Peutz-Jeghers Syndrome: Overview, Genetics, Epidemiology (original) (raw)
Overview
Peutz-Jeghers syndrome (PJS) is an autosomal dominant inherited disorder characterized by intestinal hamartomatous polyps in association with a distinct pattern of skin and mucosal macular melanin deposition. [1, 2] Patients with Peutz-Jeghers syndrome have an estimated 15-fold increased risk of developing intestinal cancer compared to the general population. [3, 4, 5, 6, 7, 8]
See the images below.
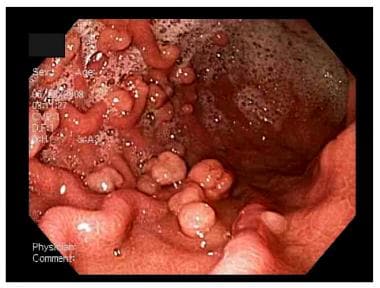
Peutz-Jeghers Syndrome. This upper endoscopy image shows multiple gastric polyps.
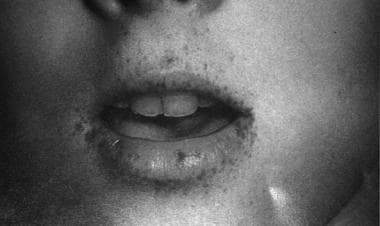
Peutz-Jeghers Syndrome. This is a facial photograph of a patient with Peutz-Jeghers syndrome. Note the mucocutaneous pigmentation that crosses the vermilion border.
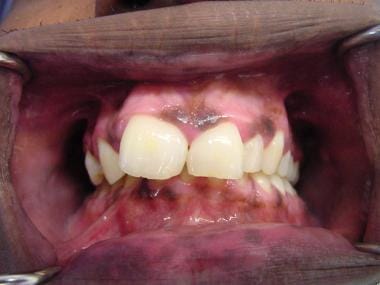
Peutz-Jeghers Syndrome. This photograph shows an oral pigmented lesion in a patient with Peutz-Jeghers syndrome.
See Clues in the Oral Cavity: Are You Missing the Diagnosis?, a Critical Images slideshow, to help identify the causes of abnormalities of the oral cavity.
The gastrointestinal polyps found in Peutz-Jeghers syndrome are typical hamartomas. Their histology is characterized by extensive smooth muscle arborization throughout the polyp. [2, 9] This may give the lesion the appearance of pseudoinvasion, because some of the epithelial cells, usually from benign glands, are surrounded by the smooth muscle (the lack of dysplasia in the polyps help to differentiate pseudoinvasion from malignancy). [2, 9]
Cancer develops in the gastrointestinal tract of patients with Peutz-Jeghers syndrome with a higher frequency than it does in the general population. [1, 7] However, this syndrome is also associated with increased breast, gynecologic, testicular, pancreatic, and thyroid papillary malignancy. [1, 2, 10] About 48% of patients with Peutz-Jeghers syndrome develop and die from cancer by age 57 years. Others may have a normal life span. The mean age at first diagnosis of cancer is 42.9 years, ± 10.2 years. [11]
During the first 3 decades of life, anemia, rectal bleeding, abdominal pain, obstruction, and/or intussusception are common complications in patients with Peutz-Jeghers syndrome. [12, 13] Nearly 50% of the patients experience an intussusception during their lifetime, most commonly in the small intestine. [14]
See the image below.
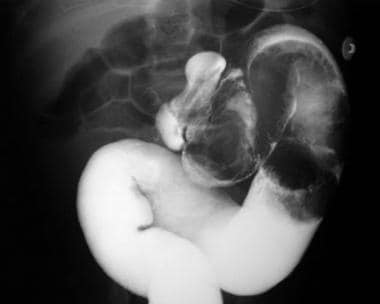
Peutz-Jeghers Syndrome. This barium enema radiograph reveals intussusception in the descending colon.
Peutz-Jeghers syndrome should be diagnosed in patients as early as possible, and genetic counseling should also be provided. [2] Many of the gastrointestinal lesions start developing early in life, even if the syndrome is not clinically apparent until the second and third decades of life. Proper screening for intestinal cancers and extraintestinal cancers should be implemented. [2, 15] (See Approach Considerations.)
Historical information
The syndrome was described in 1921 by Jan Peutz (1886-1957), a Dutch physician who noted a relationship between the intestinal polyps and the mucocutaneous macules in a Dutch family. [16] The dermatologic component had previously been reported by John McHutchinson in 1896 in identical twins, one of who subsequently died from intussusception.
Harold Jeghers (1904-1990), an American physician, is credited with the definitive descriptive reports of the syndrome when he published "Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits," in 1949, with McKusick and Katz. [17] The eponym Peutz-Jeghers syndrome was introduced by the radiologist Andre J Bruwer in 1954. [18]
![]()
Genetics
The cause of Peutz-Jeghers syndrome (PJS) in most cases (> 90%) appears to be a germline mutation of the STK11/LKB1 (serine/threonine kinase 11) tumor suppressor gene, [2, 15] located on chromosome 19p13. [1, 19]
STK11 is a tumor suppressor gene, in that its overexpression can induce a growth arrest of a cell at the G1 phase of the cell cycle and that somatic inactivation of the unaffected allele of STK11 is often observed in polyps and cancers from patients with Peutz-Jeghers syndrome.
STK11/LKB1 encodes a 433 amino acid ubiquitously expressed protein with a central catalytic domain and regulatory N- and C-terminal domains. The biologic function of LKB1 includes the regulation of downstream kinases, including adenosine monophosphate–activated protein kinase (AMPK) and the related kinases (microtube affinity-regulating kinase [MARK] 1 through MARK4 and brain-specific kinase/synapses of the amphid-defective kinase [Brsk/SAD]), which are involved in cellular metabolic regulation–stress response and cellular polarity, the latter through tubulin stabilization, tight junction formation, and E-cadherin localization. See the figure below.
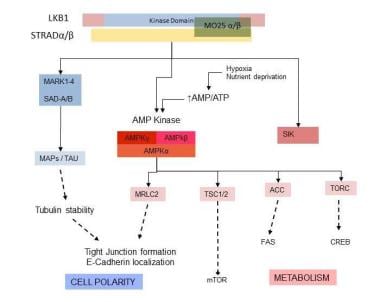
Peutz-Jeghers Syndrome. This diagram illustrates the role of STK11/LKB1 in neoplasia: regulation of cell polarity and metabolism. ACC = acetyl-CoA carboxylase; AMP/ATP = adenosine monophosphate/adenosine triphosphate; AMPK = AMP-activated protein kinase; CREB = CRE-binding protein; FAS = fatty acid synthase; MAPs = microtubule associated proteins; MARK = microtube affinity-regulating kinases; MO25 = calcium-binding protein 39; MRLC = myosin regulatory light chain; mTOR = mammalian target of rapamycin; SAD = synapses of the amphid-defective kinase; SIK = salt-induced kinase; STRAD = STE20-related kinase adaptor; TAU = tau protein; TORC = TOR complex; TSC 1/2 = tuberous sclerosis proteins 1 and 2.
There is evidence of interaction between the LKB1 pathway along with other tumor suppressor pathways p53 [20] and phosphatase and tensin homologue (PTEN). Abrogation of LKB1 function results in polyposis along with loss of heterozygosity, probably a separate process, resulting in tumorigenesis.
Penetrance of the gene mutation is variable, resulting in a spectrum of phenotypic manifestations among patients with Peutz-Jeghers syndrome (eg, inconsistent number, localization of polyps, differing presentation of the macules) and allowing for a variable presentation of cancer. [8, 21, 22, 23, 24, 25]
Data on the impact of the LKB1 mutation type and localization on disease expression are conflicting. It is believed that truncating variants in STK11 predispose to a more severe phenotype, and phenotype severity is based on an earlier onset of gastrointestinal pathology arising from the polyps (eg, intussusception , earlier onset malignancy). [1] However, a consensus does not yet exist regarding phenotype severity based on variant location.
Schumacher and colleagues reported a higher risk of malignancy with missense mutations involving the C-terminus or exons encoding for protein domains involved in substrate recognition. [26, 27] Another report described a worse prognosis with greater polyp burden and higher risk of malignancy in individuals harboring a truncating mutation of LKB, [28] whereas a different group failed to correlate the risk of (polyp-associated) intussusception with mutational characteristics. Overall opinion is divided on the usefulness of genotype-phenotype correlations in Peutz-Jeghers syndrome, and they are not, at present, routinely used in defining prognosis and management of the disease.
Mutation in the MYH11 gene may be implicated in a minority of patients without the LKB1 gene mutation. Hyperactivation of mammalian target of rapamycin (mTOR) signaling has also been associated with Peutz-Jeghers syndrome. [25]
Other genes may also play a role in Peutz-Jeghers syndrome, such as those that encode for the MARK protein, homologues of the Par 1 polarity protein that associates with LKB1. However, de Leng et al performed direct sequencing and probe amplification in 23 families with Peutz-Jeghers syndrome and were unable to identify any mutations in the MARK genes. [23] This again supports the evidence that LKB1 defects remain the major cause of Peutz-Jeghers syndrome and, although other mechanisms are involved, they remain to be elucidated. [24]
An interesting study by Tobi et al demonstrated that Adnab-9, a premalignant marker found in Paneth cells, was more common in patients with Peutz-Jeghers syndrome. [29] The authors evaluated 8 patients with Peutz-Jeghers syndrome, 8 patients with juvenile polyposis, and 36 hyperplastic polyp sections (as control subjects). The investigators found that 89% of Peutz-Jeghers syndrome polyps were labeled with Adnab-9, compared with 88% of familial juvenile polyposis sections and 11% of hyperplastic polyps. [29] This study suggested Adnab-9 labeling may identify polyps at higher risk of malignant degeneration.
Mehenni et al, reporting on the molecular and clinical characteristics of 46 families with Peutz-Jeghers syndrome, demonstrated an increase in the mutational spectrum of LKB1/STK11 allelic variants worldwide. They suggested that this new information would be helpful for clinical diagnosis and genetic counseling. [22]
Novel de novo germline mutations associated with Peutz-Jeghers syndrome and STK11 continue to be discovered. Using Sanger sequencing, Zhao et al identified a c.962_963delCC mutation in exon 8 in a Chinese patient with isolated Peutz-Jeghers syndrome who died of colon cancer. [30] This mutation caused a frameshift mutation and a premature termination at codon 358. Neither of the patient's parents nor 50 control subjects had this mutation. Similarly, in a separate report, the same investigators identified a 23-nucleotide deletion (c.426-448delCGTGCCGGAGAAGCGTTTCCCAG) in exon 3 of STK11 that caused a change of 13 codons and a truncating protein (p.S142SfsX13) in another Chinese patient. None of this patient's healthy family members nor 100 control subjects exhibited the mutation. [31]
Chiang and Chen used genomic DNA to amplify and analyze the entire sequence of STK11 in 15 Taiwanese patients with Peutz-Jeghers syndrome from 11 unrelated families and found 5 novel mutations in 8 families (exon 6, c.843 ins G; exon 8, c.2065 delete A; exon 8, c.G923A, nonsense; exon 6, c.748dupA; and mTOR c.5107dupA) in addition to three known mutations. [32] Two thirds (n = 10) of the patients developed malignancies, all diagnosed before age 40 years; half (n = 5) died of their cancers. Three families without detectable STK11 mutations had not developed neoplasms by the time of the report.
Jang et al reported the case of a 14-year-old Korean male with Peutz-Jeghers syndrome who had complete STK11 deletion and atypical symptoms. [33] The use of multiplex ligation-dependent probe amplification (MLPA) rather than direct sequencing revealed heterozygous deletions spanning exons 1-10. It was unclear whether his atypical symptoms of developmental delay, intellectual disability, and epilepsy without tuberous sclerosis were related to Peutz-Jeghers syndrome or to another cause given his apparently healthy parents and a sibling who did not exhibit any STK11 deletions. [33]
![]()
Epidemiology
Peutz-Jeghers syndrome (PJS) is rare in the United States, with a frequency of encounter from polyposis registries one tenth that of familial adenomatous polyposis. This would place the frequency between 1 case per 60,000 people and 1 case per 300,000 people.
The international frequency of Peutz-Jeghers syndrome is unknown, but given the rarity of this condition, the frequency is probably similar to that reported in the United States. Specific mutations of the STK11/LKB1 gene, however, may be more common in certain ethnic groups. For example, Zuo et al reported two new mutations in three Chinese families that had not been previously described in White families, [12, 13, 34, 35, 36] Zhao et al found one novel mutation each in two Chinese patients, [30, 31] Chiang and Chen discovered five new mutations in eight unrelated Taiwanese families, [32] and Jang et al reported complete STK11 deletion in a Korean patient. [33]
Age-related demographics
Polyps found in Peutz-Jeghers syndrome commonly present in adolescence and early adulthood. [37] One third of the affected individuals experience symptoms during the first 10 years of life. [17] The median time of first presentation with polyps is age 11-13 years; approximately 50% of individuals have experienced symptoms by age 20 years. [13]
![]()
Cancer in Peutz-Jeghers Syndrome
Peutz-Jeghers syndrome (PJS) entails a significant overall increased lifetime risk of intestinal and extraintestinal malignancy (see the Table below; see also the Approach Considerations section), as well as an increased risk of malignancy in younger individuals. The cancer risk increases with age: 1-2% risk by age 20 years, over 30% by age 50 years, and more than 80% by age 70 years. [2] Small bowel adenocarcinoma has been diagnosed in a child as young as 13 years, [38] with patients aged through 30 years harboring a 5% cumulative risk of cancer that rises to 85% by age 70 years. The overall relative risk for cancer is greater in females than in males, and it is greatest for gastrointestinal, pancreatic, and gynecologic-cervical cancers. [2, 39]
The increased risk for malignancies was also documented in another study. A systematic review that included 20 studies and 1 meta-analysis, comprising 1644 patients found 21% (n = 349) of patients developed 384 malignancies. [40] The average age for malignancy was 24 years. The most common malignancy was colorectal cancer, followed by breast and small bowel cancer as well as other less common gastrointestinal malignancies. [40]
Patients with Peutz-Jeghers syndrome appear to have an increased risk for pancreatic-biliary cancer. [41] In a study by Korsse et al that included 144 patients with Peutz-Jeghers syndrome from 61 families, 7 patients (5%) developed pancreatic cancer (median age, 54 years), and 2 patients (3%) each developed distal bile duct cancer or ampullary cancer (median age, 55 years). There was a 26% cumulative risk and 76 relative risk for pancreatic cancer at age 70 years; the cumulative risk for pancreatic-biliary cancer was 32% at age 70 years, with a relative risk of 96 (P< 0.001). [41]
Table. Risk Ratio Compared to the General Population, Lifetime Frequency, Mean Age, and Age Range of Malignancies Related to Peutz-Jeghers Syndrome [4] (Open Table in a new window)
| Organ | Risk Ratio | Frequency, % | Mean Age (Range), Years |
|---|---|---|---|
| Esophagus | 57 | 0.5 | 67 |
| Stomach | 213 | 29 | 30.1 (10-61) |
| Small Intestine | 520 | 13 | 41.7 (21-84) |
| Colon | 84 | 39 | 45.8 (27-71) |
| Pancreas | 132 | 36 | 40.8 (16-60) |
| Lung | 17 | 15 | |
| Testis | 4.5 | 9 | 8.6 (3-20) |
| Breast | 15.2 | 54 | 37 (9-48) |
| Uterus | 16 | 9 | |
| Ovary | 27 | 21 | 28 (4-57) |
| Cervix | 1.5 | 10 | 34.3 (23-54) |
In addition, other reproductive site cancers have been associated with Peutz-Jeghers syndrome, including adenoma malignum of the cervix, Sertoli cell tumors, and sex cord tumors with annular tubules. [42, 43]
Breast cancer in patients with Peutz-Jeghers syndrome significant increase, 8% at age 40 and 31% at age 60, [43] and it may also be bilateral. [6] Owing to the increased risk of pancreatic adenocarcinoma in Peutz-Jeghers syndrome, screening with endoscopic ultrasonography has emerged as a relatively new tool for early diagnosis. [44, 45]
More recent data show the following percentage lifetime risk for cancers by site in individuals with Peutz-Jeghers syndrome [15] :
- Stomach: 29%
- Small intestine: 13%
- Colon: 39%
- Pancreas: 11-36%
- Lung: 15-17%
- Breast: 45-50%
- Uterus: 9%
- Ovary: 18-21%
- Cervix: 10%
Thyroid papillary malignancy [1] as well as sinonasal adenocarcinoma [46] have also been reported in patients with Peutz-Jeghers syndrome.
![]()
Additional Complications
Gastrointestinal complications
In a series of 222 patients with Peutz-Jeghers syndrome (PJS), Utsunomiya et al noted the following distribution of presenting gastrointestinal symptoms [11] :
- Abdominal pain caused by infarction: 23% of patients
- Rectal bleeding caused by ulceration: 13.5% of patients
- Extrusion of polyp: 7% of patients
Gastric outlet obstruction can be an early presenting complaint in Peutz-Jeghers syndrome, including in the neonatal period. [47] Recurrent abdominal pain is often reported with increased frequency and intensity, as small intestinal polyp growth evolves into subtotal obstruction. Gastrointestinal hemorrhage may not be apparent and may present as iron deficiency anemia.
Polyps
Polyps in the Utsunomiya et al study patients occurred as follows:
- Small intestine: 64% of patients
- Colon: 63.2% of patients
- Stomach: 48.6% of patients
- Rectum: 32% of patients
The incidence of polyps within the small intestine is greatest in the jejunum and progressively decreases in the ileum and duodenum. [13]
Other rare reported complications include gastrointestinal obstruction and vomiting secondary to gastric polyps. [11, 48] Extraintestinal polyps are also reported, although they are rare; they include nasal polyps, [49] gall bladder polyps, [50] ureteric polyps, [51] and respiratory tract polyps. [52]
The principal causes of morbidity in Peutz-Jeghers syndrome are owing to the intestinal location of the polyps (ie, small intestine, colon, stomach). Morbidity includes small intestinal obstruction and intussusception (43%), abdominal pain (23%), hematochezia (14%), and prolapse of a colonic polyp (7%) [53] ; these typically occur in the second and third decades of life.
Intestinal obstruction can occur in about 50% of patients, and it is usually localized in the small bowel. Obstruction can be complete or incomplete and is caused by the polyp itself or by the subsequent intussusception that may occur, as seen in the images below.
Peutz-Jeghers Syndrome. This barium enema radiograph reveals intussusception in the descending colon.
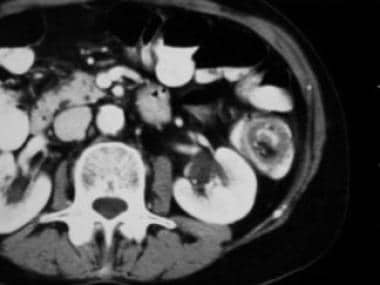
Peutz-Jeghers Syndrome. The CT scan demonstrates the classic yin-yang sign of an intussusceptum inside an intussuscipiens.
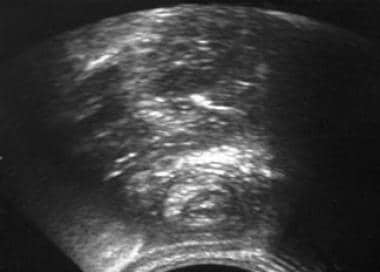
Peutz-Jeghers Syndrome. Abdominal ultrasonography reveals the classic target sign of an intussusceptum inside an intussuscipiens.
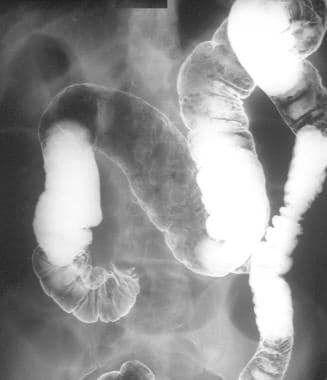
Peutz-Jeghers Syndrome. This postevacuation image is from part of a double-contrast barium enema study in a 47-year-old man presenting with features of small-bowel obstruction. The image shows a coiled-spring appearance in the region of the cecum suggestive of an intussusception. At laparotomy, an ileocecal intussusception was found in association with a carcinoid tumor of the terminal ileum.
In a retrospective study that examined the incidence of sporadic Peutz-Jeghers syndrome polyps over a 22-year period, Burkhart et al reported that although sporadic Peutz-Jeghers syndrome polyps are very rare, individuals who may have even a single Peutz-Jeghers syndrome polyp may have an accumulative lifetime risk of cancer that is similar to those with the syndrome. [54]
![]()
Patient Education
Patients with Peutz-Jeghers syndrome (PJS) should be educated about the potential symptoms of intestinal obstruction as well as instructed on the need for cancer surveillance.
In the wake of the discovery of the genetic cause(s) of Peutz-Jeghers syndrome, genetic counseling by someone knowledgeable in this disease should be provided if genetic testing is being considered, particularly as high penetrance autosomal dominant alleles have been described. Genetic counseling relays the risks, benefits, and consequences of genetic testing to the patient. It also informs the patient of the consequences of a positive, negative, or inconclusive genetic test result. In addition, genetic counseling includes information on the risk of transmission to offspring, and it may provide guidance for the patient regarding testing of other family members. [8, 55]
![]()
Patient History
Peutz-Jeghers syndrome (PJS) is characterized by the combination of pigmented lesions in the buccal mucosa and gastrointestinal polyps. The number, as well as the size and the location, of the polyps may vary from patient to patient. Isolated melanotic mucocutaneous pigmentation without gastrointestinal polyps has also been described, because of the genetic variability of the syndrome. Pigmented lesions on the extremities may fade, in time and by adulthood, although buccal mucosal lesions tend to persist. [2, 16, 17, 9]
Well-established clinical diagnostic criteria are noted for Peutz-Jeghers syndrome. One set includes the following elements: three histopathologically proven Peutz-Jeghers syndrome polyps with the classic mucocutaneous pigmentation and a positive family history. [56]
The diagnostic criteria for Peutz-Jeghers syndrome proposed by the Johns Hopkins Registry include histopathologically verified hamartomatous polyps with at least two of the following [57] :
- Small-bowel location for polyposis
- Mucocutaneous melanotic pigmentation
- Family history of Peutz-Jeghers syndrome [3]
World Health Organization (WHO) diagnostic criteria includes [58] :
- Three or more histologically confirmed Peutz-Jeghers polyps, or
- Any number of Peutz-Jeghers polyps with a family history of Peutz-Jeghers syndrome, or
- Characteristic, prominent, mucocutaneous pigmentation with a family history of Peutz-Jeghers syndrome, or
- Any number of Peutz-Jeghers polyps and characteristic, prominent, mucocutaneous pigmentation
Patient characteristics noted in the Peutz-Jeghers syndrome include [12, 13] :
- Family history of Peutz-Jeghers syndrome
- Repeated bouts of abdominal pain in patients younger than 25 years
- Unexplained intestinal bleeding in a young patient
- Prolapse of tissue from the rectum
- Menstrual irregularities in females (due to hyperestrogenism from sex cord tumors with annular tubules)
- Gynecomastia in males (possible due to the production of estrogens from Sertoli cell testicular tumors) [2, 59]
- Precocious puberty
- Gastrointestinal intussusception with bowel obstruction
- Mucocutaneous pigmentation, which may have faded
- Melena or rectal bleeding
- Hematemesis
- Weakness due to anemia
The most frequent symptom is recurrent colicky abdominal pain caused by obstruction and transient intussusceptions. [60] Melena and rectal bleeding occur less frequently, and hematemesis is uncommon. [61] Certain reports suggest that nearly 50% of the patients experience an intussusception during their lifetime, most often in the small intestine. [14]
Protein-losing enteropathy should be considered as a possible clinical manifestation or presentation of significant gastrointestinal, especially colorectal, polyp burden.
Hyperpigmented mucocutaneous macules may be present on the lips and buccal mucosa and around the mouth, eyes, and nostrils, as well as sparsely on the fingers, soles of the feet, palms, anal area, and intestinal mucosa. [2, 9] The macules are flat, blue-gray to brown-black spots 2-4 mm in size. They tend to develop by age 5 years but are rarely present at birth. Some may fade during the onset of puberty, but buccal mucosa lesions tend to persist. [2, 16, 17, 9, 62, 63]
See the images below.
Peutz-Jeghers Syndrome. This is a facial photograph of a patient with Peutz-Jeghers syndrome. Note the mucocutaneous pigmentation that crosses the vermilion border.
Peutz-Jeghers Syndrome. This photograph shows an oral pigmented lesion in a patient with Peutz-Jeghers syndrome.
![]()
Physical Examination
Mucocutaneous pigmentation and melanin spots (1-5–mm macules) are typical of patients with Peutz-Jeghers syndrome (PJS), and they are present in more than 95% of cases. They appear as small, flat, brown or dark-blue spots similar to freckles, most commonly around the mouth crossing the vermilion border (94%), nostrils, perianal area, digits, and the dorsal and volar aspects of hands and feet (62-74%). They may fade after puberty but tend to persist in the buccal mucosa. [2, 9, 17, 16, 62, 63]
Localization in the oral mucosa is typical of patients with Peutz-Jeghers syndrome and does not happen with other types of dermatologic pigmented lesions, such as common lentigo (see image below). Freckles do not localize in the buccal mucosa. An important differential diagnosis for freckling and pigmented macules involving the glans penis is PTEN-hamartoma syndrome, which is a distinct, rare, hereditary intestinal polyposis and cancer-predisposing condition.
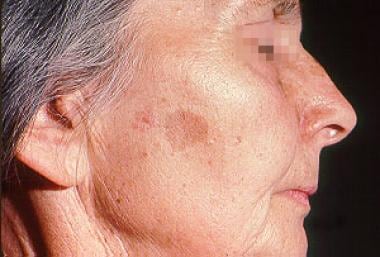
Peutz-Jeghers Syndrome. This woman has solar lentigo.
A rectal mass (rectal polyp) may be found during a rectal examination. In rare cases (7%), the polyp can prolapse outside the anus if it reaches a significant size.
Gynecomastia and growth acceleration (due to Sertoli cell tumor), as well as testicular mass, may also be noted in Peutz-Jeghers syndrome. [54, 64, 65]
![]()
Approach Considerations
2018 NCCN guidelines on genetic/familial high-risk assessment of colorectal syndromes
The National Comprehensive Cancer Network (NCCN) updated their guidelines for assessment of genetic/familial high-risk colorectal syndromes (nonpolyposis and polyposis), including Peutz-Jeghers syndrome (PJS), in July 2018. [15] Their recommendations align with those of the 2015 American College of Gastroenterology (ACG) on genetic testing and management of hereditary gastrointestinal cancer syndromes. [9]
Diagnosis
Two or more of the following features must be present for the clinical diagnosis of Peutz-Jeghers syndrome: at least two Peutz-Jeghers-type hamartomatous polyps of the gastrointestinal (GI) tract; mucocutaneous hyperpigmentation affecting the eyes, nose, mouth/lips, fingers, or genitals; a family history of Peutz-Jeghers syndrome.
Surveillance
Clinical genetic testing is available to test for mutations in the STK11 (LKB1) gene, which causes most cases of Peutz-Jeghers syndrome.
The NCCN recommends referrals to expert teams and encourages patient participation in clinical trials.
Perform a thorough evaluation for any early symptoms. Recommendations regarding approximate ages for initiation of surveillance in asymptomatic patients include:
- Breast cancer: About age 25 years
- Colon or stomach cancer: In the late teens
- Small intestinal cancer: About age 8-10 years
- Pancreatic cancer: About age 30-35 years, or 10 years younger than the earliest age of familial onset
- Ovarian/cervical/uterine cancer: About age 18-20 years
- Testicular (sex cord/Sertoli cell tumors): About age 10 years
2015 ACG guidelines on genetic testing and management of hereditary gastrointestinal cancer syndromes
The ACG released the following recommendations for the management of patients with hereditary gastrointestinal cancer syndromes—and they specifically discuss genetic testing and management of Lynch syndrome, familial adenomatous polyposis (FAP), attenuated familial adenomatous polyposis (AFAP), _MUTYH_-associated polyposis (MAP), Peutz-Jeghers syndrome (PJS), juvenile polyposis syndrome (JPS), Cowden syndrome, serrated (hyperplastic) polyposis syndrome, hereditary pancreatic cancer, and hereditary gastric cancer [9] :
- The initial assessment is the collection of a family history of cancers and premalignant gastrointestinal conditions and should provide enough information to develop a preliminary determination of the risk of a familial predisposition to cancer.
- Age at diagnosis and lineage (maternal and/or paternal) should be documented for all diagnoses, especially in first- and second-degree relatives.
- When indicated, genetic testing for a germline mutation should be done on the most informative candidate(s) identified through the family history evaluation and/or tumor analysis to confirm a diagnosis and allow for predictive testing of at-risk relatives.
- Genetic testing (including testing for STK11 mutations) should be conducted in the context of pretest and posttest genetic counseling to ensure the patient's informed decision making.
- Patients who meet clinical criteria for a syndrome as well as those with identified pathogenic germline mutations should receive appropriate surveillance measures in order to minimize their overall risk of developing syndrome-specific cancers (colon, stomach, small bowel, pancreas, breast, ovary, uterus, cervix, and testes. The risk for lung cancer is higher, but no specific recommendation has been made; therefore, consider obtaining an annual chest x-ray or CT scan in smokers.)
Laboratory Studies
Laboratory studies include [9] :
- Complete blood cell (CBC) count: The polyps of Peutz-Jeghers syndrome may ulcerate and be a source of blood loss and anemia; gastrointestinal bleeding may be massive but also microscopic, with subsequent iron deficiency, therefore, cell counts and iron studies should be monitored.
- Iron studies
- Fecal occult blood: Hemoccult should be performed to check for occult blood in the stool
- Carcinoembryonic antigen (CEA): This test has been used by some clinicians for screening and monitoring of cancer degeneration.
- Cancer antigen (CA)–19-9 and CA-125: CA-125 testing is indicated every year starting at age 18 years, and CA 19-9 is indicated every 1-2 years starting at age 25 years [57]
Genetic testing is available; however, not all families with Peutz-Jeghers syndrome map to the STK11/LKB1 locus. [21, 22, 66] Thus, a negative genetic test does not exclude the diagnosis.
![]()
Imaging Studies
Imaging studies in Peutz-Jeghers syndrome (PJS) include:
- Upper gastrointestinal endoscopy
- Capsule endoscopy
- MRI enteroclysis
- CT scanning with oral contrast medium
- CT enterography
- Colonoscopy
- Push enteroscopy
- Intraoperative enteroscopy (IOE)
- Double-balloon enteroscopy (DBE)
- Endoscopic ultrasonography (EUS)
- Endoscopic retrograde cholangiopancreatography (ERCP) [67]
- Chest radiography or CT scanning in smokers [9]
Enteroclysis and dedicated small-bowel follow-through radiographs have traditionally been used to determine the presence and the location of small intestinal polyps in individuals with Peutz-Jeghers syndrome. CT enterography is accurate at detecting small polyps, especially those larger than 1 cm in diameter. [9] Capsule endoscopy appears to be safe and as sensitive as barium enterography in the detection of significant small intestinal polyps in children with Peutz-Jeghers syndrome. [68] MRI enterography has emerged as potentially superior to capsule endoscopy in the assessment of polyp size in adult patients with Peutz-Jeghers syndrome. [69]
Imaging studies of the liver and the pancreas are indicated because of the risk of pancreatic cancer as well as of gallbladder polyps and cancer. These imaging studies may include ultrasonography and CT scanning with pancreatic details or magnetic resonance cholangiopancreatography (MRCP).
Endoscopy and wireless capsule endoscopy (WCE)
Symptoms such as abdominal pain or anemia/melena require investigation with upper and lower endoscopy. Traditionally, primary surgical resection and IOE were the only available options for treating polyps in the mid small bowel in patients with Peutz-Jeghers syndrome [70, 71, 72] ; this strategy did not allow for a preventive approach to recurrent small intestinal obstruction. WCE may improve the likelihood of detection of small intestinal polyps [73] ; however, definitive management may, given the availability of suitable expertise, depend on advanced enteroscopic techniques, including push-enteroscopy, spiral enteroscopy, and double-balloon enteroscopy with polypectomy. [74]
In a 2017 report, Belsha et al demonstrated in a small number of cases that DBE-facilitated polypectomy is an effective therapeutic option with minimal complications. [75]
Small intestinal polyps may be removed as part of a “clean-sweep” strategy for disease surveillance or because they are resulting in symptoms, including bleeding.
![]()
Histologic Findings
Polyps in Peutz-Jeghers syndrome (PJS) can reliably be differentiated from other types of polyps only by histopathology, [76] and surgical pathologists can have a critical role in recommending genetic counseling and surveillance of the patient and/or family members when the diagnosis is suspected. [77]
Smooth-muscle hyperplasia, with an elongated, arborized pattern of polyp formation towards the epithelial layer, can be seen. [9, 12, 55] This results in the formation of islands of epithelium within the underlying smooth muscle, [60] which is best appreciated in Peutz-Jeghers syndrome small intestine polyps. [54] Other distinguishing morphology includes an expanded edematous lamina propria that is chronically inflamed with dilated cystic glands filled with deeply eosinophilic mucin. [78] Thus, the polyps generally originate from the small or large bowel, are often exophytic, seldom erode, have inflamed edematous and fibrotic lamina propria and dilated mucin-filled cystic glands, and show often widespread smooth muscle proliferation. [78]
It is recommended that all cervical cytology samples from patients with Peutz-Jeghers syndrome be examined with a high level of suspicion for cervical adenocarcinoma. [79] In addition, complete surgical excision following a diagnosis of endocervical glandular hyperplasia is also recommended. [79]
See the image below.
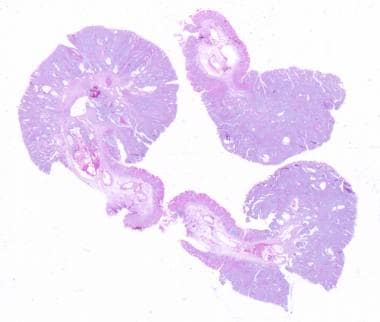
Peutz-Jeghers Syndrome. Intestinal polypoid adenomas. Tubular adenoma, low-power view. Courtesy of George H Warren, MD†.
![]()
Surgical Care
Patients with Peutz-Jeghers syndrome (PJS) usually undergo numerous surgeries during their lives. These surgeries include laparotomies and laparoscopies for both gastrointestinal involvement and extraintestinal complications. Surgical treatment of extraintestinal cancers detected through surveillance procedures is frequently required.
Laparotomy and resection may be necessary, as indicated, for small intestinal intussusception, obstruction, or persistent intestinal bleeding. Laparoscopic-assisted enteroscopy may offer a less invasive option for polyp removal.
Complications
Adhesions and intestinal obstruction or short-bowel syndrome from repeated abdominal surgeries can be limited with the use of endoscopic methods for intestinal polyp resection, such as intraoperative endoscopy and push enteroscopy.
In a study consisting of 10 children with Peutz-Jeghers syndrome, Bizzarri et al reported that single-balloon enteroscopy is effective for treating small bowel polyps in this population. [80] The investigators suggested that well-timed polypectomy may prevent polyp-related complications and the need for emergency laparotomy.
Diet
The diet is unrestricted, except for those patients who have surgical resections and or strictures, wherein avoiding food with kernels (eg, popcorn) is indicated.
![]()
Pharmacologic Management
Chemopreventive strategies for familial adenomatous polyposis (FAP) syndrome management has led to investigation into cyclooxygenase (COX) inhibitors for Peutz-Jeghers syndrome (PJS). Rossi et al demonstrated that COX-2 was highly upregulated in a murine model of Peutz-Jeghers syndrome LKB1 mutant mice. [81] Polyps recovered from patients with Peutz-Jeghers syndrome also showed a significant correlation between LKB1 staining and COX-2, suggesting COX-2 is integral to the tumorigenesis pathway in Peutz-Jeghers syndrome. [82]
A decrease in polyp burden was reported in COX-2 knockout LKB-1 mutant mice, analogous to LKB-1 mutant mice treated with celecoxib. In that report of an uncontrolled, open-labeled pilot study in humans, Udd et al noted reduced gastric polyposis in patients treated with celecoxib. [83] Celecoxib use in Peutz-Jeghers syndrome, although promising, remains to be tested and currently cannot be routinely recommended in any age group.
Given that modulation of PI3-kinase is critical to the function of STK11 and, in turn, one of the major downstream mediators of PI3-kinase signaling is mammalian target of rapamycin (mTOR), inhibition of mTOR offers potential therapeutic possibilities in chemoprevention in Peutz-Jeghers syndrome. Rapamycin has been shown to be effective in reducing polyp burden in a murine model of Peutz-Jeghers syndrome. [84] In addition, RAD001 (everolimus) has been proposed as a potential chemopreventive agent and was reportedly effective in achieving a partial remission in a patient with Peutz-Jeghers syndrome with advanced pancreatic cancer. [85] Currently, the use of mTOR inhibitors is also not recommended as standard of care in adult and pediatric patients with Peutz-Jeghers syndrome.
![]()
Consultations
Follow-up care of patients with Peutz-Jeghers syndrome (PJS) should be supervised by a primary physician familiar with this condition. Liaison with a tertiary center or registry-based team facilitates care. Gastroenterology specialist involvement, genetic consultation and counseling, as well as urologic and gynecologic consultations, are required in the management of these patients.
Psychologist referral and psychiatric consultation should be borne in mind as patients with Peutz-Jeghers syndrome are more likely to suffer from mild depression, experience a poorer mental quality of life, and are susceptible to more limitations in daily functioning owing to emotional problems and a general health perception compared with the general population. [86, 87, 88]
![]()
Patient Monitoring
Ideally, patients with Peutz-Jeghers syndrome (PJS) should be followed by a multidisciplinary team that is familiar with the syndrome. The aim of the initial consultation and continued follow-up is to educate the patient and family on the illness, outline a schema for continued disease surveillance, offer genetic counseling and, if appropriate, offer genetic testing to the extended family. Support, including identification of psychologically at-risk individuals, needs to be factored in this holistic management approach. [89] Counseling and testing of asymptomatic but at-risk individuals is directed toward limiting the likelihood of patients presenting with complications, including malignancy, inherent to their disease, as well as providing advice regarding potential preventive strategies, including cancer-surveillance measures.
Periodic surveillance and removal of larger polyps aims to reduce the likelihood of complications in Peutz-Jeghers syndrome. Hence, surveillance for gastric and small-bowel polyposis should begin at age 8-10 years and continue at 2-3–year intervals. [2, 9, 90] When small bowel polyps are present, there is broad consensus amongst quaternary referral centers that they be removed before symptoms and obstruction become evident. A regular surveillance-based, clean-sweep enteroscopy (double-balloon or intraoperative assisted and push enteroscopy) is suggested to reduce the risk of obstruction, surgical resection and, long term, the risk of short bowel syndrome. [91, 92, 93]
Follow-up care should be supervised by a gastroenterologist familiar with Peutz-Jeghers syndrome. Patients should undergo an annual complete blood cell count, as well as an annual physical examination that includes evaluation of the breasts, abdomen, pelvis, and testes. Lifelong cancer surveillance is advocated. [4]
A review by Beggs and coworkers summarized recommendations for cancer surveillance in patients with Peutz-Jeghers syndrome and pooled some published recommendations. [94, 95] Recommendations were based on literature review, cohort studies, and systematic review. Recommendations focused on upper intestinal, colorectal, pancreatic, breast, and genitourinary (reproductive) organ surveillance.
- Upper gastrointestinal tract: Annual hemoglobin concentration, esophagogastroduodenoscopy (EGD) every 2-3 years, small bowel series/enteroscopy (possible alternatives: wireless capsule endoscopy, magnetic resonance enterography) every 2 years, although there is no consensus on from what age (eg, age 10, 18, 25 years) to start
- Colorectal: Colonoscopy or flexible sigmoidoscopy and barium enema every 2-3 years from the time of first symptoms or in late teens or age 25 years onward
- Pancreatic: Annual abdominal ultrasonography or annual/every-other-year endoscopic ultrasonography from age 25-30 years onward
- Breast: Regular breast examination (monthly to 6-monthly) from age 18 years onward, mammography (or MRI) every other year (annually after age 50 years)
- Genitourinary in women: Annual pelvic examination, pelvic ultrasonography, and cervical smears; some reviews recommend serum CA-125, endometrial biopsy annually from age 20 years onward
- Genitourinary in men: Annual testicular examination; ultrasonography if symptomatic from birth
The Cancer of the Pancreas Screening (CAPS) Consortium summit addressed pancreatic cancer screening recommendations in at-risk populations, including persons with Peutz-Jeghers syndrome. [95] It recommended periodic screening of all individuals with Peutz-Jeghers syndrome from age 50 years onward. Suitable modalities for initial screening included endoscopic ultrasonography, MRI/MRCP, CT scanning, abdominal ultrasonography, and ERCP. [95] The ideal frequency for surveillance, however, was not defined.
![]()
Questions & Answers
- Daniell J, Plazzer JP, Perera A, Macrae F. An exploration of genotype-phenotype link between Peutz-Jeghers syndrome and STK11: a review. Fam Cancer. 2018 Jul. 17 (3):421-7. [QxMD MEDLINE Link].
- [Guideline] Achatz MI, Porter CC, Brugieres L, et al. Cancer screening recommendations and clinical management of inherited gastrointestinal cancer syndromes in childhood. Clin Cancer Res. 2017 Jul 1. 23 (13):e107-14. [QxMD MEDLINE Link]. [Full Text].
- Giardiello FM, Welsh SB, Hamilton SR, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987 Jun 11. 316 (24):1511-4. [QxMD MEDLINE Link].
- Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000 Dec. 119 (6):1447-53. [QxMD MEDLINE Link].
- Boardman LA, Thibodeau SN, Schaid DJ, et al. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med. 1998 Jun 1. 128 (11):896-9. [QxMD MEDLINE Link].
- Trau H, Schewach-Millet M, Fisher BK, Tsur H. Peutz-Jeghers syndrome and bilateral breast carcinoma. Cancer. 1982 Aug 15. 50 (4):788-92. [QxMD MEDLINE Link].
- Brosens LA, van Hattem WA, Jansen M, de Leng WW, Giardiello FM, Offerhaus GJ. Gastrointestinal polyposis syndromes. Curr Mol Med. 2007 Feb. 7 (1):29-46. [QxMD MEDLINE Link].
- Davidson NO. Genetic testing in colorectal cancer: who, when, how and why. Keio J Med. 2007 Mar. 56 (1):14-20. [QxMD MEDLINE Link].
- [Guideline] Syngal S, Brand RE, Church JM, et al, for the American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015 Feb. 110 (2):223-62; quiz 263. [QxMD MEDLINE Link]. [Full Text].
- Jelsig AM, van Overeem Hansen T, Gede LB, et al. Survival, surveillance, and genetics in patients with Peutz-Jeghers syndrome: A nationwide study. Clin Genet. 2023 Jul. 104(1):81-9. [QxMD MEDLINE Link]. [Full Text].
- Utsunomiya J, Gocho H, Miyanaga T, Hamaguchi E, Kashimure A. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J. 1975 Feb. 136 (2):71-82. [QxMD MEDLINE Link].
- Zbuk KM, Eng C. Hamartomatous polyposis syndromes. Nat Clin Pract Gastroenterol Hepatol. 2007 Sep. 4 (9):492-502. [QxMD MEDLINE Link].
- Gammon A, Jasperson K, Kohlmann W, Burt RW. Hamartomatous polyposis syndromes. Best Pract Res Clin Gastroenterol. 2009. 23 (2):219-31. [QxMD MEDLINE Link]. [Full Text].
- Hinds R, Philp C, Hyer W, Fell JM. Complications of childhood Peutz-Jeghers syndrome: implications for pediatric screening. J Pediatr Gastroenterol Nutr. 2004 Aug. 39 (2):219-20. [QxMD MEDLINE Link].
- [Guideline] NCCN Clinical Practice Guidelines in Oncology. Genetic/familial high-risk assessment: colorectal. V 1.2018. Available at https://www.nccn.org/professionals/physician_gls/default.aspx#genetics_colon. July 12, 2018; Accessed: October 10, 2018.
- Peutz JLA. Very remarkable case of familial polyposis of mucous membrane of intestinal tract and nasopharynx accompanied by peculiar pigmentations of skin and mucous membrane [Dutch]. Nederl Maandschr v Geneesk. 1921. 10:134-46.
- Jeghers H, McKusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med. 1949 Dec 29. 241 (26):1031-6. [QxMD MEDLINE Link].
- Bruwer A, Bargen JA, Kierland RR. Surface pigmentation and generalized intestinal polyposis; (Peutz-Jeghers syndrome). Proc Staff Meet Mayo Clin. 1954 Mar 24. 29 (6):168-71. [QxMD MEDLINE Link].
- Linhart H, Bormann F, Hutter B, Brors B, Lyko F. Genetic and epigenetic profiling of a solitary Peutz-Jeghers colon polyp. Cold Spring Harb Mol Case Stud. 2017 May. 3 (3):a001610. [QxMD MEDLINE Link]. [Full Text].
- Jiang YL, Zhao ZY, Li BR, et al. The altered activity of P53 signaling pathway by STK11 gene mutations and its cancer phenotype in Peutz-Jeghers syndrome. BMC Med Genet. 2018 Aug 9. 19 (1):141. [QxMD MEDLINE Link].
- Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998 Jan 8. 391 (6663):184-7. [QxMD MEDLINE Link].
- Mehenni H, Blouin JL, Radhakrishna U, et al. Peutz-Jeghers syndrome: confirmation of linkage to chromosome 19p13.3 and identification of a potential second locus, on 19q13.4. Am J Hum Genet. 1997 Dec. 61 (6):1327-34. [QxMD MEDLINE Link]. [Full Text].
- de Leng WW, Jansen M, Carvalho R, et al. Genetic defects underlying Peutz-Jeghers syndrome (PJS) and exclusion of the polarity-associated MARK/Par1 gene family as potential PJS candidates. Clin Genet. 2007 Dec. 72 (6):568-73. [QxMD MEDLINE Link]. [Full Text].
- Katajisto P, Vaahtomeri K, Ekman N, et al. LKB1 signaling in mesenchymal cells required for suppression of gastrointestinal polyposis. Nat Genet. 2008 Apr. 40 (4):455-9. [QxMD MEDLINE Link].
- Rosner M, Hanneder M, Siegel N, Valli A, Fuchs C, Hengstschlager M. The mTOR pathway and its role in human genetic diseases. Mutat Res. 2008 Sep-Oct. 659 (3):284-92. [QxMD MEDLINE Link].
- Schumacher V, Vogel T, Leube B, et al. STK11 genotyping and cancer risk in Peutz-Jeghers syndrome. J Med Genet. 2005 May. 42 (5):428-35. [QxMD MEDLINE Link]. [Full Text].
- Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010 Jul. 59 (7):975-86. [QxMD MEDLINE Link].
- Salloch H, Reinacher-Schick A, Schulmann K, et al. Truncating mutations in Peutz-Jeghers syndrome are associated with more polyps, surgical interventions and cancers. Int J Colorectal Dis. 2010 Jan. 25 (1):97-107. [QxMD MEDLINE Link].
- Tobi M, Kam M, Ullah N, et al. An anti-adenoma antibody, Adnab-9, may reflect the risk for neoplastic progression in familial hamartomatous polyposis syndromes. Dig Dis Sci. 2008 Mar. 53 (3):723-9. [QxMD MEDLINE Link].
- Zhao ZY, Jiang YL, Li BR, et al. Sanger sequencing in exonic regions of STK11 gene uncovers a novel de-novo germline mutation (c.962_963delCC) associated with Peutz-Jeghers syndrome and elevated cancer risk: case report of a Chinese patient. BMC Med Genet. 2017 Nov 15. 18 (1):130. [QxMD MEDLINE Link]. [Full Text].
- Zhao ZY, Jiang YL, Li BR, et al. A 23-nucleotide deletion in STK11 gene causes Peutz-Jeghers syndrome and malignancy in a Chinese patient without a positive family history. Dig Dis Sci. 2017 Nov. 62 (11):3014-20. [QxMD MEDLINE Link].
- Chiang JM, Chen TC. Clinical manifestations and STK11 germline mutations in Taiwanese patients with Peutz-Jeghers syndrome. Asian J Surg. 2018 Sep. 41 (5):480-5. [QxMD MEDLINE Link]. [Full Text].
- Jang MS, Lee YM, Ko BM, Kang G, Kim JW, Hong YH. Complete STK11 deletion and atypical symptoms in Peutz-Jeghers syndrome. Ann Lab Med. 2017 Sep. 37 (5):462-4. [QxMD MEDLINE Link].
- Chen HM, Fang JY. Genetics of the hamartomatous polyposis syndromes: a molecular review. Int J Colorectal Dis. 2009 Aug. 24 (8):865-74. [QxMD MEDLINE Link].
- Gao Y, Zhang FM, Huang S, et al. A de novo mutation of STK11 gene in a Chinese patient with Peutz-Jeghers syndrome. Dig Dis Sci. 2010 Apr. 55 (4):1032-6. [QxMD MEDLINE Link].
- Zuo YG, Xu KJ, Su B, Ho MG, Liu YH. Two novel STK11 mutations in three Chinese families with Peutz-Jeghers syndrome. Chin Med J (Engl). 2007 Jul 5. 120 (13):1183-6. [QxMD MEDLINE Link].
- Perzin KH, Fenoglio CM, Pascal RR. Tumors of the small and large intestine. In: Silverberg SG, ed. Principles and Practice of Surgical Pathology. New York, NY: John Wiley & Sons; 1983. 899-936.
- Wangler MF, Chavan R, Hicks MJ, et al. Unusually early presentation of small-bowel adenocarcinoma in a patient with Peutz-Jeghers syndrome. J Pediatr Hematol Oncol. 2013 May. 35 (4):323-8. [QxMD MEDLINE Link].
- Resta N, Pierannunzio D, Lenato GM, et al. Cancer risk associated with STK11/LKB1 germline mutations in Peutz-Jeghers syndrome patients: results of an Italian multicenter study. Dig Liver Dis. 2013 Jul. 45 (7):606-11. [QxMD MEDLINE Link].
- van Lier MG, Wagner A, Mathus-Vliegen EM, Kuipers EJ, Steyerberg EW, van Leerdam ME. High cancer risk in Peutz-Jeghers syndrome: a systematic review and surveillance recommendations. Am J Gastroenterol. 2010 Jun. 105 (6):1258-64; author reply 1265. [QxMD MEDLINE Link].
- Korsse SE, Harinck F, van Lier MG, et al. Pancreatic cancer risk in Peutz-Jeghers syndrome patients: a large cohort study and implications for surveillance. J Med Genet. 2013 Jan. 50 (1):59-64. [QxMD MEDLINE Link].
- Lim W, Hearle N, Shah B, et al. Further observations on LKB1/STK11 status and cancer risk in Peutz-Jeghers syndrome. Br J Cancer. 2003 Jul 21. 89 (2):308-13. [QxMD MEDLINE Link]. [Full Text].
- Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res. 2006 May 15. 12 (10):3209-15. [QxMD MEDLINE Link].
- Canto MI. Screening and surveillance approaches in familial pancreatic cancer. Gastrointest Endosc Clin N Am. 2008 Jul. 18 (3):535-53, x. [QxMD MEDLINE Link].
- Greenhalf W, Malats N, Nilsson M, Bartsch D, Neoptolemos J. International registries of families at high risk of pancreatic cancer. Pancreatology. 2008. 8 (6):558-65. [QxMD MEDLINE Link].
- Chiang JM, Chen TC. A Peutz-Jeghers syndrome family associated with sinonasal adenocarcinoma: 28 years follow up report. Fam Cancer. 2017 Oct. 16 (4):555-60. [QxMD MEDLINE Link].
- Burgmeier C, Schier F, Staatz G. Gastric outlet obstruction in a neonate because of Peutz-Jeghers syndrome. J Pediatr Surg. 2012 Aug. 47 (8):e1-3. [QxMD MEDLINE Link].
- Wu YK, Tsai CH, Yang JC, Hwang MH. Gastroduodenal intussusception due to Peutz-Jeghers syndrome. A case report. Hepatogastroenterology. 1994 Apr. 41 (2):134-6. [QxMD MEDLINE Link].
- de Leng WW, Westerman AM, Weterman MA, et al. Nasal polyposis in Peutz-Jeghers syndrome: a distinct histopathological and molecular genetic entity. J Clin Pathol. 2007 Apr. 60 (4):392-6. [QxMD MEDLINE Link]. [Full Text].
- Vogel T, Schumacher V, Saleh A, Trojan J, Moslein G. Extraintestinal polyps in Peutz-Jeghers syndrome: presentation of four cases and review of the literature. Deutsche Peutz-Jeghers-Studiengruppe. Int J Colorectal Dis. 2000 Apr. 15 (2):118-23. [QxMD MEDLINE Link].
- Sommerhaug RG, Mason T. Peutz-Jeghers syndrome and ureteral polyposis. JAMA. 1970 Jan 5. 211 (1):120-2. [QxMD MEDLINE Link].
- Jancu J. Peutz-Jeghers syndrome. Involvement of the gastrointestinal and upper respiratory tracts. Am J Gastroenterol. 1971 Dec. 56 (6):545-9. [QxMD MEDLINE Link].
- Rajendran R. Developmental disturbances or oral and paraoral structures. In: Rajendran A, Sivapathasundharam B, eds. Shafer's Textbook of Oral Pathology. 7th ed. New Delhi, India: Elsevier; 2014. 23.
- Burkart AL, Sheridan T, Lewin M, Fenton H, Ali NJ, Montgomery E. Do sporadic Peutz-Jeghers polyps exist? Experience of a large teaching hospital. Am J Surg Pathol. 2007 Aug. 31 (8):1209-14. [QxMD MEDLINE Link].
- Lynch HT, Lynch JF, Lynch PM, Attard T. Hereditary colorectal cancer syndromes: molecular genetics, genetic counseling, diagnosis and management. Fam Cancer. 2008. 7 (1):27-39. [QxMD MEDLINE Link].
- Launonen V. Mutations in the human LKB1/STK11 gene. Hum Mutat. 2005 Oct. 26 (4):291-7. [QxMD MEDLINE Link].
- Armstrong D, Bacon J, Viles Booker S, et al, for The Mid-Atlantic Cancer Genetics Network and The Johns Hopkins Hereditary Colorectal Cancer Program. The Johns Hopkins Guide for Patients and Families: Peutz-Jeghers Syndrome. Baltimore, MD: The Johns Hopkins Hospital; 2001. [Full Text].
- Bosman FT, Carneiro F, Hruban RH, Theise ND, eds. WHO Classification of Tumours of the Digestive System. 4th ed. Lyon, France: International Cancer Research on Cancer; 2010.
- Young S, Gooneratne S, Straus FH 2nd, Zeller WP, Bulun SE, Rosenthal IM. Feminizing Sertoli cell tumors in boys with Peutz-Jeghers syndrome. Am J Surg Pathol. 1995 Jan. 19 (1):50-8. [QxMD MEDLINE Link].
- Guillem JG, Smith AJ, Calle JP, Ruo L. Gastrointestinal polyposis syndromes. Curr Probl Surg. 1999 Apr. 36 (4):217-323. [QxMD MEDLINE Link].
- Bartholomew LG, Moore CE, Dahlin DC, Waugh JM. Intestinal polyposis associated with mucocutaneous pigmentation. Surg Gynecol Obstet. 1962 Jul. 115:1-11. [QxMD MEDLINE Link].
- Griffith CD, Bisset WH. Peutz-Jeghers syndrome. Arch Dis Child. 1980 Nov. 55 (11):866-9. [QxMD MEDLINE Link]. [Full Text].
- Dormandy TL. Gastrointestinal polyposis with mucocutaneous pigmentation (Peutz-Jeghers syndrome). N Engl J Med. 1957 Jun 20. 256 (25):1186-90; concl. [QxMD MEDLINE Link].
- O'Marcaigh AS, Ledger GA, Roche PC, Parisi JE, Zimmerman D. Aromatase expression in human germinomas with possible biological effects. J Clin Endocrinol Metab. 1995 Dec. 80 (12):3763-6. [QxMD MEDLINE Link].
- Ray LA, Billmire DF, Ferguson MJ, Eugster EA. Diagnostic conundrum of a Sertoli cell tumor in a 2-year-old girl with peripheral precocious puberty and a café-aul lait macule: a case report. Horm Res Paediatr. 2024 Apr 16. 25(4):211-4. [QxMD MEDLINE Link].
- Thakur N, Reddy DN, Rao GV, Mohankrishna P, Singh L, Chandak GR. A novel mutation in STK11 gene is associated with Peutz-Jeghers syndrome in Indian patients. BMC Med Genet. 2006 Sep 30. 7:73. [QxMD MEDLINE Link]. [Full Text].
- Hruban RH, Takaori K, Klimstra DS, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004 Aug. 28 (8):977-87. [QxMD MEDLINE Link].
- Postgate A, Hyer W, Phillips R, et al. Feasibility of video capsule endoscopy in the management of children with Peutz-Jeghers syndrome: a blinded comparison with barium enterography for the detection of small bowel polyps. J Pediatr Gastroenterol Nutr. 2009 Oct. 49 (4):417-23. [QxMD MEDLINE Link].
- Gupta A, Postgate AJ, Burling D, et al. A prospective study of MR enterography versus capsule endoscopy for the surveillance of adult patients with Peutz-Jeghers syndrome. AJR Am J Roentgenol. 2010 Jul. 195 (1):108-16. [QxMD MEDLINE Link].
- Plum N, May AD, Manner H, Ell C. [Peutz-Jeghers syndrome: endoscopic detection and treatment of small bowel polyps by double-balloon enteroscopy] [German]. Z Gastroenterol. 2007 Oct. 45 (10):1049-55. [QxMD MEDLINE Link].
- Kopacova M, Tacheci I, Rejchrt S, Bures J. Peutz-Jeghers syndrome: diagnostic and therapeutic approach. World J Gastroenterol. 2009 Nov 21. 15 (43):5397-408. [QxMD MEDLINE Link]. [Full Text].
- May A, Nachbar L, Pohl J, Ell C. Endoscopic interventions in the small bowel using double balloon enteroscopy: feasibility and limitations. Am J Gastroenterol. 2007 Mar. 102 (3):527-35. [QxMD MEDLINE Link].
- Gastineau S, Viala J, Caldari D, et al. Contribution of capsule endoscopy to Peutz-Jeghers syndrome management in children. Dig Liver Dis. 2012 Oct. 44 (10):839-43. [QxMD MEDLINE Link].
- Korsse SE, Dewint P, Kuipers EJ, van Leerdam ME. Small bowel endoscopy and Peutz-Jeghers syndrome. Best Pract Res Clin Gastroenterol. 2012 Jun. 26 (3):263-78. [QxMD MEDLINE Link].
- Belsha D, Urs A, Attard T, Thomson M. Effectiveness of double-balloon enteroscopy-facilitated polypectomy in pediatric patients with Peutz-Jeghers syndrome. J Pediatr Gastroenterol Nutr. 2017 Nov. 65 (5):500-2. [QxMD MEDLINE Link].
- Bartholomew LG, Dahlin DC, Waugh JM. Intestinal polyposis associated with mucocutaneous melanin pigmentation (Peutz-Jeghers syndrome). Proc Staff Meet Mayo Clin. 1957 Nov 27. 32 (24):675-80. [QxMD MEDLINE Link].
- Rosty C. The role of the surgical pathologist in the diagnosis of gastrointestinal polyposis syndromes. Adv Anat Pathol. 2017 Sep 8. [QxMD MEDLINE Link].
- Shaco-Levy R, Jasperson KW, Martin K, et al. Morphologic characterization of hamartomatous gastrointestinal polyps in Cowden syndrome, Peutz-Jeghers syndrome, and juvenile polyposis syndrome. Hum Pathol. 2016 Mar. 49:39-48. [QxMD MEDLINE Link].
- Meserve EE, Nucci MR. Peutz-Jeghers syndrome: pathobiology, pathologic manifestations, and suggestions for recommending genetic testing in pathology reports. Surg Pathol Clin. 2016 Jun. 9 (2):243-68. [QxMD MEDLINE Link].
- Bizzarri B, Borrelli O, de'Angelis N, et al. Management of duodenal-jejunal polyps in children with Peutz-Jeghers syndrome with single-balloon enteroscopy. J Pediatr Gastroenterol Nutr. 2014 Jul. 59 (1):49-53. [QxMD MEDLINE Link].
- Rossi DJ, Ylikorkala A, Korsisaari N, et al. Induction of cyclooxygenase-2 in a mouse model of Peutz-Jeghers polyposis. Proc Natl Acad Sci U S A. 2002 Sep 17. 99 (19):12327-32. [QxMD MEDLINE Link]. [Full Text].
- Wei C, Amos CI, Rashid A, et al. Correlation of staining for LKB1 and COX-2 in hamartomatous polyps and carcinomas from patients with Peutz-Jeghers syndrome. J Histochem Cytochem. 2003 Dec. 51 (12):1665-72. [QxMD MEDLINE Link].
- Udd L, Katajisto P, Rossi DJ, et al. Suppression of Peutz-Jeghers polyposis by inhibition of cyclooxygenase-2. Gastroenterology. 2004 Oct. 127 (4):1030-7. [QxMD MEDLINE Link].
- Wei C, Amos CI, Zhang N, Zhu J, Wang X, Frazier ML. Chemopreventive efficacy of rapamycin on Peutz-Jeghers syndrome in a mouse model. Cancer Lett. 2009 May 18. 277 (2):149-54. [QxMD MEDLINE Link].
- Kuwada SK, Burt R. A rationale for mTOR inhibitors as chemoprevention agents in Peutz-Jeghers syndrome. Fam Cancer. 2011 Sep. 10(3):469-72. [QxMD MEDLINE Link].
- Jelsig AM, Karstensen JG, Overeem Hansen TV. Progress report: Peutz-Jeghers syndrome. Fam Cancer. 2024 Mar 16. [QxMD MEDLINE Link].
- Woo A, Sadana A, Mauger DT, Baker MJ, Berk T, McGarrity TJ. Psychosocial impact of Peutz-Jeghers syndrome. Fam Cancer. 2009. 8 (1):59-65. [QxMD MEDLINE Link].
- van Lier MG, Mathus-Vliegen EM, van Leerdam ME, et al. Quality of life and psychological distress in patients with Peutz-Jeghers syndrome. Clin Genet. 2010 Sep. 78 (3):219-26. [QxMD MEDLINE Link].
- Gopie JP, Vasen HF, Tibben A. Surveillance for hereditary cancer: does the benefit outweigh the psychological burden?--A systematic review. Crit Rev Oncol Hematol. 2012 Sep. 83 (3):329-40. [QxMD MEDLINE Link].
- Tan VK, Koh PK, Loi CT, Eu KW, Tang CL. Peutz-Jeghers syndrome: data from the Singapore Polyposis Registry and a shifting paradigm in management. Ann Acad Med Singapore. 2010 Jan. 39 (1):17-21. [QxMD MEDLINE Link].
- Chen TH, Lin WP, Su MY, et al. Balloon-assisted enteroscopy with prophylactic polypectomy for Peutz-Jeghers syndrome: experience in Taiwan. Dig Dis Sci. 2011 May. 56 (5):1472-5. [QxMD MEDLINE Link].
- Sakamoto H, Yamamoto H, Hayashi Y, et al. Nonsurgical management of small-bowel polyps in Peutz-Jeghers syndrome with extensive polypectomy by using double-balloon endoscopy. Gastrointest Endosc. 2011 Aug. 74 (2):328-33. [QxMD MEDLINE Link].
- Oncel M, Remzi FH, Church JM, Connor JT, Fazio VW. Benefits of 'clean sweep' in Peutz-Jeghers patients. Colorectal Dis. 2004 Sep. 6 (5):332-5. [QxMD MEDLINE Link].
- Beggs AD, Latchford AR, Vasen HF, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010 Jul. 59 (7):975-86. [QxMD MEDLINE Link].
- Canto MI, Harinck F, Hruban RH, et al, for the International Cancer of Pancreas Screening (CAPS) Consortium. International Cancer of the Pancreas Screening (CAPS) Consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2013 Mar. 62 (3):339-47. [QxMD MEDLINE Link]. [Full Text].
- Shaco-Levy R, Jasperson KW, Martin K, et al. Morphologic characterization of hamartomatous gastrointestinal polyps in Cowden syndrome, Peutz-Jeghers syndrome, and juvenile polyposis syndrome. Hum Pathol. 2016 Mar. 49:39-48. [QxMD MEDLINE Link].
Author
Coauthor(s)
Burt Cagir, MD, FACS Associate Regional Dean and Professor of Surgery, Geisinger Commonwealth School of Medicine; Director, General Surgery Residency Program, Executive Director, Donald Guthrie Foundation for Research and Education, Guthrie Robert Packer Hospital; Medical Director, Guthrie/RPH Skills and Simulation Lab; Associate in Surgery, Guthrie Robert Packer Hospital and Corning Hospital
Burt Cagir, MD, FACS is a member of the following medical societies: American College of Surgeons, Association of Program Directors in Surgery, Society for Surgery of the Alimentary Tract
Disclosure: Nothing to disclose.
Chief Editor
Additional Contributors
Seth Septer, DO Pediatric Gastroenterologist, Children’s Mercy Hospital Clinics; Assistant Professor, University of Missouri-Kansas City School of Medicine
Disclosure: Nothing to disclose.
Acknowledgements
Ruchir Agrawal, MD Chief, Allergy and Immunology, Aurora Sheboygan Clinic
Ruchir Agrawal, MD, is a member of the following medical societies: American Academy of Allergy Asthma and Immunology, American Academy of Pediatrics, American College of Allergy, Asthma and Immunology, and American Medical Association
Disclosure: Nothing to disclose.
Vivek Bansal Armed Forces Medical College, India
Disclosure: Nothing to disclose.
John M Carethers, MD Professor of Medicine, Chief, Division of Gastroenterology, Department of Medicine, University of California, San Diego, School of Medicine
John M Carethers, MD is a member of the following medical societies: Alpha Omega Alpha, American Association for Cancer Research, American College of Gastroenterology, American College of Physicians, and American Gastroenterological Association
Disclosure: Nothing to disclose.
Carmen Cuffari, MD Associate Professor, Department of Pediatrics, Division of Gastroenterology/Nutrition, Johns Hopkins University School of Medicine
Carmen Cuffari, MD is a member of the following medical societies: American College of Gastroenterology, American Gastroenterological Association, North American Society for Pediatric Gastroenterology, Hepatology and Nutrition, and Royal College of Physicians and Surgeons of Canada
Disclosure: Nothing to disclose.
Andrea Duchini, MD Associate Professor of Medicine and Surgery, Director of Hepatology, University of Texas Medical Branch School of Medicine; Medical Director of Liver Transplantation, Department of Surgery, The Methodist Hospital
Andrea Duchini, MD is a member of the following medical societies: American College of Physicians, American Gastroenterological Association, American Society for Gastrointestinal Endoscopy, and International Liver Transplantation Society
Disclosure: Nothing to disclose.
Robert J Fingerote, MD, MSc, FRCPC Consultant, Clinical Evaluation Division, Biologic and Gene Therapies, Directorate Health Canada; Consulting Staff, Department of Medicine, Division of Gastroenterology, York Central Hospital, Ontario
Robert J Fingerote, MD, MSc, FRCPC is a member of the following medical societies: American Association for the Study of Liver Diseases, American Gastroenterological Association, Canadian Medical Association, Ontario Medical Association, and Royal College of Physicians and Surgeons of Canada
Disclosure: Nothing to disclose.
Rohit Kohli, MBBS, MS Assistant Professor of Pediatrics, Division of Gastroenterology, Hepatology and Nutrition, Cincinnati Children's Hospital Medical Center
Rohit Kohli, MBBS, MS is a member of the following medical societies: American Association for the Study of Liver Diseases, American Gastroenterological Association, American Society of Transplantation, International Pediatric Transplant Association, and North American Society for Pediatric Gastroenterology, Hepatology and Nutrition
Disclosure: Johnson and Johnson Grant/research funds Consulting
Sandeep Mukherjee, MB, BCh, MPH, FRCPC Associate Professor, Department of Internal Medicine, Section of Gastroenterology and Hepatology, University of Nebraska Medical Center; Consulting Staff, Section of Gastroenterology and Hepatology, Veteran Affairs Medical Center
Sandeep Mukherjee, MB, BCh, MPH, FRCPC is a member of the following medical societies: Royal College of Physicians and Surgeons of Canada
Disclosure: Merck Honoraria Speaking and teaching; Ikaria Pharmaceuticals Honoraria Board membership
Francisco Talavera, PharmD, PhD Adjunct Assistant Professor, University of Nebraska Medical Center College of Pharmacy; Editor-in-Chief, Medscape Drug Reference
Disclosure: Medscape Salary Employment
Jorge H Vargas, MD Professor of Pediatrics and Clinical Professor of Pediatric Gastroenterology, University of California, Los Angeles, David Geffen School of Medicine; Consulting Physician, Department of Pediatrics, University of California at Los Angeles Health System
Jorge H Vargas, MD is a member of the following medical societies: American Liver Foundation, American Society for Gastrointestinal Endoscopy, American Society for Parenteral and Enteral Nutrition, Latin American Society of Pediatric Gastroenterology, Hepatology & Nutrition, and North American Society for Pediatric Gastroenterology and Nutrition
Disclosure: Nothing to disclose.
Mary L Windle, PharmD Adjunct Associate Professor, University of Nebraska Medical Center College of Pharmacy; Editor-in-Chief, Medscape Drug Reference
Disclosure: Nothing to disclose.