Neuroblastoma Workup: Laboratory Studies, Imaging Studies, Diagnostic Procedures (original) (raw)
Laboratory Studies
General laboratory studies should be routinely obtained in children suspected of having neuroblastoma. Results are as follows:
- A complete blood cell count (CBC) should be obtained to determine if the child has anemia, which typically does not occur until the tumor has become widely disseminated; in patients with overwhelming bone marrow involvement, thrombocytopenia may also be present
- Once dissemination occurs, abnormalities in findings of coagulation studies (prothrombin time [PT], activated partial thromboplastin time [aPTT]) may occur secondary to liver involvement
- The erythrocyte sedimentation rate, a nonspecific acute-phase reactant, is elevated in classic neuroblastoma
Elevated metabolic catecholamine by-products can be detected in the urine of patients with neuroblastoma. The presence of these by-products serves as useful inclusion criteria when the diagnosis of neuroblastoma is being considered.
Phenylalanine and tyrosine are catecholamine precursors, which are converted through a sequence of enzymatic events to dihydroxyphenylalanine (DOPA), dopamine, norepinephrine, and epinephrine. DOPA and dopamine are metabolized into their final product, homovanillic acid (HVA), while norepinephrine and epinephrine are metabolized into vanillylmandelic acid (VMA).
Ninety percent of neuroblastoma tumors secrete these by-products. This fact becomes clinically relevant because children with dedifferentiated tumors excrete higher levels of HVA than VMA. This occurs because dedifferentiated tumors have lost the final enzymatic pathway that converts HVA to VMA. A low VMA-to-HVA ratio is consistent with a poorly differentiated tumor and indicative of a poor prognosis.
Neuroblastoma cells lack the enzyme that converts norepinephrine to epinephrine. Despite this fact, elevated levels of norepinephrine are not identified in the serum of patients with neuroblastoma. This might be explained by at least two processes: (1) norepinephrine may be catabolized within the tumor; and/or (2) tyrosine hydrolase, the initial enzyme in catecholamine synthesis, is subject to a negative feedback loop by norepinephrine.
A LaBrosse VMA spot test may be used to screen patients in certain institutions. It is economical but has low sensitivity and specificity.
High-performance liquid chromatography has a much lower false-positive rate and is more sensitive than the LaBrosse VMA spot test, but it is more expensive and is therefore often used only to confirm a positive result on a spot test.
Nonspecific tumor markers can be identified in patients with neuroblastoma. [20] Neuron-specific enolase (NSE), lactate dehydrogenase (LDH), and ferritin are markers useful in the identification of active disease, as well as in prognostication. Approximately 96% of patients with metastatic neuroblastomas demonstrate an elevated NSE level, which has been associated with a poor prognosis.
![]()
Imaging Studies
Radiographic assessment is recommended in all infants and children with an abdominal mass. The standard diagnostic imaging modalities include the following:
- Plain abdominal radiography (kidneys, ureters, bladder [KUB])
- Renal/bladder ultrasonography
- Bone scintigraphy
- Computed tomography (CT) or magnetic resonance imaging (MRI)
KUB most commonly reveals finely stippled calcifications of the abdomen or posterior mediastinum.
Renal/bladder ultrasonography improves the diagnostic evaluation and is probably the single best imaging modality to obtain. Ultrasonography is noninvasive and provides relevant information regarding the laterality, consistency, and size of the mass.
Abdominal CT scanning or MRI usually follows ultrasonography. Both of these studies are more invasive, in that they require general sedation for young children. The benefit is that they enhance the ultrasonographic findings by providing information about regional lymph nodes, vessel invasion, and distant metastatic disease. See the images below.
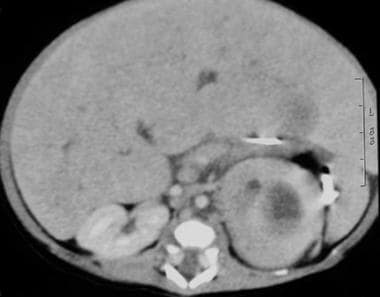
CT scan in a 2-week-old boy noted to have an abdominal mass on a prenatal sonogram. This postnatal abdominal CT scan revealed a left suprarenal mass with mass effect of the spleen.
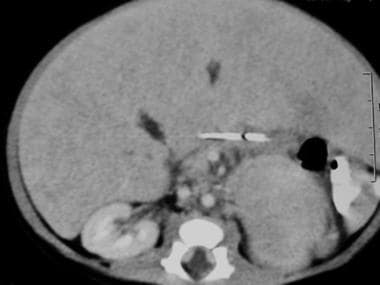
Abdominal CT scan in a 2-week-old boy noted to have an abdominal mass on a prenatal sonogram. A postnatal abdominal CT scan revealed a left suprarenal mass with mass effect of the spleen (see the previous image). This abdominal CT scan represents a more caudal view. Note the very large left mass with central necrosis. The mass effect of the spleen is apparent.
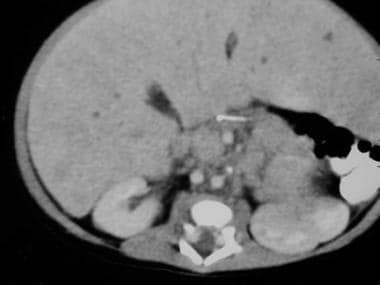
A 2-week-old boy is noted to have an abdominal mass on prenatal ultrasound. A postnatal abdominal CT scan revealed a left suprarenal mass with mass effect of the spleen (see the first image above). A more caudal view revealed the very large left mass with central necrosis (see the second image above). This is a more caudal view of the CT scan than in the previous 2 images. The left kidney comes into view, as it is inferiorly displaced and laterally rotated by the large superior neuroblastoma.
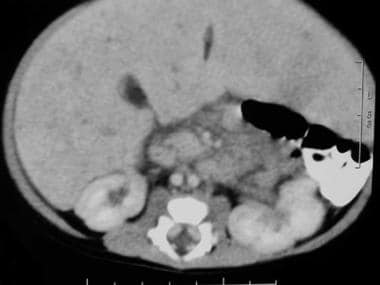
Bulky lymph nodes just medial to the left kidney.
Bone scintigraphy and a skeletal survey to detect cortical bone disease are helpful in the diagnosis of neuroblastoma. Metaiodobenzylguanidine (MIBG) is a compound taken up by catecholaminergic cells that competes for uptake even in neuroblastoma cells. In this way, MIBG is quite sensitive and specific in the detection of metastasis to bones and soft tissue, with highest sensitivity (91-97%) in the detection of bone deposits. [21]
Bone scintigraphy using 99Tc diphosphonate and a skeletal bone survey to detect cortical bone disease are essential if MIBG scintigraphy results are negative in the bone. MIBG is recommended for re-assessment both during and after therapy in high-risk patients with MIBG-avid disease at diagnosis.
Expression of somatostatin (SS) receptors has been described in neuroblastoma cell lines and tumors. Studies have shown that these tumors can be successfully targeted with radioactive SS analogs as a method of detection. Currently, the indication for scanning with radio-labeled SS analog in children with neuroblastoma is not well-defined because this method is less sensitive than MIBG scanning (64% vs 94%). However, because the presence of neuroblastoma SS receptors is associated with favorable clinical and biological prognostic factors, radio-labeled SS analog could provide valuable information. In fact, improved survival has been found in patients with SS receptor–positive neuroblastoma. However, more studies need to be performed to confirm the benefits of SS receptor scanning.
![]()
Diagnostic Procedures
Biopsy is the sine qua non in the diagnostic evaluation of neuroblastoma. To confirm the diagnosis of neuroblastoma, histologic evidence of neural origin or differentiation is required. Samples of tumor tissue can be viewed via light or electron microscopy or via immunohistochemistry. Although open surgical biopsy has traditionally been advocated, Mullassery and colleagues reported that image-guided needle biopsy can also yield adequate tissue samples. [22]
Another option is to sample bone marrow, a frequent metastatic site for neuroblastoma. The literature is confusing in terms of the number of bone marrow aspirates or biopsies needed to diagnose neuroblastoma. An international committee on neuroblastoma staging has recommended obtaining two bone marrow aspirates and two biopsies, one from each posterior iliac crest.
The issue of biopsies might become obsolete because immunocytology of marrow aspirates may offer the single best source of diagnostic information. A large body of published work has addressed the use of immunocytochemical and polymerase chain reaction (PCR)–based technologies to detect neuroblastoma cells and neuroblastoma-specific transcripts such as tyrosine hydroxylase and disialoganglioside (GD2) synthase in marrow or blood samples. [23] This method is used to assess minimal disease during the course of treatment. Although these techniques can greatly increase sensitivity, whether this increased sensitivity provides prognostic information about the likelihood of relapse is still unclear.
![]()
Histologic Findings
The three distinct histologic patterns of the neurocristopathies include neuroblastoma, ganglioneuroblastoma, and ganglioneuroma. They represent a spectrum of maturation and dedifferentiation. The typical neuroblastoma is characterized by small uniform cells that contain dense hyperchromatic nuclei and scant cytoplasm. A neuritic process called neuropil is pathognomonic of all except the most primitive neuroblastoma. Homer-Wright pseudorosettes are clusters of neuroblasts surrounding areas of eosinophilic neuropil and are observed in 15-50% of patients. If identified, they are diagnostic of neuroblastoma.
The minimal diagnostic criteria for diagnosing neuroblastoma have been established by an international group of conferees and corresponding participants. These criteria include (1) unequivocal pathologic diagnosis or (2) unequivocal bone marrow (syncytia) and elevated levels of urinary catecholamine metabolic by-products. Both of these diagnostic criteria require a histopathologic diagnosis.
Molecular pathogenesis
The cancer genes most commonly altered in adult carcinogenesis (eg, TP53, CDKN2A, ras) are rarely aberrant in neuroblastoma. _TP53_-inactivating mutations are uncommon in primary tumors due to neuroblastoma, although they have been documented in cell lines among patients with relapsing neuroblastoma. [24] Thus, with the exception of MYCN, major pathways of human neoplasia do not seem to be deregulated. Indeed, the only reliable genetically engineered murine model of neuroblastoma results from targeted overexpression of human MYCN copy DNA to the murine neural crest.
Activating mutations in the tyrosine kinase domain of the anaplastic lymphoma kinase (ALK) gene occur in the majority of hereditary neuroblastoma cases, and also in some sporadic cases of advanced neuroblastoma. Subclonal ALK mutations can be present at diagnosis, with subsequent clonal expansion at relapse. [25]
![]()
Staging
At least six different staging systems for neuroblastoma exist. Historically, each staging system represents a temporal improvement in the understanding of the tumor. However, the presence of so many systems has not only confounded the literature but also complicated the comparison of studies between institutions. In 1988, the International Neuroblastoma Staging System (INSS) was established to provide a uniform staging system. [26]
The INSS is a clinical, radiographic, and surgical appraisal of children with neuroblastoma. The system combines many of the most important diagnostic criteria from each of the staging systems and includes initial distribution and surgical resectability of the tumor.
Specific requirements to stage neuroblastoma include the following:
- Bone marrow aspirates and biopsy samples
- Body CT scan (excluding head, if not clinically indicated)
- Bone scan
- MIBG scintigraphy
Arabic numerals are used to distinguish INSS staging from other systems. INSS stages are as follows:
- Stage 1 is characterized by a localized tumor with complete gross excision, with or without microscopic residual disease; representative ipsilateral lymph nodes that test negative for tumor are present microscopically (nodes attached to and removed with the primary tumor may test positive)
- Stage 2A is characterized by a localized tumor with incomplete gross excision; representative ipsilateral nonadherent lymph nodes that test negative for tumor are present microscopically
- Stage 2B is characterized by a localized tumor with or without complete gross excision, with ipsilateral nonadherent lymph nodes that test positive for tumor; enlarged contralateral lymph nodes must test negative microscopically
- Stage 3 is an unresectable unilateral tumor infiltrating across the midline, with or without regional lymph node involvement; a localized unilateral tumor with contralateral regional lymph node involvement; or a midline tumor with bilateral extension by infiltration (unresectable) or by lymph node involvement (the midline is defined as the vertebral column; tumors that originate on one side and cross the midline must infiltrate to or beyond the opposite side of the vertebral column)
- Stage 4 is any primary tumor disseminated to distant lymph nodes, bone, bone marrow, liver, skin, and/or other organs (except as defined for stage 4S)
- Stage 4S (S = special) occurs in infants younger than 12 months and is characterized by a localized primary tumor (as defined for stage 1, 2A, or 2B), with dissemination limited to skin, liver, and/or bone marrow
Other features of stage 4S include the following:
- Marrow involvement should be minimal (ie, < 10% of total nucleated cells identified as malignant via bone biopsy or bone marrow aspirate); more extensive bone marrow involvement is considered stage 4 disease
- The results of the MIBG scan (if performed) should be negative for disease in the bone marrow.
Stage 4S is the most unusual group, comprising approximately 5% of patients with neuroblastoma. All else being equal, these children would normally be classified as having stage 1 or 2 disease; however, disease in this special group of infants almost always spontaneously regresses. Nonetheless, infants younger than 2 months frequently develop extensive and rapidly progressive intrahepatic expansion of neuroblastoma that can result in respiratory compromise. The 5-year survival rate in patients with stage 4S disease is 75%.
Stage for stage, infants with neuroblastoma have a better prognosis than older children. In fact, statistically, age is the most significant clinical prognosticator for neuroblastoma. Forty percent of infants (< 1 y) have localized neuroblastoma, compared with 20% of children older than 1 year. Additionally, nearly 70% of children older than 1 year have disseminated neuroblastoma, compared with less than 25% of infants.
The INSS is a postsurgical staging system. In 2009, the International Neuroblastoma Risk Group published a pretreatment risk classification system, the International Neuroblastoma Risk Group Staging System (INRGSS). [27] This system classifies neuroblastomas as locoregional (L), based on the absence or presence of one or more of 20 image-defined risk factors (IDRFs), or metastatic (M). Stages are as follows:
- L1 – Localized tumor not involving vital structures and confined to one body compartment
- L2 – Locoregional tumor with presence of one or more IDRFs
- M – Distant metastatic disease (except stage MS)
- MS – Metastatic disease in children younger than 18 months with metastases confined to skin, liver, and/or bone marrow
![]()
Risk Groups
The Children’s Oncology Group (COG) uses the major prognostic factors, including those based on imaging studies, to place children into three risk groups: low, intermediate, and high. [28] COG risk groups are used to guide treatment selection.
In the 2006 version, COG defines low-risk group criteria as follows:
- Stage 1 disease
- Stage 2A or 2B disease, patient age younger than 12 months
- Stage 2A or 2B disease, no extra copies of the MYCN gene, resection ≥50%
- Stage 4S disease, patient age < 12 months, no extra copies of _MYCN_, hyperdiploid (DNA index > 1), favorable histology, asymptomatic
Intermediate-risk group criteria are as follows:
- Stage 2A or 2B disease, age ≤12 years, no extra copies of MYCN, biopsy or resection < 50%
- Stage 3 disease, age < 18 months, no extra copies of MYCN, any DNA ploidy or histology
- Stage 3 disease, age 18 months–12 years, no extra copies of MYCN, favorable histology
- Stage 4 disease, age < 12 months, no extra copies of MYCN
- Stage 4 disease, age 12 to < 18 months, no extra copies of _MYCN,_ DNA index > 1,favorable histology
- Stage 4S disease, age < 12 months, no extra copies of MYCN, symptomatic
- Stage 4S disease, age < 12 months, no extra copies of MYCN, DNA index = 1, asymptomatic or symptomatic
- Stage 4S disease, age < 12 months, no extra copies of MYCN, unfavorable histology, asymptomatic or symptomatic
- Stage 4S disease, age < 12 months; MYCN status, DNA ploidy, and histology missing; too sick for biopsy
High-risk group criteria are as follows:
- Stage 2A or 2B disease, any age, extra copies of MYCN, any DNA ploidy or histology, any degree of resection
- Stage 3 disease, any age, extra copies of MYCN
- Stage 3 disease, age ≥ 18 months, no extra copies of MYCN, unfavorable histology
- Stage 4 disease, age < 12 months, extra copies of MYCN
- Stage 4 disease, age 12 to < 18 months, extra copies of MYCN
- Stage 4 disease, age 12 to < 18 months, any MYCN status, DNA index = 1, any histology
- Stage 4 disease, age 12 to < 18 months, any MYCN status or DNA ploidy, unfavorable histology
- Stage 4 disease; age ≥ 18 months; any MYCN status, DNA ploidy, or histology
- Stage 4S disease, age < 12 months; extra copies of MYCN; any DNA ploidy or histology; asymptomatic or symptomatic
The most recent revision of the COG risk group classification, published in 2021, uses the International Neuroblastoma Risk Group Staging System (INRGSS; see Staging) and International Neuroblastoma Pathology Classification (INPC) histology and incorporates loss or gain of a portion of a chromosome arm at 1p or 11q, termed segmental chromosome aberrations (SCA), as an additional genomic biomarker. [19]
Low-risk neuroblastoma criteria comprise the following:
- INRGSS L1; no MYCN amplification; any SCA, DNA ploidy, or INPC
- INRGSS L1; MYCN amplification; any SCA, DNA ploidy, or INPC; gross total surgical resection (otherwise this qualifies as high risk)
Intermediate-risk neuroblastoma criteria comprise the following:
- INRGSS L2, age < 18 months, no _MYCN_ amplification, SCA absent, DNA pIoidy index > 1, favorable INPC histology
- INRGSS L2; age < 18 months; no MYCN amplification; any SCA, DNA ploidy, or INPC
- INRGSS L2, age 18 months to 5 years, no MYCN amplification, any SCA or DNA ploidy, favorable INPC histology
- INRGSS L2, age ≥5 years, no MYCN amplification, any SCA or DNA ploidy, unfavorable INPC histology but histologic differentiation
High-risk neuroblastoma criteria comprise the following:
- INRGSS L2; MYCN amplification; any SCA, DNA ploidy, or INPC
- INRGSS L2, age 18 months to 5 years, no MYCN amplification, any SCA or DNA ploidy, unfavorable INPC histology
- INRGSS L2, age ≥5 years, no MYCN amplification, any SCA or DNA ploidy, unfavorable INPC histology (undifferentiated or poorly differentiated)
![]()
- Key Statistics About Neuroblastoma. American Cancer Society. Available at https://www.cancer.org/cancer/neuroblastoma/about/key-statistics.html. April 28, 2021; Accessed: January 31, 2024.
- PDQ Pediatric Treatment Editorial Board. Neuroblastoma Treatment (PDQ®): Health Professional Version. August 22, 2023. [QxMD MEDLINE Link]. [Full Text].
- Irwin MS, Park JR. Neuroblastoma: Paradigm for Precision Medicine. Pediatr Clin North Am. 2015 Feb. 62(1):225-256. [QxMD MEDLINE Link].
- Virchow R. Hyperplasie der Zirbel und der Nebennieren. Die Krankhaften Geschwulste. Vol 2: 1864-65.
- Marchand F. Beitrage zur Kenntniss der normalen und pathologischen Anatomie der Glandula carotica und der Nebennieren. Festschrift fur Ruduloph. Vichows Arch. 1891. 5:578.
- Herxheimer G. Ueber Turmoren des Nebennierenmarkes, insbesondere das Neuroblastoma sympaticum. Beitr Pathol Anat. 1914. 57:112.
- Cushing H, Wolback SB. The transformation of a malignant paravertebral sympathicoblastoma into a benign ganglioneuoma. Am J Pathol. 1927. 3:203.
- Everson TC, Cole WH. Spontaneous regression of neuroblastoma. Everson TC, Cole WH, eds. Spontaneous Regression of Cancer. Philadelphia, Pa: WB Saunders; 1966. 88.
- Mason GA, Hart-Mercer J, Millar EJ, Strang LB, Wynne NA. Adrenaline-secreting neuroblastoma in an infant. Lancet. 1957 Aug 17. 273(6990):322-5. [QxMD MEDLINE Link].
- Beckwith JB, Perrin EV. In situ neuroblastomas: A contribution to the natural history of neural crest tumors. Am J Pathol. 1963 Dec. 43:1089-104. [QxMD MEDLINE Link].
- Knudson AG Jr, Strong LC. Mutation and cancer: neuroblastoma and pheochromocytoma. Am J Hum Genet. 1972 Sep. 24(5):514-32. [QxMD MEDLINE Link].
- Bosse KR, Maris JM. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer. 2016 Jan 1. 122 (1):20-33. [QxMD MEDLINE Link]. [Full Text].
- Powers JT, Tsanov KM, Pearson DS, Roels F, Spina CS, Ebright R, et al. Multiple mechanisms disrupt the let-7 microRNA family in neuroblastoma. Nature. 2016 Jul 14. 535 (7611):246-51. [QxMD MEDLINE Link]. [Full Text].
- Bosse KR, Raman P, Zhu Z, et al. Identification of GPC2 as an Oncoprotein and Candidate Immunotherapeutic Target in High-Risk Neuroblastoma. Cancer Cell. 2017 Sep 11. 32 (3):295-309.e12. [QxMD MEDLINE Link]. [Full Text].
- Barr EK, Applebaum MA. Genetic Predisposition to Neuroblastoma. Children (Basel). 2018 Aug 31. 5 (9):[QxMD MEDLINE Link].
- Mallepalli S, Gupta MK, Vadde R. Neuroblastoma: An Updated Review on Biology and Treatment. Curr Drug Metab. 2019. 20 (13):1014-1022. [QxMD MEDLINE Link].
- Shimada H, Chatten J, Newton WA Jr, et al. Histopathologic prognostic factors in neuroblastic tumors: definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. J Natl Cancer Inst. 1984 Aug. 73(2):405-16. [QxMD MEDLINE Link].
- Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009 Jan 10. 27 (2):289-97. [QxMD MEDLINE Link]. [Full Text].
- Irwin MS, Naranjo A, Zhang FF, Cohn SL, London WB, Gastier-Foster JM, et al. Revised Neuroblastoma Risk Classification System: A Report From the Children's Oncology Group. J Clin Oncol. 2021 Oct 10. 39 (29):3229-3241. [QxMD MEDLINE Link]. [Full Text].
- Stigliani S, Coco S, Moretti S, Oberthuer A, Fischer M, Theissen J, et al. High genomic instability predicts survival in metastatic high-risk neuroblastoma. Neoplasia. 2012 Sep. 14(9):823-32. [QxMD MEDLINE Link]. [Full Text].
- Rufini V, Calcagni ML, Baum RP. Imaging of neuroendocrine tumors. Semin Nucl Med. 2006 Jul. 36(3):228-47. [QxMD MEDLINE Link].
- Mullassery D, Sharma V, Salim A, Jawaid WB, Pizer BL, Abernethy LJ, et al. Open versus needle biopsy in diagnosing neuroblastoma. J Pediatr Surg. 2014 Oct. 49(10):1505-7. [QxMD MEDLINE Link].
- Beiske K, Burchill SA, Cheung IY, Hiyama E, Seeger RC, Cohn SL, et al. Consensus criteria for sensitive detection of minimal neuroblastoma cells in bone marrow, blood and stem cell preparations by immunocytology and QRT-PCR: recommendations by the International Neuroblastoma Risk Group Task Force. Br J Cancer. 2009 May 19. 100 (10):1627-37. [QxMD MEDLINE Link]. [Full Text].
- Van Maerken T, Vandesompele J, Rihani A, De Paepe A, Speleman F. Escape from p53-mediated tumor surveillance in neuroblastoma: switching off the p14(ARF)-MDM2-p53 axis. Cell Death Differ. 2009 Dec. 16(12):1563-72. [QxMD MEDLINE Link].
- Schleiermacher G, Javanmardi N, Bernard V, Leroy Q, Cappo J, Rio Frio T, et al. Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol. 2014 Sep 1. 32(25):2727-34. [QxMD MEDLINE Link].
- Brodeur GM, Seeger RC, Barrett A, et al. International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol. 1988 Dec. 6(12):1874-81. [QxMD MEDLINE Link].
- Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol. 2009 Jan 10. 27 (2):298-303. [QxMD MEDLINE Link]. [Full Text].
- Neuroblastoma Risk Groups. American Cancer Society. Available at https://www.cancer.org/cancer/types/neuroblastoma/detection-diagnosis-staging/risk-groups.html. April 28, 2021; Accessed: December 18, 2023.
- Nitschke R, Smith EI, Shochat S, et al. Localized neuroblastoma treated by surgery: a Pediatric Oncology Group Study. J Clin Oncol. 1988 Aug. 6(8):1271-9. [QxMD MEDLINE Link].
- Suzuki M, Kushner BH, Kramer K, Basu EM, Roberts SS, Hammond WJ, et al. Treatment and outcome of adult-onset neuroblastoma. Int J Cancer. 2018 Sep 1. 143 (5):1249-1258. [QxMD MEDLINE Link]. [Full Text].
- Herd F, Basta NO, McNally RJQ, Tweddle DA. A systematic review of re-induction chemotherapy for children with relapsed high-risk neuroblastoma. Eur J Cancer. 2019 Feb 26. 111:50-58. [QxMD MEDLINE Link]. [Full Text].
- Gains J, Mandeville H, Cork N, Brock P, Gaze M. Ten challenges in the management of neuroblastoma. Future Oncol. 2012 Jul. 8(7):839-58. [QxMD MEDLINE Link].
- Ladenstein R, Pötschger U, Pearson ADJ, et al. Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): an international, randomised, multi-arm, open-label, phase 3 trial. Lancet Oncol. 2017 Apr. 18 (4):500-514. [QxMD MEDLINE Link].
- Rujkijyanont P, Photia A, Traivaree C, Monsereenusorn C, Anurathapan U, Seksarn P, et al. Clinical outcomes and prognostic factors to predict treatment response in high risk neuroblastoma patients receiving topotecan and cyclophosphamide containing induction regimen: a prospective multicenter study. BMC Cancer. 2019 Oct 16. 19 (1):961. [QxMD MEDLINE Link]. [Full Text].
- Berthold F, Faldum A, Ernst A, et al. Extended induction chemotherapy does not improve the outcome for high-risk neuroblastoma patients: results of the randomized open-label GPOH trial NB2004-HR. Ann Oncol. 2020 Mar. 31 (3):422-429. [QxMD MEDLINE Link]. [Full Text].
- Bayeva N, Coll E, Piskareva O. Differentiating Neuroblastoma: A Systematic Review of the Retinoic Acid, Its Derivatives, and Synergistic Interactions. J Pers Med. 2021 Mar 16. 11 (3):[QxMD MEDLINE Link]. [Full Text].
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010 Sep 30. 363(14):1324-34. [QxMD MEDLINE Link].
- FDA grants accelerated approval to naxitamab for high-risk neuroblastoma in bone or bone marrow. Food and Drug Administration. Available at https://www.fda.gov/drugs/drug-approvals-and-databases/fda-grants-accelerated-approval-naxitamab-high-risk-neuroblastoma-bone-or-bone-marrow. November 27, 2020; Accessed: December 18, 2023.
- FDA approves eflornithine for adult and pediatric patients with high-risk neuroblastoma. U.S. Food & Drug Administration. Available at https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-eflornithine-adult-and-pediatric-patients-high-risk-neuroblastoma. December 14, 2023; Accessed: January 31, 2024.
- Oesterheld J, Ferguson W, Kraveka JM, Bergendahl G, Clinch T, Lorenzi E, et al. Eflornithine as Postimmunotherapy Maintenance in High-Risk Neuroblastoma: Externally Controlled, Propensity Score-Matched Survival Outcome Comparisons. J Clin Oncol. 2023 Oct 26. JCO2202875. [QxMD MEDLINE Link]. [Full Text].
- Mastrangelo S, Romano A, Attinà G, Maurizi P, Ruggiero A. Timing and chemotherapy association for 131-I-MIBG treatment in high-risk neuroblastoma. Biochem Pharmacol. 2023 Oct. 216:115802. [QxMD MEDLINE Link]. [Full Text].
- Buckley SE, Chittenden SJ, Saran FH, Meller ST, Flux GD. Whole-body dosimetry for individualized treatment planning of 131I-MIBG radionuclide therapy for neuroblastoma. J Nucl Med. 2009 Sep. 50(9):1518-24. [QxMD MEDLINE Link].
- Pai Panandiker AS, Beltran C, Billups CA, McGregor LM, Furman WL, Davidoff AM. Intensity modulated radiation therapy provides excellent local control in high-risk abdominal neuroblastoma. Pediatr Blood Cancer. 2012 Sep 28. [QxMD MEDLINE Link].
- Russo C, Cohn SL, Petruzzi MJ, de Alarcon PA. Long-term neurologic outcome in children with opsoclonus-myoclonus associated with neuroblastoma: a report from the Pediatric Oncology Group. Med Pediatr Oncol. 1997 Apr. 28(4):284-8. [QxMD MEDLINE Link].
- de Alarcon PA, Matthay KK, London WB, Naranjo A, Tenney SC, Panzer JA, et al. Intravenous immunoglobulin with prednisone and risk-adapted chemotherapy for children with opsoclonus myoclonus ataxia syndrome associated with neuroblastoma (ANBL00P3): a randomised, open-label, phase 3 trial. Lancet Child Adolesc Health. 2018 Jan. 2 (1):25-34. [QxMD MEDLINE Link]. [Full Text].
- Nuchtern JG, London WB, Barnewolt CE, Naranjo A, McGrady PW, Geiger JD, et al. A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: a Children's Oncology Group study. Ann Surg. 2012 Oct. 256 (4):573-80. [QxMD MEDLINE Link].
Author
Byron D Joyner, MD, MPA Vice Dean for Graduate Medical Education and Designated Institutional Official Professor, Department of Urology, University of Washington School of Medicine; Pediatric Urologist, Seattle Children's Hospital
Byron D Joyner, MD, MPA is a member of the following medical societies: American Academy of Pediatrics, Society of University Urologists, Washington State Medical Association, Society of Urology Chairpersons and Program Directors, American College of Surgeons, American Urological Association, The Society of Federal Health Professionals (AMSUS), Massachusetts Medical Society
Disclosure: Nothing to disclose.
Specialty Editor Board
Francisco Talavera, PharmD, PhD Adjunct Assistant Professor, University of Nebraska Medical Center College of Pharmacy; Editor-in-Chief, Medscape Drug Reference
Disclosure: Received salary from Medscape for employment. for: Medscape.
Chief Editor
Brian H Kopell, MD Associate Professor, Department of Neurosurgery, Icahn School of Medicine at Mount Sinai; Director, Center for Neuromodulation, Co-Director, The Bonnie and Tom Strauss Movement Disorders Center, Department of Neurosurgery, Mount Sinai Health System
Brian H Kopell, MD is a member of the following medical societies: Alpha Omega Alpha, American Association of Neurological Surgeons, American Society for Stereotactic and Functional Neurosurgery, Congress of Neurological Surgeons, International Parkinson and Movement Disorder Society, North American Neuromodulation Society
Disclosure: Received income in an amount equal to or greater than $250 from: Medtronic; Abbott Neuromodulation; Turing Medical.
Acknowledgements
Natalya Lopushnyan Yale University School of Medicine
Disclosure: Nothing to disclose.