Nephronophthisis (original) (raw)
Introduction
Nephronophthisis (NPH), an autosomal recessive disorder initially described in 1945 by Smith and Graham and in 1951 by Fanconi, is a chronic tubulointerstitial nephritis that uniformly progresses to end-stage renal disease (ESRD) [1, 2]. With regard to the age of onset for ESRD, three clinical variants have been described: infantile, juvenile, and adolescent forms [3]. Of these, juvenile NPH is the most common, which accounts for 5–10% of cases of ESRD in children. In the past, NPH and medullary cystic kidney disease (MCKD) were considered in the same complex. Whereas these disorders share a number of clinical as well as histological features (tubular basement membrane disintegration, tubular cyst formation, and tubulointerstitial inflammation and fibrosis) [4–6], MCKD is distinct from NPH by its autosomal dominant inheritance and by the late onset of renal failure after the fourth decade of life [7]. In this review, we only consider NPH.
Juvenile nephronophthisis
Juvenile NPH is an uncommon condition that affects girls and boys equally. The incidence is approximately 0.13 for 10,000 live births in Finland, whereas in Canada, it is 1 per 50,000 live births and in United States 9 per 8.3 million [8–10]. The disorder has been reported worldwide. The first symptoms generally develop around 4–6 years of age. Polyuria and polydipsia related to a reduced urinary concentrating ability and loss of sodium conservation occurs early in the course of the disease, whereas glomerular filtration rate (GFR) remains normal [11]. Decreased urinary concentrating defect is demonstrated by a low urinary osmolarity (<400 mosm/kg in the first urine sample in the morning), which does not increase after desmopressin acetate administration [5]. Urinary sodium wasting may be responsible for hyponatremia and hypovolemia in cases of decreased sodium intake. Decreased growth velocity related to chronic dehydration and later to renal insufficiency results in growth retardation. Hematuria and proteinuria are absent or minimal. Blood pressure is normal before the onset of renal failure.
Renal insufficiency is often present when the diagnosis is made. Late symptoms are related to the progressive renal insufficiency and include anemia, metabolic acidosis, nausea, anorexia, and weakness. ESRD develops at a mean age of about 13 years but can also occurs in some rare cases much later during adulthood [12, 13].
Renal ultrasound may be normal, with normal-sized kidneys, but renal parenchymal hyperechogenicity and loss of corticomedullary differentiation are often observed. At later stages, small cysts are present in the medulla [14, 15]. Renal biopsy shows severe tubular damage on light microscopy. Groups of atrophic tubules with thickened basement membranes alternate with groups of dilated or collapsed tubules. Homogeneous or multilayered thickening of tubular basement membranes is prominent, but disintegration of the basement membrane can also occur (Fig. 1). Abrupt transition from one abnormality to another is highly suggestive of juvenile NPH [16]. These various changes in the tubular basement membrane, although nonspecific, occur in NPH more extensively than in any kidney disorders with abnormal tubules. There is moderate to massive interstitial fibrosis with few inflammatory cells. The glomeruli are often normal, although secondary sclerosis is observed in advanced disease.
Fig. 1
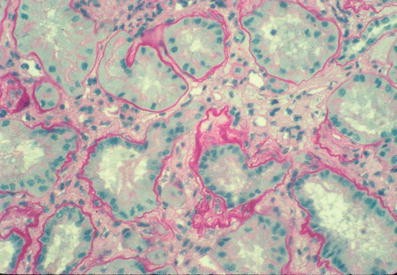
Renal histology of nephronophthisis. Cross-section of kidney showing diffuse interstitial fibrosis and various tubular changes. Some tubules are collapsed, others are surrounded by thickened tubular basement membranes. Note the laminated and wrinkled appearance of some tubular basement membrane segments as well as the abrupt attenuation of others in the same tubule (Light microscopy; magnification ×360; from Marie-Claire Gubler)
Adolescent nephronophthisis
This form of autosomal recessive NPH has been called the adolescent form following the identification of the NPHP3 gene in a large Venezuelan family in which ESRD occurred at a mean age of 19 years [17]. However, there is no clear correlation between the age at ESRD and the genotype, as some patients with an NPHP3 mutation progress to ESRD before 10 years of age, whereas ESRD occurs in adulthood in some patients with NPHP1 deletion. Histological lesions are similar to those observed in juvenile NPH.
Infantile nephronophthisis
A chronic autosomal recessive tubulointerstitial nephritis with cortical microcyts progressing to ESRD before 2 years of age was initially described by Gagnadoux et al. [18]. The disease differs from juvenile NPH not only by its early onset but also by the histopathologic features. Whereas cystic dilatations of the collecting ducts are seen in these patients, the typical changes in the tubular basement membranes seen in juvenile NPH are usually absent. Ultrasonography usually shows moderately enlarged kidneys. Severe hypertension is common.
Associated disorders
In 10–20% of cases, extrarenal symptoms are present, in particular, retinitis pigmentosa (RP) [Senior-Løken syndrome (SLS)], cerebellar ataxia [Joubert syndrome (JS)], oculomotor apraxia type Cogan, mental retardation, bone anomalies and hepatic fibrosis. Situs inversus and ventricular cardiac septal defect are associated in some patients with the infantile form (Table 1).
Table 1 Genetic heterogeneity and overlap of nephronophthisis (NPH), Senior-Løken, Joubert, and Meckel-Gruber syndromes
SLS, in which tapetoretinal degeneration (also known as RP) accompanies juvenile NPH is seen in approximately 10–15% percent of cases. Initially, Senior and Løken described patients with early and severe visual impairment resembling Leber congenital amaurosis [19, 20], but the syndrome has thereafter been extended to all patients with NPH and degenerative retinopathy. In the late-onset form, children may develop night blindness followed by complete visual impairment in the following years. Electroretinogram (ERG) shows complete extinction before RP may be observed by funduscopic examination (Fig. 2). Some patients have only an attenuated ERG, but visual acuity is normal. RP has been observed in association with mutations in most NPHP genes (except NPHP7), but whereas RP is always present and severe in patients with NPHP5 and NPHP6 mutations, the symptoms are in general mild in patients with mutations in the other NPHP genes. Georges et al. reported on four patients, from three different families, with RP responsible for severe visual impairment during childhood who developed chronic interstitial nephritis with histological lesions characteristic of NPH and renal failure only between 42 and 56 years of age [21]. No NPHP1 deletion was found in these patients, but the other genes were not analyzed.
Fig. 2
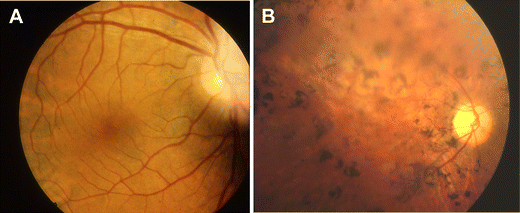
Retinitis pigmentosa. Ophtalmoscopic examinations of a control subject (a) and an affected individual (b) showing typical retinitis pigmentosa fundus characterized by very thin retinal vessels, retinal pigment epithelium atrophy, abnormal pigmentary migrations, and pallor of the optic disk
Nephrocystin proteins encoded by NPHP4, 5, 6, and 8 genes have been shown to localize to the connecting cilia between the inner and outer segments of the photoreceptors. All components necessary for assembly, maintenance, and turnover of the outer segment where the phototransduction takes place are synthesized in the cell body and are transported through the connecting cilia. Proteins implicated in other syndromes with retinal degeneration, such as Bardet-Biedl [22], Alstrom [23] or Usher [24] syndromes, have been shown to be localized to the connecting cilia where they are probably involved in the transport of phototransduction proteins, i.e. rhodopsin. Perturbation of these transporters leads to degeneration of the photoreceptor [25].
JS is an autosomal recessive neurological disorder that associates congenital hypotonia evolving into cerebellar ataxia, developmental delay, oculomotor apraxia, and abnormal breathing pattern during the first month of life. JS is characterized by a complex cerebellar and brainstem malformation, the so-called “molar tooth sign” (MTS) observed by magnetic resonance imaging (MRI) (Fig. 3). JS can be associated with juvenile NPH and/or retinal involvement (JS type B). To date, three gene loci have been mapped to 9q34.3 (JBTS1), 11p11.2-q12.3 (JBTS2), and 6q23 (JBTS3), and mutations in the AHI1 (Abelson helper integration site) gene have been identified in _JBTS3_-linked families with a pure cerebellar phenotype [26–28].
Fig. 3
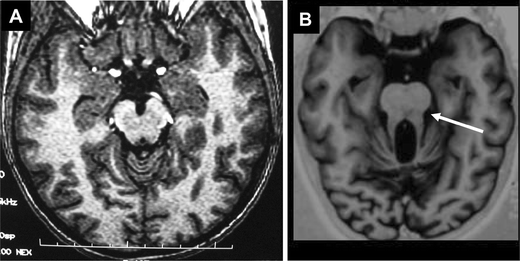
Molar tooth sign on brain magnetic resonance imaging (MRI). Brain MRI axial image at the level of the superior cerebellar peduncles of a control subject (a) and an affected individual (b) showing abnormally increased depth of the interpeduncular fossa, narrowing of the midbrain tegmentum, and thickening of the superior cerebellar peduncles, all of which contribute to the radiologic feature known as the molar tooth sign (white arrow)
NPHP1 homozygous deletions (identical to the deletions observed in patients with isolated NPH) have been identified in a small percentage of patients with a mild neurological form of JS and NPH (and RP in one case) [29, 30]. A recent survey of 56 families with NPHP1 deletion revealed that 5 (8.9%) had JS-related disorders with a variable phenotype. Most of them had no mental retardation, the characteristic MTS aspect was present in only one family, and thickened superior peduncles was the only malformation on brain MRI in two other families, whereas it was unremarkable in one family [29]. Recently, mutations in the NPHP6/CEP290 [31, 32] and NPHP8/RPGRIP1L [33, 34] genes have been found in patients with JS. They were associated with severe RP in the first case, whereas there was no or only mild retinopathy in patients with RPGRIP1L mutations. In a series of 28 patients with NPH and at least one JS-related neurological symptom, Tory et al. found NPHP1 and NPHP6 homozygous or compound heterozygous mutations in 13 (46%) [35].
Meckel-Gruber syndrome (MKS) is an autosomal recessive lethal disorder characterized by central nervous system malformation (typically occipital encephalocele), postaxial polydactyly, cystic kidney dysplasia, and ductal proliferation in the portal area of the liver [34, 36]. Interestingly, mutations in the NPHP6, NPHP8, and MKS3 genes have been found in patients with MKS as well as in patients with JS, suggesting that these two conditions represent a broad spectrum of the same underlying disorder [34, 36, 37]. As with other genes implicated in cystic kidney diseases, most mutated proteins responsible for MKS and JS have been shown to be localized to kidney primary cilia, further suggesting a connection between these syndromes.
Bone anomalies can lead to phalangeal cone-shaped epiphyses, which are usually associated with other extrarenal manifestations (Saldino-Mainzer syndrome) [38]. Other skeletal dysplasia are associated in different syndromes. Hepatic involvement may be characterized by hepatosplenomegaly and portal fibrosis, with no or only mild bile duct proliferation [39–41]. Mutations of the NPHP3 gene were reported in affected members with hepatic fibrosis and NPH from one family [17].
Situs inversus has been reported in a patient with infantile NPH and mutation of the NPHP2 gene [42]. This patient also had a cardiac ventricular septal defect (Table 2).
Table 2 Extrarenal manifestations in nephronophthisis
Several other syndromes that feature NPH have been described, such as Jeune [43], Ellis van Creveld (chondroectodermal dysplasia) [44], RHYNS (retinitis pigmentosa, hypopituitarism, and skeletal dysplasia) [45], Alstrom [46], Sensenbrenner (cranioectodermal dysplasia) [47, 48], and Arima-Dekaban [49] syndromes (Table 3). The description of these rare syndromes is beyond the scope of this paper.
Table 3 Syndromes featuring nephronophthisis or associated with mutations of NPHP genes
Genetics
Positional cloning and candidate-gene approaches led to the identification of causative genes. To date, mutations in eight different genes (NPHP1, 3, 4, 5, 6, 7, 8, and 9) have been identified in juvenile NPH, whereas in the infantile form, mutations have been found in the NPHP2 gene [8] (Fig. 4 and Table 2).
Fig. 4
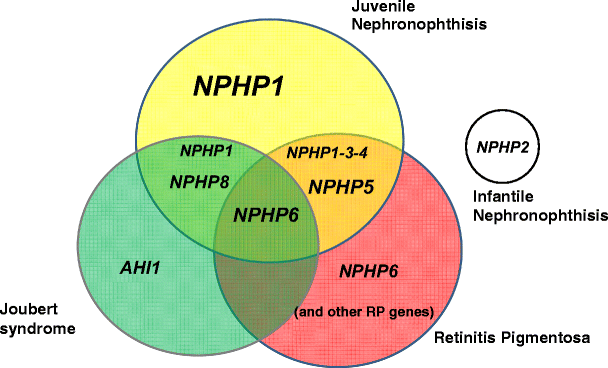
Genes implicated in nephronophthisis, retinitis, and Joubert syndrome. Different associations between these syndromes and the corresponding mutated genes. The most frequently mutated genes are indicated in large characters. NPHP7 and NPHP9 genes are not represented because of their low mutation rate. Extrarenal disorders associated with mutation in the NPHP2 gene are not indicated. Two patients with NPHP8 mutations and mild retinitis have been reported [34, 74]
NPHP1 gene
In 1993, the first gene responsible for juvenile NPH was localized on chromosome 2q13 by positional cloning in consanguineous families [50]. Homozygous deletions of about 250 kb in the 2q13 region were initially detected in 70% of patients [51, 52] and allowed identification of the responsible gene, NPHP1, in 1997. NPHP1 contains 20 exons and encodes for a protein, named nephrocystin or nephrocystin-1, that has an src homology 3 (SH3) and coil-coiled domains that interact with proteins (including products of other NPHP genes) [53, 54]. Nephrocystin and its partners are localized at the cell–cell junction (adherens junction) and at the cell–matrix interface (focal adhesion), suggesting important functions in maintaining tubular epithelium (Fig. 5) [55, 56]. Moreover, nephrocystin-1 is also localized at the primary cilia-like proteins associated with other cystic kidney diseases, such as polycystins. The detection of homozygous mutations by polymerase chain reaction (PCR) amplification permits fast and accurate diagnosis of the disease without the need for renal biopsy. In large series of patients with a presumptive diagnosis of NPH based mainly on clinical and radiological data, NPHP1 homozygous deletion is present in 20–40% of the cases ([8] and personal data). Heterozygous deletions are found in 6% of patients, with concomitant point mutation of the NPHP1 gene on the second allele (personal data). Whereas most patients with NPHP1 deletions or mutations have no extrarenal symptoms, a moderate form of retinal degeneration [57] or JS has been reported in some cases [29, 30, 35, 57].
Fig. 5
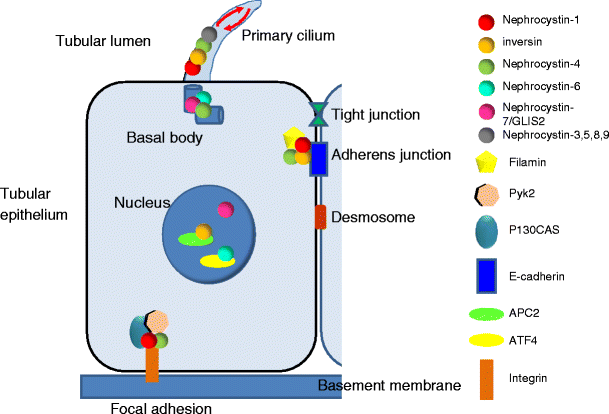
Schematic representation of the tubular epithelium and subcellular localization of the nephrocystin proteins. Nephrocystins localize to different subcellular compartments within the cell in a cell-cycle-dependent manner. Most nephrocystins interact with one another, forming a nephrocystin complex. In polarized renal tubular cells, all nephrocystin proteins localized to the primary cilia at the base of the cilium (basal body) and in a punctate pattern along the ciliary axoneme, suggesting their transport along the microtubule system (red arrows). Nephrocystin-1, nephrocystin-4, and inversin also localize to the cell–cell junctions and interact with focal adhesion proteins (p130Cas, Pyk2). Nephrocystins also associate with proteins associated directly with the microtubular and actin cytoskeleton (tubulins, tensin, and filamin). These localizations suggest a role for nephrocystins in modulating the cytoskeleton and maintaining epithelial-cell polarity. During cell cycle, nephrocystins localize to the centrosome. Moreover, during cell division, inversin, nephrocystin-4 and nephrocystin-6 localize to the mitotic spindle. In addition, inversin and nephrocystin-6 bind the anaphase-promoting complex (APC2) and activating transcription factor (ATF4), respectively, suggesting their potential involvement in cell-cycle regulation. The transcription factor nephrocystin-7/GLIS2 localized to both nucleus and primary cilia, as did the other NPHP proteins in renal epithelial cells
NPHP2 gene
Mutations in the NPHP2 (INVS) gene are responsible for the infantile form of NPH. This gene, located on chromosome 9q31, encodes inversin, a protein that is critical for normal left–right patterning in the vertebrate embryo [42, 58]. In proximal tubular cells, inversin is associated with nephrocystin-1 and with components of microtubule cytoskeleton [59]. It is localized to different subcellular compartments, including nuclei, cytoplasm, and cell–cell junction [60]. Its interaction with APC2 suggests that inversin might play a role during the cell cycle [61]. Knockout mice for the INVS gene show large renal cysts, altered left–right laterality (situs inversus), and hepatobiliary-duct malformations.
NPHP3 gene
NPHP3 located on chromosome 3q22 was initially mapped in a large family of NPH patients from Venezuela [62]. Mutations in NPHP3 were described in families with renal disease alone, as well as in families with renal disease associated with hepatic fibrosis or retinal degeneration [17]. Age at onset of ESRD was highly variable in patients with NPHP3 mutations from 4 to 37 years of age. Recently, two NPH patients with ESRD at 4 years of age were found to carry NPHP3 mutations [63]. In another recently published by Bergmann et al., NPHP3 mutations were found in patients with a broad clinical spectrum of early embryonic patterning defects comprising situs inversus, polydactyly, central nervous system malformations, structural heart defects, preauricular fistulas and multicystic kidneys with perinatal death in some cases [64]. NPHP3 encodes a 1,330 amino acid protein with a tubulin-tyrosine ligase domain that interacts with nephrocystin. Interestingly, the pcy mouse, a spontaneously occurring renal cystic disease model that closely resembles NPH, harbors a homozygous missense mutation in the mouse NPHP3 ortholog (Nphp3) that most likely causes the kidney phenotype [17]. Recent observations that the _pcy_-associated renal cystic disease is amenable to treatment with a vasopressin-2 receptor antagonist [65] opens new perspectives for potential therapeutic strategies in NPH, for which no effective treatment is available.
NPHP4 gene
The NPHP4 gene located on chromosome 1p36 encodes a 1,426 amino acid protein called nephrocystin-4/nephroretinin [66]. Nephrocystin-4 interacts with nephrocystin-1 and is probably involved in the same intracellular signaling pathway [66]. Interestingly, nephrocystin-4 is conserved in the nematode Caenorhabditis elegans, which exhibits male mating phenotype defect upon Nphp4 knockdown, a phenotype also observed with orthologs of polycystic kidney disease genes. Direct sequencing of the NPHP4 gene has been performed in 250 individuals, 190 with isolated NPH, 50 with RP and ten with oculomotor apraxia [67]. Twenty-three different NPHP4 mutations were found in 26 (10%) unrelated patients (13 with isolated NPH, eight with RP, and two with oculomotor apraxia). Of note, nephrocystin-4 localizes to the connecting cilium of photoreceptor cells and interacts with RPGRIP1 [RP guanosine riphosphatase (GTPase) regulator interacting protein 1], a component of cone and rod photoreceptors that is mutated in patients with Leber amaurosis [68].
NPHP5 gene
In contrast to the previous NPHP genes, mutations in the IQCB1 gene, now referred to as NPHP5, were reported only in patients with NPH in combination with severe retinal degeneration and early blindness—SLS [25]. NPHP5 mutations, involving both alleles in all cases, were found in 16 of 92 patients with early onset RP [25]. NPHP5 encodes an IQ-domain protein called IQCB1 or nephrocystin-5 that is expressed in connecting cilia of photoreceptors, where it is associated with calmodulin and retinitis pigmentosa GTPase regulator (RPGR). Nephrocystin-5 is also present in the primary cilia of renal epithelial cells [25].
NPHP6 gene
The NPHP6 gene, also known as CEP290, encodes a centrosomal protein that activates ATF4, a transcription factor involved in the control of the cell cycle. Thirteen different mutations in the NPHP6 gene were initially reported in 12 families with JS [31]. Most of the patients had congenital blindness or severe visual defect in the first years of life. In two families (patients aged 3, 9.5, 10, and 15 years), there was no renal disease. Interestingly, the NPHP6 gene was thereafter reported to be mutated in more than 20% of patients with severe congenital blindness but no renal involvement (Leber congenital amaurosis) [69, 70]. A hypomorphic mutation (c.2991+1655A>G) that creates a strong splice-donor site and inserts a cryptic exon in the CEP290 ribonucleic acid (RNA) was detected in 16 (21%) of 76 unrelated patients with blindness but without clinical signs of renal disease. Moreover, these patients had no neurological symptoms typical of JS and had normal cognitive function [69]. Another group has confirmed that mutations in the NPHP6 gene is the most common cause of Leber amaurosis [70]. Given the broad spectrum of the phenotype associated with NPHP6 mutations, Helou et al. screened this gene in 99 patients with cerebellar ataxia (JS), 75 patients with RP, and 21 patients with isolated NPH and found mutations in seven, two, and one case, respectively [71]. In four patients, only single heterozygous mutations were found, and in one of them, an additional heterozygous NPHP4 missense mutation was present, arguing for a digenic inheritance. A genome-wide linkage scan in families with MKS led to the identification of NPHP6 mutations in some patients [36].
NPHP7 gene
The NPHP7 gene, also known as the GLIS2 gene, contains six coding exons and encodes a Kruppel-like zinc-finger transcription factor, which has been found mutated in one consanguineous Oji-Cree Canadian family with isolated NPH in three children who developed ESRD by 8 years of age. This gene seems to be very rarely involved, as no other mutation was found in a cohort of 470 individuals with NPH-like phenotypes [72]. The kidneys of mice with a targeted disruption of the Glis2 gene are atrophic, with fibrosis starting at 8 weeks of age. Apoptosis is increased in renal tubular cells, whereas cell proliferation is not. Interestingly, the genes promoting epithelial–mesenchymal transition and fibrosis are up-regulated in the absence of Glis2.
NPHP8 gene
Mutations in a novel gene on chromosome 16, RPGRIP1L, have been found in patients with JS and in fetuses with MKS [34]. Interestingly, Delous et al. reported that MKS fetuses carried two truncating mutations, whereas JS patients carried missense mutations and/or one truncating mutation, suggesting a genotype–phenotype correlation [34]. RPGRIP1L is a cytosolic protein that colocalizes at the basal bodies, centrosomes, and primary cilia in renal tubular cells with nephrocystin-4 and nephrocystin-6 [34]. Interestingly, missense mutations found in JS patients decrease the interaction of RPGRIP1L with nephrocystin-4 without affecting its localization, suggesting that a defect in this association may contribute to the phenotype [33, 34]. In the same lines, NPHP4 missense mutations known to cause NPH with RP also disrupt this interaction [33]. Recently, it was shown that Rpgrip1l participates in sonic hedgehog (Shh) signaling and by this means plays a critical role in patterning of the developing neural tube and limb through the cilium [73]. The RPGRIP1L gene was thereafter analyzed in a cohort of 56 patients with JS. RPGRIP1L mutations were identified in five kindreds, including six individuals (8%). Of note, patients with RPGRIP1L mutations had normal retina, except two patients out of 12 with moderate visual impairment [34, 74]. Additional clinical symptoms were present in some patients with RPGRIP1L mutations, such as liver fibrosis, postaxial polydactyly, pituitary agenesis, and partial growth hormone deficiency. These last findings indicate a possible overlap with RHYNS syndrome [34, 74].
NPHP9
The jck cystic kidney mouse model is associated with a mutation in the Nek8 gene [75]. Analysis of the NEK8 gene in a cohort of 588 patients with NPH led to the identification of three missense mutations in patients with isolated NPH [76]. In one patient, an additional mutation in the NPHP5 gene was also present. Interestingly, mutant forms of NEK8 showed defects in ciliary localization.
Genetic heterogeneity and oligogenism
Overall, NPHP1 to NPHP9 mutations have been reported in cases of juvenile NPH with or without extrarenal symptoms, except for mutations in NPHP2 that have been found only in patients with infantile NPH. NPHP1 mutations were found in ~20% to 40% of cases [8], whereas mutations in the other genes seem to account for a very low percentage of cases (Table 1). Analysis of the NPHP1, NPHP3, and NPHP4 genes in a cohort of 94 different families have shown that a mutation or a deletion in one of these three genes was identified in 44 (47%) patients [77]. Interestingly, in six families, three mutations in two NPHP genes were found, whereas two mutations in two NPHP genes (NPHP3 and NPHP4) were found in another kindred. Finally, a single mutation in one of these three genes was discovered in nine other patients [77]. In the same lines, Tory et al. found that some patients with NPH and at least one JS-related neurological symptom had both an NPHP1 deletion and either a heterozygous NPHP6 or AHI1 mutation [35]. These findings demonstrate that similar to the inheritance patterns described in Bardet-Biedl syndrome (BBS), NPH, at least in some patients, follows a digenic or oligogenic inheritance with heterozygous mutations in different genes in the same patients. Sequencing of all the known NPH genes in a large cohort of patients will be necessary to validate this model and appreciate the mutation load that could account for the severity of the nephropathy as well as the extrarenal symptoms.
The cilia connection
Cilia are present in almost all cells in the organism and act as sensory organelles that connect visual, mechanosensory, odorant or other stimuli to cell-cycle, control of epithelial architecture, or other yet unknown processes. The finding that polycystin-1 and polycystin-2—the proteins responsible for autosomal dominant polycystic kidney disease [78] and proteins involved in other cystic kidney diseases (BBS [22], oro-facio-digital syndrome [79])—were present in cilia in renal tubular cells indicates a possible connection between this organelle and cyst formation. It has been suggested that the primary cilium senses fluid movement in the renal tubules. The presence of cystoproteins (such as nephrocystins) in cilia of various organs (photoreceptor, ependymal cells, cholangiocytes, chondrocytes) explains the multiple organ involvement in some patients with NPH. To illustrate this unifying theory, the term ciliopathies has been coined to design all the syndromes related to dysfunction of ciliary proteins. For more information on this subject, the readers are referred to an excellent recent review [80].
Conclusions
In conclusion, and from a practical point of view, the diagnosis of NPH should be considered if a child presents with polyuria, urinary sodium loss, growth failure, renal insufficiency without hematuria or proteinuria, normal blood pressure, and normal-sized kidneys without dilatation of the urinary tract. These patients should be screened for homozygous or heterozygous NPHP1 deletion, which is found in 20–40% of cases. In the absence of such deletion, renal biopsy may be proposed to confirm the diagnosis. At present, screening for mutation in all the other NPHP genes is not routinely performed due to the low frequency of detected mutations and the high cost of the procedure. Patients with associated disorders should be considered separately. In case of severe RP, the NPHP5 gene may be screened for mutations, whereas in patients with neurological symptoms such as cerebellar ataxia, the NPHP6 and NPHP8 genes should be analyzed first. In case of early onset tubulointerstitial nephritis with cortical microcysts, the NPHP2 gene should be screened for mutation (Fig. 6).
Fig. 6
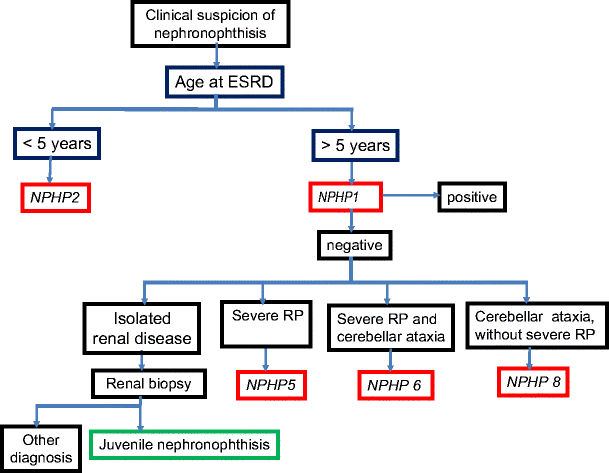
Decision algorithm for genetic analyses when nephronophthisis is suspected on clinical and radiological basis. At first, NPHP1 or NPHP2 genes should be screened for mutations, depending on the age at onset of end-stage renal disease. The other genes are analyzed in function of extrarenal symptoms. NPHP3, NPHP4, NPHP7, and NPHP9 genes are currently not sequenced for diagnostic purposes because of their low mutation rate. RP retinitis pigmentosa
References
- Fanconi G, Hanhart E, von Albertini A, Uhlinger E, Dolivo G, Prader A (1951) Familial, juvenile nephronophthisis (idiopathic parenchymal contracted kidney). Helv Paediatr Acta 6:1–49
CAS PubMed Google Scholar - Smith C, Graham J (1945) Congenital medullary cysts with severe refractory anemia. Am J Dis Child 69:369–377
Google Scholar - Saunier S, Salomon R, Antignac C (2005) Nephronophthisis. Curr Opin Genet Dev 15:324–331
CAS PubMed Google Scholar - Caridi G, Dagnino M, Gusmano R, Ginevri F, Murer L, Ghio L, Piaggio G, Ciardi MR, Perfumo F, Ghiggeri GM (2000) Clinical and molecular heterogeneity of juvenile nephronophthisis in Italy: insights from molecular screening. Am J Kidney Dis 35:44–51
CAS PubMed Google Scholar - Hildebrandt F, Waldherr R, Kutt R, Brandis M (1992) The nephronophthisis complex: clinical and genetic aspects. Clin Investig 70:802–808
CAS PubMed Google Scholar - Waldherr R, Lennert T, Weber HP, Fodisch HJ, Scharer K (1982) The nephronophthisis complex. A clinicopathologic study in children. Virchows Arch A Pathol Anat Histol 394:235–225
CAS PubMed Google Scholar - Stavrou C, Koptides M, Tombazos C, Psara E, Patsias C, Zouvani I, Kyriacou K, Hildebrandt F, Christofides T, Pierides A, Deltas CC (2002) Autosomal-dominant medullary cystic kidney disease type 1: clinical and molecular findings in six large Cypriot families. Kidney Int 62:1385–1394
PubMed Google Scholar - Hildebrandt F, Zhou W (2007) Nephronophthisis-associated ciliopathies. J Am Soc Nephrol 18:1855–1871
CAS PubMed Google Scholar - Pistor K, Olbing H, Scharer K (1985) Children with chronic renal failure in the Federal Republic of Germany: I. Epidemiology, modes of treatment, survival. Arbeitsgemeinschaft fur Pädiatrische Nephrologie. Clin Nephrol 23:272–277
CAS PubMed Google Scholar - Potter DE, Holliday MA, Piel CF, Feduska NJ, Belzer FO, Salvatierra O Jr (1980) Treatment of end-stage renal disease in children: a 15-year experience. Kidney Int 18:103–109
CAS PubMed Google Scholar - Gusmano R, Ghiggeri GM, Caridi G (1998) Nephronophthisis-medullary cystic disease: clinical and genetic aspects. J Nephrol 11:224–228
CAS PubMed Google Scholar - Bollee G, Fakhouri F, Karras A, Noel LH, Salomon R, Servais A, Lesavre P, Moriniere V, Antignac C, Hummel A (2006) Nephronophthisis related to homozygous NPHP1 gene deletion as a cause of chronic renal failure in adults. Nephrol Dial Transplant 21:2660–2663
PubMed Google Scholar - Hildebrandt F, Strahm B, Nothwang HG, Gretz N, Schnieders B, Singh-Sawhney I, Kutt R, Vollmer M, Brandis M (1997) Molecular genetic identification of families with juvenile nephronophthisis type 1: rate of progression to renal failure. APN Study Group. Arbeitsgemeinschaft fur Pädiatrische Nephrologie. Kidney Int 51:261–269
CAS PubMed Google Scholar - Aguilera A, Rivera M, Gallego N, Nogueira J, Ortuno J (1997) Sonographic appearance of the juvenile nephronophthisis-cystic renal medulla complex. Nephrol Dial Transplant 12:625–626
CAS PubMed Google Scholar - Blowey DL, Querfeld U, Geary D, Warady BA, Alon U (1996) Ultrasound findings in juvenile nephronophthisis. Pediatr Nephrol 10:22–24
CAS PubMed Google Scholar - Zollinger HU, Mihatsch MJ, Edefonti A, Gaboardi F, Imbasciati E, Lennert T (1980) Nephronophthisis (medullary cystic disease of the kidney). A study using electron microscopy, immunofluorescence, and a review of the morphological findings. Helv Paediatr Acta 35:509–530
CAS PubMed Google Scholar - Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, Wolf MT, Sasmaz G, Trauer U, Reinhardt R, Sudbrak R, Antignac C, Gretz N, Walz G, Schermer B, Benzing T, Hildebrandt F, Omran H (2003) Mutations in a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet 34:455–459
CAS PubMed Google Scholar - Gagnadoux MF, Bacri JL, Broyer M, Habib R (1989) Infantile chronic tubulo-interstitial nephritis with cortical microcysts: variant of nephronophthisis or new disease entity? Pediatr Nephrol 3:50–55
CAS PubMed Google Scholar - Løken AC, Hanssen O, Halvorsen S, Jolster NJ (1961) Hereditary renal dysplasia and blindness. Acta Paediatr 50:177–184
PubMed Google Scholar - Senior B, Friedmann AI, Braudo JL (1961) Juvenile familial nephropathy with tapetoretinal degeneration. A new oculorenal dystrophy. Am J Ophthalmol 52:625–633
CAS PubMed Google Scholar - Georges B, Cosyns JP, Dahan K, Snyers B, Carlier B, Loute G, Pirson Y (2000) Late-onset renal failure in Senior-Loken syndrome. Am J Kidney Dis 36:1271–1275
CAS PubMed Google Scholar - Tobin JL, Beales PL (2007) Bardet-Biedl syndrome: beyond the cilium. Pediatr Nephrol 22:926–936
PubMed PubMed Central Google Scholar - Marshall JD, Hinman EG, Collin GB, Beck S, Cerqueira R, Maffei P, Milan G, Zhang W, Wilson DI, Hearn T, Tavares P, Vettor R, Veronese C, Martin M, So WV, Nishina PM, Naggert JK (2007) Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alstrom syndrome. Hum Mutat 28:1114–1123
CAS PubMed Google Scholar - Liu X, Bulgakov OV, Darrow KN, Pawlyk B, Adamian M, Liberman MC, Li T (2007) Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc Natl Acad Sci USA 104:4413–4418
CAS PubMed PubMed Central Google Scholar - Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, Muerb U, O’Toole JF, Helou J, Attanasio M, Utsch B, Sayer JA, Lillo C, Jimeno D, Coucke P, De Paepe A, Reinhardt R, Klages S, Tsuda M, Kawakami I, Kusakabe T, Omran H, Imm A, Tippens M, Raymond PA, Hill J, Beales P, He S, Kispert A, Margolis B, Williams DS, Swaroop A, Hildebrandt F (2005) Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet 37:282–288
CAS PubMed Google Scholar - Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, Gleeson JG (2004) Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet 75:979–987
CAS PubMed PubMed Central Google Scholar - Lagier-Tourenne C, Boltshauser E, Breivik N, Gribaa M, Betard C, Barbot C, Koenig M (2004) Homozygosity mapping of a third Joubert syndrome locus to 6q23. J Med Genet 41:273–277
CAS PubMed PubMed Central Google Scholar - Utsch B, Sayer JA, Attanasio M, Pereira RR, Eccles M, Hennies HC, Otto EA, Hildebrandt F (2006) Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol 21:32–35
PubMed Google Scholar - Caridi G, Dagnino M, Rossi A, Valente EM, Bertini E, Fazzi E, Emma F, Murer L, Verrina E, Ghiggeri GM (2006) Nephronophthisis type 1 deletion syndrome with neurological symptoms: prevalence and significance of the association. Kidney Int 70:1342–1347
CAS PubMed Google Scholar - Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DW, McDonald R, Eddy A, Chance PF, Glass IA (2004) The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet 75:82–91
CAS PubMed PubMed Central Google Scholar - Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, Utsch B, Khanna H, Liu Y, Drummond I, Kawakami I, Kusakabe T, Tsuda M, Ma L, Lee H, Larson RG, Allen SJ, Wilkinson CJ, Nigg EA, Shou C, Lillo C, Williams DS, Hoppe B, Kemper MJ, Neuhaus T, Parisi MA, Glass IA, Petry M, Kispert A, Gloy J, Ganner A, Walz G, Zhu X, Goldman D, Nurnberg P, Swaroop A, Leroux MR, Hildebrandt F (2006) The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet 38:674–681
CAS PubMed Google Scholar - Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, Fazzi E, Signorini S, Louie CM, Bellacchio E, Bertini E, Dallapiccola B, Gleeson JG (2006) Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet 38:623–625
CAS PubMed Google Scholar - Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Marker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R (2007) Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet 39:882–888
CAS PubMed Google Scholar - Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, Gubler MC, Martinovic J, Encha-Razavi F, Boddaert N, Gonzales M, Macher MA, Nivet H, Champion G, Bertheleme JP, Niaudet P, McDonald F, Hildebrandt F, Johnson CA, Vekemans M, Antignac C, Ruther U, Schneider-Maunoury S, Attie-Bitach T, Saunier S (2007) The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet 39:875–881
CAS PubMed Google Scholar - Tory K, Lacoste T, Burglen L, Moriniere V, Boddaert N, Macher MA, Llanas B, Nivet H, Bensman A, Niaudet P, Antignac C, Salomon R, Saunier S (2007) High NPHP1 and NPHP6 mutation rate in patients with Joubert syndrome and nephronophthisis: potential epistatic effect of NPHP6 and AHI1 mutations in patients with NPHP1 mutations. J Am Soc Nephrol 18:1566–1575
CAS PubMed Google Scholar - Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, Munnich A, Lyonnet S, Salomon R, Encha-Razavi F, Gubler MC, Boddaert N, de Lonlay P, Johnson CA, Vekemans M, Antignac C, Attie-Bitach T (2007) The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet 80:186–194
CAS PubMed Google Scholar - Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC, Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, Rattenberry E, Esculpavit C, Toutain A, Moraine C, Parent P, Marcorelles P, Dauge MC, Roume J, Le Merrer M, Meiner V, Meir K, Menez F, Beaufrere AM, Francannet C, Tantau J, Sinico M, Dumez Y, MacDonald F, Munnich A, Lyonnet S, Gubler MC, Genin E, Johnson CA, Vekemans M, Encha-Razavi F, Attie-Bitach T (2007) Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet 81:170–179
CAS PubMed PubMed Central Google Scholar - Mainzer F, Saldino RM, Ozonoff MB, Minagi H (1970) Familial nephropathy associated with retinitis pigmentosa, cerebellar ataxia and skeletal abnormalities. Am J Med 49:556–562
CAS PubMed Google Scholar - Boichis H, Passwell J, David R, Miller H (1973) Congenital hepatic fibrosis and nephronophthisis. A family study. Q J Med 42:221–233
CAS PubMed Google Scholar - Delaney V, Mullaney J, Bourke E (1978) Juvenile nephronophthisis, congenital hepatic fibrosis and retinal hypoplasia in twins. Q J Med 47:281–290
CAS PubMed Google Scholar - Witzleben CL, Sharp AR (1982) “Nephronophthisis-congenital hepatic fibrosis”: an additional hepatorenal disorder. Hum Pathol 13:728–733
CAS PubMed Google Scholar - Otto EA, Schermer B, Obara T, O’Toole JF, Hiller KS, Mueller AM, Ruf RG, Hoefele J, Beekmann F, Landau D, Foreman JW, Goodship JA, Strachan T, Kispert A, Wolf MT, Gagnadoux MF, Nivet H, Antignac C, Walz G, Drummond IA, Benzing T, Hildebrandt F (2003) Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet 34:413–420
CAS PubMed PubMed Central Google Scholar - Donaldson MD, Warner AA, Trompeter RS, Haycock GB, Chantler C (1985) Familial juvenile nephronophthisis, Jeune’s syndrome, and associated disorders. Arch Dis Child 60:426–434
CAS PubMed PubMed Central Google Scholar - Moudgil A, Bagga A, Kamil ES, Rimoin DL, Lachman RS, Cohen AH, Jordan SC (1998) Nephronophthisis associated with Ellis-van Creveld syndrome. Pediatr Nephrol 12:20–22
CAS PubMed Google Scholar - Di Rocco M, Picco P, Arslanian A, Restagno G, Perfumo F, Buoncompagni A, Gattorno M, Borrone C (1997) Retinitis pigmentosa, hypopituitarism, nephronophthisis, and mild skeletal dysplasia (RHYNS): a new syndrome? Am J Med Genet 73:1–4
PubMed Google Scholar - Marshall JD, Bronson RT, Collin GB, Nordstrom AD, Maffei P, Paisey RB, Carey C, Macdermott S, Russell-Eggitt I, Shea SE, Davis J, Beck S, Shatirishvili G, Mihai CM, Hoeltzenbein M, Pozzan GB, Hopkinson I, Sicolo N, Naggert JK, Nishina PM (2005) New Alstrom syndrome phenotypes based on the evaluation of 182 cases. Arch Intern Med 165:675–683
PubMed Google Scholar - Costet C, Betis F, Berard E, Tsimaratos M, Sigaudy S, Antignac C, Gastaud P (2000) Pigmentosum retinis and tubulo-interstitial nephronophtisis in Sensenbrenner syndrome: a case report. J Fr Ophtalmol 23:158–160
CAS PubMed Google Scholar - Tsimaratos M, Berard E, Sigaudy S, Almahana T, Delarue A, Roquelaure B, Costet C, Antignac C, Gubler MC, Picon G, Philip N, Sarles J (1997) Chronic renal failure and cranioectodermal dysplasia: a further step. Pediatr Nephrol 11:785–786
CAS PubMed Google Scholar - Matsuzaka T, Sakuragawa N, Nakayama H, Sugai K, Kohno Y, Arima M (1986) Cerebro-oculo-hepato-renal syndrome (Arima’ syndrome): a distinct clinicopathological entity. J Child Neurol 1:338–346
CAS PubMed Google Scholar - Antignac C, Arduy CH, Beckmann JS, Benessy F, Gros F, Medhioub M, Hildebrandt F, Dufier JL, Kleinknecht C, Broyer M, Weissenbach J, Habib R, Cohen D (1993) A gene for familial juvenile nephronophthisis (recessive medullary cystic kidney disease) maps to chromosome 2p. Nat Genet 3:342–345
CAS PubMed Google Scholar - Konrad M, Saunier S, Heidet L, Silbermann F, Benessy F, Calado J, Le Paslier D, Broyer M, Gubler MC, Antignac C (1996) Large homozygous deletions of the 2q13 region are a major cause of juvenile nephronophthisis. Hum Mol Genet 5:367–371
CAS PubMed Google Scholar - Saunier S, Calado J, Benessy F, Silbermann F, Heilig R, Weissenbach J, Antignac C (2000) Characterization of the NPHP1 locus: mutational mechanism involved in deletions in familial juvenile nephronophthisis. Am J Hum Genet 66:778–789
CAS PubMed PubMed Central Google Scholar - Hildebrandt F, Otto E, Rensing C, Nothwang HG, Vollmer M, Adolphs J, Hanusch H, Brandis M (1997) A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat Genet 17:149–153
CAS PubMed Google Scholar - Saunier S, Calado J, Heilig R, Silbermann F, Benessy F, Morin G, Konrad M, Broyer M, Gubler MC, Weissenbach J, Antignac C (1997) A novel gene that encodes a protein with a putative src homology 3 domain is a candidate gene for familial juvenile nephronophthisis. Hum Mol Genet 6:2317–2323
CAS PubMed Google Scholar - Donaldson JC, Dise RS, Ritchie MD, Hanks SK (2002) Nephrocystin-conserved domains involved in targeting to epithelial cell-cell junctions, interaction with filamins, and establishing cell polarity. J Biol Chem 277:29028–29035
CAS PubMed Google Scholar - Mollet G, Silbermann F, Delous M, Salomon R, Antignac C, Saunier S (2005) Characterization of the nephrocystin/nephrocystin-4 complex and subcellular localization of nephrocystin-4 to primary cilia and centrosomes. Hum Mol Genet 14:645–656
CAS PubMed Google Scholar - Betz R, Rensing C, Otto E, Mincheva A, Zehnder D, Lichter P, Hildebrandt F (2000) Children with ocular motor apraxia type Cogan carry deletions in the gene (NPHP1) for juvenile nephronophthisis. J Pediatr 136:828–831
CAS PubMed Google Scholar - Eley L, Turnpenny L, Yates LM, Craighead AS, Morgan D, Whistler C, Goodship JA, Strachan T (2004) A perspective on inversin. Cell Biol Int 28:119–124
CAS PubMed Google Scholar - Nurnberger J, Kribben A, Opazo Saez A, Heusch G, Philipp T, Phillips CL (2004) The Invs gene encodes a microtubule-associated protein. J Am Soc Nephrol 15:1700–1710
PubMed Google Scholar - Nurnberger J, Kavapurackal R, Zhang SJ, Opazo Saez A, Heusch G, Philipp T, Pietruck F, Kribben A (2006) Differential tissue distribution of the Invs gene product inversin. Cell Tissue Res 323:147–155
PubMed Google Scholar - Morgan D, Eley L, Sayer J, Strachan T, Yates LM, Craighead AS, Goodship JA (2002) Expression analyses and interaction with the anaphase promoting complex protein Apc2 suggest a role for inversin in primary cilia and involvement in the cell cycle. Hum Mol Genet 11:3345–3350
CAS PubMed Google Scholar - Omran H, Fernandez C, Jung M, Haffner K, Fargier B, Villaquiran A, Waldherr R, Gretz N, Brandis M, Ruschendorf F, Reis A, Hildebrandt F (2000) Identification of a new gene locus for adolescent nephronophthisis, on chromosome 3q22 in a large Venezuelan pedigree. Am J Hum Genet 66:118–127
CAS PubMed Google Scholar - Otto EA, Helou J, Allen SJ, O'Toole JF, Wise EL, Ashraf S, Attanasio M, Zhou W, Wolf MT, Hildebrandt F (2008) Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Hum Mutat 29:418–426
Google Scholar - Bergmann C, Fliegauf M, Brüchle NO, Frank V, Olbrich H, Kirschner J, Schermer B, Schmedding I, Kispert A, Kränzlin B, Nürnberg G, Becker C, Grimm T, Girschick G, Lynch SA, Kelehan P, Senderek J, Neuhaus TJ, Stallmach T, Zentgraf H, Nürnberg P, Gretz N, Lo C, Lienkamp S, Schäfer T, Walz G, Benzing T, Zerres K, Omran H (2008) Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet 82:959-970
CAS PubMed PubMed Central Google Scholar - Gattone VH 2nd, Wang X, Harris PC, Torres VE (2003) Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med 9:1323–1326
CAS PubMed Google Scholar - Mollet G, Salomon R, Gribouval O, Silbermann F, Bacq D, Landthaler G, Milford D, Nayir A, Rizzoni G, Antignac C, Saunier S (2002) The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat Genet 32:300–305
CAS PubMed Google Scholar - Hoefele J, Sudbrak R, Reinhardt R, Lehrack S, Hennig S, Imm A, Muerb U, Utsch B, Attanasio M, O’Toole JF, Otto E, Hildebrandt F (2005) Mutational analysis of the NPHP4 gene in 250 patients with nephronophthisis. Hum Mutat 25:411
PubMed Google Scholar - Roepman R, Letteboer SJ, Arts HH, van Beersum SE, Lu X, Krieger E, Ferreira PA, Cremers FP (2005) Interaction of nephrocystin-4 and RPGRIP1 is disrupted by nephronophthisis or Leber congenital amaurosis-associated mutations. Proc Natl Acad Sci USA 102:18520–18525
CAS PubMed PubMed Central Google Scholar - den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, Hoyng CB, van den Born LI, Rohrschneider K, Cremers FP (2006) Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet 79:556–561
CAS PubMed PubMed Central Google Scholar - Perrault I, Delphin N, Hanein S, Gerber S, Dufier JL, Roche O, Defoort-Dhellemmes S, Dollfus H, Fazzi E, Munnich A, Kaplan J, Rozet JM (2007) Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat 28:416
PubMed Google Scholar - Helou J, Otto EA, Attanasio M, Allen SJ, Parisi MA, Glass I, Utsch B, Hashmi S, Fazzi E, Omran H, O’Toole JF, Sayer JA, Hildebrandt F (2007) Mutation analysis of NPHP6/CEP290 in patients with Joubert syndrome and Senior-Loken syndrome. J Med Genet 44:657–663
CAS PubMed PubMed Central Google Scholar - Attanasio M, Uhlenhaut NH, Sousa VH, O’Toole JF, Otto E, Anlag K, Klugmann C, Treier AC, Helou J, Sayer JA, Seelow D, Nurnberg G, Becker C, Chudley AE, Nurnberg P, Hildebrandt F, Treier M (2007) Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet 39:1018–1024
CAS PubMed Google Scholar - Vierkotten J, Dildrop R, Peters T, Wang B, Ruther U (2007) Ftm is a novel basal body protein of cilia involved in Shh signalling. Development 134:2569–2577
CAS PubMed Google Scholar - Wolf MT, Saunier S, O’Toole JF, Wanner N, Groshong T, Attanasio M, Salomon R, Stallmach T, Sayer JA, Waldherr R, Griebel M, Oh J, Neuhaus TJ, Josefiak U, Antignac C, Otto EA, Hildebrandt F (2007) Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int 72:1520–1526
CAS PubMed Google Scholar - Liu S, Lu W, Obara T, Kuida S, Lehoczky J, Dewar K, Drummond IA, Beier DR (2002) A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development 129:5839–5846
CAS PubMed Google Scholar - Otto EA, Trapp ML, Schultheiss UT, Helou J, Quarmby LM, Hildebrandt F (2008) NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J Am Soc Nephrol 19:587–592
CAS PubMed PubMed Central Google Scholar - Hoefele J, Wolf MT, O’Toole JF, Otto EA, Schultheiss U, Deschenes G, Attanasio M, Utsch B, Antignac C, Hildebrandt F (2007) Evidence of oligogenic inheritance in nephronophthisis. J Am Soc Nephrol 18:2789–2795
CAS PubMed Google Scholar - Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet 33:129–137
CAS PubMed Google Scholar - Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA (2004) OFD1 is a centrosomal/basal body protein expressed during mesenchymal-epithelial transition in human nephrogenesis. J Am Soc Nephrol 15:2556–2568
CAS PubMed Google Scholar - Fliegauf M, Benzing T, Omran H (2007) When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Bio 8:880–893
CAS Google Scholar