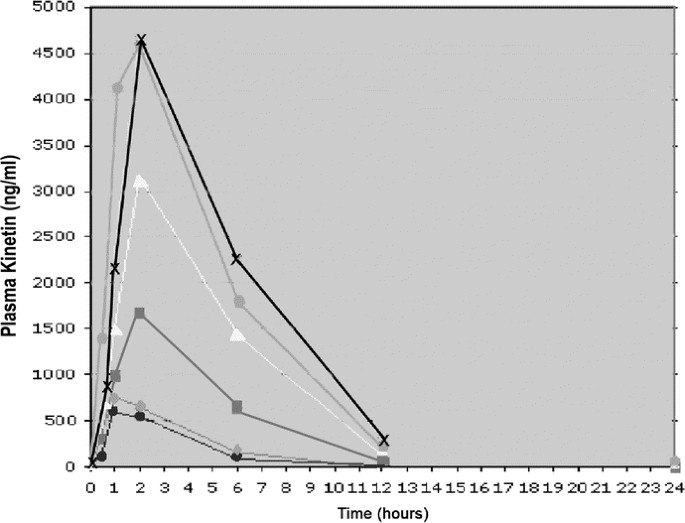
Plasma kinetin (ng/mL) vs. time (h): This graph plots the plasma kinetin level (y axis) against time (x axis) by cohort (• = cohort 1, ▴ = cohort 2, ▪ = cohort 3, ▵ = cohort 4,  = cohort 5, X = cohort 6). As dose increased in each cohort, plasma levels increased accordingly. Although the true _C_max cannot be estimated, one can observe that plasma kinetin levels are almost undetectable by 12 h in all dosing groups.
= cohort 5, X = cohort 6). As dose increased in each cohort, plasma levels increased accordingly. Although the true _C_max cannot be estimated, one can observe that plasma kinetin levels are almost undetectable by 12 h in all dosing groups.
Reliability of repeated IKBKAP mRNA measurements.
For each subject, two baseline blood samples were obtained for measurement of IKBKAP mRNA that were at least 2 h apart and at most 7 days apart. These replicate baseline measurements of IKBKAP mRNA levels were highly reproducible (estimated ICC = 0.81, p < 0.001).
Association between kinetin dose and difference between baseline and day 8 IKBKAP mRNA level.
Table 3 displays median IKBKAP mRNA levels achieved by 8 d, and the median change from baseline IKBKAP mRNA levels at day 8, by dosing cohort. For the 26 subjects who completed eight consecutive days of single daily dose administration of kinetin in the fasting state, there was a median increase per cohort in the expression of IKBKAP mRNA in peripheral blood leukocytes.
Table 3 Median IKBKAP mRNA levels achieved at day 8, and the median change from baseline IKBKAP mRNA levels at 8 days, by dosing cohort (includes only patients completing 8 days of kinetin dosing)
The scatterplot displayed in Figure 2a shows the linear association between log plasma kinetin levels achieved at 8 d and the log change in IKBKAP mRNA levels from baseline level to the 8-d level; the regression is displayed with 95% confidence intervals. Data were transformed using natural logarithms. Patients in dosing groups 1 through 5 (fasting) were included in this analysis. Log plasma kinetin levels and log changes in IKBKAP mRNA levels (r = 0.51, p = 0.007) are significantly associated.
Figure 2
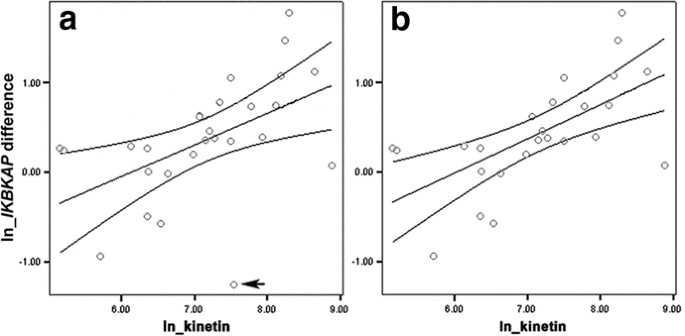
(a and b): Scatterplots/linear regressions: The scatterplots with the estimated linear regression line and 95% confidence intervals in Fig. 2a show the association between the logarithmic (log or ln) plasma kinetin levels and the log change in IKBKAP mRNA from baseline level to the 8 d level. (2a) All subjects in cohorts 1 through 5 (all in the fasting state) are included; the regression equation is: ln(IKBKAP diff + 1) = −2.17 + 0.35 × ln kinetin, where IKBKAP diff is the difference between IKBKAP mRNA level at baseline and at 8 d, and kinetin is the kinetin level achieved at day 8. The outlier is marked with an arrow. (2b) The outlier has been removed; the regression equation is: ln(IKBKAP diff + 1) = −2.31 + 0.38 × ln kinetin. Note that the confidence intervals are much narrower, indicating a stronger association (r = 0.63, p < 0.001).
The subject shown in the scatterplot (Fig. 2a) with a log kinetin value of approximately 7.5 and a negative log change in IKBKAP mRNA level is an outlier with a standardized residual greater than 2. When the analysis is performed excluding this subject (Fig. 2b), the association between the log kinetin and the log change from baseline IKBKAP is considerably stronger (r = 0.63, p < 0.001). A kinetin level of 2150 ng/mL is considered an effective concentration, based on extrapolation from cell culture studies (12). If this kinetin level is reached at 8 d, we expect that the corresponding increase from baseline in IKBKAP mRNA level by 8 d would be 0.88 (IKBKAP mRNA levels are expressed as relative levels); 95% confidence interval is 0.51–1.33.
Adverse events.
The most commonly reported side effect was nausea; the frequency increased as doses escalated and was not altered by administering with food in cohort 6. Although two subjects experienced transient headaches, there were no long-term neurologic sequelae. There were no cardiovascular effects or weight changes, nor changes in hemogram, chemistry, liver, and renal laboratory values in any of the 29 participants.
Four subjects withdrew from the study because of adverse effects; all withdrawals occurred after the first dose and were from the higher dose cohorts 4, 5, and 6, and all reported effects lasted less than 24 h (Table 4).
Table 4 Adverse effects and drop outs (highest grade reported)
Comparison of IKBKAP mRNA levels at 8 d when kinetin is administered with and without food.
The median IKBKAP mRNA levels at 8 d for subjects who took the highest kinetin dose in the fasting (cohort 5) and in the fed state (cohort 6) were 5.25 (interquartile range [IQR] = 1.89) and 5.30 (IQR = 1.20), respectively. There was no difference in IKBKAP mRNA levels at 8 d between these two groups. The median change from the baseline IKBKAP mRNA level for subjects taking kinetin in the fasting state was 1.10 (IQR = 1.17) and in the fed state was 1.43 (IQR = 0.83). There was no difference in the change in IKBKAP mRNA levels from baseline at 8 d between the two groups.
DISCUSSION
This article addresses several fundamental issues relevant to kinetin's role as a potential therapeutic agent for individuals affected with FD. We confirm that oral administration of kinetin results in plasma levels equivalent to cell culture concentrations reported to affect IKBKAP mRNA splicing (12). Furthermore, we demonstrate that after 8 d of single daily dosing, normal splicing of IKBKAP is increased and is dependent on kinetin dose and plasma level.
This is the first demonstration of in vivo modification of IKBKAP splicing. The measurement of exon 20 inclusion as a direct marker for splicing alteration has been proven (9,12,13). The misspliced message is targeted for NMD, however, our previous molecular studies have shown that blocking NMD does not alter the effect of kinetin, which targets mRNA splicing directly due to the presence of a specific “kinetin response element” at the end of IKBKAP exon 20 (12,13). Furthermore, kinetin modifies splicing of other genes, other than the IKBKAP gene, including a neurofibromatosis mutant (13). Splicing mutations are prevalent in a variety of disorders including Marfan syndrome, cystic fibrosis, retinoblastoma, tuberous sclerosis, neurofibromatosis, breast cancer, and spinal muscular atrophy. In fact, carriers and patients with spinal muscular atrophy increase their SMN transcript and protein levels after treatment with valproate, which may prove to be an effective treatment (19). On the basis of the SMN studies and our current findings, we hypothesize that pharmacologic treatments can be developed that target a particular type of mRNA splicing mutation, and that these agents will modify mRNA splicing and increase protein levels in humans. This proof-of-principle experiment not only supports moving on to clinical trials in FD patients, but also promotes wider exploration of splicing defects and alternative splicing as a target for therapy. mRNA splicing mutations are unique in that they frequently lead to variable expression of disease, because the gene retains the ability to code for normal protein. Therefore, treatment of such disorders does not necessarily require replacing gene products that are missing, or designing methods to obliterate dominant-negative effects; instead the gene can be targeted directly and expression increased by improving mRNA splicing. This exciting mechanism for the treatment of genetic disease is illustrated by this study.
Kinetin follows first-order elimination. Administration with food did not alter the pharmacokinetic profile. The most common adverse event was mild-moderate nausea. Gastrointestinal symptoms may be decreased by diet or dosing modifications or concomitant use of antiemetics. These concerns will be addressed in future clinical trials. Within 2 h of the initial dose at 23.5 mg/kg, 11 of 12 subjects (92%) had kinetin plasma levels greater than or equal to 2150 ng/mL, which is equivalent to 10 μM, the minimum concentration observed to affect IKBKAP splicing in vitro (12). Furthermore, there is a linear association between log plasma kinetin level and log change from baseline in IKBKAP mRNA. What remains to be determined is whether IKBKAP mRNA expression is similarly affected in neural tissues where missplicing is most profound. This is the major limitation to our study, and animal models will be critical to address this question (13).
Although we cannot directly measure splicing changes in human brain, the CNS distribution of kinetin after acute oral administration in rats (Sitek studies No. 0849-M208 and No. 0849-1721.4, SITEK Research Laboratories, 2005–2007), target levels of kinetin were achieved in both blood and brain tissue. Thus, we are optimistic that kinetin will alter neuronal splicing. As with any treatment for a neurologic disease, the true test of efficacy will require careful clinical studies of suitable biomarkers in patients, because direct sampling of neuronal tissue is not possible. Our demonstrated ability to measure splicing changes in peripheral blood provides direct in vivo evidence that kinetin is targeting the defective gene, and this demonstration of potential efficacy is stronger than what exists for many drugs on the market today.
In conclusion, kinetin restores exon 20 inclusion in heterozygotes of the common FD mutation after 8 d of treatment. Although induction of nerve growth in FD patients is an unrealistic expectation, it may be possible to halt the relentless disease progression and neurodegenerative changes in FD via up-regulation of IKAP/ELP-1 (20). Although long-term studies in FD subjects will be necessary to determine whether splicing modification and subsequent up-regulation of IKAP/ELP-1 protein will translate into changes in objective measures of autonomic and sensory function, this mechanism offers great promise as a new, unexplored direction for pharmacologic intervention in disease.
Abbreviations
FD:
familial dysautonomia
IKAP/ELP-1:
IκB kinase–associated protein/elongator protein 1
IKBKAP:
IκB kinase–associated protein gene
IQR:
interquartile range
References
- Axelrod FB 2004 Familial dysautonomia. Muscle Nerve 29: 352–363
Article Google Scholar - Axelrod FB 2002 Hereditary sensory and autonomic neuropathies: familial dysautonomia and other HSANs. Clin Auton Res 1: I2–I14
Article Google Scholar - Axelrod FB, Goldberg JD, Ye XY, Maayan C 2002 Survival in familial dysautonomia: impact of early intervention. J Pediatr 141: 518–523
Article Google Scholar - Anderson SL, Coli R, Daly IW, Kichula EA, Rork MJ, Volpi SA, Ekstein J, Rubin BY 2001 Familial dysautonomia is caused by mutations of the IKAP gene. Am J Hum Genet 68: 753–758
Article CAS Google Scholar - Slaugenhaupt SA, Blumenfeld A, Gill SP, Leyne M, Mull J, Cuajungco MP, Liebert CB, Chadwick B, Idelson M, Reznik L, Robbins C, Makalowska I, Brownstein M, Krappmann D, Scheidereit C, Maayan C, Axelrod FB, Gusella JF 2001 Tissue-specific expression of a slicing mutation in the IKBKAP gene causes familial dysautonomia. Am J Hum Genet 68: 598–605
Article CAS Google Scholar - Leyne M, Mull J, Gill SP, Cuajungco MP, Oddoux C, Blumenfeld A, Maayan C, Gusella JF, Axelrod FB, Slaugenhaupt SA 2003 Identification of the first non-Jewish mutation in familial Dysautonomia. Am J Med Genet A 118A: 305–308
Article Google Scholar - Close P, Hawkes N, Cornez I, Creppe C, Lambert CA, Rogister B, Siebenlist U, Merville MP, Slaugenhaupt SA, Bours V, Svejstrup JQ, Chariot A 2006 Transcription impairment and cell migration defects in elongator-depleted cells: implication for familial dysautonomia. Mol Cell 22: 521–531
Article CAS Google Scholar - Cuajungco MP, Leyne M, Gill SP, Mull J, Lu W, Zagzag D, Axelrod FB, Gusella JF, Maayan Ch, Slaugenhaupt SA 2003 Tissue-specific reduction in splicing efficiency of IKBKAP due to the major mutation associated with Familial Dysautonomia. Am J Hum Genet 72: 749–758
Article CAS Google Scholar - Gold-von Simson G, Leyne M, Mull J, Rolnitzky LM, Goldberg JD, Berlin D, Axelrod FB, Slaugenhaupt SA 2008 IKBKAP mRNA in peripheral blood leukocytes: a molecular marker of gene expression and splicing in familial dysautonomia. Pediatr Res 63: 186–190
Article CAS Google Scholar - Anderson SL, Qiu J, Rubin BY 2003 Tocotrienols induce IKBKAP expression: a possible therapy for familial dysautonomia. Biochem Biophys Res Commun 306: 303–309
Article CAS Google Scholar - Anderson SL, Qiu J, Rubin BY 2003 EGCG corrects aberrant splicing of IKAP mRNA in cells from patients with familial dysautonomia. Biochem Biophys Res Commun 310: 627–633
Article CAS Google Scholar - Slaugenhaupt SA, Mull J, Leyne M, Cuajungco MP, Gill SP, Hims MM, Quintero F, Axelrod FB, Gusella JF 2004 Rescue of a human mRNA splicing defect by the plant cytokinin kinetin. Hum Mol Genet 13: 429–436
Article CAS Google Scholar - Hims MM, Ibrahim EC, Leyne M, Mull J, Liu L, Lazaro C, Shetty RS, Gill S, Gusella JF, Reed R, Slaugenhaupt SA 2007 Therapeutic potential and mechanism of kinetin as a treatment for the human splicing disease familial dysautonomia. J Mol Med 85: 149–161
Article CAS Google Scholar - Stanley N, Muthiah PT, Geib SJ 2003 N6-furfuryladenine (kinetin) hydrochloride. Acta Crystallogr C 59: o27–o29
Article Google Scholar - Soriano-Garcia M, Parthsarathy R 1975 Structure-activity relationship of cytokinins: crystal structure and conformation of 6-furfurylaminopurine (kinetin). Biochem Biophys Res Commun 64: 1062–1068
Article CAS Google Scholar - Ge L, Yong JW, Goh NK, Chia LS, Tan SN, Ong ES 2005 Identification of kinetin and kinetin riboside in coconut (Cocos nucifera L.) water using a combined approach of liquid chromatography-tandem mass spectrometry, high performance liquid chromatography and capillary electrophoresis. J Chromatogr B Analyt Technol Biomed Life Sci 829: 26–34
Article CAS Google Scholar - Omura GA 2003 Modified Fibonacci search. J Clin Oncol 21: 3177
Article Google Scholar - Livak KJ, Schmittgen TD 2001 Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25: 402–408
Article CAS Google Scholar - Brichta L, Holker I, Huag K, Klockgether T, Wirth B 2006 In vivo activation of SMN in spinal muscular atrophy carriers and patients treated with valproate. Ann Neurol 59: 970–975
Article CAS Google Scholar - Luft FC 2007 Better days are coming for Riley-Day patients. J Mol Med 85: 99–101
Article CAS Google Scholar
Author information
Author notes
- Susan A Slaugenhaupt and Felicia B Axelrod: S.A.S. and F.B.A. are cosenior authors for this study.
Authors and Affiliations
- Department of Pediatrics, New York University School of Medicine, New York, 10016, New York
Gabrielle Gold-von Simson, Andrei Voustianiouk & Felicia B Axelrod - Division of Biostatistics, New York University School of Medicine, New York, 10016, New York
Judith D Goldberg & Linda M Rolnitzky - Department of Neurology (Genetics), Center for Human Genetic Research, Massachusetts General Hospital, Boston, 02114, Massachusetts
James Mull, Maire Leyne & Susan A Slaugenhaupt - Department of Neurology (Genetics), Harvard Medical School, Boston, 02115, Massachusetts
James Mull, Maire Leyne & Susan A Slaugenhaupt
Authors
- Gabrielle Gold-von Simson
You can also search for this author inPubMed Google Scholar - Judith D Goldberg
You can also search for this author inPubMed Google Scholar - Linda M Rolnitzky
You can also search for this author inPubMed Google Scholar - James Mull
You can also search for this author inPubMed Google Scholar - Maire Leyne
You can also search for this author inPubMed Google Scholar - Andrei Voustianiouk
You can also search for this author inPubMed Google Scholar - Susan A Slaugenhaupt
You can also search for this author inPubMed Google Scholar - Felicia B Axelrod
You can also search for this author inPubMed Google Scholar
Corresponding author
Correspondence toGabrielle Gold-von Simson.
Additional information
This work was supported, in part, by grant M01 RR-00096 (NYU, Bellevue GCRC) from the General Clinical Research Centers Program of the National Center for Research Resources, National Institutes of Health, by grant R01:36326 (S.A.S.) from the National Institute of Neurological Disorders and Stroke, National Institutes of Health, and also by the Dysautonomia Foundation, New York, NY.
Rights and permissions
About this article
Cite this article
Gold-von Simson, G., Goldberg, J., Rolnitzky, L. et al. Kinetin in Familial Dysautonomia Carriers: Implications for a New Therapeutic Strategy Targeting mRNA Splicing.Pediatr Res 65, 341–346 (2009). https://doi.org/10.1203/PDR.0b013e318194fd52
- Received: 28 July 2008
- Accepted: 01 October 2008
- Issue Date: March 2009
- DOI: https://doi.org/10.1203/PDR.0b013e318194fd52